
Protofibrillar Amyloid Beta Modulation of Recombinant hCaV2.2 (N-Type) Voltage-Gated Channels




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Protofibrillar Assembly of Amyloid
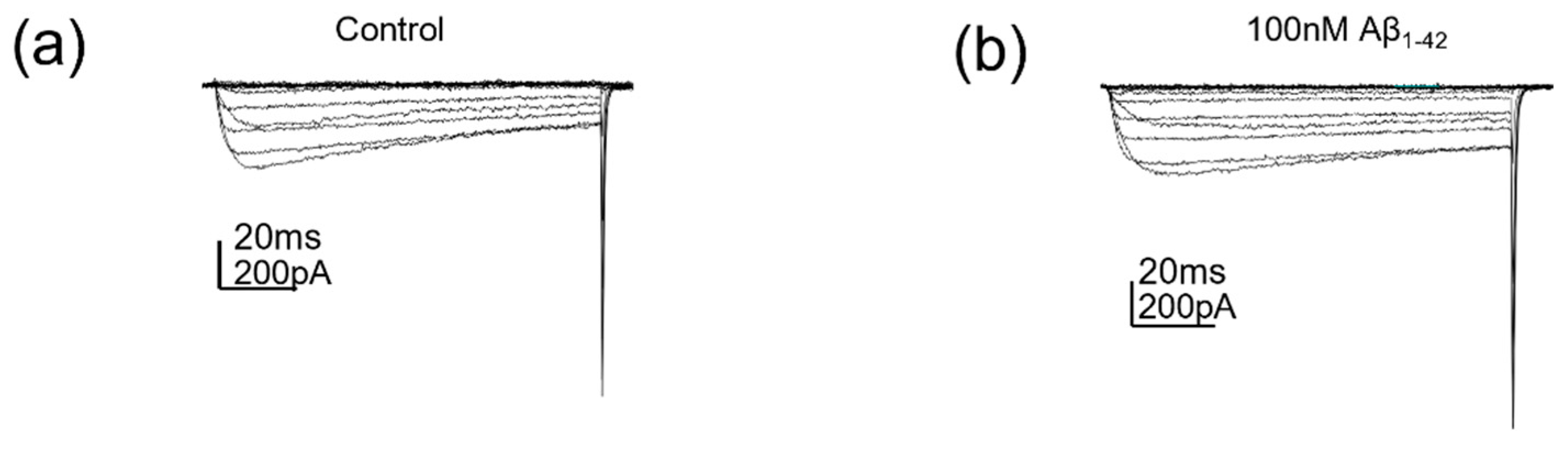
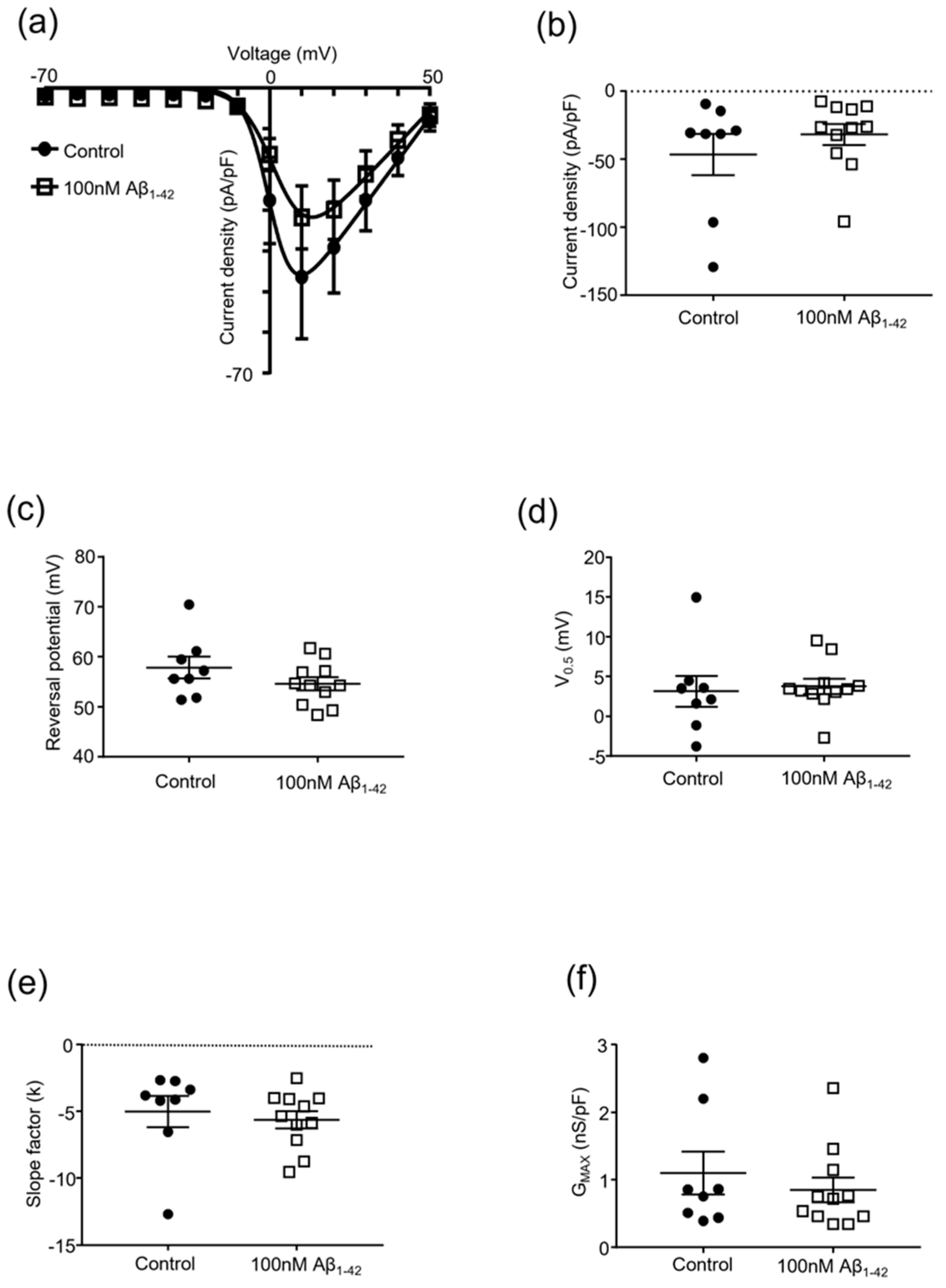
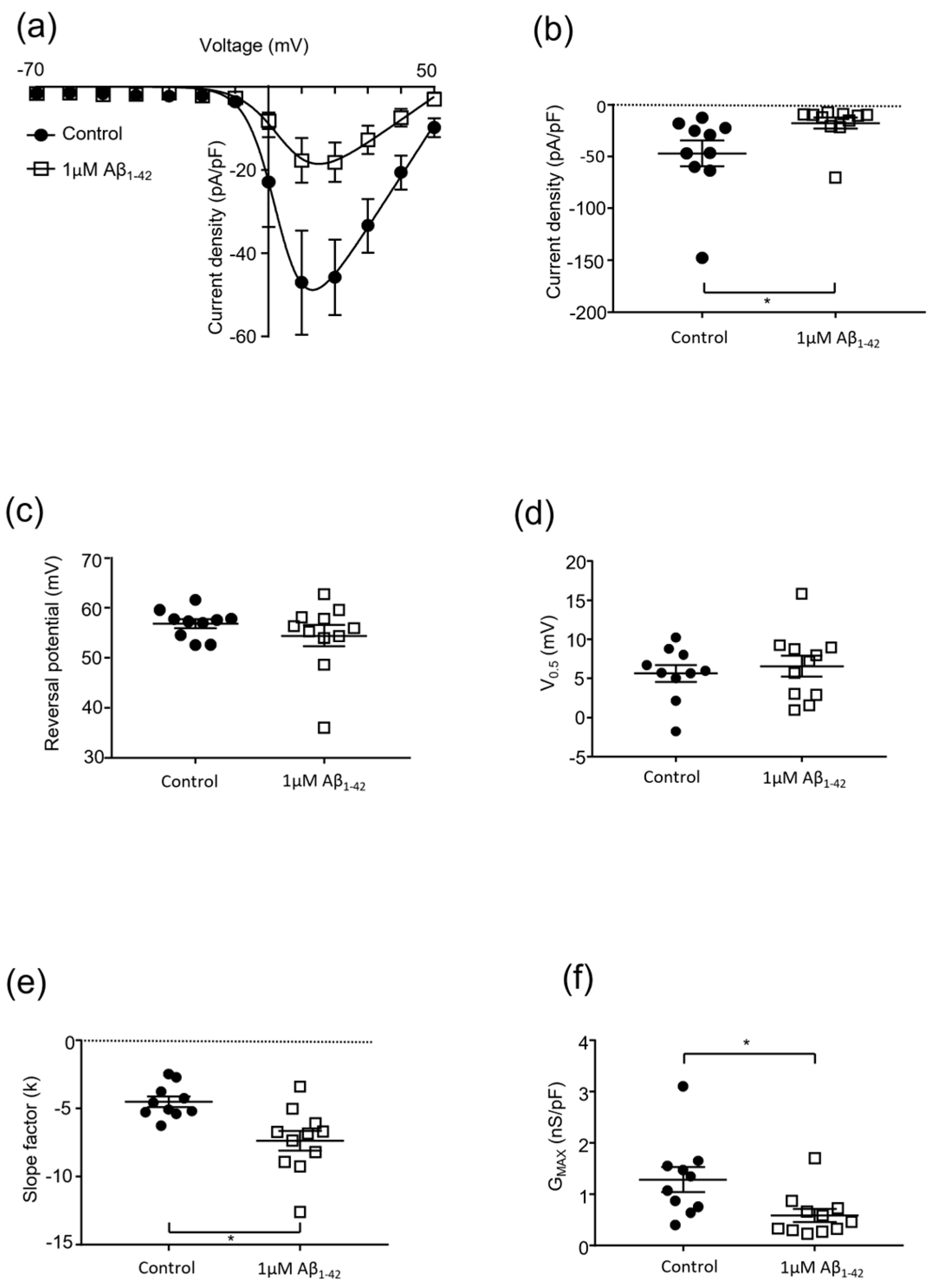
2.2. Protofibrillar Amyloid Beta Modulation of CaV2.2 Currents
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Aβ Assembly and Characterisation
4.3. Transmission Electron Microscopy
4.4. Electrophysiology
4.5. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science (1979) 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Decourt, B.; Boumelhem, F.; Pope, E.D.; Shi, J.; Mari, Z.; Sabbagh, M.N. Critical Appraisal of Amyloid Lowering Agents in AD. Curr. Neurol. Neurosci. Rep. 2021, 21, 39. [Google Scholar] [CrossRef] [PubMed]
- Dawkins, E.; Small, D.H. Insights into the Physiological Function of the β-Amyloid Precursor Protein: Beyond Alzheimer’s Disease. J. Neurochem. 2014, 129, 756–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brothers, H.M.; Gosztyla, M.L.; Robinson, S.R. The Physiological Roles of Amyloid-β Peptide Hint at New Ways to Treat Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishop, G.M.; Robinson, S.R. Physiological Roles of Amyloid-β and Implications for Its Removal in Alzheimer’s Disease. Drugs Aging 2004, 21, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Plant, L.D.; Webster, N.J.; Boyle, J.P.; Ramsden, M.; Freir, D.B.; Peers, C.; Pearson, H.A. Amyloid β Peptide as a Physiological Modulator of Neuronal ‘A’-Type K+ Current. Neurobiol. Aging 2006, 27, 1673–1683. [Google Scholar] [CrossRef]
- Yamamoto, K.; Yamamoto, R.; Kato, N. Amyloid β and Amyloid Precursor Protein Synergistically Suppress Large-Conductance Calcium-Activated Potassium Channel in Cortical Neurons. Front. Aging Neurosci. 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Zamponi, G.W.; Striessnig, J.; Koschak, A.; Dolphin, A.C. The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential. Pharm. Rev. 2015, 67, 821–870. [Google Scholar] [CrossRef] [Green Version]
- Mezler, M.; Barghorn, S.; Schoemaker, H.; Gross, G.; Nimmrich, V. A β-Amyloid Oligomer Directly Modulates P/Q-Type Calcium Currents in Xenopus Oocytes. Br. J. Pharm. 2012, 165, 1572–1583. [Google Scholar] [CrossRef] [Green Version]
- Hermann, D.; Mezler, M.; Müller, M.K.; Wicke, K.; Gross, G.; Draguhn, A.; Bruehl, C.; Nimmrich, V. Synthetic Aβ Oligomers (Aβ(1-42) Globulomer) Modulate Presynaptic Calcium Currents: Prevention of Aβ-Induced Synaptic Deficits by Calcium Channel Blockers. Eur. J. Pharm. 2013, 702, 44–55. [Google Scholar] [CrossRef]
- Nimmrich, V.; Grimm, C.; Draguhn, A.; Barghorn, S.; Lehmann, A.; Schoemaker, H.; Hillen, H.; Gross, G.; Ebert, U.; Bruehl, C. Amyloid β Oligomers (Aβ1-42 Globulomer) Suppress Spontaneous Synaptic Activity by Inhibition of P/Q-Type Calcium Currents. J. Neurosci. 2008, 28, 788–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, K.; Shinohara, S.; Yagami, T.; Asakura, K.; Kawasaki, K. Amyloid β Protein Potentiates Ca2+ Influx through L-Type Voltage- Sensitive Ca2+ Channels: A Possible Involvement of Free Radicals. J. Neurochem. 1997, 68, 265–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Sivaji, S.; Chiang, M.C.; Ali, H.; Zukowski, M.; Ali, S.; Kennedy, B.; Sklyar, A.; Cheng, A.; Guo, Z.; et al. Amyloid Beta Peptides Block New Synapse Assembly by Nogo Receptor-Mediated Inhibition of T-Type Calcium Channels. Neuron 2017, 96, 355–372.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, J.H.; Pike, C.J.; Cotman, C.W. Rapid Communication: Ca2+ Channel Blockers Attenuate β-Amyloid Peptide Toxicity to Cortical Neurons in Culture. J. Neurochem. 1994, 62, 372–375. [Google Scholar] [CrossRef]
- Morin, A.; Mouzon, B.; Ferguson, S.; Paris, D.; Browning, M.; Stewart, W.; Mullan, M.; Crawford, F. Nilvadipine Suppresses Inflammation via Inhibition of P-SYK and Restores Spatial Memory Deficits in a Mouse Model of Repetitive Mild TBI. Acta Neuropathol. Commun. 2020, 8, 166. [Google Scholar] [CrossRef]
- Paris, D.; Quadros, A.; Humphrey, J.; Patel, N.; Crescentini, R.; Crawford, F.; Mullan, M. Nilvadipine Antagonizes Both Aβ Vasoactivity in Isolated Arteries, and the Reduced Cerebral Blood Flow in APPsw Transgenic Mice. Brain Res. 2004, 999, 53–61. [Google Scholar] [CrossRef]
- Hussain, S.; Singh, A.; Rahman, S.O.; Habib, A.; Najmi, A.K. Calcium Channel Blocker Use Reduces Incident Dementia Risk in Elderly Hypertensive Patients: A Meta-Analysis of Prospective Studies. Neurosci. Lett. 2018, 671, 120–127. [Google Scholar] [CrossRef]
- Yasar, S.; Corrada, M.; Brookmeyer, R.; Kawas, C. Calcium Channel Blockers and Risk of AD: The Baltimore Longitudinal Study of Aging. Neurobiol. Aging 2005, 26, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Forette, F.; Seux, M.L.; Staessen, J.A.; Thijs, L.; Birkenhäger, W.H.; Babarskiene, M.R.; Babeanu, S.; Bossini, A.; Gil-Extremera, B.; Girerd, X.; et al. Prevention of Dementia in Randomised Double-Blind Placebo-Controlled Systolic Hypertension in Europe (Syst-Eur) Trial. Lancet 1998, 352, 1347–1351. [Google Scholar] [CrossRef]
- Lawlor, B.; Segurado, R.; Kennelly, S.; Olde Rikkert, M.G.M.; Howard, R.; Pasquier, F.; Börjesson-Hanson, A.; Tsolaki, M.; Lucca, U.; Molloy, D.W.; et al. Nilvadipine in Mild to Moderate Alzheimer Disease: A Randomised Controlled Trial. PLoS Med. 2018, 15, e1002660. [Google Scholar] [CrossRef]
- Stevens, E.B.; Stephens, G.J. Recent Advances in Targeting Ion Channels to Treat Chronic Pain. Br. J. Pharm. 2018, 175, 2133–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, S.; Fisher, D.W.; Yu, T.; Dong, H. The Link between Chronic Pain and Alzheimer’s Disease. J. Neuroinflammation 2019, 16, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumaradev, S.; Fayosse, A.; Dugravot, A.; Dumurgier, J.; Roux, C.; Kivimäki, M.; Singh-Manoux, A.; Sabia, S. Timeline of Pain before Dementia Diagnosis: A 27-Year Follow-up Study. Pain 2021, 162, 1578–1585. [Google Scholar] [CrossRef] [PubMed]
- Mochida, S. Presynaptic Calcium Channels. Neurosci. Res. 2018, 127, 33–44. [Google Scholar] [CrossRef] [Green Version]
- Catterall, W.A.; Few, A.P. Calcium Channel Regulation and Presynaptic Plasticity. Neuron 2008, 59, 882–901. [Google Scholar] [CrossRef] [Green Version]
- Ryan, T.M.; Caine, J.; Mertens, H.D.T.; Kirby, N.; Nigro, J.; Breheney, K.; Waddington, L.J.; Streltsov, V.A.; Curtain, C.; Masters, C.L.; et al. Ammonium Hydroxide Treatment of Aβ Produces an Aggregate Free Solution Suitable for Biophysical and Cell Culture Characterization. PeerJ 2013, 1, e73. [Google Scholar] [CrossRef] [Green Version]
- Poncer, J.C.; McKinney, R.A.; Gähwiler, B.H.; Thompson, S.M. Either N- or P-Type Calcium Channels Mediate GABA Release at Distinct Hippocampal Inhibitory Synapses. Neuron 1997, 18, 463–472. [Google Scholar] [CrossRef] [Green Version]
- Scholz, K.P.; Miller, R.J. Developmental Changes in Presynaptic Calcium Channels Coupled to Glutamate Release in Cultured Rat Hippocampal Neurons. J. Neurosci. 1995, 15, 4612–4617. [Google Scholar] [CrossRef] [Green Version]
- Huntula, S.; Saegusa, H.; Wang, X.; Zong, S.; Tanabe, T. Involvement of N-Type Ca2+ Channel in Microglial Activation and Its Implications to Aging-Induced Exaggerated Cytokine Response. Cell Calcium 2019, 82, 102059. [Google Scholar] [CrossRef]
- Saegusa, H.; Tanabe, T. N-Type Voltage-Dependent Ca2+ Channel in Non-Excitable Microglial Cells in Mice Is Involved in the Pathophysiology of Neuropathic Pain. Biochem. Biophys. Res. Commun. 2014, 450, 142–147. [Google Scholar] [CrossRef]
- Chiou, W.F. Effect of Aβ Exposure on the MRNA Expression Patterns of Voltage-Sensitive Calcium Channel A1 Subunits (A1A–A1D) in Human SK-N-SH Neuroblastoma. Neurochem. Int. 2006, 49, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Rhim, H. Effects of Amyloid-β Peptides on Voltage-Gated L-Type Ca v1.2 and Ca v1.3 Ca2+ Channels. Mol. Cells 2011, 32, 289–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Yang, H.; He, T.; Zhang, L.; Liu, C. Post-Translational Modification of Cav1.2 and Its Role in Neurodegenerative Diseases. Front. Pharm. 2022, 12, 3825. [Google Scholar] [CrossRef]
- Hettiarachchi, N.T.N.; Dallas, M.M.L.; Al-Owais, M.M.M.; Griffiths, H.H.H.; Hooper, N.M.N.; Scragg, J.J.L.; Boyle, J.P.J.; Peers, C. Heme Oxygenase-1 Protects against Alzheimer’s Amyloid-β(1-42)-Induced Toxicity via Carbon Monoxide Production. Cell Death Dis. 2014, 5, e1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, W.Y.; Wang, R.; Liu, Y.; Jin, H.; Zhao, Z.W.; Wang, Y.L.; Li, H.Y.; Zhang, X.; Ni, J.X. Chronic Monoarthritis Pain Accelerates the Processes of Cognitive Impairment and Increases the NMDAR Subunits NR2B in CA3 of Hippocampus from 5-Month-Old Transgenic APP/PS1 Mice. Front. Aging Neurosci. 2017, 9, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husebo, B.S.; Achterberg, W.; Flo, E. Identifying and Managing Pain in People with Alzheimer’s Disease and Other Types of Dementia: A Systematic Review. CNS Drugs 2016, 30, 481–497. [Google Scholar] [CrossRef] [Green Version]
- Corbett, A.; Husebo, B.; Malcangio, M.; Staniland, A.; Cohen-Mansfield, J.; Aarsland, D.; Ballard, C. Assessment and Treatment of Pain in People with Dementia. Nat. Rev. Neurol. 2012, 8, 264–274. [Google Scholar] [CrossRef]
- Ezzati, A.; Wang, C.; Katz, M.J.; Derby, C.A.; Zammit, A.R.; Zimmerman, M.E.; Pavlovic, J.M.; Sliwinski, M.J.; Lipton, R.B. The Temporal Relationship between Pain Intensity and Pain Interference and Incident Dementia. Curr. Alzheimer Res. 2018, 16, 109–115. [Google Scholar] [CrossRef]
- Schroeder, C.; Doering, C.; Zamponi, G.; Lewis, R. N-Type Calcium Channel Blockers: Novel Therapeutics for the Treatment of Pain. Med. Chem. 2006, 2, 535–543. [Google Scholar] [CrossRef]
- LaFerla, F.M. Calcium Dyshomeostasis and Intracellular Signalling in Alzheimer’s Disease. Nat. Rev. Neurosci. 2002, 3, 862–872. [Google Scholar] [CrossRef]
- Park, J.F.; Luo, Z.D. Calcium Channel Functions in Pain Processing. Channels 2010, 4, 510–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, R.; Montagut-Bordas, C.; Dickenson, A.H. Calcium Channel Modulation as a Target in Chronic Pain Control. Br. J. Pharm. 2018, 175, 2173–2184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Fu, A.K.Y.; Ip, N.Y. Synaptic Dysfunction in Alzheimer’s Disease: Mechanisms and Therapeutic Strategies. Pharm. 2019, 195, 186–198. [Google Scholar] [CrossRef] [PubMed]
- McDavid, S.; Currie, K.P.M. G-Proteins Modulate Cumulative Inactivation of N-Type (Cav2.2) Calcium Channels. J. Neurosci. 2006, 26, 13373–13383. [Google Scholar] [CrossRef] [Green Version]
- Vogl, C.; Tanifuji, S.; Danis, B.; Daniels, V.; Foerch, P.; Wolff, C.; Whalley, B.J.; Mochida, S.; Stephens, G.J. Synaptic Vesicle Glycoprotein 2A Modulates Vesicular Release and Calcium Channel Function at Peripheral Sympathetic Synapses. Eur. J. Neurosci. 2015, 41, 398–409. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaisis, E.; Thei, L.J.; Stephens, G.J.; Dallas, M.L. Protofibrillar Amyloid Beta Modulation of Recombinant hCaV2.2 (N-Type) Voltage-Gated Channels. Pharmaceuticals 2022, 15, 1459. https://doi.org/10.3390/ph15121459
Kaisis E, Thei LJ, Stephens GJ, Dallas ML. Protofibrillar Amyloid Beta Modulation of Recombinant hCaV2.2 (N-Type) Voltage-Gated Channels. Pharmaceuticals. 2022; 15(12):1459. https://doi.org/10.3390/ph15121459
Chicago/Turabian StyleKaisis, Eleni, Laura J. Thei, Gary J. Stephens, and Mark L. Dallas. 2022. "Protofibrillar Amyloid Beta Modulation of Recombinant hCaV2.2 (N-Type) Voltage-Gated Channels" Pharmaceuticals 15, no. 12: 1459. https://doi.org/10.3390/ph15121459