
A Class I HDAC Inhibitor BG45 Alleviates Cognitive Impairment through the CaMKII/ITPKA/Ca2+ Signaling Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
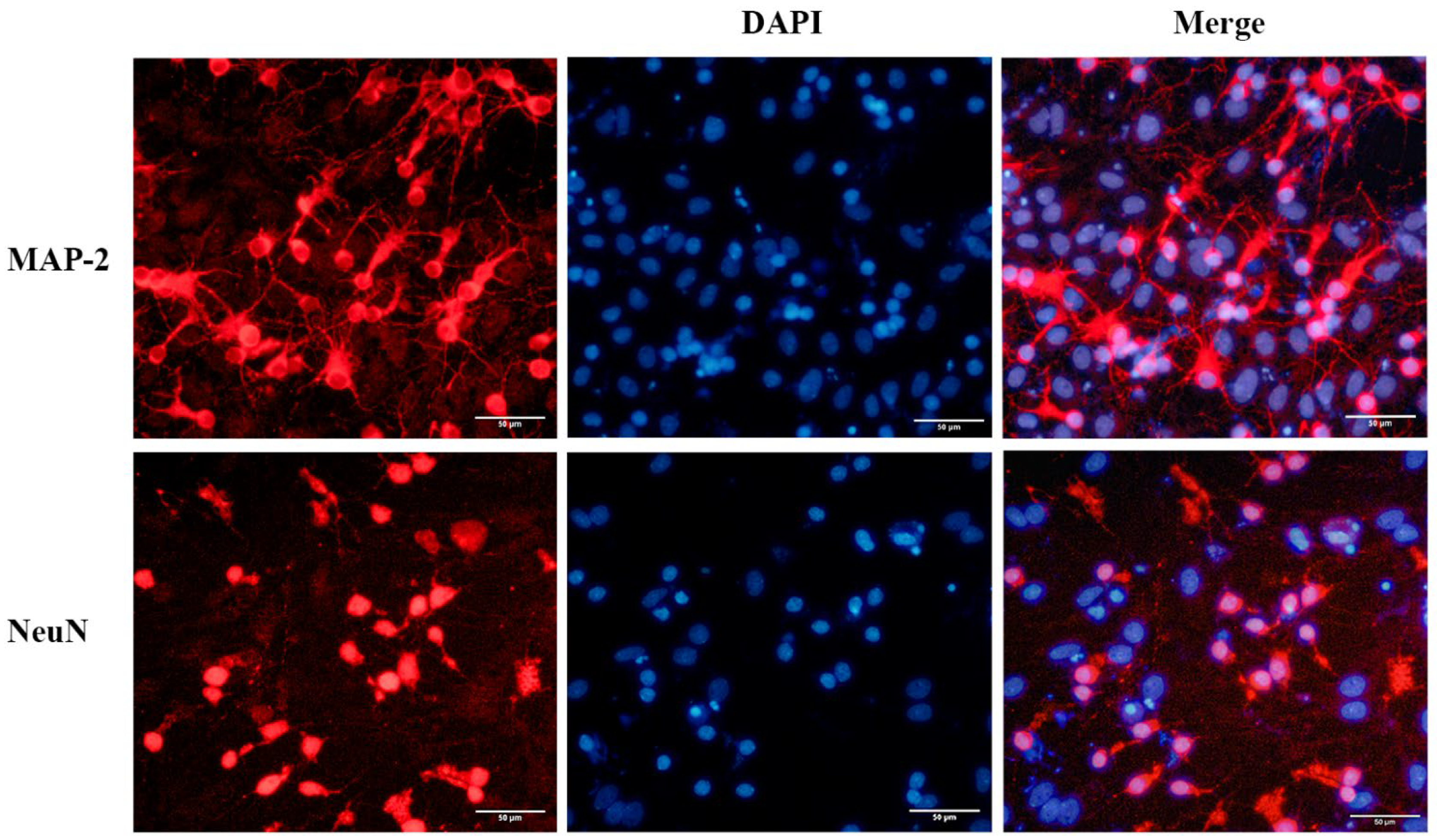
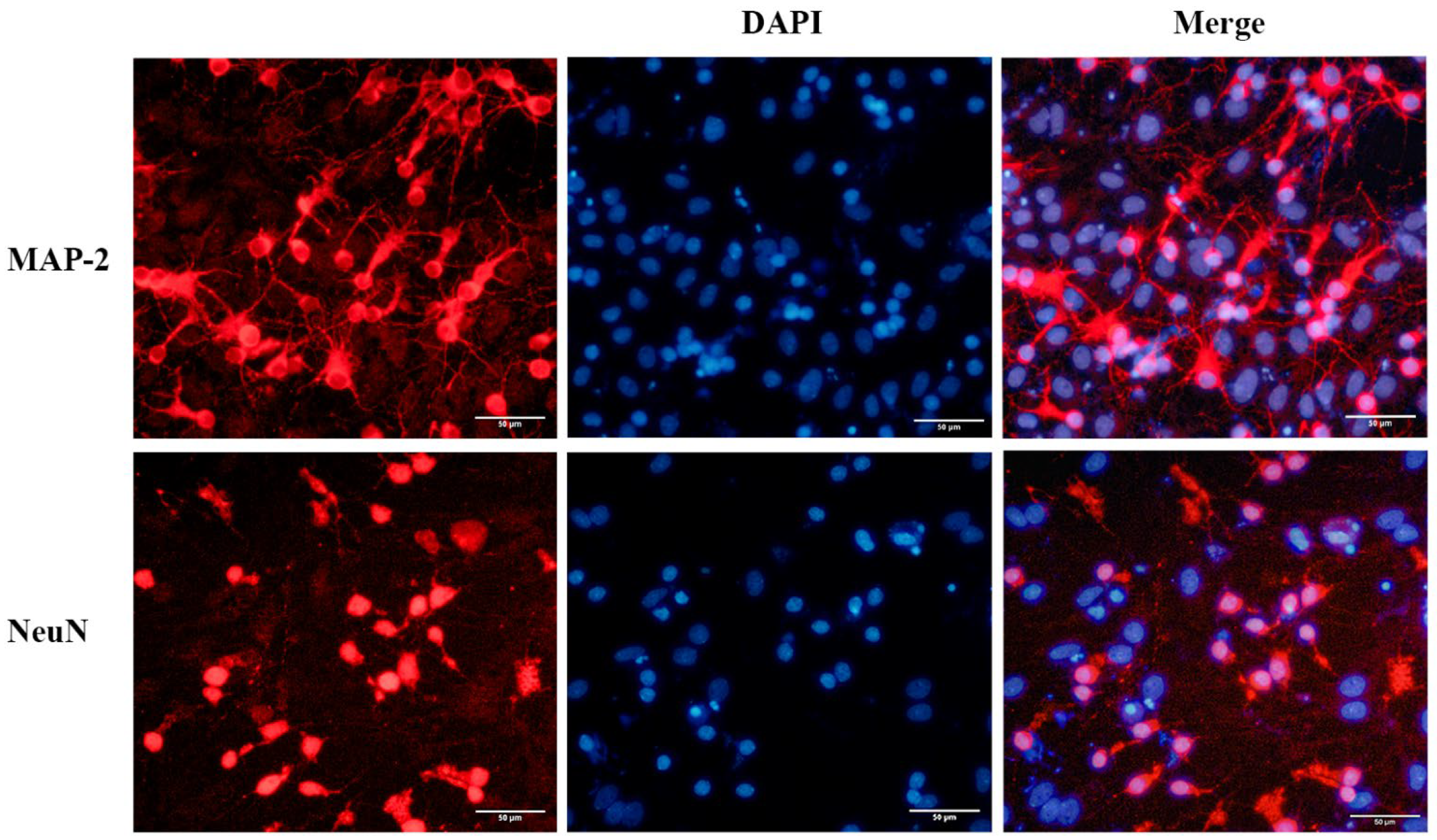
2.1. Identification and Purity of the Primary Cortical Neurons from APP/PS1 Transgenic Mice
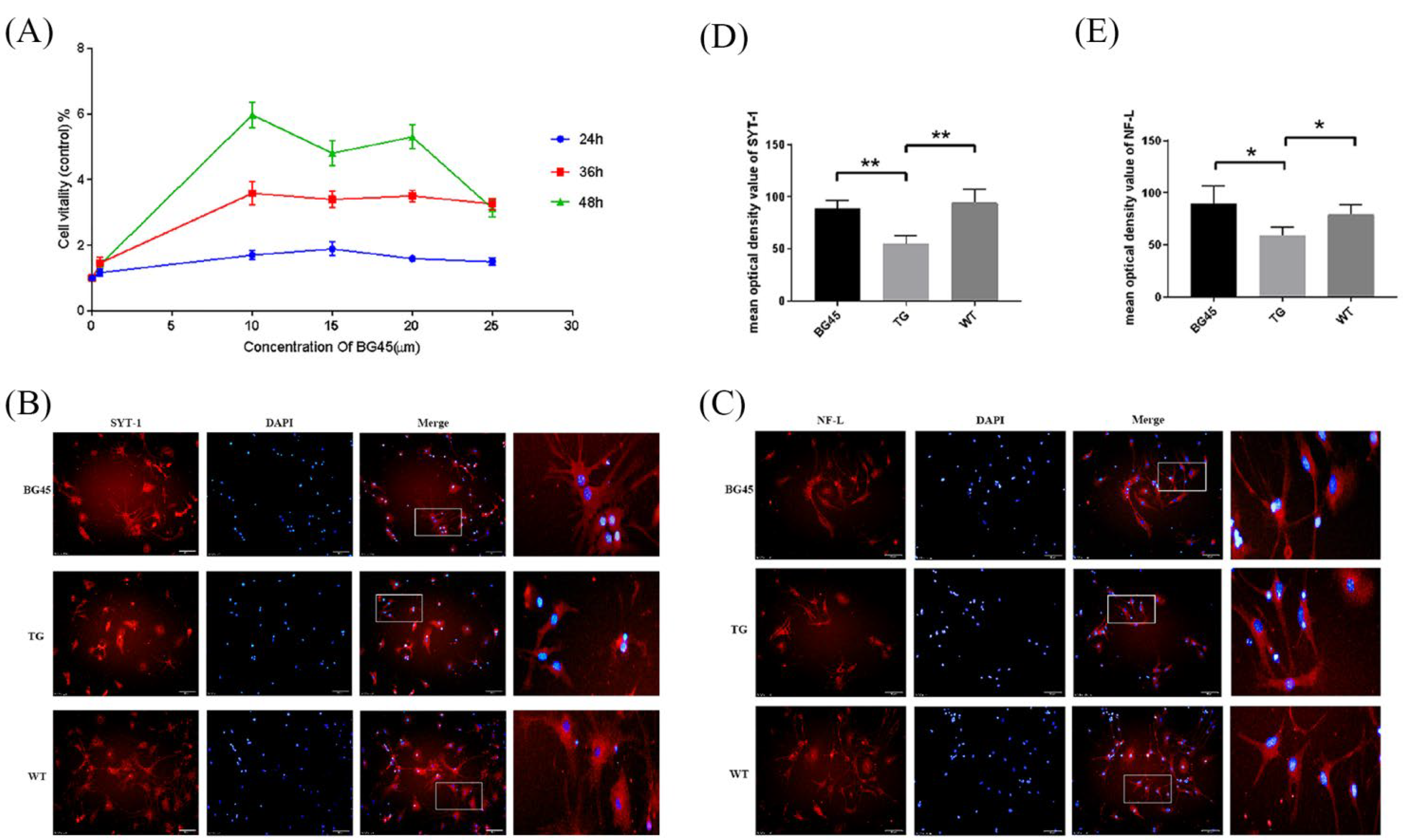
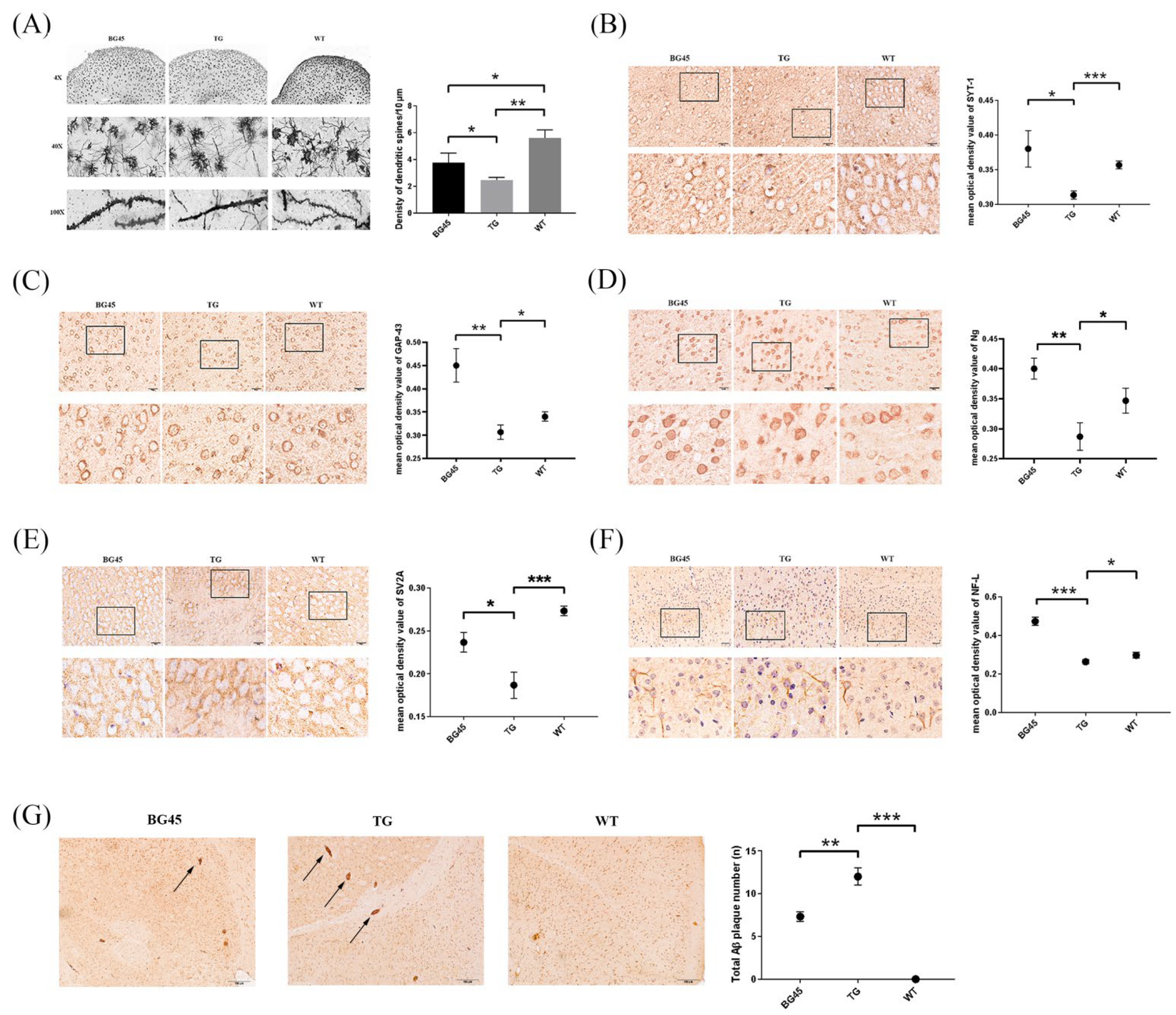
2.2. BG45 Increased the Expression Levels of SYT-1 and NF-L in Primary Cultured Cortical Neurons
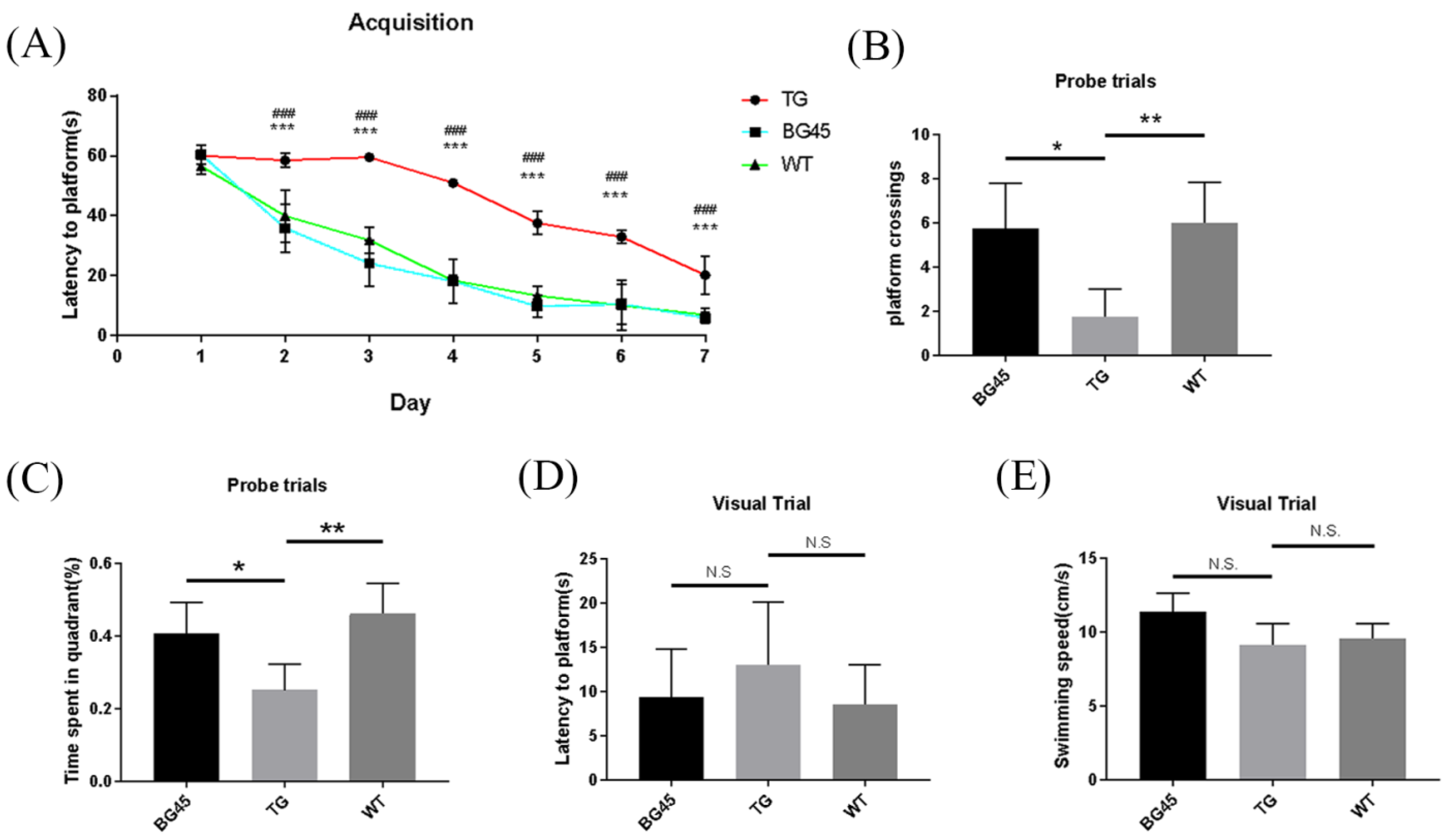
2.3. BG45 Improved Spatial Memory and Learning Ability in APP/PS1 Mice
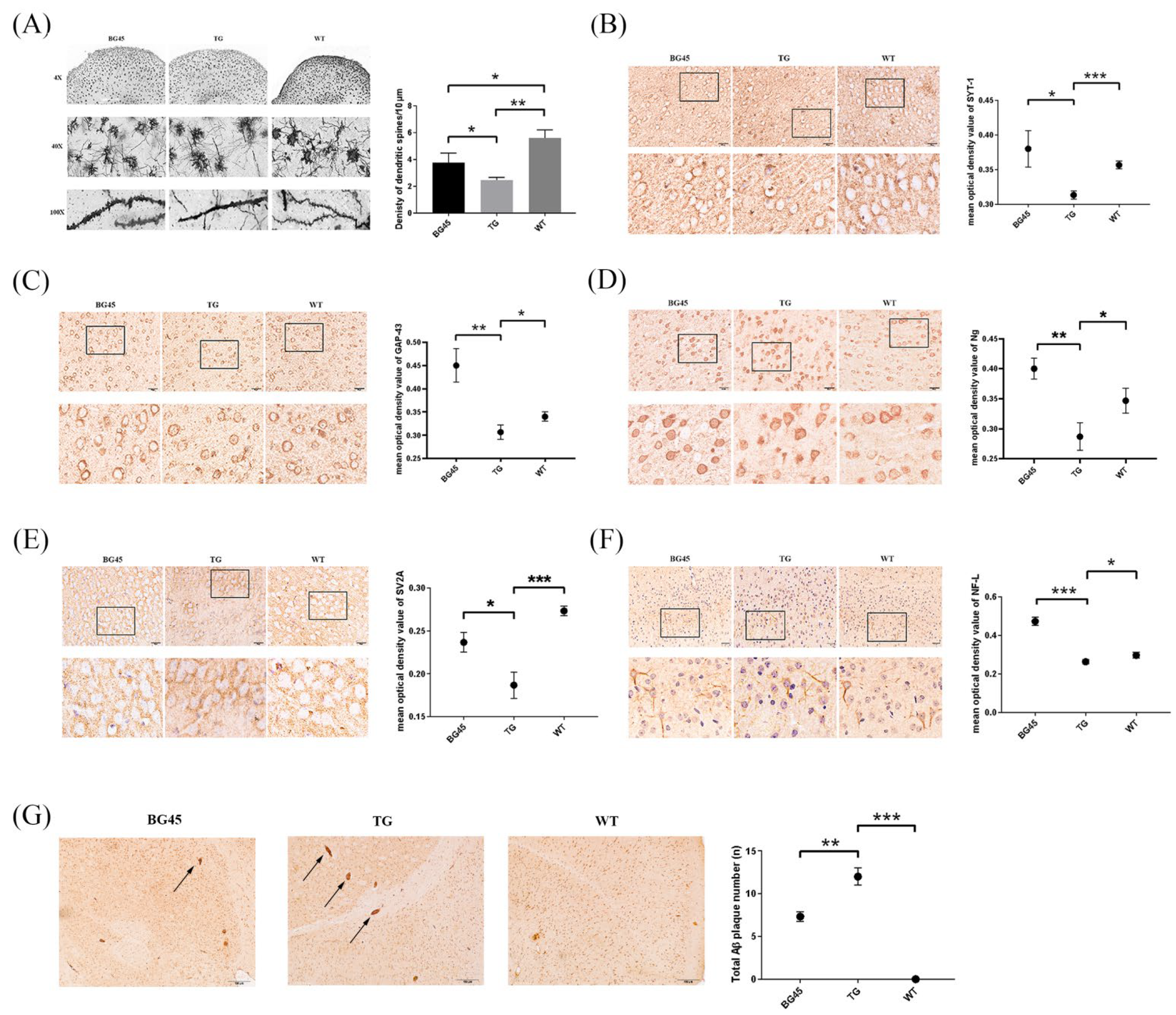
2.4. Effects of BG45 on Dendritic Spines, the Expression of Synapse-Related Proteins and the Accumulation of Aβ in the Cortex of APP/PS1 Mice
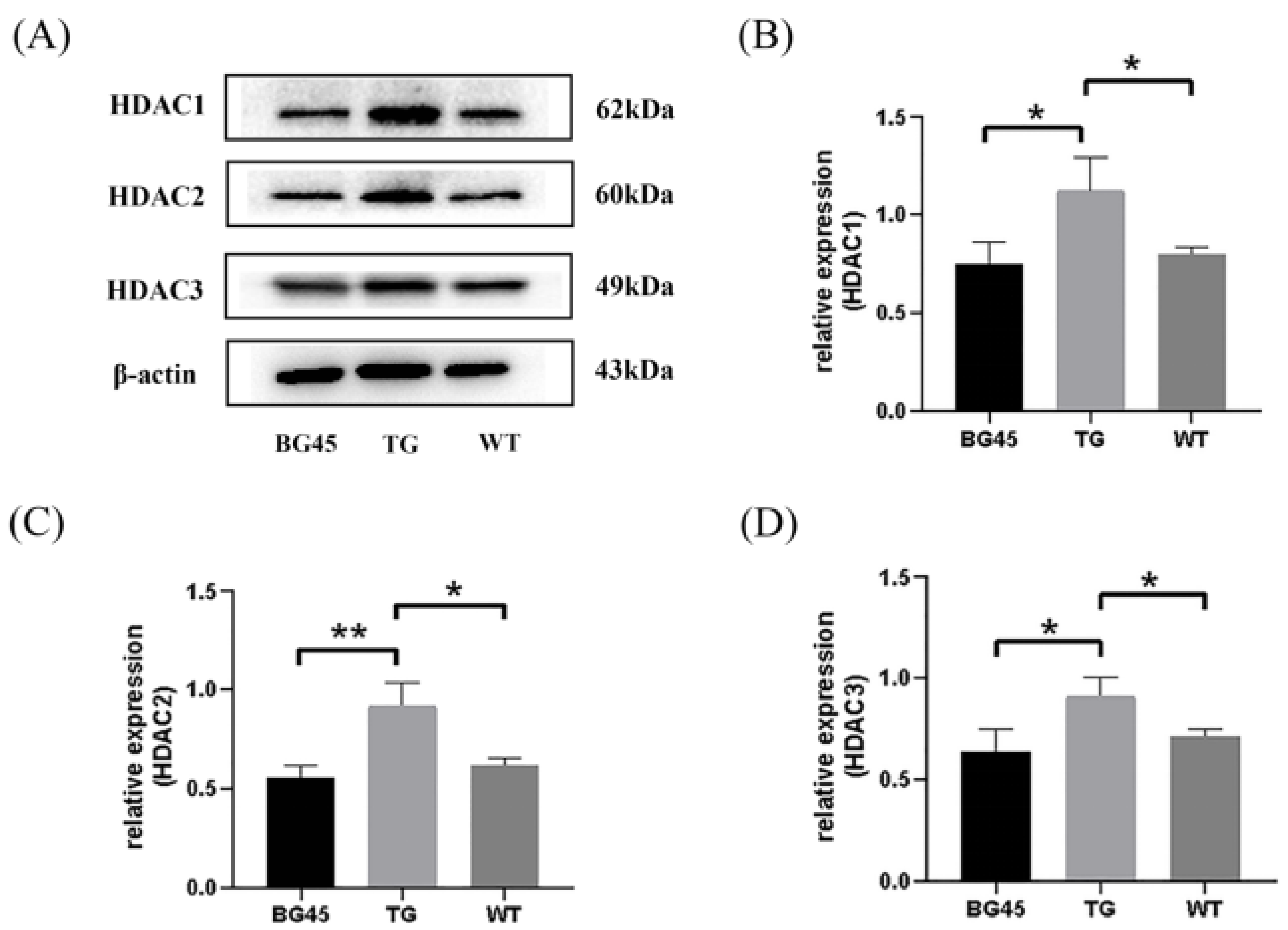
2.5. BG45 Decreased the Protein Expression Levels of HDAC1, HDAC2 and HDAC3 in the Cortex of APP/PS1 Mice
2.6. Profiling of Differentially Expressed Proteins in the Prefrontal Cortex
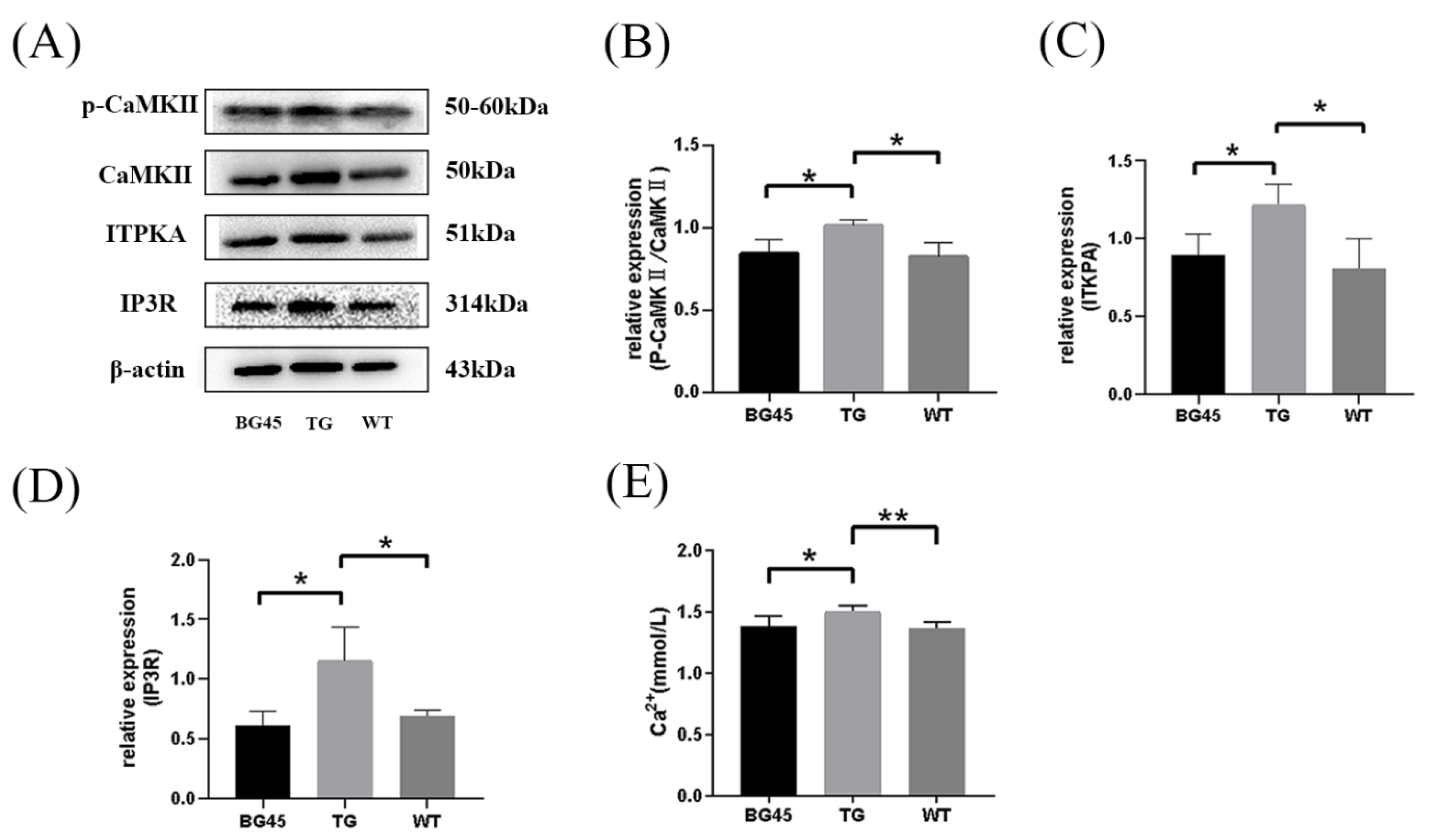
2.7. BG45 Influenced the CaMKII/ITPKA/Ca2+ Pathway In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Cultures and Treatments
4.2. CCK-8 Assay
4.3. Immunohistochemistry
4.4. Animals and Drug Administration
4.5. Morris Water Maze
4.6. Golgi-Cox Staining
4.7. Proteome Analysis of the Prefrontal Cortex of Mice
4.7.1. Sample Preparation
4.7.2. Instruments and Analytical Conditions
4.7.3. Bioinformatics Analysis
4.8. Western Blotting
4.9. Ca2+ Assay
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2017, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and futuredirections. Alzheimer’s Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Cleary, J.P.; Walsh, D.M.; Hofmeister, J.J.; Shankar, G.M.; Kuskowski, M.A.; Selkoe, D.J.; Ashe, K.H. Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nat. Neurosci. 2005, 8, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Toward a comprehensive theory for Alzheimer’s disease. Hypothesis: Alzheimer’s disease is caused by the cerebral accumulation and cytotoxicity of amyloid beta-protein. Ann. N. Y. Acad. Sci. 2010, 924, 17–25. [Google Scholar] [CrossRef] [PubMed]
- John, A.; Reddy, P.H. Journal Pre-proof Synaptic Basis of Alzheimer’s Disease: Focus on Synaptic Amyloid Beta, P-Tau and Mitochondria. Ageing Res. Rev. 2020, 65, 101208. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, G.; Martorana, A.; Caltagirone, C. Transcranial magnetic stimulation: Emerging biomarkers and novel therapeutics in Alzheimer’s disease. Neurosci. Lett. 2019, 719, 134355. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; Zetterberg, H. Biomarkers for Alzheimer’s disease: Current status and prospects for the future. J. Intern. Med. 2018, 284, 643–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, S.J.; Bachstetter, A.D.; Nelson, P.T.; Schmitt, F.A.; Van Eldik, L.J. Using mice to model Alzheimer’s dementia: An overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front. Genet. 2014, 5, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkovic, M.N.; Strac, D.S.; Tudor, L.; Konjevod, M.; Erjavec, G.N.; Pivac, N. Catechol-O-methyltransferase, Cognition and Alzheimer’s Disease. Curr. Alzheimer Res. 2018, 15, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Rein, B. Mechanisms of synaptic transmission dysregulation in the prefrontal cortex: Pathophysiological implications. Mol. Psychiatry 2021, 27, 445–465. [Google Scholar] [CrossRef]
- Yang, S.S.; Zhang, R.; Wang, G.; Zhang, Y.F. The development prospection of HDAC inhibitors as a potential therapeutic direction in Alzheimer’s disease. Transl. Neurodegener. 2017, 6, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowak-Imialek, M.; Wrenzycki, C.; Herrmann, D.; Lagutina, I.; Niemann, H. 258 messenger rna expression patterns of histone modification genes in bovine embryos derived from different origins. Reprod. Fertil. Dev. 2006, 18, 236–237. [Google Scholar] [CrossRef]
- Gupta, R.; Ambasta, R.K.; Kumar, P. Identification of novel class I and class IIb histone deacetylase inhibitor for Alzheimer’s disease therapeutics. Life Sci. 2020, 256, 117912. [Google Scholar] [CrossRef]
- Fischer, A. Targeting histone-modifications in Alzheimer’s disease. What is the evidence that this s a promising therapeutic avenue? Neuropharmacology 2014, 80, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Kilgore, M.; Miller, C.A.; Fass, D.M.; Hennig, K.M.; Haggarty, S.J.; Sweatt, J.D.; Rumbaugh, G. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 2010, 35, 870–880. [Google Scholar] [CrossRef]
- Cuadrado-Tejedor, M.; Garcia-Barroso, C.; Sanzhez-Arias, J.; Mederos, S.; Rabal, O.; Ugarte, A.; Franco, R.; Pascual-Lucas, M.; Segura, V.; Perea, G. Concomitant histone deacetylase and phosphodiesterase 5 inhibition synergistically prevents the disruption in synaptic plasticity and it reverses cognitive impairment in a mouse model of Alzheimer’s disease. Clin. Epigenet. 2015, 7, 1–108. [Google Scholar] [CrossRef] [Green Version]
- Ishizuka, Y.; Shimizu, H.; Takagi, E.; Kato, M.; Yamagata, H.; Mikuni, M.; Shirao, T. Histone deacetylase mediates the decrease in drebrin cluster density induced by amyloid beta oligomers. Neurochem. Int. 2014, 76, 114–121. [Google Scholar] [CrossRef]
- Han, Y.; Chen, L.; Guo, Y.; Wang, C.; Ma, H. Class I HDAC Inhibitor Improves Synaptic Proteins and Repairs Cytoskeleton Through Regulating Synapse-Related Genes In vitro and In vivo. Front. Aging Neurosci. 2021, 12, 619866. [Google Scholar] [CrossRef]
- Gupta, R.; Ambasta, R.K.; Kumar, P. Pharmacological intervention of histone deacetylase enzymes in the neurodegenerative disorders. Life Sci. 2020, 243, 117278. [Google Scholar] [CrossRef]
- Courtney, N.A.; Bao, H.; Briguglio, J.S.; Chapman, E.R. Synaptotagmin 1 clamps synaptic vesicle fusion in mammalian neurons independent of complexin. Nat. Commun. 2019, 10, 4076. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.-l.; Qiao, P.-f.; Yan, N.; Gao, D.; Liu, M.-j.; Yan, Y. Hydrogen sulfide selectively inhibits γ-secretase activity and decreases mitochondrial Aβ production in neurons from APP/PS1 transgenic mice. Neurochem. Res. 2016, 41, 1145–1159. [Google Scholar] [CrossRef]
- Huang, H.J.; Huang, H.Y.; Hsieh-Li, H.M. MGCD 0103, a selective histone deacetylase inhibitor, coameliorates oligomeric Aβ25-35-induced anxiety and cognitive deficits in a mouse model. CNS Neurosci. Ther. 2019, 25, 175–186. [Google Scholar] [CrossRef] [Green Version]
- Ullah, N.; Maaiden, E.; Uddin, M.S.; Ashraf, G.M. Synaptotagmin-1: A Multi-Functional Protein that Mediates Vesicle Docking, Priming, and Fusion. Curr. Protein Pept. Sci. 2021, 22, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Chapman, E.R. A Ca2+ Sensor for Exocytosis. Trends Neuroences 2018, 41, 327–330. [Google Scholar] [CrossRef]
- Zhao, Y.; Arceneaux, L.; Culicchia, F.; Lukiw, W.J. Neurofilament Light (NF-L) Chain Protein from a Highly Polymerized Structural Component of the Neuronal Cytoskeleton to a Neurodegenerative Disease Biomarker in the Periphery. HSOA J. Alzheimer’s Neurodegener. Dis. 2021, 7, 056. [Google Scholar]
- Kasai, H.; Matsuzaki, M.; Noguchi, J.; Yasumatsu, N.; Nakahara, H. Structure–stability–function relationships of dendritic spines. Trends Neurosci. 2003, 26, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Holmes, S.E.; Scheinost, D.; Finnema, S.J.; Naganawa, M.; Davis, M.T.; Dellagioia, N.; Nabulsi, N.; Matuskey, D.; Angarita, G.A.; Pietrzak, R.H. Lower synaptic density is associated with depression severity and network alterations. Nat. Commun. 2019, 10, 1529. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.C.; Koleske, A.J. Mechanisms of synapse and dendrite maintenance and their disruption in psychiatric and neurodegenerative disorders. Annu. Rev. Neurosci. 2010, 33, 349–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacor, P.N.; Buniel, M.C.; Furlow, P.W.; Clemente, A.S.; Klein, W.L. Aβ Oligomer-Induced Aberrations in Synapse Composition, Shape, and Density Provide a Molecular Basis for Loss of Connectivity in Alzheimer’s Disease. J. Neurosci. 2007, 27, 796–807. [Google Scholar] [CrossRef] [Green Version]
- Walsh, D.M.; Selkoe, D.J. Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron 2004, 44, 181–193. [Google Scholar] [CrossRef]
- Aigner, L.; Arber, S.; Kapfhammer, J.P.; Laux, T.; Schneider, C.; Botteri, F.; Brenner, H.R.; Caroni, P. Overexpression of the neural growth-associated protein GAP-43 induces nerve sprouting in the adult nervous system of transgenic mice. Cell 1995, 83, 269. [Google Scholar] [CrossRef] [Green Version]
- Camporesi, E.; Nilsson, J.; Brinkmalm, A.; Becker, B.; Zetterberg, H. Fluid Biomarkers for Synaptic Dysfunction and Loss. Biomark. Insights 2020, 15, 117727192095031. [Google Scholar] [CrossRef]
- Cai, Z.; Li, S.; Matuskey, D.; Nabulsi, N.; Huang, Y. PET imaging of synaptic density: A new tool for investigation of neuropsychiatric diseases. Neurosci. Lett. 2019, 691, 44–50. [Google Scholar] [CrossRef]
- Xiang, Y.; Xin, J.; Le, W.; Yang, Y. Neurogranin: A Potential Biomarker of Neurological and Mental Diseases. Front. Aging Neurosci. 2020, 12, 584743. [Google Scholar] [CrossRef]
- Zhong, L.; Cherry, T.; Bies, C.E.; Florence, M.A.; Gerges, N.Z. Neurogranin enhances synaptic strength through its interaction with calmodulin. EMBO J. 2009, 28, 3027–3039. [Google Scholar] [CrossRef] [Green Version]
- Höglund, K.; Schussler, N.; Kvartsberg, H.; Smailovic, U.; Brinkmalm, G.; Liman, V.; Becker, B.; Zetterberg, H.; Cedazo-Minguez, A.; Janelidze, S. Cerebrospinal fluid neurogranin in an inducible mouse model of neurodegeneration: A translatable marker of synaptic degeneration. Neurobiol. Dis. 2020, 134, 104645. [Google Scholar] [CrossRef] [PubMed]
- Winston, C.N.; Goetzl, E.J.; Akers, J.C.; Carter, B.S.; Rockenstein, E.M.; Galasko, D.; Masliah, E.; Rissman, R.A. Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. Alzheimers Dement. Diagn. Assess. Dis. Monit. 2016, 3, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Atluri, V.S.R.; Tiwari, S.; Rodriguez, M.; Kaushik, A.; Yndart, A.; Kolishetti, N.; Yatham, M.; Nair, M. Inhibition of Amyloid-Beta production, associated neuroinflammation, and Histone Deacetylase 2-mediated epigenetic modifications prevent neuropathology in Alzheimer’s disease in vitro Model. Front. Aging Neurosci. 2020, 11, 342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heacock, A.M.; Seguin, E.B.; Agranoff, B.W. Developmental and Regional Studies of the Metabolism of Inositol 1,4,5-Trisphosphate in Rat Brain. J. Neurochem. 2010, 54, 1405–1411. [Google Scholar] [CrossRef] [Green Version]
- Mailleux, P.; Takazawa, K.; Erneux, C.; Vanderhaeghen, J.J. Inositol 1,4,5-trisphosphate 3-kinase mRNA: High levels in the rat hippocampal CA1 pyramidal and dentate gyrus granule cells and in cerebellar Purkinje cells. J. Neurochem. 2010, 56, 345–347. [Google Scholar] [CrossRef]
- Kim, I.H.; Park, S.K.; Sun, W.; Kang, Y.; Kim, H.T.; Kim, H. Spatial learning enhances the expression of inositol 1,4,5-trisphosphate 3-kinase A in the hippocampal formation of rat. Brain Res. Mol. Brain Res. 2004, 124, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Communi, D.; Vanweyenberg, V.; Erneux, C. D-myo-inositol 1,4,5-trisphosphate 3-kinase A is activated by receptor activation through a calcium:calmodulin-dependent protein kinase II phosphorylation mechanism. EMBO J. 1997, 16, 1943–1952. [Google Scholar] [CrossRef] [Green Version]
- Windhorst, S.; Song, K.; Gazdar, A.F. Inositol-1,4,5-trisphosphate 3-kinase-A (ITPKA) is frequently over-expressed and functions as an oncogene in several tumor types. Biochem. Pharmacol. 2017, 137, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Irvine, R.F.; Moor, R.M. Micro-injection of inositol 1,3,4,5-tetrakisphosphate activates sea urchin eggs by a mechanism dependent on external Ca2+. Biochem. J. 1986, 240, 917–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, A.P.; Gallacher, D.V.; Irvine, R.F.; Petersen, O.H. Synergism of inositol trisphosphate and tetrakisphosphate in activating Ca2+-dependent K+ channels. Nature 1987, 330, 653. [Google Scholar] [CrossRef] [PubMed]
- Neve, R.L.; Coopersmith, R.; Mcphie, D.L.; Santeufemio, C.; Lynn, S.D. The Neuronal Growth-Associated Protein GAP-43 Interacts with Rabaptin-5 and Participates in Endocytosis. J. Neurosci. Off. J. Soc. Neurosci. 1998, 18, 7757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.; Ryu, J.K. Models of synaptotagmin-1 to trigger Ca2+-dependent vesicle fusion. FEBS Lett. 2018, 592, 3480–3492. [Google Scholar] [CrossRef] [Green Version]
- Bradberry, M.M.; Chapman, E.R. All-optical monitoring of excitation–secretion coupling demonstrates that SV2A functions downstream of evoked Ca2+ entry. J. Physiol. 2022, 600, 645–654. [Google Scholar] [CrossRef]
- Díez-Guerra, F.J. Neurogranin, a link between calcium/calmodulin and protein kinase C signaling in synaptic plasticity. IUBMB Life 2010, 62, 597–606. [Google Scholar] [CrossRef]
- Ozaki, H.; Katoh, T.; Nakagawa, R.; Ishihara, Y.; Sueyoshi, N.; Kameshita, I.; Taniguchi, T.; Hirano, T.; Yamazaki, T.; Ishida, A. Ca(2+)/calmodulin-dependent protein kinase phosphatase (CaMKP/PPM1F) interacts with neurofilament L and inhibits its filament association. Biochem. Biophys. Res. Commun. 2016, 477, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Bettegazzi, B.; Sebastian Monasor, L.; Bellani, S.; Codazzi, F.; Restelli, L.M.; Colombo, A.V.; Deigendesch, N.; Frank, S.; Saito, T.; Saido, T.C.; et al. Casein Kinase 2 dependent phosphorylation of eIF4B regulates BACE1 expression in Alzheimer’s disease. Cell Death Dis. 2021, 12, 769. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Chen, L.; Liu, J.; Chen, J.; Wang, C.; Guo, Y.; Yu, X.; Zhang, C.; Chu, H.; Ma, H. A Class I HDAC Inhibitor Rescues Synaptic Damage and Neuron Loss in APP-Transfected Cells and APP/PS1 Mice through the GRIP1/AMPA Pathway. Molecules 2022, 27, 4160. [Google Scholar] [CrossRef] [PubMed]
- Du, F. Golgi-Cox Staining of Neuronal Dendrites and Dendritic Spines with FD Rapid GolgiStain™ Kit. Curr. Protoc. Neurosci. 2019, 88, e69. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Zhang, C.; Wang, J.; Huang, Y.; Shen, D.; Hu, Y.; Chu, H.; Yu, X.; Zhang, L.; Ma, H. A Class I HDAC Inhibitor BG45 Alleviates Cognitive Impairment through the CaMKII/ITPKA/Ca2+ Signaling Pathway. Pharmaceuticals 2022, 15, 1481. https://doi.org/10.3390/ph15121481
Liu J, Zhang C, Wang J, Huang Y, Shen D, Hu Y, Chu H, Yu X, Zhang L, Ma H. A Class I HDAC Inhibitor BG45 Alleviates Cognitive Impairment through the CaMKII/ITPKA/Ca2+ Signaling Pathway. Pharmaceuticals. 2022; 15(12):1481. https://doi.org/10.3390/ph15121481
Chicago/Turabian StyleLiu, Jingyun, Chenghong Zhang, Jiale Wang, Yufei Huang, Di Shen, Yingqiu Hu, Haiying Chu, Xuebin Yu, Liyuan Zhang, and Haiying Ma. 2022. "A Class I HDAC Inhibitor BG45 Alleviates Cognitive Impairment through the CaMKII/ITPKA/Ca2+ Signaling Pathway" Pharmaceuticals 15, no. 12: 1481. https://doi.org/10.3390/ph15121481