
Integration of Transcriptome and Exome Genotyping Identifies Significant Variants with Autism Spectrum Disorder

 , , , ,
, , , ,  and
and 
Abstract
1. Introduction
2. Results
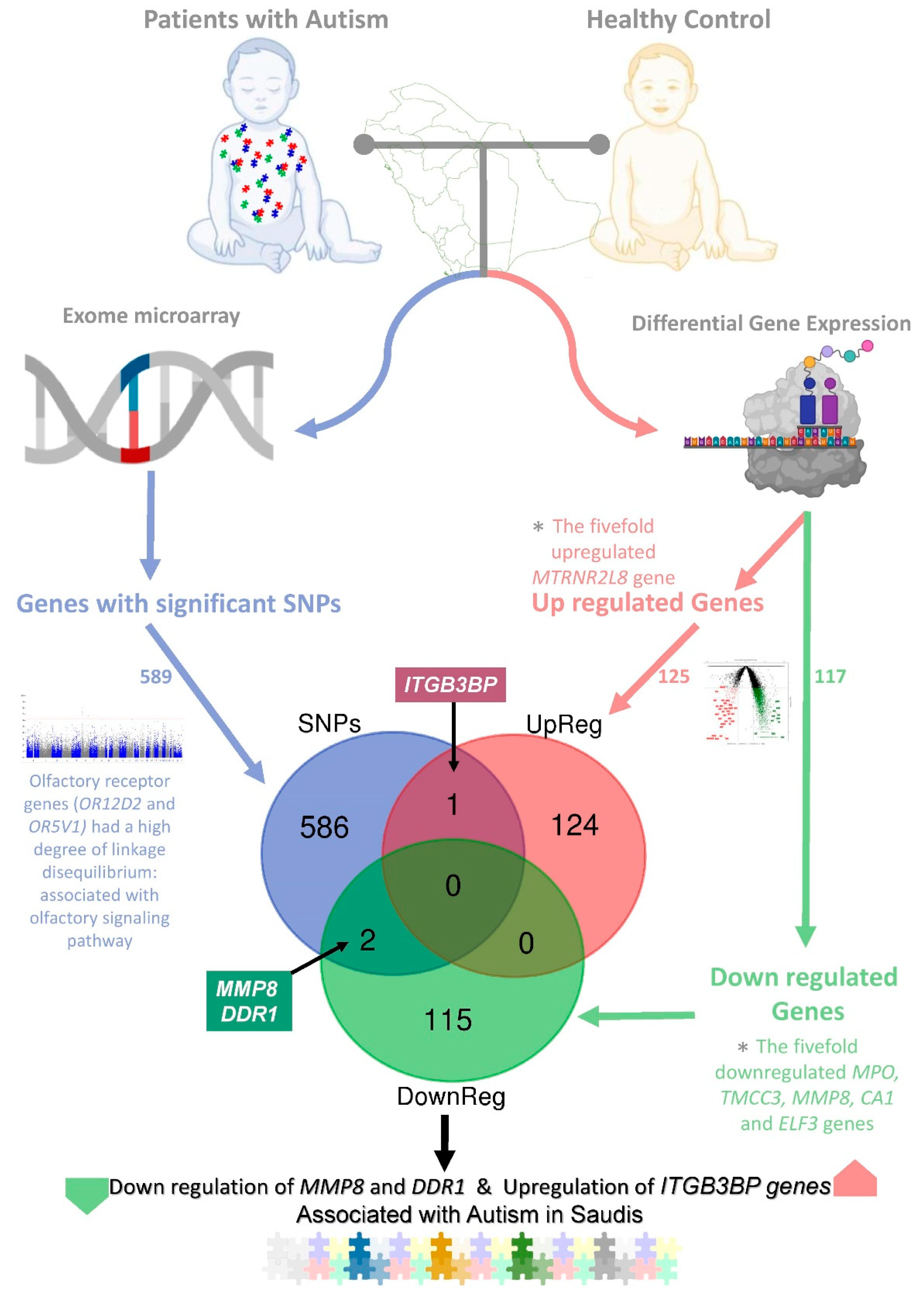
2.1. RNA Sequencing and Differentially Expressed Genes
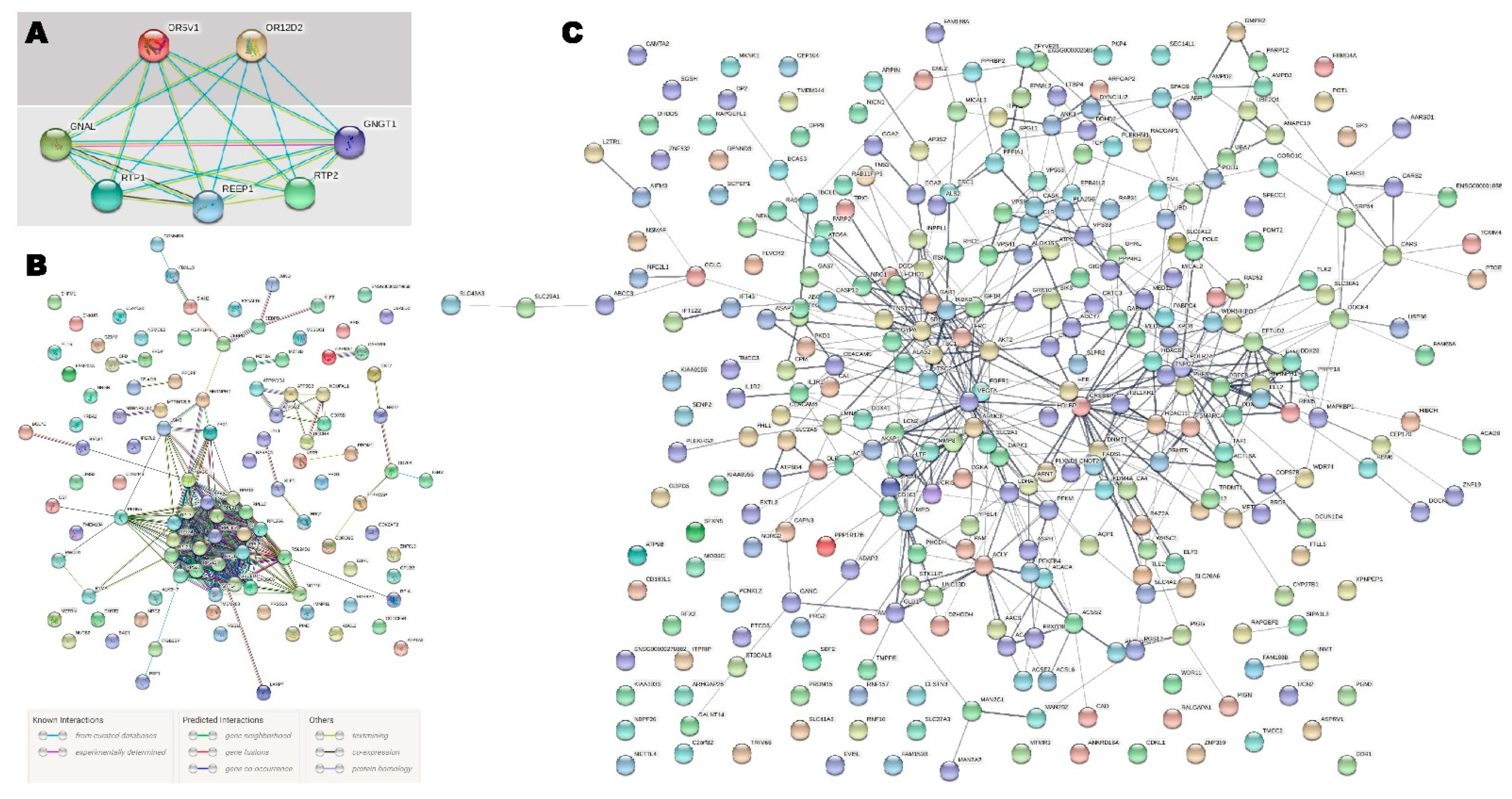
2.2. Pathway Enrichment Analysis
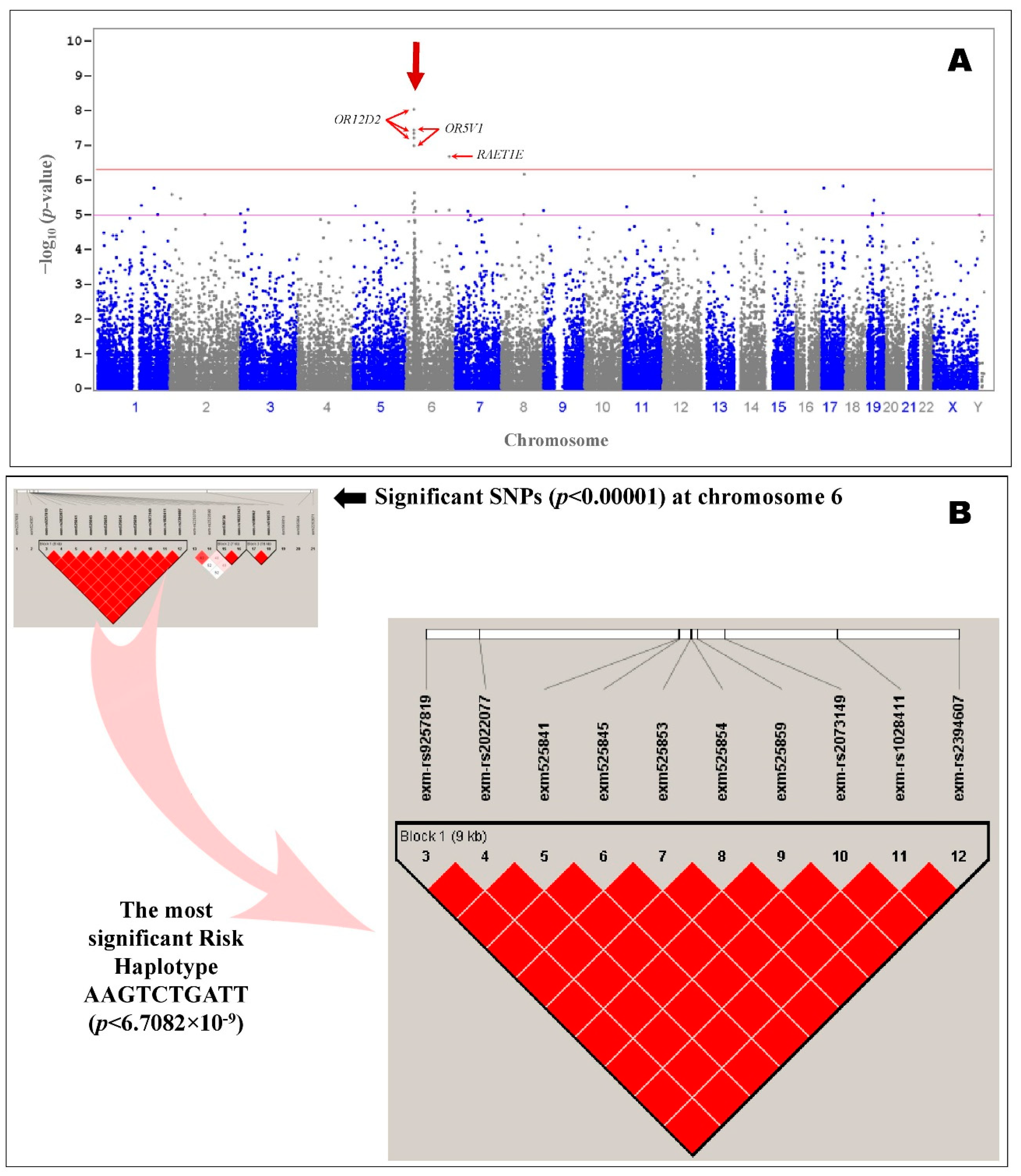
2.3. Integration of Transcriptome and Exome Genotyping
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. DNA Extraction and Exome Array
4.3. Genotyping and Functional Analysis
4.4. Whole RNA Sequencing and Differentially Expressed Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Almandil, N.; Alkuroud, D.; AbdulAzeez, S.; AlSulaiman, A.; Elaissari, A.; Borgio, J. Environmental and Genetic Factors in Autism Spectrum Disorders: Special Emphasis on Data from Arabian Studies. Int. J. Environ. Res. Public Health 2019, 16, 658. [Google Scholar] [CrossRef] [PubMed]
- Cheroni, C.; Caporale, N.; Testa, G. Autism Spectrum Disorder at the Crossroad between Genes and Environment: Contributions, Convergences, and Interactions in ASD Developmental Pathophysiology. Mol. Autism 2020, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yu, S.; Fu, Y.; Li, X. Synaptic Proteins and Receptors Defects in Autism Spectrum Disorders. Front. Cell. Neurosci. 2014, 8, 276. [Google Scholar] [CrossRef] [PubMed]
- Freitag, C.M. The Genetics of Autistic Disorders and Its Clinical Relevance: A Review of the Literature. Mol. Psychiatry 2007, 12, 2–22. [Google Scholar] [CrossRef]
- Freitag, C.M.; Staal, W.; Klauck, S.M.; Duketis, E.; Waltes, R. Genetics of Autistic Disorders: Review and Clinical Implications. Eur. Child Adolesc. Psychiatry 2010, 19, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Tick, B.; Bolton, P.; Happé, F.; Rutter, M.; Rijsdijk, F. Heritability of Autism Spectrum Disorders: A Meta-Analysis of Twin Studies. J. Child Psychol. Psychiatry 2016, 57, 585–595. [Google Scholar] [CrossRef]
- Baranek, G.T.; Boyd, B.A.; Poe, M.D.; David, F.J.; Watson, L.R. Hyperresponsive Sensory Patterns in Young Children with Autism, Developmental Delay, and Typical Development. Am. J. Ment. Retard 2007, 112, 233. [Google Scholar] [CrossRef]
- Boyd, B.A.; Baranek, G.T.; Sideris, J.; Poe, M.D.; Watson, L.R.; Patten, E.; Miller, H. Sensory Features and Repetitive Behaviors in Children with Autism and Developmental Delays. Autism Res. 2010, 3, 78–87. [Google Scholar] [CrossRef]
- Watson, L.R.; Patten, E.; Baranek, G.T.; Poe, M.; Boyd, B.A.; Freuler, A.; Lorenzi, J. Differential Associations between Sensory Response Patterns and Language, Social, and Communication Measures in Children with Autism or Other Developmental Disabilities. J. Speech Lang. Heart Res. 2011, 54, 1562–1576. [Google Scholar] [CrossRef]
- Ning, L.F.; Yu, Y.Q.; GuoJi, E.T.; Kou, C.G.; Wu, Y.H.; Shi, J.P.; Ai, L.Z.; Yu, Q. Meta-Analysis of Differentially Expressed Genes in Autism Based on Gene Expression Data. Genet. Mol. Res. 2015, 14, 2146–2155. [Google Scholar] [CrossRef]
- Rahman, M.R.; Petralia, M.C.; Ciurleo, R.; Bramanti, A.; Fagone, P.; Shahjaman, M.; Wu, L.; Sun, Y.; Turanli, B.; Arga, K.Y.; et al. Comprehensive Analysis of RNA-Seq Gene Expression Profiling of Brain Transcriptomes Reveals Novel Genes, Regulators, and Pathways in Autism Spectrum Disorder. Brain Sci. 2020, 10, 747. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Deng, L.; Jia, Q.; Huang, S.; Gu, J.; Zhou, F.; Gao, M.; Sun, X.; Feng, C.; Fan, G. DbMDEGA: A Database for Meta-Analysis of Differentially Expressed Genes in Autism Spectrum Disorder. BMC Bioinform. 2017, 18, 494. [Google Scholar] [CrossRef] [PubMed]
- Huyghe, J.R.; Jackson, A.U.; Fogarty, M.P.; Buchkovich, M.L.; Stančáková, A.; Stringham, H.M.; Sim, X.; Yang, L.; Fuchsberger, C.; Cederberg, H.; et al. Exome Array Analysis Identifies New Loci and Low-Frequency Variants Influencing Insulin Processing and Secretion. Nat. Genet. 2013, 45, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; He, J.; Zhao, S.; Wu, H.; Zhong, X.; Sheng, Q.; Samuels, D.C.; Shyr, Y.; Long, J. Illumina Human Exome Genotyping Array Clustering and Quality Control. Nat. Protoc. 2014, 9, 2643–2662. [Google Scholar] [CrossRef]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential Analysis of Gene Regulation at Transcript Resolution with RNA-Seq. Nat. Biotechnol. 2013, 31, 46–53. [Google Scholar] [CrossRef]
- Al-Mubarak, B.; Abouelhoda, M.; Omar, A.; AlDhalaan, H.; Aldosari, M.; Nester, M.; Alshamrani, H.A.; El-Kalioby, M.; Goljan, E.; Albar, R.; et al. Whole Exome Sequencing Reveals Inherited and de Novo Variants in Autism Spectrum Disorder: A Trio Study from Saudi Families. Sci. Rep. 2017, 7, 5679. [Google Scholar] [CrossRef]
- Papatheodorou, I.; Fonseca, N.A.; Keays, M.; Tang, Y.A.; Barrera, E.; Bazant, W.; Burke, M.; Füllgrabe, A.; Fuentes, A.M.-P.; George, N.; et al. Expression Atlas: Gene and Protein Expression across Multiple Studies and Organisms. Nucleic Acids Res. 2018, 46, D246–D251. [Google Scholar] [CrossRef]
- The GTEx Consortium; Ardlie, K.G.; Deluca, D.S.; Segrè, A.V.; Sullivan, T.J.; Young, T.R.; Gelfand, E.T.; Trowbridge, C.A.; Maller, J.B.; Tukiainen, T. The Genotype-Tissue Expression (GTEx) Pilot Analysis: Multitissue Gene Regulation in Humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef]
- Rozenkrantz, L.; Zachor, D.; Heller, I.; Plotkin, A.; Weissbrod, A.; Snitz, K.; Secundo, L.; Sobel, N. A Mechanistic Link between Olfaction and Autism Spectrum Disorder. Curr. Biol. 2015, 25, 1904–1910. [Google Scholar] [CrossRef]
- Schecklmann, M.; Schwenck, C.; Taurines, R.; Freitag, C.; Warnke, A.; Gerlach, M.; Romanos, M. A Systematic Review on Olfaction in Child and Adolescent Psychiatric Disorders. J. Neur. Transm. 2013, 120, 121–130. [Google Scholar] [CrossRef]
- Berko, E.R.; Suzuki, M.; Beren, F.; Lemetre, C.; Alaimo, C.M.; Calder, R.B.; Ballaban-Gil, K.; Gounder, B.; Kampf, K.; Kirschen, J.; et al. Mosaic Epigenetic Dysregulation of Ectodermal Cells in Autism Spectrum Disorder. PLoS Genet. 2014, 10, e1004402. [Google Scholar] [CrossRef] [PubMed]
- Tonacci, A.; Billeci, L.; Tartarisco, G.; Ruta, L.; Muratori, F.; Pioggia, G.; Gangemi, S. Olfaction in Autism Spectrum Disorders: A Systematic Review. Child Neuropsychol. 2017, 23, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Hatami, M.; Karamghadiri, N.; Mohaghegh, H.; Yoosefee, S.; Karimipoor, M.; Hadjighasem, M.; Ananloo, E.S. Neuregulin-1 Gene and Schizophrenia, and Its Negative Symptoms in an Iranian Population. Iran. J. Psychiatry Behav. Sci. 2016, 11. [Google Scholar] [CrossRef]
- Hawi, Z.; Cummins, T.D.R.; Tong, J.; Johnson, B.; Lau, R.; Samarrai, W.; Bellgrove, M.A. The Molecular Genetic Architecture of Attention Deficit Hyperactivity Disorder. Mol. Psychiatry 2015, 20, 289–297. [Google Scholar] [CrossRef]
- Jungerius, B.J.; Bakker, S.C.; Monsuur, A.J.; Sinke, R.J.; Kahn, R.S.; Wijmenga, C. Is MYO9B the Missing Link between Schizophrenia and Celiac Disease? Am. J. Med. Genet. 2008, 147B, 351–355. [Google Scholar] [CrossRef]
- Kerner, B.; Lambert, C.G.; Muthén, B.O. Genome-Wide Association Study in Bipolar Patients Stratified by Co-Morbidity. PLoS ONE 2011, 6, e28477. [Google Scholar] [CrossRef]
- Latiano, A.; Palmieri, O.; Valvano, M.R.; D’Incà, R.; Caprilli, R.; Cucchiara, S.; Sturniolo, G.C.; Bossa, F.; Andriulli, A.; Annese, V. The Association of MYO9B Gene in Italian Patients with Inflammatory Bowel Diseases: MYO9B gene in inflammatory bowel disease ibd. Aliment. Pharmacol. Ther. 2007, 27, 241–248. [Google Scholar] [CrossRef]
- Lü, M.; Xia, B.; Ge, L.; Li, Y.; Zhao, J.; Chen, F.; Zhou, F.; Zhang, X.; Tan, J. Role of Major Histocompatibility Complex Class I-Related Molecules A*A5·1 Allele in Ulcerative Colitis in Chinese Patients. Immunology 2009, 128, e230–e236. [Google Scholar] [CrossRef]
- Naj, A.C.; Beecham, G.W.; Martin, E.R.; Gallins, P.J.; Powell, E.H.; Konidari, I.; Whitehead, P.L.; Cai, G.; Haroutunian, V.; Scott, W.K.; et al. Dementia Revealed: Novel Chromosome 6 Locus for Late-Onset Alzheimer Disease Provides Genetic Evidence for Folate-Pathway Abnormalities. PLoS Genet. 2010, 6, e1001130. [Google Scholar] [CrossRef]
- Orchard, T.R.; Dhar, A.; Simmons, J.D.; Vaughan, R.; Welsh, K.I.; Jewell, D.P. MHC Class I Chain-like Gene A (MICA) and Its Associations with Inflammatory Bowel Disease and Peripheral Arthropathy: MICA in IBD. Clin. Exp. Immunol. 2001, 126, 437–440. [Google Scholar] [CrossRef]
- Pal, D.K.; Evgrafov, O.V.; Tabares, P.; Zhang, F.; Durner, M.; Greenberg, D.A. BRD2 (RING3) Is a Probable Major Susceptibility Gene for Common Juvenile Myoclonic Epilepsy. Am. J. Hum. Genet. 2003, 73, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Ren, R.-J.; Wang, L.-L.; Fang, R.; Liu, L.-H.; Wang, Y.; Tang, H.-D.; Deng, Y.-L.; Xu, W.; Wang, G.; Chen, S.-D. The MTHFD1L Gene Rs11754661 Marker Is Associated with Susceptibility to Alzheimer’s Disease in the Chinese Han Population. J. Neurol. Sci. 2011, 308, 32–34. [Google Scholar] [CrossRef] [PubMed]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’ayan, A. The Harmonizome: A Collection of Processed Datasets Gathered to Serve and Mine Knowledge about Genes and Proteins. Database 2016, 2016, baw100. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, E.; Alizadeh, B.Z.; Valdigem, G.; Ortego-Centeno, N.; Jiménez-Alonso, J.; de Ramón, E.; García, A.; López-Nevot, M.A.; Wijmenga, C.; Martín, J.; et al. MYO9B Gene Polymorphisms Are Associated with Autoimmune Diseases in Spanish Population. Hum. Immunol. 2007, 68, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Aragam, N.; Li, X.; Villla, E.C.; Wang, L.; Briones, D.; Petty, L.; Posada, Y.; Arana, T.B.; Cruz, G.; et al. BCL9 and C9orf5 Are Associated with Negative Symptoms in Schizophrenia: Meta-Analysis of Two Genome-Wide Association Studies. PLoS ONE 2013, 8, e51674. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A Comprehensive Gene Set Enrichment Analysis Web Server 2016 Update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Vinet, É.; Pineau, C.A.; Clarke, A.E.; Scott, S.; Fombonne, É.; Joseph, L.; Platt, R.W.; Bernatsky, S. Increased Risk of Autism Spectrum Disorders in Children Born to Women with Systemic Lupus Erythematosus: Results from a Large Population-Based Cohort: ASD RISK IN SLE OFFSPRING. Arthritis Rheumatol. 2015, 67, 3201–3208. [Google Scholar] [CrossRef]
- Gaillard, I.; Rouquier, S.; Giorgi, D. Olfactory Receptors. Cell Mol. Life Sci. 2004, 61, 456–469. [Google Scholar] [CrossRef]
- Mesallam, T.A.; Farahat, M.; Shoeib, R.; Alharethy, S.; Alshahwan, A.; Murry, T.; Malki, K.H. Acute Effects of Inhaling Oud Incense on Voice of Saudi Adults. Ann. Saudi Med. 2015, 35, 111–119. [Google Scholar] [CrossRef]
- Liu, L.; Lei, J.; Sanders, S.J.; Willsey, A.J.; Kou, Y.; Cicek, A.E.; Klei, L.; Lu, C.; He, X.; Li, M.; et al. DAWN: A Framework to Identify Autism Genes and Subnetworks Using Gene Expression and Genetics. Mol. Autism 2014, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Cotney, J.; Muhle, R.A.; Sanders, S.J.; Liu, L.; Willsey, A.J.; Niu, W.; Liu, W.; Klei, L.; Lei, J.; Yin, J.; et al. The Autism-Associated Chromatin Modifier CHD8 Regulates Other Autism Risk Genes during Human Neurodevelopment. Nat. Commun. 2015, 6, 6404. [Google Scholar] [CrossRef]
- Ahmad, E. Differential Gene Expression in the Prenatal Brain of Cyclooxygenase-1 and 2 Knockout Male Mice-A Model System of Autism Spectrum Disorders; York University: Toronto, ON, Canada, 2017. [Google Scholar]
- Choi, Y.K.; Vasudevan, A. Mechanistic Insights into Autocrine and Paracrine Roles of Endothelial GABA Signaling in the Embryonic Forebrain. Sci. Rep. 2019, 9, 16256. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, K.J.; Feltrin, A.S.; Barbosa, A.R.; Jaffe, A.E.; Collado-Torres, L.; Burke, E.E.; Shin, J.H.; Ulrich, W.S.; Deep-Soboslay, A.; Tao, R.; et al. Genetic and Environmental Regulation of Caudate Nucleus Transcriptome: Insight into Schizophrenia Risk and the Dopamine System. Psychiatry Clin. Psychol. 2020. [Google Scholar] [CrossRef]
- Hoerder-Suabedissen, A.; Oeschger, F.M.; Krishnan, M.L.; Belgard, T.G.; Wang, W.Z.; Lee, S.; Webber, C.; Petretto, E.; Edwards, A.D.; Molnár, Z. Expression Profiling of Mouse Subplate Reveals a Dynamic Gene Network and Disease Association with Autism and Schizophrenia. Proc. Natl. Acad. Sci. USA 2013, 110, 3555–3560. [Google Scholar] [CrossRef] [PubMed]
- Roig, B.; Virgos, C.; Franco, N.; Martorell, L.; Valero, J.; Costas, J.; Carracedo, A.; Labad, A.; Vilella, E. The Discoidin Domain Receptor 1 as a Novel Susceptibility Gene for Schizophrenia. Mol. Psychiatry 2007, 12, 833–841. [Google Scholar] [CrossRef][Green Version]
- Kim, S.E.; Kim, H.-N.; Yun, Y.-J.; Heo, S.G.; Cho, J.; Kwon, M.-J.; Chang, Y.; Ryu, S.; Shin, H.; Shin, C.; et al. Meta-Analysis of Genome-Wide SNP- and Pathway-Based Associations for Facets of Neuroticism. J. Hum. Genet. 2017, 62, 903–909. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Karam, C.S.; Jones, S.K.; Javitch, J.A. Come Fly with Me: An Overview of Dopamine Receptors in Drosophila melanogaster. Basic Clin. Pharmacol. Toxicol. 2020, 126, 56–65. [Google Scholar] [CrossRef]
- Pham, H.T.N.; Tran, H.N.; Le, X.T.; Do, H.T.; Nguyen, T.T.; Le Nguyen, C.; Yoshida, H.; Yamaguchi, M.; William, F.R.; Matsumoto, K.; et al. Tseng Mitigates Phenotypic Characteristics of Human Autism Spectrum Disorders in a Drosophila Melanogaster Rugose Mutant. Neurochem. Res. 2021, 46, 1995–2007. [Google Scholar] [CrossRef]
- Vilella, E.; Gas, C.; Garcia-Ruiz, B.; Rivera, F.J. Expression of DDR1 in the CNS and in Myelinating Oligodendrocytes. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2019, 1866, 118483. [Google Scholar] [CrossRef]
- Franco-Pons, N.; Virgos, C.; Vogel, W.F.; Ureña, J.M.; Soriano, E.; del Rio, J.A.; Vilella, E. Expression of Discoidin Domain Receptor 1 during Mouse Brain Development Follows the Progress of Myelination. Neuroscience 2006, 140, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Song, G.G.; Kim, J.-H.; Lee, Y.H. Genome-Wide Pathway Analysis in Major Depressive Disorder. J. Mol. Neurosci. 2013, 51, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Näkki, A.; Rodriguez-Fontenla, C.; Gonzalez, A.; Harilainen, A.; Leino-Arjas, P.; Heliövaara, M.; Eriksson, J.G.; Tallroth, K.; Videman, T.; Kaprio, J.; et al. Association Study of MMP8 Gene in Osteoarthritis. Connect. Tissue Res. 2016, 57, 44–52. [Google Scholar] [CrossRef]
- Du, J.; Jin, T.; Cao, Y.; Chen, J.; Guo, Y.; Sun, M.; Li, J.; Zhang, X.; Wang, G.; Wang, J. Association between Genetic Polymorphisms of MMP8 and the Risk of Steroid-Induced Osteonecrosis of the Femoral Head in the Population of Northern China. Medicine 2016, 95, e4794. [Google Scholar] [CrossRef] [PubMed]
- Kader, A.K.; Liu, J.; Shao, L.; Dinney, C.P.; Lin, J.; Wang, Y.; Gu, J.; Grossman, H.B.; Wu, X. Matrix Metalloproteinase Polymorphisms Are Associated with Bladder Cancer Invasiveness. Clin. Cancer Res. 2007, 13, 2614–2620. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Liu, J.; Jin, L.; Jiang, Y. Polymorphisms in Matrix Metalloproteinases 2, 3, and 8 Increase Recurrence and Mortality Risk by Regulating Enzyme Activity in Gastric Adenocarcinoma. Oncotarget 2017, 8, 105971–105983. [Google Scholar] [CrossRef]
- Jin, C.; Lee, Y.; Kang, H.; Jeong, K.; Park, J.; Zhang, Y.; Kang, H.R.; Ma, R.; Seong, H.; Kim, Y.; et al. Increased Ribosomal Protein Levels and Protein Synthesis in the Striatal Synaptosome of Shank3-Overexpressing Transgenic Mice. Mol. Brain 2021, 14, 39. [Google Scholar] [CrossRef]
- Zoghbi, H.Y.; Bear, M.F. Synaptic Dysfunction in Neurodevelopmental Disorders Associated with Autism and Intellectual Disabilities. Cold Spring Harb. Perspect. Biol. 2012, 4, a009886. [Google Scholar] [CrossRef]
- Wang, L.; Pang, K.; Han, K.; Adamski, C.J.; Wang, W.; He, L.; Lai, J.K.; Bondar, V.V.; Duman, J.G.; Richman, R.; et al. An Autism-Linked Missense Mutation in SHANK3 Reveals the Modularity of Shank3 Function. Mol. Psychiatry 2020, 25, 2534–2555. [Google Scholar] [CrossRef]
- Jin, C.; Kim, S.; Kang, H.; Yun, K.N.; Lee, Y.; Zhang, Y.; Kim, Y.; Kim, J.Y.; Han, K. Shank3 Regulates Striatal Synaptic Abundance of Cyld, a Deubiquitinase Specific for Lys63-linked Polyubiquitin Chains. J. Neurochem. 2019, 150, 776–786. [Google Scholar] [CrossRef]
- Glusman, G.; Caballero, J.; Mauldin, D.E.; Hood, L.; Roach, J.C. Kaviar: An Accessible System for Testing SNV Novelty. Bioinformatics 2011, 27, 3216–3217. [Google Scholar] [CrossRef]
- Dayem Ullah, A.Z.; Lemoine, N.R.; Chelala, C. SNPnexus: A Web Server for Functional Annotation of Novel and Publicly Known Genetic Variants (2012 Update). Nucleic Acids Res. 2012, 40, W65–W70. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and Visualization of LD and Haplotype Maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING Database in 2017: Quality-Controlled Protein–Protein Association Networks, Made Broadly Accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Pathan, M.; Keerthikumar, S.; Chisanga, D.; Alessandro, R.; Ang, C.; Askenase, P.; Batagov, A.O.; Benito-Martin, A.; Camussi, G.; Clayton, A.; et al. A Novel Community Driven Software for Functional Enrichment Analysis of Extracellular Vesicles Data. J. Extracell. Vesicles 2017, 6, 1321455. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for Gene List Enrichment Analysis and Candidate Gene Prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.No | SNP ID | CHR | BP | MA | MAF | CHISQ | P | OR (L95–U95) | Gene | AA | Case, Control Frequencies | HWpval |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | rs2073149 | 6 | 29365423 | A | 0.5795 | 33.14 | 8.57 × 10−9 | 0.2367 (0.143–0.3918) | OR12D2 | A | 0.754, 0.424 | 0.0563 |
| 2 | rs2073153 | 6 | 29364835 | G | 0.5465 | 30.5 | 3.34 × 10−8 | 0.2489 (0.15–0.4131) | OR12D2 | T | 0.769, 0.456 | 0.0388 |
| 3 | rs2073151 | 6 | 29364951 | A | 0.5398 | 30.1 | 4.10 × 10−8 | 0.2498 (0.1501–0.4155) | OR12D2 | G | 0.773, 0.457 | 0.0504 |
| 4 | rs2394607 | 6 | 29369519 | C | 0.5682 | 30.07 | 4.18 × 10−8 | 0.2586 (0.1576–0.4241) | OR5V1 | T | 0.746, 0.438 | 0.1677 |
| 5 | rs9257819 | 6 | 29360183 | C | 0.5398 | 29.55 | 5.46 × 10−8 | 0.2558 (0.1545–0.4235) | OR5V1 | A | 0.769, 0.462 | 0.0394 |
| 6 | rs9257834 | 6 | 29364615 | T | 0.5398 | 29.55 | 5.46 × 10−8 | 0.2558 (0.1545–0.4235) | OR12D2 | G | 0.769, 0.462 | 0.0301 |
| 7 | rs4987411 | 6 | 29364643 | C | 0.5398 | 29.55 | 5.46 × 10−8 | 0.2558 (0.1545–0.4235) | OR12D2 | T | 0.769, 0.462 | 0.0301 |
| 8 | rs2073154 | 6 | 29364815 | G | 0.5398 | 29.55 | 5.46 × 10−8 | 0.2558 (0.1545–0.4235) | OR12D2 | C | 0.769, 0.462 | 0.0394 |
| 9 | rs1028411 | 6 | 29367399 | C | 0.5398 | 29.55 | 5.46 × 10−8 | 0.2558 (0.1545–0.4235) | OR5V1 | T | 0.769, 0.462 | 0.0394 |
| 10 | rs2022077 | 6 | 29361124 | A | 0.5398 | 28.55 | 9.14 × 10−8 | 0.261 (0.1575–0.4325) | OR5V1 | A | 0.766, 0.466 | 0.0475 |
| 11 | rs9383583 | 6 | 150212003 | T | 0.2898 | 27.09 | 1.94 × 10−7 | 0.1395 (0.06093–0.3193) | RAET1E | C | 0.946, 0.719 | 0.2627 |
| 12 | rs28703878 | 8 | 79417222 | G | 0.233 | 24.82 | 6.30 × 10−7 | 3.396 (2.08–5.544) | LOC105375911 | G | 0.508, 0.255 | 0.1448 |
| 13 | rs7963027 | 12 | 108894909 | C | 0.6118 | 24.58 | 7.15 × 10−7 | 0.3029 (0.1874–0.4894) | T | 0.677, 0.436 | 0.9318 | |
| 14 | rs571264 | 17 | 74878259 | A | 0.3693 | 23.29 | 1.39 × 10−6 | 0.2397 (0.1307–0.4394) | MGAT5B | G | 0.877, 0.667 | 0.0024 |
| 15 | rs62637606 | 17 | 8172506 | G | 0.0511 | 23.09 | 1.55 × 10−6 | 5.81 (2.656–12.71) | PFAS | G | 0.238, 0.062 | 1 |
| 16 | rs2523590 | 6 | 31327064 | G | 0.4059 | 22.43 | 2.18 × 10−6 | 0.2661 (0.1511–0.4689) | T | 0.846, 0.619 | 0.037 | |
| 17 | rs6741819 | 2 | 7147973 | T | 0.4602 | 22.27 | 2.37 × 10−6 | 0.2932 (0.174–0.4942) | RNF144A | C | 0.800, 0.576 | 0.7589 |
| 18 | rs888096 | 2 | 37603801 | G | 0.3466 | 21.74 | 3.12 × 10−6 | 3.016 (1.885–4.828) | LOC107985868 | G | 0.615, 0.371 | 1 |
| 19 | rs2253705 | 6 | 30900094 | A | 0.125 | 21.38 | 3.76 × 10−6 | 3.706 (2.086–6.583) | SFTA2 | T | 0.346, 0.129 | 1 |
| 20 | rs6911487 | 6 | 23774487 | A | 0.358 | 21.1 | 4.37 × 10−6 | 2.965 (1.853–4.743) | A | 0.623, 0.386 | 1 | |
| 21 | rs4902780 | 14 | 70591661 | C | 0.5795 | 20.96 | 4.69 × 10−6 | 0.3342 (0.2077–0.5379) | SLC8A3 | T | 0.685, 0.414 | 0.0077 |
| 22 | rs267733 | 1 | 150958836 | G | 0.0804 | 20.9 | 4.84 × 10−6 | 4.377 (2.245–8.535) | ANXA9 | G | 0.277, 0.120 | 0.8843 |
| 23 | rs7729273 | 5 | 7228047 | T | 0.0454 | 20.8 | 5.10 × 10−6 | 5.765 (2.53–13.13) | T | 0.215, 0.057 | 1 | |
| 24 | rs2132517 | 11 | 10791983 | A | 0.25 | 20.71 | 5.33 × 10−6 | 0.1707 (0.07411–0.3933) | CTR9 | G | 0.946, 0.776 | 0.4454 |
| 25 | rs580962 | 6 | 32925692 | G | 0.5852 | 20.61 | 5.63 × 10−6 | 0.3383 (0.2105–0.5436) | HLA-DMA | T | 0.677, 0.457 | 1 |
| 26 | rs516535 | 6 | 32942302 | C | 0.5852 | 20.61 | 5.63 × 10−6 | 0.3383 (0.2105–0.5436) | BRD2 | A | 0.677, 0.462 | 1 |
| 27 | rs9266825 | 6 | 31382882 | A | 0.4659 | 20.38 | 6.35 × 10−6 | 0.3147 (0.1885–0.5253) | MICA | C | 0.785, 0.571 | 0.0328 |
| 28 | rs10223421 | 6 | 31390055 | T | 0.4659 | 20.38 | 6.35 × 10−6 | 0.3147 (0.1885–0.5253) | HCP5 | G | 0.785, 0.571 | 0.0328 |
| 29 | rs9854207 | 3 | 27614316 | C | 0.25 | 20.38 | 6.36 × 10−6 | 3 (1.848–4.869) | C | 0.500, 0.262 | 0.09 | |
| 30 | rs505358 | 6 | 151327848 | A | 0.5909 | 20.27 | 6.72 × 10−6 | 0.3422 (0.2132–0.5492) | MTHFD1L | G | 0.669, 0.452 | 0.0046 |
| 31 | rs10970979 | 9 | 334337 | G | 0.2045 | 20.23 | 6.86 × 10−6 | 3.122 (1.883–5.176) | DOCK8 | G | 0.445, 0.214 | 0.3033 |
| 32 | rs1802437 | 7 | 44874113 | T | 0.5698 | 20.12 | 7.29 × 10−6 | 0.3336 (0.2051–0.5428) | H2AFV | C | 0.694, 0.436 | 0.6251 |
| 33 | rs941920 | 14 | 91739081 | A | 0.233 | 20.07 | 7.47 × 10−6 | 0.1593 (0.06538–0.3883) | CCDC88C | G | 0.954, 0.793 | 0.2034 |
| 34 | rs7168069 | 15 | 68624396 | A | 0.233 | 20.07 | 7.47 × 10−6 | 0.1593 (0.06538–0.3883) | ITGA11 | C | 0.954, 0.795 | 0.4658 |
| 35 | rs62620225 | 6 | 28117331 | T | 0.1591 | 19.98 | 7.84 × 10−6 | 0.04097 (0.005498–0.3054) | ZKSCAN8 | C | 0.992, 0.833 | 0.0025 |
| 36 | rs1545620 | 19 | 17303774 | T | 0.358 | 19.91 | 8.14 × 10−6 | 2.87 (1.796–4.586) | MYO9B | T | 0.615, 0.362 | 0.0766 |
| 37 | rs7507442 | 19 | 53278953 | A | 0.6193 | 19.88 | 8.24 × 10−6 | 0.3481 (0.2176–0.5567) | ZNF600 | G | 0.638, 0.429 | 0.083 |
| 38 | rs2727943 | 3 | 1897973 | T | 0.2184 | 19.84 | 8.44 × 10−6 | 0.1432 (0.05462–0.3752) | C | 0.962, 0.782 | 0.4488 | |
| 39 | rs1507765 | 1 | 207535246 | A | 0.5795 | 19.74 | 8.87 × 10−6 | 0.3463 (0.2155–0.5562) | CD55 | C | 0.677, 0.462 | 0.2045 |
| 40 | rs906998 | 8 | 78530715 | T | 0.5795 | 19.74 | 8.87 × 10−6 | 0.3463 (0.2155–0.5562) | C | 0.677, 0.443 | 0.4206 | |
| 41 | rs6542573 | 2 | 120986872 | T | 0.3864 | 19.73 | 8.91 × 10−6 | 0.2888 (0.1641–0.508) | C | 0.846, 0.629 | 0.0841 | |
| 42 | rs2633350 | 19 | 16808183 | C | 0.4148 | 19.65 | 9.32 × 10−6 | 0.3033 (0.1765–0.521) | T | 0.823, 0.614 | 0.9978 | |
| 43 | rs1012036 | 7 | 52472450 | T | 0.3663 | 19.58 | 9.66 × 10−6 | 0.2781 (0.1547–0.4998) | CDC14C | C | 0.862, 0.660 | 0.0124 |
| Block | Haplotype | Frequency | Case, Control Ratio Counts | Case, Control Frequencies | Chi-Square | p-value |
|---|---|---|---|---|---|---|
| Block 1 | AAGTCTGATT | 0.547 | 97.0:33.0, 89.0:121.0 | 0.746, 0.424 | 33.618 | 6.71 × 10−9 ** |
| CTTCGGATGC | 0.421 | 30.0:100.0, 113.0:97.0 | 0.231, 0.538 | 31.12 | 2.43 × 10−8 * | |
| AAGTCTGTTC | 0.024 | 3.0:127.0, 5.0:205.0 | 0.023, 0.024 | 0.002 | 0.9655 | |
| Block 2 | CG | 0.653 | 102.0:28.0, 120.0:90.0 | 0.785, 0.571 | 16.104 | 6.00 × 10−5 ** |
| AT | 0.347 | 28.0:102.0, 90.0:120.0 | 0.215, 0.429 | 16.104 | 6.00 × 10−5 * | |
| Block 3 | TA | 0.541 | 88.0:42.0, 96.0:114.0 | 0.677, 0.457 | 15.62 | 7.74 × 10−5 ** |
| CG | 0.459 | 42.0:88.0, 114.0:96.0 | 0.323, 0.543 | 15.62 | 7.74 × 10−5 * |
| Gene | Locus | FPKM Control | FPKM Case | log2 (Fold Change) | p-value | q-value | Ensemble Gene ID | Gene Description |
|---|---|---|---|---|---|---|---|---|
| RPS12 | 6:132814568–132817564 | 325.7 | 2218.21 | 2.76778 | 0.00005 | 0.004411 | ENSG00000112306 | ribosomal protein S12 |
| HNRNPH2 | X:101390823–101414133 | 79.1408 | 1227.64 | 3.95532 | 0.00005 | 0.004411 | ENSG00000126945 | heterogeneous nuclear ribonucleoprotein H2 |
| KRT8 | 12:52897186–52952906 | 0.834316 | 9.49503 | 3.50851 | 0.00005 | 0.004411 | ENSG00000170421 | keratin 8 |
| RPL36A | X:101390823–101414133 | 79.1408 | 1227.64 | 3.95532 | 0.00005 | 0.004411 | ENSG00000241343 | ribosomal protein L36a |
| MTRNR2L8 | 11:10507893–10509186 | 4.3216 | 253.717 | 5.87551 | 0.00005 | 0.004411 | ENSG00000255823 | MT-RNR2 like 8 |
| RPL36A-HNRNPH2 | X:101390823–101414133 | 79.1408 | 1227.64 | 3.95532 | 0.00005 | 0.004411 | ENSG00000257529 | RPL36A-HNRNPH2 readthrough |
| RPL26 | 17:8356901–8383213 | 88.4494 | 682.892 | 2.94873 | 0.00025 | 0.01343 | ENSG00000161970 | ribosomal protein L26 |
| KRBA2 | 17:8356901–8383213 | 88.4494 | 682.892 | 2.94873 | 0.00025 | 0.01343 | ENSG00000184619 | KRAB-A domain containing 2 |
| AC135178.3 | 17:8356901–8383213 | 88.4494 | 682.892 | 2.94873 | 0.00025 | 0.01343 | ENSG00000263809 | novel protein |
| DBI | 2:119366920–119372560 | 19.2423 | 82.5386 | 2.10079 | 0.0004 | 0.017644 | ENSG00000155368 | diazepam binding inhibitor, acyl-CoA binding protein |
| PROM1 | 4:15960244–16084378 | 29.4262 | 128.084 | 2.12192 | 0.0005 | 0.019545 | ENSG00000007062 | prominin 1 |
| FGFBP2 | 4:15960244–16084378 | 29.4262 | 128.084 | 2.12192 | 0.0005 | 0.019545 | ENSG00000137441 | fibroblast growth factor binding protein 2 |
| PPDPF | 20:63520764–63522206 | 43.8331 | 377.291 | 3.10559 | 0.0008 | 0.025989 | ENSG00000125534 | pancreatic progenitor cell differentiation and proliferation factor |
| IGHG3 | 14:105764502–105771405 | 26.0445 | 179.176 | 2.78233 | 0.00085 | 0.027065 | ENSG00000211897 | immunoglobulin heavy constant gamma 3 (G3m marker) |
| CFD | 19:859663–863641 | 57.7465 | 285.781 | 2.30711 | 0.00115 | 0.032814 | ENSG00000197766 | complement factor D |
| IMMP1L | 11:31369839–31509645 | 3.40717 | 39.2517 | 3.52611 | 0.0015 | 0.038464 | ENSG00000148950 | inner mitochondrial membrane peptidase subunit 1 |
| BAG1 | 9:33218364–33264720 | 91.759 | 363.23 | 1.98496 | 0.00155 | 0.039018 | ENSG00000107262 | BAG cochaperone 1 |
| COX5B | 2:97646061–97648383 | 31.6214 | 128.267 | 2.02018 | 0.00165 | 0.040067 | ENSG00000135940 | cytochrome c oxidase subunit 5B |
| PDZK1IP1 | 1:47183581–47191044 | 41.5874 | 174.343 | 2.06771 | 0.0023 | 0.046077 | ENSG00000162366 | PDZK1 interacting protein 1 |
| HBQ1 | 16:180458–181179 | 25.0227 | 198.741 | 2.98958 | 0.00245 | 0.047885 | ENSG00000086506 | hemoglobin subunit theta 1 |
| UQCRB | 8:96222946–96239149 | 13.8377 | 99.6279 | 2.84795 | 0.0025 | 0.048566 | ENSG00000156467 | ubiquinol-cytochrome c reductase binding protein |
| Gene | Locus | FPKM Control | FPKM Case | log2 (Fold Change) | p-value | q-value | Ensemble Gene ID | Gene Description |
|---|---|---|---|---|---|---|---|---|
| MPO | 17:58269854–58280935 | 74.2902 | 1.24471 | −5.89929 | 0.00005 | 0.0044 | ENSG00000005381 | myeloperoxidase |
| TMCC3 | 12:94567121–94650557 | 377.371 | 6.56036 | −5.84606 | 0.00005 | 0.0044 | ENSG00000057704 | transmembrane and coiled-coil domain family 3 |
| CEACAM6 | 19:41708584–41786893 | 10.139 | 0.665306 | −3.92976 | 0.00005 | 0.0044 | ENSG00000086548 | CEA cell adhesion molecule 6 |
| PLEKHG2 | 19:39412668–39428415 | 65.41 | 4.86538 | −3.74889 | 0.00005 | 0.0044 | ENSG00000090924 | pleckstrin homology and RhoGEF domain containing G2 |
| CEACAM5 | 19:41708584–41786893 | 10.139 | 0.665306 | −3.92976 | 0.00005 | 0.0044 | ENSG00000105388 | CEA cell adhesion molecule 5 |
| GRB10 | 7:50590062–50793462 | 10.5363 | 0.725095 | −3.86106 | 0.00005 | 0.0044 | ENSG00000106070 | growth factor receptor bound protein 10 |
| DDX5 | 17:64498253–64662307 | 4447.04 | 280.637 | −3.98607 | 0.00005 | 0.0044 | ENSG00000108654 | DEAD-box helicase 5 |
| CARS1 | 11:3000921–3064707 | 210.99 | 15.4503 | −3.77147 | 0.00005 | 0.0044 | ENSG00000110619 | cysteinyl-tRNA synthetase 1 |
| MMP8 | 11:102711795–102727050 | 25.7828 | 0.665084 | −5.27673 | 0.00005 | 0.0044 | ENSG00000118113 | matrix metallopeptidase 8 |
| AKAP1 | 17:57085091–57121346 | 53.8928 | 4.44175 | −3.60089 | 0.00005 | 0.0044 | ENSG00000121057 | A-kinase anchoring protein 1 |
| TMCC2 | 1:205227945–205285632 | 96.9766 | 5.46508 | −4.14932 | 0.00005 | 0.0044 | ENSG00000133069 | transmembrane and coiled-coil domain family 2 |
| CA1 | 8:85327607–85481493 | 521.489 | 11.6803 | −5.48049 | 0.00005 | 0.0044 | ENSG00000133742 | carbonic anhydrase 1 |
| ACE | 17:63477060–63521848 | 7.07334 | 0.510574 | −3.7922 | 0.00005 | 0.0044 | ENSG00000159640 | angiotensin I converting enzyme |
| ELF3 | 1:201982371–202017183 | 9.77011 | 0.261935 | −5.22109 | 0.00005 | 0.0044 | ENSG00000163435 | E74 like ETS transcription factor 3 |
| YPEL4 | 11:57638023–57661865 | 15.1177 | 0.851345 | −4.15035 | 0.00005 | 0.0044 | ENSG00000166793 | yippee like 4 |
| OLR1 | 12:10158300–10191801 | 6.43025 | 0.410073 | −3.97092 | 0.00005 | 0.0044 | ENSG00000173391 | oxidized low-density lipoprotein receptor 1 |
| POLE | 12:132623752–132687376 | 46.5435 | 4.11377 | −3.50005 | 0.00005 | 0.0044 | ENSG00000177084 | DNA polymerase epsilon, catalytic subunit |
| DDX41 | 5:177511576–177516961 | 597.042 | 27.017 | −4.46589 | 0.00005 | 0.0044 | ENSG00000183258 | DEAD-box helicase 41 |
| AC113554.1 | 17:63477060–63521848 | 7.07334 | 0.510574 | −3.7922 | 0.00005 | 0.0044 | ENSG00000264813 | novel protein |
| AC243967.1 | 19:41708584–41786893 | 10.139 | 0.665306 | −3.92976 | 0.00005 | 0.0044 | ENSG00000267881 | novel protein, readthrough between CEACAM5-CEACAM6 |
| KEGG Pathway ID | Term Description | OGC | BGC | Strength | False Discovery Rate | Matching Proteins in Your Network (Labels) |
|---|---|---|---|---|---|---|
| Upregulated genes | ||||||
| hsa03010 | Ribosome | 17 | 130 | 1.37 | 3.53E-16 | RPS12, RPL35, RSL24D1, RPL13, RPL7, RPL21, RPS27, RPL11, RPL24, RPL34, RPL36A, RPS24, RPL29, RPL35A, RPL23, RPL26, RPS15 |
| hsa00190 | Oxidative phosphorylation | 6 | 131 | 0.91 | 0.0041 | COX5B, ATP5G3, ATP6V1G1, ATP5G2, NDUFA11, UQCRB |
| hsa03060 | Protein export | 3 | 23 | 1.37 | 0.0093 | SRP14, IMMP1L, SEC61G |
| hsa05012 | Parkinson’s disease | 5 | 142 | 0.8 | 0.0242 | COX5B, ATP5G3, ATP5G2, NDUFA11, UQCRB |
| hsa05010 | Alzheimer’s disease | 5 | 168 | 0.73 | 0.0394 | COX5B, ATP5G3, ATP5G2, NDUFA11, UQCRB |
| Downregulated genes | ||||||
| hsa04066 | HIF-1 signaling pathway | 9 | 98 | 0.76 | 0.0115 | CREBBP, IGF1R, ARNT, TFRC, MKNK1, AKT2, SLC2A1, LDHA, VEGFA |
| hsa00620 | Pyruvate metabolism | 5 | 39 | 0.91 | 0.0243 | ACSS2, ACACB, ALDH3A2, LDHA, ACACA |
| hsa00640 | Propanoate metabolism | 5 | 32 | 0.99 | 0.0243 | ACSS2, ACACB, HIBCH, LDHA, ACACA |
| hsa01100 | Metabolic pathways | 37 | 1250 | 0.27 | 0.0243 | EXTL3, CYP27B1,GCLC, ACSS2, ACLY, AMPD2, POMT2,ATP6V0A1, CAD, POLG, AMT, ACAD8, GLB1, SGSH, POLE, GANC, DGKA, ALAS2, PLA2G6, ACACB, PFKM, ALDH3A2, PIGN, DNMT1, HIBCH, PHGDH, ACSL6, BPGM, AMPD3, MTMR3, EARS2, PGM3, LDHA, MAN2A2, ALOX15, POLR2A, ACACA |
| hsa04015 | Rap1 signaling pathway | 11 | 203 | 0.53 | 0.0243 | SIPA1L3, FARP2, RAPGEF2, IGF1R, SRC, AKT2, ADCY7, RASGRP3, FGFR1, DOCK4, VEGFA |
| hsa04152 | AMPK signaling pathway | 9 | 120 | 0.67 | 0.0243 | TSC2, PFKFB4, IGF1R, TSC1, ACACB, PFKM, CRTC2, AKT2, ACACA |
| hsa04922 | Glucagon signaling pathway | 8 | 100 | 0.7 | 0.0243 | CREBBP, ACACB, CRTC2, ITPR2, AKT2, SLC2A1, LDHA, ACACA |
| hsa05205 | Proteoglycans in cancer | 11 | 195 | 0.55 | 0.0243 | DDX5, GAB1, IGF1R, ANK3, PTCH1, SRC, ITPR2, AKT2, FGFR1, PPP1R12B, VEGFA |
| hsa05211 | Renal cell carcinoma | 6 | 68 | 0.74 | 0.0289 | CREBBP, GAB1, ARNT, AKT2, SLC2A1, VEGFA |
| hsa00061 | Fatty acid biosynthesis | 3 | 12 | 1.2 | 0.0368 | ACACB, ACSL6, ACACA |
| hsa04910 | Insulin signaling pathway | 8 | 134 | 0.57 | 0.0401 | TSC2, INPPL1, TSC1, ACACB, MKNK1, AKT2, IKBKB, ACACA |
| hsa01521 | EGFR tyrosine kinase inhibitor resistance | 6 | 78 | 0.68 | 0.0417 | GAB1, IGF1R, SRC, AKT2, NRG1, VEGFA |
| Parameter | Control Group n = 132 | Autism Group n = 70 | p-value |
|---|---|---|---|
| Age (year) | 8.04 ± 3.01 | 7.56 ± 3.68 | 0.1955 |
| Gender | F = 71; M = 61 | F = 23; M = 47 | - |
| Weight (kg) | 30.34 ± 13.36 | 26.40 ± 12.04 | 0.0582 |
| Height (cm) | 121.59 ± 22.01 | 121.68 ± 21.58 | 0.4913 |
| Body mass index | 19.93 ± 4.53 | 16.80 ± 1.78 | <0.00001 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almandil, N.B.; AlSulaiman, A.; Aldakeel, S.A.; Alkuroud, D.N.; Aljofi, H.E.; Alzahrani, S.; Al-mana, A.; Alfuraih, A.A.; Alabdali, M.; Alkhamis, F.A.; et al. Integration of Transcriptome and Exome Genotyping Identifies Significant Variants with Autism Spectrum Disorder. Pharmaceuticals 2022, 15, 158. https://doi.org/10.3390/ph15020158
Almandil NB, AlSulaiman A, Aldakeel SA, Alkuroud DN, Aljofi HE, Alzahrani S, Al-mana A, Alfuraih AA, Alabdali M, Alkhamis FA, et al. Integration of Transcriptome and Exome Genotyping Identifies Significant Variants with Autism Spectrum Disorder. Pharmaceuticals. 2022; 15(2):158. https://doi.org/10.3390/ph15020158
Chicago/Turabian StyleAlmandil, Noor B., Abdulla AlSulaiman, Sumayh A. Aldakeel, Deem N. Alkuroud, Halah Egal Aljofi, Safah Alzahrani, Aishah Al-mana, Asma A. Alfuraih, Majed Alabdali, Fahd A. Alkhamis, and et al. 2022. "Integration of Transcriptome and Exome Genotyping Identifies Significant Variants with Autism Spectrum Disorder" Pharmaceuticals 15, no. 2: 158. https://doi.org/10.3390/ph15020158
APA StyleAlmandil, N. B., AlSulaiman, A., Aldakeel, S. A., Alkuroud, D. N., Aljofi, H. E., Alzahrani, S., Al-mana, A., Alfuraih, A. A., Alabdali, M., Alkhamis, F. A., AbdulAzeez, S., & Borgio, J. F. (2022). Integration of Transcriptome and Exome Genotyping Identifies Significant Variants with Autism Spectrum Disorder. Pharmaceuticals, 15(2), 158. https://doi.org/10.3390/ph15020158