
Pharmacological Activities of Ginkgolic Acids in Relation to Autophagy

Abstract
:1. Introduction
2. Pharmacological Effects of Ginkgolic Acids
2.1. Antidiabetics
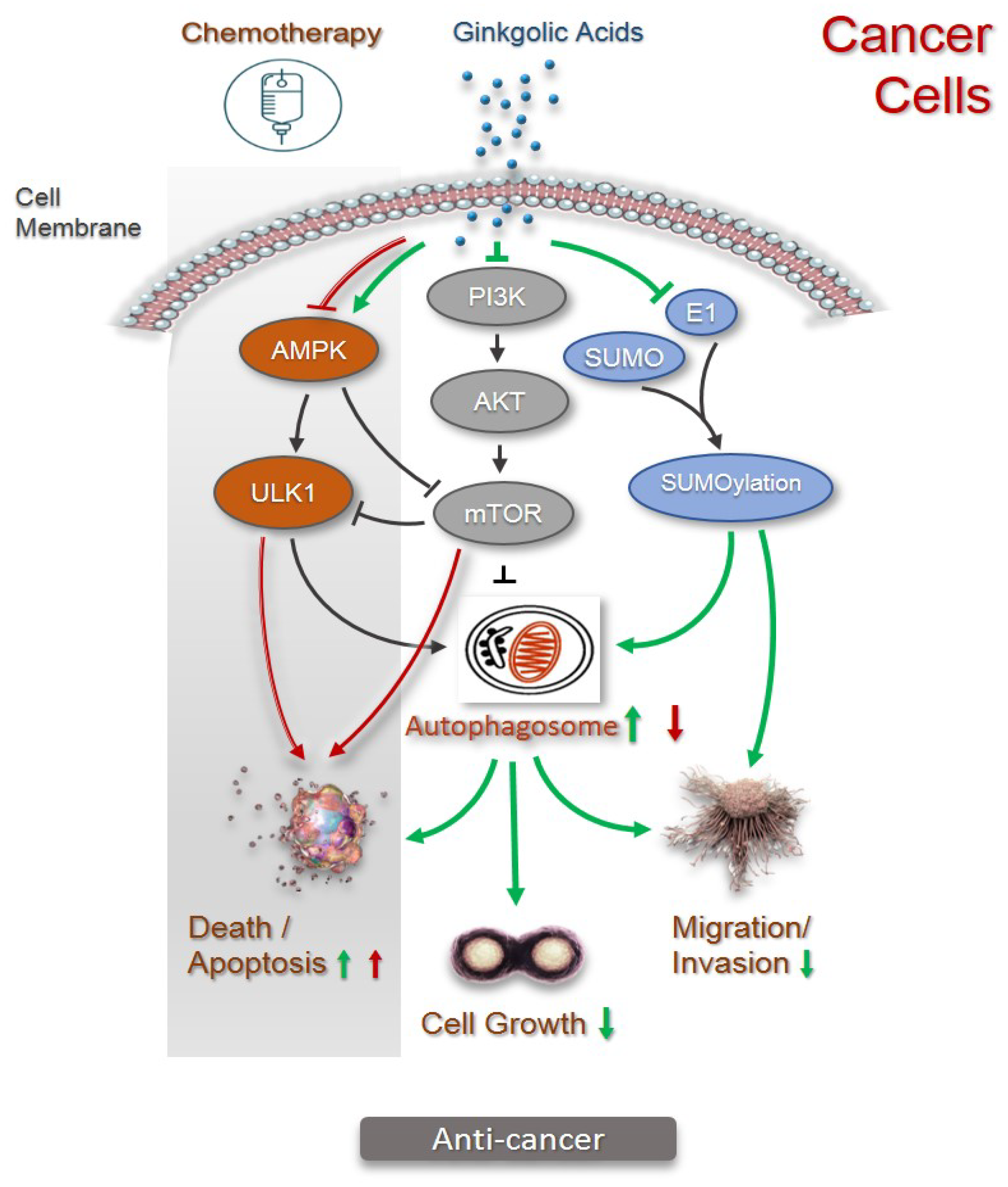
2.2. Anticancer
2.3. Antimicrobials, Anti-Virus, and Anti-Inflammation
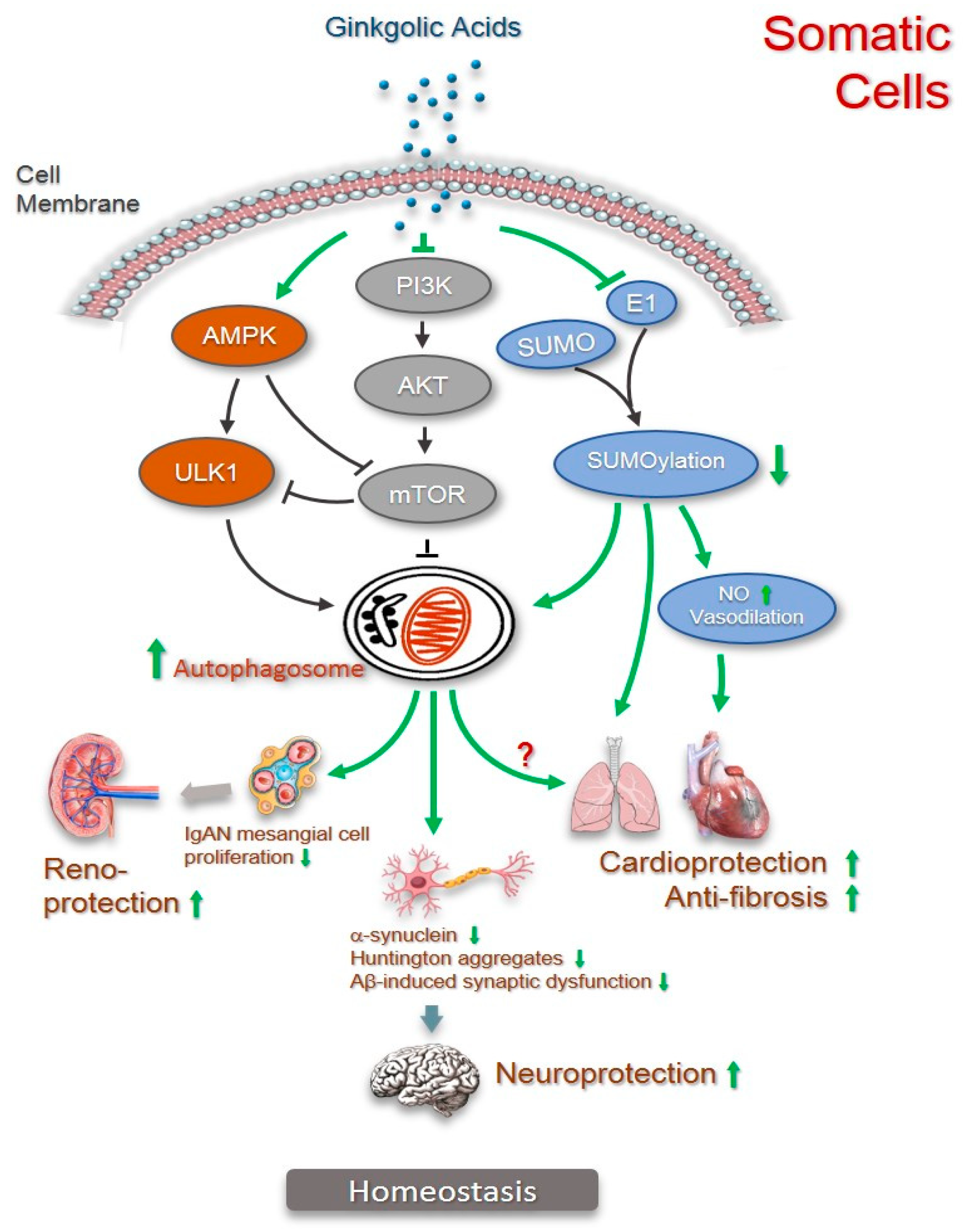
2.4. Anti-Fibrosis
2.5. Cardiovascular Protection
2.6. Renoprotection
2.7. Neuroprotection
3. Autophagy in Pharmacological Activities of Ginkgolic Acids
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gong, W.; Chen, C.; Dobes, C.; Fu, C.-X.; Koch, M.A. Phylogeography of a Living Fossil: Pleistocene Glaciations Forced Ginkgo Biloba L. (Ginkgoaceae) into Two Refuge Areas in China with Limited Subsequent Postglacial Expansion. Mol. Phylogenet. Evol. 2008, 48, 1094–1105. [Google Scholar] [CrossRef]
- Crane, P.R. An Evolutionary and Cultural Biography of Ginkgo. Plants People Planet 2019, 1, 32–37. [Google Scholar] [CrossRef]
- Noor-E-Tabassum; Das, R.; Lami, M.S.; Chakraborty, A.J.; Mitra, S.; Tallei, T.E.; Idroes, R.; Mohamed, A.A.-R.; Hossain, M.J.; Dhama, K.; et al. Ginkgo Biloba: A Treasure of Functional Phytochemicals with Multimedicinal Applications. Evid. Based Complement. Alternat. Med. 2022, 2022, 8288818. [Google Scholar] [CrossRef]
- Singh, B.; Kaur, P.; Gopichand; Singh, R.D.; Ahuja, P.S. Biology and Chemistry of Ginkgo Biloba. Fitoterapia 2008, 79, 401–418. [Google Scholar] [CrossRef]
- Tang, C.Q.; Yang, Y.; Ohsawa, M.; Yi, S.-R.; Momohara, A.; Su, W.-H.; Wang, H.-C.; Zhang, Z.-Y.; Peng, M.-C.; Wu, Z.-L. Evidence for the Persistence of Wild Ginkgo Biloba (Ginkgoaceae) Populations in the Dalou Mountains, Southwestern China. Am. J. Bot. 2012, 99, 1408–1414. [Google Scholar] [CrossRef] [Green Version]
- Šamec, D.; Karalija, E.; Dahija, S.; Hassan, S.T.S. Biflavonoids: Important Contributions to the Health Benefits of Ginkgo (Ginkgo Biloba L.). Plants 2022, 11, 1381. [Google Scholar] [CrossRef] [PubMed]
- Mohanta, T.K.; Occhipinti, A.; Atsbaha Zebelo, S.; Foti, M.; Fliegmann, J.; Bossi, S.; Maffei, M.E.; Bertea, C.M. Ginkgo Biloba Responds to Herbivory by Activating Early Signaling and Direct Defenses. PLoS ONE 2012, 7, e32822. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Bernart, M.W.; Nolan, G.S.; Lin, L.; Lindenmaier, M.P. High-Performance Liquid Chromatography-Electrospray Ionization-Mass Spectrometry Study of Ginkgolic Acid in the Leaves and Fruits of the Ginkgo Tree (Ginkgo Biloba). J. Chromatogr. Sci. 2000, 38, 169–173. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Martínez, M.; González-González, M.; Martagón, A.J.; Hlavinka, V.; Willson, R.C.; Rito-Palomares, M. Recent Developments in Biomarkers for Diagnosis and Screening of Type 2 Diabetes Mellitus. Curr. Diabetes Rep. 2022, 22, 95–115. [Google Scholar] [CrossRef]
- Coughlan, K.A.; Valentine, R.J.; Ruderman, N.B.; Saha, A.K. AMPK Activation: A Therapeutic Target for Type 2 Diabetes? Diabetes Metab. Syndr. Obes. Targets Ther. 2014, 7, 241–253. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, S.; Tao, R.; Wei, D.; Chen, L.; Shen, W.; Yu, Z.-H.; Wang, L.; Jones, D.R.; Dong, X.C.; et al. A Highly Selective and Potent PTP-MEG2 Inhibitor with Therapeutic Potential for Type 2 Diabetes. J. Am. Chem. Soc. 2012, 134, 18116–18124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Dembski, M.; Yang, Q.; Yang, D.; Moriarty, A.; Tayber, O.; Chen, H.; Kapeller, R.; Tartaglia, L.A. Dual Specificity Mitogen-Activated Protein (MAP) Kinase Phosphatase-4 Plays a Potential Role in Insulin Resistance. J. Biol. Chem. 2003, 278, 30187–30192. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.-Y.; Lee, J.H.; Kwon, S.J.; Kang, H.J.; Chung, S.J. Ginkgolic Acid as a Dual-Targeting Inhibitor for Protein Tyrosine Phosphatases Relevant to Insulin Resistance. Bioorg. Chem. 2018, 81, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Tran, K.B.; Lang, J.J.; Compton, K.; Xu, R.; Acheson, A.R.; Henrikson, H.J.; Kocarnik, J.M.; Penberthy, L.; Aali, A.; Abbas, Q.; et al. The Global Burden of Cancer Attributable to Risk Factors, 2010–2019: A Systematic Analysis for the Global Burden of Disease Study 2019. Lancet 2022, 400, 563–591. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Li, X.; Du, W.; Feng, Y.; Kong, X.; Li, Y.; Xiao, L.; Zhang, P. Antitumor Effects of Ginkgolic Acid in Human Cancer Cell Occur via Cell Cycle Arrest and Decrease the Bcl-2/Bax Ratio to Induce Apoptosis. Chemotherapy 2010, 56, 393–402. [Google Scholar] [CrossRef]
- Liang, J.-R.; Yang, H. Ginkgolic Acid (GA) Suppresses Gastric Cancer Growth by Inducing Apoptosis and Suppressing STAT3/JAK2 Signaling Regulated by ROS. Biomed. Pharmacother. 2020, 125, 109585. [Google Scholar] [CrossRef]
- Xiao, Y.; Li, F.; Zheng, A.; Chen, Q.; Chen, F.; Cheng, X.; Tao, Z. Ginkgolic Acid Suppresses Nasopharyngeal Carcinoma Growth by Inducing Apoptosis and Inhibiting AKT/NF-ΚB Signaling. J. Med. Food 2021, 24, 806–816. [Google Scholar] [CrossRef]
- Baek, S.; Lee, J.; Kim, C.; Ko, J.-H.; Ryu, S.-H.; Lee, S.-G.; Yang, W.; Um, J.-Y.; Chinnathambi, A.; Alharbi, S.; et al. Ginkgolic Acid C 17:1, Derived from Ginkgo Biloba Leaves, Suppresses Constitutive and Inducible STAT3 Activation through Induction of PTEN and SHP-1 Tyrosine Phosphatase. Molecules 2017, 22, 276. [Google Scholar] [CrossRef]
- Fei, Z.; Yu, Y.; Xiang, M.; Luo, F. Ginkgolic Acid (GA) Inhibits the Growth of OCa by Inhibiting LncRNA MALAT1/JAK2 Axis. Evid. Based Complement. Alternat. Med. 2021, 2021, 5481271. [Google Scholar] [CrossRef]
- Ma, J.; Duan, W.; Han, S.; Lei, J.; Xu, Q.; Chen, X.; Jiang, Z.; Nan, L.; Li, J.; Chen, K.; et al. Ginkgolic Acid Suppresses the Development of Pancreatic Cancer by Inhibiting Pathways Driving Lipogenesis. Oncotarget 2015, 6, 20993–21003. [Google Scholar] [CrossRef]
- Qiao, L.; Zheng, J.; Jin, X.; Wei, G.; Wang, G.; Sun, X.; Li, X. Ginkgolic Acid Inhibits the Invasiveness of Colon Cancer Cells through AMPK Activation. Oncol. Lett. 2017, 14, 5831–5838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Liu, J.; Yang, X.; Dong, Y.; Liu, Y.; Chen, M. Ginkgol C17:1 Inhibits Tumor Growth by Blunting the EGF- PI3K/Akt Signaling Pathway. J. Biomed. Res. 2017, 31, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Na, N.; Sheng, H.; Feng, B.; Wang, H.; Zhu, P.; Zhang, W.; Zhang, M.; Deng, Z. Ginkgolic Acid Inhibits the Growth of Renal Cell Carcinoma Cells via Inactivation of the EGFR Signaling Pathway. Exp. Ther. Med. 2020, 19, 2949–2956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.-M.; Wang, Y.-F.; Li, Y.-Y.; Ma, H.-L. Thermal Stability of Ginkgolic Acids from Ginkgo Biloba and the Effects of Ginkgol C17:1 on the Apoptosis and Migration of SMMC7721 Cells. Fitoterapia 2014, 98, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-M.; Yeh, E.T.H. SUMO: From Bench to Bedside. Physiol. Rev. 2020, 100, 1599–1619. [Google Scholar] [CrossRef]
- Fukuda, I.; Ito, A.; Hirai, G.; Nishimura, S.; Kawasaki, H.; Saitoh, H.; Kimura, K.; Sodeoka, M.; Yoshida, M. Ginkgolic Acid Inhibits Protein SUMOylation by Blocking Formation of the E1-SUMO Intermediate. Chem. Biol. 2009, 16, 133–140. [Google Scholar] [CrossRef]
- Hamdoun, S.; Efferth, T. Ginkgolic Acids Inhibit Migration in Breast Cancer Cells by Inhibition of NEMO Sumoylation and NF-ΚB Activity. Oncotarget 2017, 8, 35103–35115. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.; Yuan, Y.; Ren, H.; Yue, H.; Xu, B.; Qian, J. SUMOylation Regulates Rb Hyperphosphorylation and Inactivation in Uveal Melanoma. Cancer Sci. 2022, 113, 622–633. [Google Scholar] [CrossRef]
- Liu, K.; Wang, X.; Li, D.; Xu, D.; Li, D.; Lv, Z.; Zhao, D.; Chu, W.-F.; Wang, X.-F. Ginkgolic Acid, a SUMO-1 Inhibitor, Inhibits the Progression of Oral Squamous Cell Carcinoma by Alleviating SUMOylation of SMAD4. Mol. Ther. Oncolytics 2020, 16, 86–99. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Wang, M.; Ruan, Y.; Tan, J.; Wang, H.; Yang, T.; Li, J.; Zhou, Q. Ginkgolic Acids Impair Mitochondrial Function by Decreasing Mitochondrial Biogenesis and Promoting FUNDC1-Dependent Mitophagy. J. Agric. Food Chem. 2019, 67, 10097–10106. [Google Scholar] [CrossRef]
- Lorente, M.; García-Casas, A.; Salvador, N.; Martínez-López, A.; Gabicagogeascoa, E.; Velasco, G.; López-Palomar, L.; Castillo-Lluva, S. Inhibiting SUMO1-Mediated SUMOylation Induces Autophagy-Mediated Cancer Cell Death and Reduces Tumour Cell Invasion via RAC1. J. Cell Sci. 2019, 132, jcs.234120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhaesebroeck, B.; Stephens, L.; Hawkins, P. PI3K Signalling: The Path to Discovery and Understanding. Nat. Rev. Mol. Cell Biol. 2012, 13, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Gao, N.; Zhang, Z.; Jiang, B.-H.; Shi, X. Role of PI3K/AKT/MTOR Signaling in the Cell Cycle Progression of Human Prostate Cancer. Biochem. Biophys. Res. Commun. 2003, 310, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Hay, N. The Akt-MTOR Tango and Its Relevance to Cancer. Cancer Cell 2005, 8, 179–183. [Google Scholar] [CrossRef] [Green Version]
- Baek, S.H.; Ko, J.-H.; Lee, J.H.; Kim, C.; Lee, H.; Nam, D.; Lee, J.; Lee, S.-G.; Yang, W.M.; Um, J.-Y.; et al. Ginkgolic Acid Inhibits Invasion and Migration and TGF-β-Induced EMT of Lung Cancer Cells Through PI3K/Akt/MTOR Inactivation. J. Cell. Physiol. 2017, 232, 346–354. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, B.; Zhang, L.; Cong, X.; Liu, Z.; Hu, Y.; Zhang, J.; Hu, H. Ginkgolic Acid Induces Interplay between Apoptosis and Autophagy Regulated by ROS Generation in Colon Cancer. Biochem. Biophys. Res. Commun. 2018, 498, 246–253. [Google Scholar] [CrossRef]
- Zhou, L.; Li, S.; Sun, J. Ginkgolic Acid Induces Apoptosis and Autophagy of Endometrial Carcinoma Cells via Inhibiting PI3K/Akt/MTOR Pathway In Vivo and In Vitro. Hum. Exp. Toxicol. 2021, 40, 2156–2164. [Google Scholar] [CrossRef]
- Li, Y.; Liu, J.; Liu, Y.; Yang, X.; Huang, B.; Chen, M. Inhibitory Effect of Ginkgol C17:1 on the Biological Behavior of Tumor Cells. Oncol. Lett. 2017, 13, 1873–1879. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Li, Y.; Yang, X.; Dong, Y.; Wu, J.; Chen, M. Effects of Ginkgol C17:1 on Cisplatin-Induced Autophagy and Apoptosis in HepG2 Cells. Oncol. Lett. 2018, 15, 1021–1029. [Google Scholar] [CrossRef] [Green Version]
- Amaravadi, R.K.; Thompson, C.B. The Roles of Therapy-Induced Autophagy and Necrosis in Cancer Treatment. Clin. Cancer Res. 2007, 13, 7271–7279. [Google Scholar] [CrossRef]
- Bao, L.; Jaramillo, M.C.; Zhang, Z.; Zheng, Y.; Yao, M.; Zhang, D.D.; Yi, X. Induction of Autophagy Contributes to Cisplatin Resistance in Human Ovarian Cancer Cells. Mol. Med. Rep. 2015, 11, 91–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Shao, Z.; Xiong, L.; Yang, S. Inhibition of Autophagy Enhances Cisplatin-Induced Apoptosis in the MG63 Human Osteosarcoma Cell Line. Oncol. Lett. 2015, 10, 2941–2946. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhang, X.; Yang, X.; Liu, J.; Li, L.; Ma, W.; Chen, M. Differential Effects of Ginkgol C17:1 on Cisplatin-induced Cytotoxicity: Protecting Human Normal L02 Hepatocytes versus Sensitizing Human Hepatoma HepG2 Cells. Oncol. Lett. 2019, 17, 3181–3190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chassagne, F.; Huang, X.; Lyles, J.T.; Quave, C.L. Validation of a 16th Century Traditional Chinese Medicine Use of Ginkgo Biloba as a Topical Antimicrobial. Front. Microbiol. 2019, 10, 775. [Google Scholar] [CrossRef]
- Lee, J.-H.; Kim, Y.-G.; Ryu, S.Y.; Cho, M.H.; Lee, J. Ginkgolic Acids and Ginkgo Biloba Extract Inhibit Escherichia Coli O157:H7 and Staphylococcus Aureus Biofilm Formation. Int. J. Food Microbiol. 2014, 174, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, S.; Hayes, D.; Wozniak, D.J. Cystic Fibrosis and Pseudomonas Aeruginosa: The Host-Microbe Interface. Clin. Microbiol. Rev. 2019, 32, e00138-18. [Google Scholar] [CrossRef] [PubMed]
- Tahrioui, A. Membrane-Interactive Compounds From Pistacia Lentiscus L. Thwart Pseudomonas Aeruginosa Virulence. Front. Microbiol. 2020, 11, 15. [Google Scholar]
- Hua, Z.; Wu, C.; Fan, G.; Tang, Z.; Cao, F. The Antibacterial Activity and Mechanism of Ginkgolic Acid C15:1. BMC Biotechnol. 2017, 17, 5. [Google Scholar] [CrossRef] [Green Version]
- Lü, J.-M.; Yan, S.; Jamaluddin, S.; Weakley, S.M.; Liang, Z.; Siwak, E.B.; Yao, Q.; Chen, C. Ginkgolic Acid Inhibits HIV Protease Activity and HIV Infection In Vitro. Med. Sci. Monit. 2012, 18, BR293–BR298. [Google Scholar] [CrossRef] [Green Version]
- Borenstein, R.; Hanson, B.A.; Markosyan, R.M.; Gallo, E.S.; Narasipura, S.D.; Bhutta, M.; Shechter, O.; Lurain, N.S.; Cohen, F.S.; Al-Harthi, L.; et al. Ginkgolic Acid Inhibits Fusion of Enveloped Viruses. Sci. Rep. 2020, 10, 4746. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Zhu, G.-H.; Wang, H.-N.; Hu, Q.; Chen, L.-L.; Guan, X.-Q.; Li, H.-L.; Chen, H.-Z.; Tang, H.; Ge, G.-B. Discovery of Naturally Occurring Inhibitors against SARS-CoV-2 3CLpro from Ginkgo Biloba Leaves via Large-Scale Screening. Fitoterapia 2021, 152, 104909. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yan, J. Protective Effect of Ginkgolic Acid in Attenuating LDL Induced Inflammation Human Peripheral Blood Mononuclear Cells via Altering the NF-ΚB Signaling Pathway. Front. Pharmacol. 2019, 10, 1241. [Google Scholar] [CrossRef] [PubMed]
- Qiu, F.; Dong, C.; Liu, Y.; Shao, X.; Huang, D.; Han, Y.; Wang, B.; Liu, Y.; Huo, R.; Paulo, P.; et al. Pharmacological Inhibition of SUMO-1 with Ginkgolic Acid Alleviates Cardiac Fibrosis Induced by Myocardial Infarction in Mice. Toxicol. Appl. Pharmacol. 2018, 345, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic Pulmonary Fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef] [PubMed]
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and Safety of Nintedanib in Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, L.H.; de Andrade, J.A.; Zibrak, J.D.; Padilla, M.L.; Albera, C.; Nathan, S.D.; Wijsenbeek, M.S.; Stauffer, J.L.; Kirchgaessler, K.-U.; Costabel, U. Pirfenidone Safety and Adverse Event Management in Idiopathic Pulmonary Fibrosis. Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2017, 26, 170057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Bian, X.; Zhang, C.; Wu, Z.; Huang, N.; Yang, J.; Jin, W.; Feng, Z.; Li, D.; Huo, X.; et al. Ginkgolic Acid Improves Bleomycin-Induced Pulmonary Fibrosis by Inhibiting SMAD4 SUMOylation. Oxid. Med. Cell. Longev. 2022, 2022, 8002566. [Google Scholar] [CrossRef] [PubMed]
- Sarmah, N.; Nauli, A.M.; Ally, A.; Nauli, S.M. Interactions among Endothelial Nitric Oxide Synthase, Cardiovascular System, and Nociception during Physiological and Pathophysiological States. Molecules 2022, 27, 2835. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-B.; Ruan, C.-C.; Chen, D.-R.; Zhang, K.; Yan, C.; Gao, P.-J. Activating Transcription Factor 3 SUMOylation Is Involved in Angiotensin II-Induced Endothelial Cell Inflammation and Dysfunction. J. Mol. Cell. Cardiol. 2016, 92, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, R.J.; Julian, B.A. IgA Nephropathy. N. Engl. J. Med. 2013, 368, 2402–2414. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, G.P.; Shah, S.V. Autophagy in Acute Kidney Injury. Kidney Int. 2016, 89, 779–791. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Liu, Y.; Chen, G.; He, L.; Tang, C.; Wang, C.; Yang, D.; Li, H.; Dong, Z.; Liu, H. Rapamycin Enhances Repressed Autophagy and Attenuates Aggressive Progression in a Rat Model of IgA Nephropathy. Am. J. Nephrol. 2017, 45, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Wei, Q.; Su, Y.; Dong, Z. SUMOylation Occurs in Acute Kidney Injury and Plays a Cytoprotective Role. Biochim. Biophys. Acta 2015, 1852, 482–489. [Google Scholar] [CrossRef] [Green Version]
- Tossidou, I.; Himmelseher, E.; Teng, B.; Haller, H.; Schiffer, M. SUMOylation Determines Turnover and Localization of Nephrin at the Plasma Membrane. Kidney Int. 2014, 86, 1161–1173. [Google Scholar] [CrossRef] [Green Version]
- Tan, X.; Liu, Y.; Liu, D.; Tang, X.; Xia, M.; Chen, G.; He, L.; Zhu, X.; Liu, H. SUMO1 Promotes Mesangial Cell Proliferation Through Inhibiting Autophagy in a Cell Model of IgA Nephropathy. Front. Med. 2022, 9, 834164. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.; Wielsch, B.; Boltze, J. The Role of SUMOylation in Cerebral Hypoxia and Ischemia. Neurochem. Int. 2017, 107, 66–77. [Google Scholar] [CrossRef]
- Fei, E.; Jia, N.; Yan, M.; Ying, Z.; Sun, Q.; Wang, H.; Zhang, T.; Ma, X.; Ding, H.; Yao, X.; et al. SUMO-1 Modification Increases Human SOD1 Stability and Aggregation. Biochem. Biophys. Res. Commun. 2006, 347, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.; Sixt, K.M.; Barrow, R.; Snyder, S.H. Rhes, a Striatal Specific Protein, Mediates Mutant-Huntingtin Cytotoxicity. Science 2009, 324, 1327–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.M.; Jang, W.H.; Quezado, M.M.; Oh, Y.; Chung, K.C.; Junn, E.; Mouradian, M.M. Proteasome Inhibition Induces α-Synuclein SUMOylation and Aggregate Formation. J. Neurol. Sci. 2011, 307, 157–161. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, J.G.; Gareau, J.R.; Ochaba, J.; Song, W.; Raskó, T.; Reverter, D.; Lee, J.; Monteys, A.M.; Pallos, J.; Mee, L.; et al. SUMO-2 and PIAS1 Modulate Insoluble Mutant Huntingtin Protein Accumulation. Cell Rep. 2013, 4, 362–375. [Google Scholar] [CrossRef]
- Luo, H.-B.; Xia, Y.-Y.; Shu, X.-J.; Liu, Z.-C.; Feng, Y.; Liu, X.-H.; Yu, G.; Yin, G.; Xiong, Y.-S.; Zeng, K.; et al. SUMOylation at K340 Inhibits Tau Degradation through Deregulating Its Phosphorylation and Ubiquitination. Proc. Natl. Acad. Sci. USA 2014, 111, 16586–16591. [Google Scholar] [CrossRef] [Green Version]
- Ochaba, J.; Monteys, A.M.; O’Rourke, J.G.; Reidling, J.C.; Steffan, J.S.; Davidson, B.L.; Thompson, L.M. PIAS1 Regulates Mutant Huntingtin Accumulation and Huntington’s Disease-Associated Phenotypes In Vivo. Neuron 2016, 90, 507–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arendt, T. Synaptic Degeneration in Alzheimer’s Disease. Acta Neuropathol. 2009, 118, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Mango, D.; Nisticò, R. Ginkgolic Acid Protects against Aβ-Induced Synaptic Dysfunction in the Hippocampus. Front. Pharmacol. 2016, 7, 7. [Google Scholar] [CrossRef] [Green Version]
- Lee, L.; Dale, E.; Staniszewski, A.; Zhang, H.; Saeed, F.; Sakurai, M.; Fa’, M.; Orozco, I.; Michelassi, F.; Akpan, N.; et al. Regulation of Synaptic Plasticity and Cognition by SUMO in Normal Physiology and Alzheimer’s Disease. Sci. Rep. 2014, 4, 7190. [Google Scholar] [CrossRef] [Green Version]
- Nisticò, R.; Ferraina, C.; Marconi, V.; Blandini, F.; Negri, L.; Egebjerg, J.; Feligioni, M. Age-Related Changes of Protein SUMOylation Balance in the AβPP Tg2576 Mouse Model of Alzheimer’s Disease. Front. Pharmacol. 2014, 5, 63. [Google Scholar] [CrossRef] [Green Version]
- Rott, R.; Szargel, R.; Shani, V.; Hamza, H.; Savyon, M.; Abd Elghani, F.; Bandopadhyay, R.; Engelender, S. SUMOylation and Ubiquitination Reciprocally Regulate α-Synuclein Degradation and Pathological Aggregation. Proc. Natl. Acad. Sci. USA 2017, 114, 13176–13181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijayakumaran, S.; Nakamura, Y.; Henley, J.M.; Pountney, D.L. Ginkgolic Acid Promotes Autophagy-Dependent Clearance of Intracellular Alpha-Synuclein Aggregates. Mol. Cell. Neurosci. 2019, 101, 103416. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Jarquín, U.N.; Sharma, M.; Zhou, W.; Shahani, N.; Subramaniam, S. Deletion of SUMO1 Attenuates Behavioral and Anatomical Deficits by Regulating Autophagic Activities in Huntington Disease. Proc. Natl. Acad. Sci. USA 2022, 119, e2107187119. [Google Scholar] [CrossRef] [PubMed]
- White, E. Deconvoluting the Context-Dependent Role for Autophagy in Cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Sridhar, S.; Botbol, Y.; Macian, F.; Cuervo, A.M. Autophagy and Disease: Always Two Sides to a Problem. J. Pathol. 2012, 226, 255–273. [Google Scholar] [CrossRef] [Green Version]
- Ohsumi, Y. Historical Landmarks of Autophagy Research. Cell Res. 2014, 24, 9–23. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.-Z.; Harrison-Findik, D.D. Autophagy and Cancer. World J. Biol. Chem. 2013, 4, 64. [Google Scholar] [CrossRef] [PubMed]
- Vitto, V.A.M.; Bianchin, S.; Zolondick, A.A.; Pellielo, G.; Rimessi, A.; Chianese, D.; Yang, H.; Carbone, M.; Pinton, P.; Giorgi, C.; et al. Molecular Mechanisms of Autophagy in Cancer Development, Progression, and Therapy. Biomedicines 2022, 10, 1596. [Google Scholar] [CrossRef]
- Ghavami, S.; Yeganeh, B.; Stelmack, G.L.; Kashani, H.H.; Sharma, P.; Cunnington, R.; Rattan, S.; Bathe, K.; Klonisch, T.; Dixon, I.M.C.; et al. Apoptosis, Autophagy and ER Stress in Mevalonate Cascade Inhibition-Induced Cell Death of Human Atrial Fibroblasts. Cell Death Dis. 2012, 3, e330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between Apoptosis, Necrosis and Autophagy. Biochim. Biophys. Acta 2013, 1833, 3448–3459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Kang, J.; Fu, C. The Independence of and Associations among Apoptosis, Autophagy, and Necrosis. Signal Transduct. Target. Ther. 2018, 3, 18. [Google Scholar] [CrossRef] [Green Version]
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent Insights into the Function of Autophagy in Cancer. Genes Dev. 2016, 30, 1913–1930. [Google Scholar] [CrossRef]
- Tsai, J.-P.; Lee, C.-H.; Ying, T.-H.; Lin, C.-L.; Lin, C.-L.; Hsueh, J.-T.; Hsieh, Y.-H. Licochalcone A Induces Autophagy through PI3K/Akt/MTOR Inactivation and Autophagy Suppression Enhances Licochalcone A-Induced Apoptosis of Human Cervical Cancer Cells. Oncotarget 2015, 6, 28851–28866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Martín, P.; Saito, T.; Komatsu, M. P62/SQSTM 1: ‘Jack of All Trades’ in Health and Cancer. FEBS J. 2019, 286, 8–23. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Xie, M.; Zhao, F.; Li, J.; Fan, C.; Zheng, H.; Wei, Z.; Ci, X.; Zhang, S. Daphnetin Triggers ROS-Induced Cell Death and Induces Cytoprotective Autophagy by Modulating the AMPK/Akt/MTOR Pathway in Ovarian Cancer. Phytomedicine 2021, 82, 153465. [Google Scholar] [CrossRef]
- Buchser, W.J.; Laskow, T.C.; Pavlik, P.J.; Lin, H.-M.; Lotze, M.T. Cell-Mediated Autophagy Promotes Cancer Cell Survival. Cancer Res. 2012, 72, 2970–2979. [Google Scholar] [CrossRef] [Green Version]
- Mulcahy Levy, J.M.; Thorburn, A. Autophagy in Cancer: Moving from Understanding Mechanism to Improving Therapy Responses in Patients. Cell Death Differ. 2020, 27, 843–857. [Google Scholar] [CrossRef]
- Sethy, C.; Kundu, C.N. 5-Fluorouracil (5-FU) Resistance and the New Strategy to Enhance the Sensitivity against Cancer: Implication of DNA Repair Inhibition. Biomed. Pharmacother. Biomed. Pharmacother. 2021, 137, 111285. [Google Scholar] [CrossRef]
- Gu, J.; Li, Z.; Zhou, J.; Sun, Z.; Bai, C. Response Prediction to Oxaliplatin plus 5-fluorouracil Chemotherapy in Patients with Colorectal Cancer Using a Four-protein Immunohistochemical Model. Oncol. Lett. 2019, 18, 2091–2101. [Google Scholar] [CrossRef] [Green Version]
- Inoue, D.; Kubo, H.; Taguchi, K.; Suzuki, T.; Komatsu, M.; Motohashi, H.; Yamamoto, M. Inducible Disruption of Autophagy in the Lung Causes Airway Hyper-Responsiveness. Biochem. Biophys. Res. Commun. 2011, 405, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Yang, D.-S.; Lee, J.-H. Neurodegenerative Lysosomal Disorders: A Continuum from Development to Late Age. Autophagy 2008, 4, 590–599. [Google Scholar] [CrossRef]
- Lee, J.-H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal Proteolysis and Autophagy Require Presenilin 1 and Are Disrupted by Alzheimer-Related PS1 Mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef] [Green Version]
- Fleming, A.; Noda, T.; Yoshimori, T.; Rubinsztein, D.C. Chemical Modulators of Autophagy as Biological Probes and Potential Therapeutics. Nat. Chem. Biol. 2011, 7, 9–17. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ginkgolic Acids | Cancer Cell Types | Actions and Mechanisms | References |
|---|---|---|---|
| C15:1 | Human tongue squamous Tac8113 | Inhibit cell growth via reduction in the Bcl-2/Bax ratio and stimulation of caspase-3 activity; suppress tumor growth in mouse xenografts | Ref. [15] |
| C17:1 | Human gastric BGC-823, SGC-7901, MGC-803 and AGS | Suppress cancer growth by inducing apoptosis and suppressing STAT3/JAK2 signaling regulated by ROS, both in vitro and in mouse xenografts | Ref. [16] |
| C15:1 | Human nasopharyngeal 5-8F, CNE2 and NP69 | Suppress cancer growth by inducing apoptosis through inhibition of AKT/NF-κB signaling, both in vitro and in mouse xenografts; synergistically suppress cancer with 5-FU | Ref. [17] |
| C17:1 | Human multiple myeloma U266ata | Promote apoptosis via suppressing STAT3/JAK2 signaling | Ref. [18] |
| C15:1 | Human ovary SKOV3 and CAOV3 | Inhibit proliferation and migration through suppressing lncRNA MALAT1/JAK2 axis activity, and suppress tumor growth in mouse xenografts | Ref. [19] |
| C15:1 | Human pancreas Panc-1 and BxPC-3 | Suppress colony formation, migration, invasion, and lipogenesis through activating AMPK signaling; inhibit tumor growth in mouse xenografts | Ref. [20] |
| C15:1 | Human colon SW480 | Reduce proliferation, migration, and invasion through stimulating AMPK signaling | Ref. [21] |
| C17:1 | Human liver hepatoblastoma HepG2 | Reduce proliferation, migration, and invasion via inhibiting EGF-induced activation of PI3K/Akt signaling pathways; inhibit tumor growth in mouse xenografts | Ref. [22] |
| C17:1 | Human kidney 786-O and A498 | Suppress proliferation and invasion via inactivating epidermal growth factor receptor (EGFR) signaling pathway; inhibit tumor growth in mouse xenografts | Ref. [23] |
| C13:0, C15:1, C17:1 | Human liver SMMC7721 | Induce apoptosis via caspases-3 activity, upregulate Bax expression; inhibit migration | Ref. [24] |
| C13:0 | Human breast MCF-7 and MDA-MB-231 | Inhibit migration by inhibition of NEMO SUMOylation and NF-κB activity | Ref. [27] |
| C15:1 | Human uveal melanoma OCM3, OMM2.3 and Mel285 | Induce apoptosis through inhibition of SUMOylation; inhibit tumor growth in mouse xenografts | Ref. [28] |
| C15:1 | Human tongue squamous Tac8113 and Cal-27 | Induce apoptosis and suppress invasion through inhibition of TGF-β1-induced enhancement of SUMOylation of SMAD4; inhibit tumor growth in mouse xenografts | Ref. [29] |
| C15:1 | Human cervical carcinoma derived HeLa | Impair mitochondrial function by decreasing mitochondrial biogenesis, promote mitophagy | Ref. [30] |
| C15:1 | Human breast MCF7, MDA-MB-231 and BT474; human prostate LnCap and 22Rv1 | In vitro and in vivo inhibition of SUMO1-mediated SUMOylation, induce autophagy-mediated cancer cell death and reduce invasion via RAC1; inhibit tumor growth in mouse xenografts | Ref. [31] |
| C15:1 | Human lung A549 and H1299 | Inhibit viability, invasion, and migration and TGF-β-induced epithelial–mesenchymal transition (EMT) by inactivating PI3K/Akt/mTOR signaling pathway | Ref. [35] |
| C15:1 | Human colon SW480 | Cause G0/G1 phase cell arrest, trigger intrinsic apoptosis and autophagy modulated by ROS generation | Ref. [36] |
| C15:1 | Human endometrial Ishikawa and HEC-1-B | Induce apoptosis and autophagy via inhibiting PI3K/Akt/mTOR pathway in vivo and in vitro; inhibit tumor growth in mouse xenografts | Ref. [37] |
| C17:1 | Human liver hepatoblastoma HepG2 | Suppress viability, migration, and invasion by inhibiting the activation of the mitogen-activated protein kinase/MMP, Rho/Rho-associated protein kinase and PI3K/Akt signaling pathways; inhibit tumor growth in mouse xenografts | Ref. [38] |
| C17:1 | Human liver hepatoblastoma HepG2 | Inhibit cisplatin-induced autophagy via AMPK/ULK1 signaling and increase cisplatin-induced apoptosis via the PI3K/Akt/mTOR pathway | Ref. [39] |
| C17:1 | Human liver hepatoblastoma HepG2 | Inhibit cisplatin-induced autophagy via AMPK/ULK1 signaling and increase cisplatin-induced apoptosis via the PI3K/Akt/mTOR pathway in cancer cells, activate autophagy and reverse cisplatin-induced apoptosis in normal hepatocytes; synergistic cancer suppression with cisplatin | Ref. [43] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, Y.; Ding, Z.; Xu, J.; Li, Y.; Chen, M. Pharmacological Activities of Ginkgolic Acids in Relation to Autophagy. Pharmaceuticals 2022, 15, 1469. https://doi.org/10.3390/ph15121469
Ding Y, Ding Z, Xu J, Li Y, Chen M. Pharmacological Activities of Ginkgolic Acids in Relation to Autophagy. Pharmaceuticals. 2022; 15(12):1469. https://doi.org/10.3390/ph15121469
Chicago/Turabian StyleDing, Yuan, Zheheng Ding, Jin Xu, Yueying Li, and Min Chen. 2022. "Pharmacological Activities of Ginkgolic Acids in Relation to Autophagy" Pharmaceuticals 15, no. 12: 1469. https://doi.org/10.3390/ph15121469