
Identification of Novel Diarylpyrimidines as Potent HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors by Exploring the Primer Grip Region

 ,
,  ,
, 
Abstract
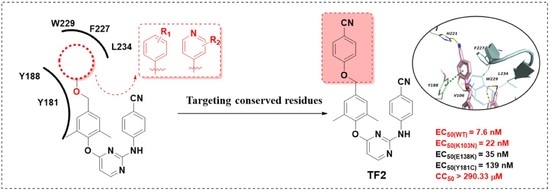
:
1. Introduction
2. Result and Discussion
2.1. Chemistry
2.2. Biological Evaluation
2.3. Molecular Dynamics Simulation Studies
3. Materials and Methods
3.1. Synthesis of Compounds
3.1.1. General Synthesis Procedure for the Preparation of TF1-TF16
3.1.2. Preparation Method of TF17
3.2. In Vitro Anti-HIV Assay
3.3. HIV-1 RT Inhibition Assay
with RT and inhibitors]/[O.D. value with RT and inhibitors − O.D. value
without RT and inhibitors].
3.4. MD Simulation Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fauci, A.S.; Lane, H.C. Four Decades of HIV/AIDS–Much Accomplished, Much to Do. N. Engl. J. Med. 2020, 383, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Xavier Ruiz, F.; Arnold, E. Evolving understanding of HIV-1 reverse transcriptase structure, function, inhibition, and resistance. Curr. Opin. Struct. Biol. 2020, 61, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Martinez, S.E.; Bauman, J.D.; Arnold, E. HIV-1 reverse transcriptase complex with DNA and nevirapine reveals non-nucleoside inhibition mechanism. Nat. Struct. Mol. Biol. 2012, 19, 253–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- UNAIDS. Global HIV & AIDS statistics—Fact sheet. Available online: https://www.unaids.org/en/resources/fact-sheet (accessed on 28 October 2022).
- Kang, D.; Sun, Y.; Feng, D.; Gao, S.; Wang, Z.; Jing, L.; Zhang, T.; Jiang, X.; Lin, H.; De Clercq, E.; et al. Development of Novel Dihydrofuro[3,4-d] pyrimidine Derivatives as HIV-1 NNRTIs to Overcome the Highly Resistant Mutant Strains F227L/V106A and K103N/Y181C. J. Med. Chem. 2022, 65, 2458–2470. [Google Scholar] [CrossRef] [PubMed]
- Battini, L.; Bollini, M. Challenges and approaches in the discovery of human immunodeficiency virus type-1 non-nucleoside reverse transcriptase inhibitors. Med. Res. Rev. 2019, 39, 1235–1273. [Google Scholar] [CrossRef]
- Vanangamudi, M.; Kurup, S.; Namasivayam, V. Non-nucleoside reverse transcriptase inhibitors (NNRTIs): A brief overview of clinically approved drugs and combination regimens. Curr. Opin. Pharmacol. 2020, 54, 179–187. [Google Scholar] [CrossRef]
- London, R.E. HIV-1 Reverse Transcriptase: A Metamorphic Protein with Three Stable States. Structure 2019, 27, 420–426. [Google Scholar] [CrossRef] [Green Version]
- Schauer, G.; Leuba, S.; Sluis-Cremer, N. Biophysical Insights into the Inhibitory Mechanism of Non-Nucleoside HIV-1 Reverse Transcriptase Inhibitors. Biomolecules 2013, 3, 889–904. [Google Scholar] [CrossRef] [Green Version]
- Shirvani, P.; Fassihi, A.; Saghaie, L. Recent Advances in the Design and Development of Non-nucleoside Reverse Transcriptase Inhibitor Scaffolds. ChemMedChem 2019, 14, 52–77. [Google Scholar] [CrossRef]
- Putz, M.V.; Dudaș, N.A.; Isvoran, A. Double Variational Binding—(SMILES) Conformational Analysis by Docking Mechanisms for Anti-HIV Pyrimidine Ligands. Int. J. Mol. Sci. 2015, 16, 19553–19601. [Google Scholar] [CrossRef]
- Al-Salama, Z.T. Elsulfavirine: First Global Approval. Drugs 2017, 77, 1811–1816. [Google Scholar] [CrossRef]
- National Medical Products Administration approves ainovirine tablets. Available online: https://www.nmpa.gov.cn/yaowen/ypjgyw/20210628162213161.html (accessed on 28 July 2022).
- Adams, J.; Patel, N.; Mankaryous, N.; Tadros, M.; Miller, C.D. Nonnucleoside reverse transcriptase inhibitor resistance and the role of the second-generation agents. Ann. Pharmacother. 2010, 44, 157–165. [Google Scholar] [CrossRef]
- Gu, S.X.; Zhu, Y.Y.; Wang, C.; Wang, H.F.; Liu, G.Y.; Cao, S.; Huang, L. Recent discoveries in HIV-1 reverse transcriptase inhibitors. Curr. Opin. Pharmacol. 2020, 54, 166–172. [Google Scholar] [CrossRef]
- Iyidogan, P.; Anderson, K.S. Current Perspectives on HIV-1 Antiretroviral Drug Resistance. Viruses 2014, 6, 4095–4139. [Google Scholar] [CrossRef] [Green Version]
- Himmel, D.M.; Arnold, E. Non-Nucleoside Reverse Transcriptase Inhibitors Join Forces with Integrase Inhibitors to Combat HIV. Pharmaceuticals 2020, 13, 122. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.C.; Pauly, G.T.; Rai, G.; Patel, D.; Bauman, J.D.; Baker, H.L.; Das, K.; Schneider, J.P.; Maloney, D.J.; Arnold, E.; et al. A comparison of the ability of rilpivirine (TMC278) and selected analogues to inhibit clinically relevant HIV-1 reverse transcriptase mutants. Retrovirology 2012, 9, 99. [Google Scholar] [CrossRef] [Green Version]
- Das, K.; Arnold, E. HIV-1 reverse transcriptase and antiviral drug resistance. Part 1. Curr. Opin. Virol. 2013, 3, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.; Flores, J.A.; Kirby, K.A.; Neogi, U.; Sonnerborg, A.; Hachiya, A.; Das, K.; Arnold, E.; McArthur, C.; Parniak, M.; et al. Drug Resistance in Non-B Subtype HIV-1: Impact of HIV-1 Reverse Transcriptase Inhibitors. Viruses 2014, 6, 3535–3562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esnouf, R.; Ren, J.; Ross, C.; Jones, Y.; Stammers, D.; Stuart, D. Mechanism of inhibition of HIV-1 reverse transcriptase by non-nucleoside inhibitors. Nat. Struct. Biol. 1995, 2, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Esnouf, R.; Garman, E.; Somers, D.; Ross, C.; Kirby, I.; Keeling, J.; Darby, G.; Jones, Y.; Stuart, D.; et al. High resolution structures of HIV-1 RT from four RT-inhibitor complexes. Nat. Struct. Biol. 1995, 2, 293–302. [Google Scholar] [CrossRef]
- Fu, Z.; Zhang, T.; Zhou, Z.; Kang, D.; Sun, L.; Gao, S.; Cherukupalli, S.; De Clercq, E.; Pannecouque, C.; Liu, X.; et al. Exploiting the hydrophobic channel of the NNIBP: Discovery of novel diarylpyrimidines as HIV-1 NNRTIs against wild-type and K103N mutant viruses. Bioorganic Med. Chem. 2021, 42, 116239. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Marchand, B.; Kirby, K.A.; Michailidis, E.; Sarafianos, S.G. Structural Aspects of Drug Resistance and Inhibition of HIV-1 Reverse Transcriptase. Viruses 2010, 2, 606–638. [Google Scholar] [CrossRef] [Green Version]
- Vijayan, R.S.; Arnold, E.; Das, K. Molecular dynamics study of HIV-1 RT-DNA-nevirapine complexes explains NNRTI inhibition and resistance by connection mutations. Proteins 2014, 82, 815–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Zhuang, C.; Chen, F. Druggability modification strategies of the diarylpyrimidine-type non-nucleoside reverse transcriptase inhibitors. Med. Res. Rev. 2021, 41, 1255–1290. [Google Scholar] [CrossRef]
- Lansdon, E.B.; Brendza, K.M.; Hung, M.; Wang, R.; Mukund, S.; Jin, D.; Birkus, G.; Kutty, N.; Liu, X. Crystal structures of HIV-1 reverse transcriptase with etravirine (TMC125) and rilpivirine (TMC278): Implications for drug design. J. Med. Chem. 2010, 53, 4295–4299. [Google Scholar] [CrossRef]
- Huang, B.; Ginex, T.; Luque, F.J.; Jiang, X.; Gao, P.; Zhang, J.; Kang, D.; Daelemans, D.; De Clercq, E.; Pannecouque, C.; et al. Structure-Based Design and Discovery of Pyridyl-Bearing Fused Bicyclic HIV-1 Inhibitors: Synthesis, Biological Characterization, and Molecular Modeling Studies. J. Med. Chem. 2021, 64, 13604–13621. [Google Scholar] [CrossRef]
- Gu, S.X.; Lu, H.H.; Liu, G.Y.; Ju, X.L.; Zhu, Y.Y. Advances in diarylpyrimidines and related analogues as HIV-1 nonnucleoside reverse transcriptase inhibitors. Eur. J. Med. Chem. 2018, 158, 371–392. [Google Scholar] [CrossRef] [PubMed]
- Jacobo-Molina, A.; Ding, J.; Nanni, R.G.; Clark, A.D., Jr.; Lu, X.; Tantillo, C.; Williams, R.L.; Kamer, G.; Ferris, A.L.; Clark, P.; et al. Crystal structure of human immunodeficiency virus type 1 reverse transcriptase complexed with double-stranded DNA at 3.0 A resolution shows bent DNA. Proc. Natl. Acad. Sci. USA 1993, 90, 6320–6324. [Google Scholar] [CrossRef] [Green Version]
- Powell, M.D.; Ghosh, M.; Jacques, P.S.; Howard, K.J.; Le Grice, S.F.; Levin, J.G. Alanine-scanning mutations in the "primer grip" of p66 HIV-1 reverse transcriptase result in selective loss of RNA priming activity. J. Biol. Chem. 1997, 272, 13262–13269. [Google Scholar] [CrossRef] [Green Version]
- Stanford University. NNRTI Resistance Notes. Available online: https://hivdb.stanford.edu/dr-summary/resistance-notes/NNRTI/ (accessed on 28 June 2022).
- Cilento, M.E.; Kirby, K.A.; Sarafianos, S.G. Avoiding Drug Resistance in HIV Reverse Transcriptase. Chem. Rev. 2021, 121, 3271–3296. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, T.; Wu, G.; Kang, D.; Fu, Z.; Wang, Z.; De Clercq, E.; Pannecouque, C.; Zhan, P.; Liu, X. Targeting the hydrophobic channel of NNIBP: Discovery of novel 1,2,3-triazole-derived diarylpyrimidines as novel HIV-1 NNRTIs with high potency against wild-type and K103N mutant virus. Org. Biomol. Chem. 2019, 17, 3202–3217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhou, Z.; Zalloum, W.A.; Wang, Z.; Fu, Z.; Cherukupalli, S.; Feng, D.; Sun, Y.; Gao, S.; De Clercq, E.; et al. Design, synthesis, and antiviral evaluation of novel piperidine-substituted arylpyrimidines as HIV-1 NNRTIs by exploring the hydrophobic channel of NNIBP. Bioorganic Chem. 2021, 116, 105353. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Liu, T.; Kang, D.; Huo, Z.; Wu, G.; Daelemans, D.; De Clercq, E.; Pannecouque, C.; Zhan, P.; Liu, X. Discovery of novel diarylpyrimidines as potent HIV-1 NNRTIs by investigating the chemical space of a less explored "hydrophobic channel". Org. Biomol. Chem. 2018, 16, 1014–1028. [Google Scholar] [CrossRef]
- Gustafson, K.R.; Cardellina, J.H., 2nd; Fuller, R.W.; Weislow, O.S.; Kiser, R.F.; Snader, K.M.; Patterson, G.M.; Boyd, M.R. AIDS-antiviral sulfolipids from cyanobacteria (blue-green algae). J. Natl. Cancer Inst. 1989, 81, 1254–1258. [Google Scholar] [CrossRef]
- Suzuki, K.; Craddock, B.P.; Okamoto, N.; Kano, T.; Steigbigel, R.T. Poly A-linked colorimetric microtiter plate assay for HIV reverse transcriptase. J. Virol. Methods 1993, 44, 189–198. [Google Scholar] [CrossRef]
- Carroll, S.S.; Olsen, D.B.; Bennett, C.D.; Gotlib, L.; Graham, D.J.; Condra, J.H.; Stern, A.M.; Shafer, J.A.; Kuo, L.C. Inhibition of HIV-1 reverse transcriptase by pyridinone derivatives. Potency, binding characteristics, and effect of template sequence. J. Biol. Chem. 1993, 268, 276–281. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]
- Ylilauri, M.; Pentikäinen, O.T. MMGBSA as a tool to understand the binding affinities of filamin-peptide interactions. J. Chem. Inf. Model. 2013, 53, 2626–2633. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Compounds | R | EC50 a | CC50 b (nM) | SI c (IIIB) | |
| IIIB (nM) | ROD (nM) | ||||
| TF1 |  | 17.1 ± 2.3 | >295,865.0 | >295,865.0 | >17,326.7 |
| TF2 |  | 7.6 ± 0.6 | >279,329.6 | >279,329.6 | >36,610.9 |
| TF3 |  | 24.5 ± 4.1 | >5522.3 | 5522.3 ± 296.6 | 225.2 |
| TF4 |  | 7.8 ± 1.0 | >5293.0 | 5293.0 ± 355.6 | 674.4 |
| TF5 |  | 21.7 ± 3.6 | >253,718.1 | 253,718.1 ± 5720.2 | 11,681.3 |
| TF6 |  | 35.3 ± 4.1 | >6896.1 | 6896.1 ± 249.1 | 195.6 |
| TF7 |  | 25.5 ± 8.4 | >8152.2 | 8152.2 ± 547.6 | 319.6 |
| TF8 |  | 42.8 ± 5.6 | >2917.9 | 2917.9 ± 271.5 | 68.1 |
| TF9 |  | 30.5 ± 4.0 | >3458.6 | 3458.6 ± 375.4 | 113.3 |
| TF10 |  | 32.2 ± 3.3 | >4804.6 | 4804.6 ± 341.7 | 149.1 |
| TF11 |  | 199.0 ± 13.0 | >14,827.6 | 14,827.6 ± 919.6 | 74.5 |
| TF12 |  | 7.8 ± 0.8 | >2462.8 | 2462.8 ± 197.9 | 316.9 |
| TF13 |  | 19.4 ± 3.4 | >4772.8 | 4772.8 ± 1561.8 | 246.4 |
| TF14 |  | 23.6 ± 3.8 | >24,438.7 | 24,438.7 ± 6675.8 | 1034.6 |
| TF15 |  | 23.1 ± 3.1 | >215,001.1 | 215,001.1 ± 20,337.8 | 9303.7 |
| TF16 |  | 45.3 ± 14.1 | >6196.6 | 6196.6 ± 220.4 | 136.7 |
| TF17 |  | 32.5 ± 1.7 | >5489.9 | 5489.9 ± 328.0 | 168.8 |
| AZT | —— | 27.3 ± 3.5 | 23.3 ± 2.1 | >7483.6 | >274.0 |
| NVP | —— | 122.6 ± 7.3 | - | >15,020.7 | >122.5 |
| EFV | —— | 2.4 ± 0.3 | - | >6335.5 | >2689.1 |
| ETR | —— | 2.9 ± 0.4 | - | >4594.6 | >1578.9 |
| RPVd | —— | 1.00 ± 0.27 | - | 3.98 (µM) | 3989 |
| Compounds | EC50 a (nM) | ||||||
|---|---|---|---|---|---|---|---|
| L100I | K103N | Y181C | Y188L | E138K | F227L + V106A | RES056 | |
| TF1 | 221.3 ± 53.5 | 50.7 ± 9.6 | 515.3 ± 60.4 | 1519.8 ± 92.1 | 76.5 ± 9.4 | 1710.6 ± 581.4 | 6853.1 ± 1172.2 |
| TF2 | 117.0 ± 49.5 | 28.1 ± 7.1 | 139.3 ± 34.9 | 1390.3 ± 118.8 | 44.0 ± 2.9 | 1136.6 ± 346.5 | > 279,329.6 |
| TF3 | 116.0 ± 5.8 | 48.4 ± 7.5 | 245.0 ± 39.6 | 894.7 ± 90.2 | 87.7 ± 8.5 | 755.4 ± 155.6 | 1543.5 ± 102.5 |
| TF4 | 314.9 ± 30.7 | 75.9 ± 13.9 | 482.3 ± 42.6 | > 5296.1 | 103.1 ± 6.8 | 1279.5 ± 303.3 | > 5296.1 |
| TF5 | 126.1 ± 12.6 | 44.5 ± 4.9 | 267.8 ± 28.9 | 963.7 ± 98.1 | 65.3 ± 10.0 | 1406.4 ± 569.6 | > 253,718.1 |
| TF6 | 156.0 ± 16.8 | 91.2 ± 14.4 | 386.7 ± 87.4 | 1391.7 ± 83.6 | 149.7 ± 0.7 | 1096.7 ± 281.1 | > 6896.1 |
| TF7 | 451.9 ± 70.5 | 164.1 ± 5.5 | 790.6 ± 62.9 | > 8152.2 | 180.7 ± 10.7 | 1224.1 ± 179.3 | > 8152.2 |
| TF8 | 328.9 ± 56.7 | 110.2 ± 4.6 | 667.2 ± 78.0 | 885.5 ± 20.1 | 147.5 ± 4.6 | 853.3 ± 81.4 | 1418.6 ± 9.7 |
| TF9 | 308.1 ± 46.3 | 119.6 ± 18.2 | 499.2 ± 44.6 | 1095.3 ± 95.7 | 147.1 ± 12.0 | 999.8 ± 29.6 | 1586.6 ± 87.5 |
| TF10 | 634.6 ± 28.2 | 158.4 ± 18.6 | 765.6 ± 22.5 | > 4804.6 | 178.8 ± 26.9 | 1487.4 ± 33.9 | > 4804.6 |
| TF11 | 9574.0 ± 22.2 | 444.1 ± 68.6 | > 14,827.6 | > 14,827.6 | ≥ 3024.0 | > 14,827.6 | > 14,827.6 |
| TF12 | 245.6 ± 54.6 | 34.7 ± 8.8 | 384.5 ± 75.5 | ≥ 1560.8 | 46.2 ± 7.1 | 742.4 ± 145.1 | > 2462.8 |
| TF13 | 125.3 ± 5.9 | 28.0 ± 2.5 | 265.2 ± 24.6 | 1053.3 ± 66.8 | 57.7 ± 11.6 | 1650.5 ± 19.9 | 1182.9 ± 68.2 |
| TF14 | 185.1 ± 11.6 | 36.7 ± 3.7 | 285.5 ± 52.1 | 773.6 ± 39.2 | 67.4 ± 8.1 | 999.4 ± 125.8 | 1192.8 ± 182.2 |
| TF15 | 105.2 ± 15.2 | 46.3 ± 9.7 | 195.9 ± 25.4 | 1079.6 ± 99.4 | 50.0 ± 4.4 | 1655.9 ± 448.3 | 1801.7 ± 68.5 |
| TF16 | 715.2 ± 183.5 | 131.7 ± 28.4 | 1274.9 ± 153.7 | > 6196.6 | 245.5 ± 30.4 | ≥ 1618.2 | > 6196.6 |
| TF17 | 226.5 ± 28.9 | 65.3 ± 7.6 | 552.5 ± 62.5 | 905.1 ± 147.4 | 151.9 ± 10.3 | 747.1 ± 72.2 | > 5.490 |
| AZT | 10.0 ± 1.2 | 22.8 ± 1.1 | 25.1 ± 3.0 | 19.4 ± 2.9 | 30.1 ± 3.0 | 14.8 ± 1.1 | 25.1 ± 1.8 |
| NVP | 1856.7 ± 364.6 | 7495.1 ± 866.8 | 10,113.7 ± 850.9 | ≥ 13,781.4 | 149.4 ± 12.0 | > 15,020.7 | > 15,020.7 |
| EFV | 35.0 ± 4.7 | 95.1 ± 9.1 | 5.5 ± 0.5 | 239.6 ± 30.4 | 4.4 ± 0.3 | 200.8 ± 17.3 | 183.8 ± 22.0 |
| ETR | 5.9 ± 1.0 | 3.0 ± 0.3 | 11.1 ± 1.3 | 18.1 ± 3.2 | 6.3 ± 1.2 | 20.4 ± 7.8 | 29.5 ± 4.8 |
| RPVb | 1.54 ± 0.00 | 1.31 ± 0.36 | 4.73 ± 0.48 | 79.4 ± 0.77 | 5.75 ± 0.11 | 5.75 ± 0.11 | 10.7 ± 7.96 |
| Compounds | Resistance Folds a | Compds | Resistance Folds a |
|---|---|---|---|
| K103N | K103N | ||
| TF1 | 3.00 | TF10 | 4.94 |
| TF2 | 3.68 | TF11 | 2.23 |
| TF3 | 1.92 | TF12 | 4.49 |
| TF4 | 9.74 | TF13 | 1.47 |
| TF5 | 2.00 | TF14 | 1.54 |
| TF6 | 2.60 | TF15 | 2.00 |
| TF7 | 6.31 | TF16 | 2.93 |
| TF8 | 2.56 | TF17 | 1.97 |
| TF9 | 3.87 | EFV | 47.5 |
| Compds | IC50 a (μM) | Compds | IC50 a (μM) |
|---|---|---|---|
| TF1 | 0.062 ± 0.000 | TF11 | 0.483 ± 0.03 |
| TF2 | 0.055 ± 0.001 | TF12 | 0.038 ± 0.004 |
| TF3 | 0.060 ± 0.000 | TF13 | 0.059 ± 0.001 |
| TF4 | 0.050 ± 0.002 | TF14 | 0.041 ± 0.003 |
| TF5 | 0.054 ± 0.004 | TF15 | 0.063 ± 0.001 |
| TF6 | 0.052 ± 0.004 | TF16 | 0.160 ± 0.001 |
| TF7 | 0.073 ± 0.003 | TF17 | 0.057 ± 0.003 |
| TF8 | 0.041 ± 0.001 | NVP | 0.568 ± 0.063 |
| TF9 | 0.053 ± 0.000 | EFV | 0.013 ± 0.001 |
| TF10 | 0.055 ± 0.000 | ETR | 0.011 ± 0.000 b |
| Binding Energies a (kcal/mol) | TF2 → NNIBP (WT) | TF2 → NNIBP (K103N) |
|---|---|---|
| ΔGHbond | −1.1 ± 0.3 | −0.9 ± 0.3 |
| ΔGbind | −113.8 ± 3.8 | −109.0 ± 4.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.; Zhou, Z.; Zhao, F.; Sang, Z.; De Clercq, E.; Pannecouque, C.; Kang, D.; Zhan, P.; Liu, X. Identification of Novel Diarylpyrimidines as Potent HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors by Exploring the Primer Grip Region. Pharmaceuticals 2022, 15, 1438. https://doi.org/10.3390/ph15111438
Zhang T, Zhou Z, Zhao F, Sang Z, De Clercq E, Pannecouque C, Kang D, Zhan P, Liu X. Identification of Novel Diarylpyrimidines as Potent HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors by Exploring the Primer Grip Region. Pharmaceuticals. 2022; 15(11):1438. https://doi.org/10.3390/ph15111438
Chicago/Turabian StyleZhang, Tao, Zhongxia Zhou, Fabao Zhao, Zihao Sang, Erik De Clercq, Christophe Pannecouque, Dongwei Kang, Peng Zhan, and Xinyong Liu. 2022. "Identification of Novel Diarylpyrimidines as Potent HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors by Exploring the Primer Grip Region" Pharmaceuticals 15, no. 11: 1438. https://doi.org/10.3390/ph15111438