
JAK2 in Myeloproliferative Neoplasms: Still a Protagonist

Abstract
:1. Introduction
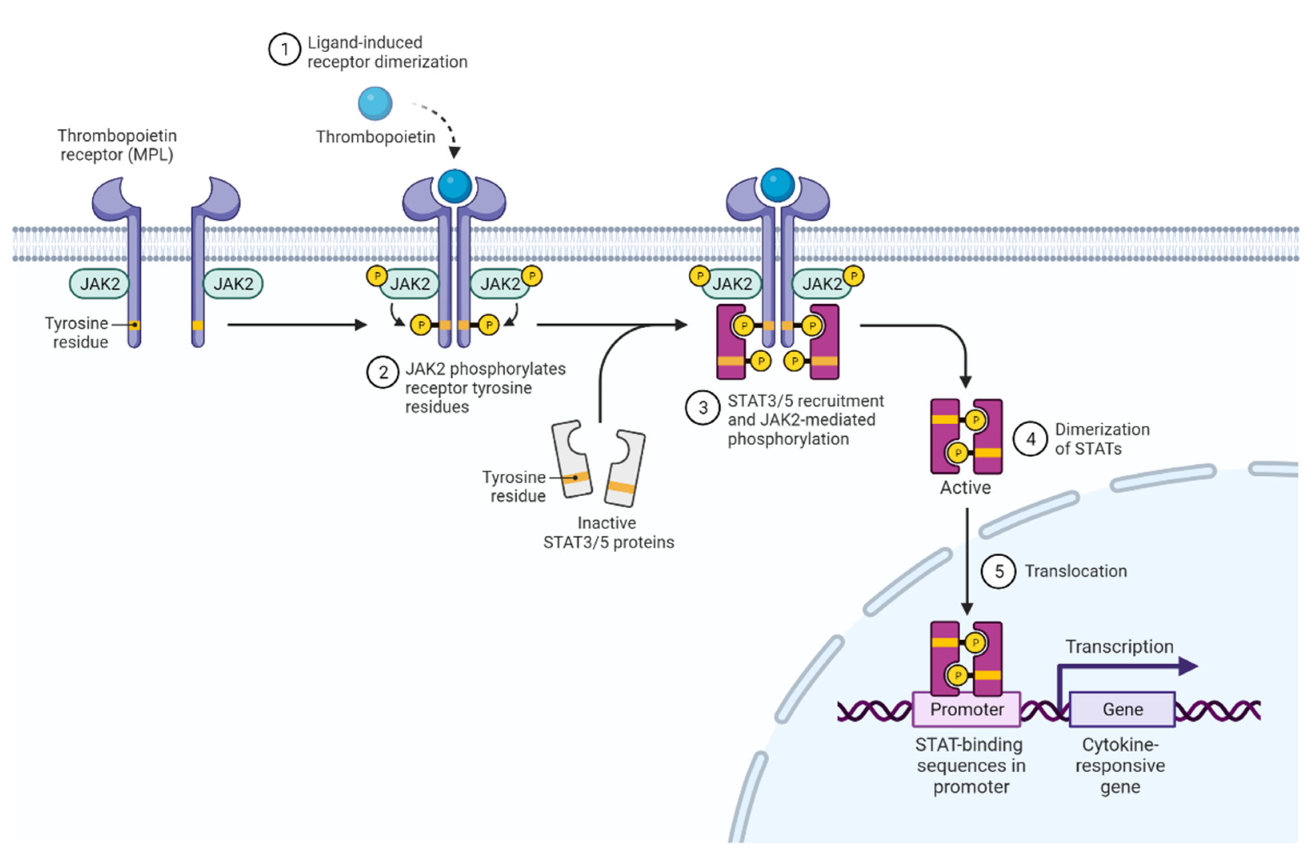
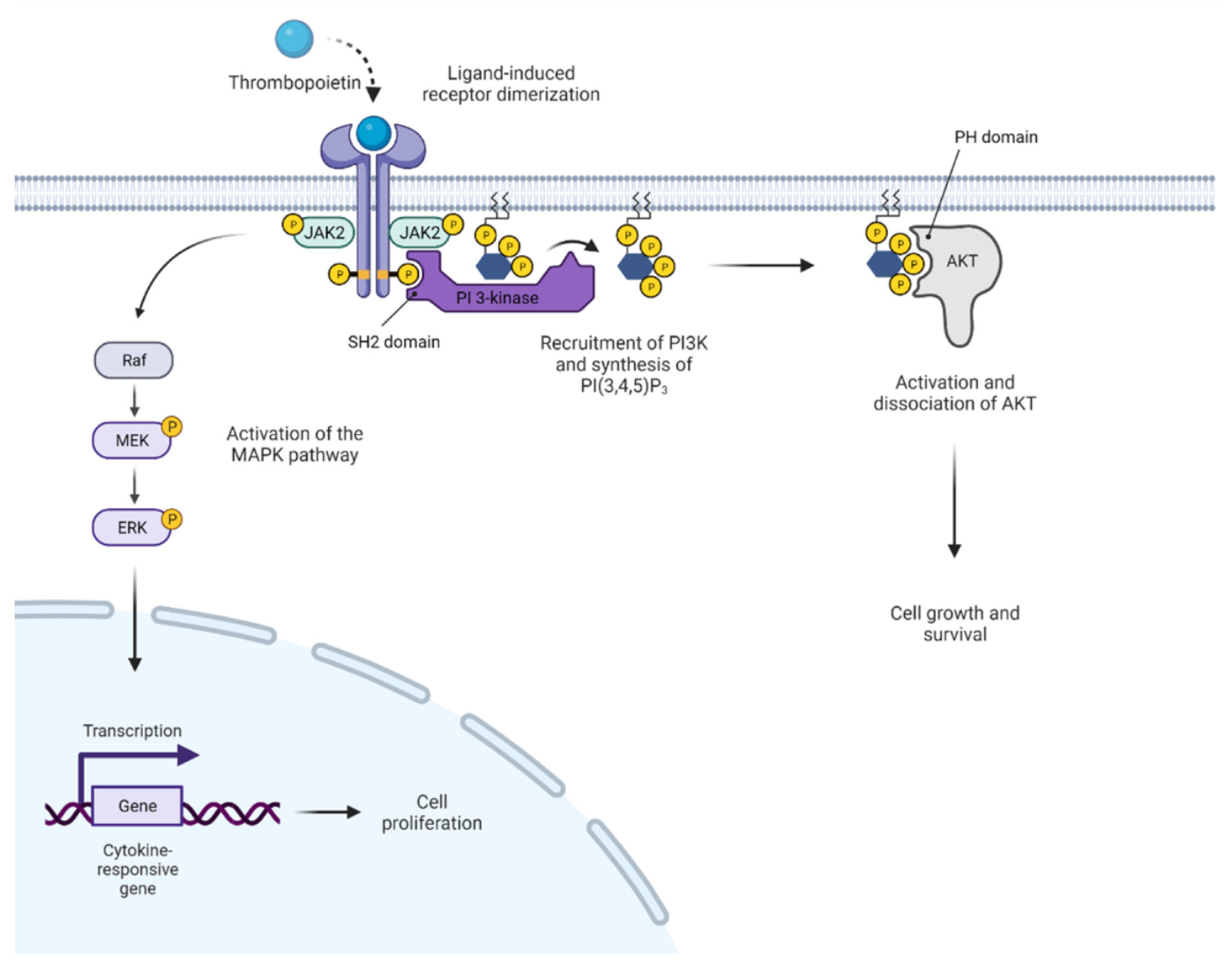
2. JAK2 Structure and Signaling
3. Activated JAK2 Signaling in MPN
4. Clinical Presentation of MPN
5. Clinical Benefit of JAK Inhibitor Therapy in MPN
6. Limitations of JAK Inhibitor Therapy in MPN
7. Potential Future Avenues for JAK2 Inhibitor Development and Alternative Therapeutic Approaches
8. JAK2 V617F in Clonal Hematopoiesis of Indeterminate Potential
9. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hammarén, H.M.; Virtanen, A.T.; Raivola, J.; Silvennoinen, O. The Regulation of JAKs in Cytokine Signaling and Its Breakdown in Disease. Cytokine 2019, 118, 48–63. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.C.; Levine, R.L. Molecular Pathways: Molecular Basis for Sensitivity and Resistance to JAK Kinase Inhibitors. Clin. Cancer Res. 2014, 20, 2051–2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skoda, R.C.; Duek, A.; Grisouard, J. Pathogenesis of Myeloproliferative Neoplasms. Exp. Hematol. 2015, 43, 599–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Shea, J.J.; Plenge, R. JAK and STAT Signaling Molecules in Immunoregulation and Immune-Mediated Disease. Immunity 2012, 36, 542–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNally, R.; Toms, A.V.; Eck, M.J. Crystal Structure of the FERM-SH2 Module of Human Jak2. PLoS ONE 2016, 11, e0156218. [Google Scholar] [CrossRef] [PubMed]
- Raivola, J.; Haikarainen, T.; Abraham, B.G.; Silvennoinen, O. Janus Kinases in Leukemia. Cancers 2021, 13, 800. [Google Scholar] [CrossRef]
- Wallweber, H.J.A.; Tam, C.; Franke, Y.; Starovasnik, M.A.; Lupardus, P.J. Structural Basis of Recognition of Interferon-α Receptor by Tyrosine Kinase 2. Nat. Struct. Mol. Biol. 2014, 21, 443–448. [Google Scholar] [CrossRef] [Green Version]
- Bandaranayake, R.M.; Ungureanu, D.; Shan, Y.; Shaw, D.E.; Silvennoinen, O.; Hubbard, S.R. Crystal Structures of the JAK2 Pseudokinase Domain and the Pathogenic Mutant V617F. Nat. Struct. Mol. Biol. 2012, 19, 754–759. [Google Scholar] [CrossRef] [Green Version]
- Saharinen, P.; Takaluoma, K.; Silvennoinen, O. Regulation of the Jak2 Tyrosine Kinase by Its Pseudokinase Domain. Mol. Cell. Biol. 2000, 20, 3387–3395. [Google Scholar] [CrossRef]
- Argetsinger, L.S.; Kouadio, J.-L.K.; Steen, H.; Stensballe, A.; Jensen, O.N.; Carter-Su, C. Autophosphorylation of JAK2 on Tyrosines 221 and 570 Regulates Its Activity. Mol. Cell. Biol. 2004, 24, 4955–4967. [Google Scholar] [CrossRef] [Green Version]
- Mazurkiewicz-Munoz, A.M.; Argetsinger, L.S.; Kouadio, J.-L.K.; Stensballe, A.; Jensen, O.N.; Cline, J.M.; Carter-Su, C. Phosphorylation of JAK2 at Serine 523: A Negative Regulator of JAK2 That Is Stimulated by GrowthHormone and Epidermal Growth Factor. Mol. Cell. Biol. 2006, 26, 4052–4062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babon, J.J.; Lucet, I.S.; Murphy, J.M.; Nicola, N.A.; Varghese, L.N. The Molecular Regulation of Janus Kinase (JAK) Activation. Biochem. J. 2014, 462, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villarino, A.V.; Kanno, Y.; O’Shea, J.J. Mechanisms and Consequences of Jak-STAT Signaling in the Immune System. Nat. Immunol. 2017, 18, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Pan, W.; Liu, S.; Shen, Z.; Xu, Y.; Hu, L. ERK/MAPK Signalling Pathway and Tumorigenesis (Review). Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nepstad, I.; Hatfield, K.J.; Grønningsæter, I.S.; Reikvam, H. The PI3K-AKT-MTOR Signaling Pathway in Human Acute Myeloid Leukemia (AML) Cells. Int. J. Mol. Sci. 2020, 21, 2907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varghese, L.N.; Ungureanu, D.; Liau, N.P.D.; Young, S.N.; Laktyushin, A.; Hammaren, H.; Lucet, I.S.; Nicola, N.A.; Silvennoinen, O.; Babon, J.J.; et al. Mechanistic Insights into Activation and SOCS3-Mediated Inhibition of Myeloproliferative Neoplasm-Associated JAK2 Mutants from Biochemical and Structural Analyses. Biochem. J. 2014, 458, 395–405. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Qu, C.K. Protein Tyrosine Phosphatases in the JAK/STAT Pathway. Front. Biosci. 2008, 13, 4925–4932. [Google Scholar] [CrossRef] [Green Version]
- Maslah, N.; Cassinat, B.; Verger, E.; Kiladjian, J.J.; Velazquez, L. The Role of LNK/SH2B3 Genetic Alterations in Myeloproliferative Neoplasms and Other Hematological Disorders. Leukemia 2017, 31, 1661–1670. [Google Scholar] [CrossRef]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.P.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating Mutation in the Tyrosine Kinase JAK2 in Polycythemia Vera, Essential Thrombocythemia, and Myeloid Metaplasia with Myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A Gain-of-Function Mutation of JAK2 in Myeloproliferative Disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [Green Version]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired Mutation of the Tyrosine Kinase JAK2 in Human Myeloproliferative Disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- Ugo, V.; James, C.; Vainchenker, W. A Unique Clonal JAK2 Mutation Leading to Constitutive Signalling Causes Polycythaemia Vera. Medicine/Sciences 2005, 21, 669–670. [Google Scholar] [CrossRef] [Green Version]
- Shan, Y.; Gnanasambandan, K.; Ungureanu, D.; Kim, E.T.; Hammarén, H.; Yamashita, K.; Silvennoinen, O.; Shaw, D.E.; Hubbard, S.R. Molecular Basis for Pseudokinase-Dependent Autoinhibition of JAK2 Tyrosine Kinase. Nat. Struct. Mol. Biol. 2014, 21, 579–584. [Google Scholar] [CrossRef] [Green Version]
- Szybinski, J.; Meyer, S.C. Genetics of Myeloproliferative Neoplasms. Hematol. Oncol. Clin. N. Am. 2021, 35, 217–236. [Google Scholar] [CrossRef]
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. JAK2 Exon 12 Mutations in Polycythemia Vera and Idiopathic Erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Passamonti, F.; Elena, C.; Schnittger, S.; Skoda, R.C.; Green, A.R.; Girodon, F.; Kiladjian, J.J.; McMullin, M.F.; Ruggeri, M.; Besses, C.; et al. Molecular and Clinical Features of the Myeloproliferative Neoplasm Associated with JAK2 Exon 12 Mutations. Blood 2011, 117, 2813–2816. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A. Primary Myelofibrosis: 2021 Update on Diagnosis, Risk-Stratification and Management. Am. J. Hematol. 2021, 96, 145–162. [Google Scholar] [CrossRef]
- Vainchenker, W.; Kralovics, R. Genetic Basis and Molecular Pathophysiology of Classical Myeloproliferative Neoplasms. Blood 2017, 129, 667–679. [Google Scholar] [CrossRef] [Green Version]
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I.; et al. MPLW515L Is a Novel Somatic Activating Mutation in Myelofibrosis with Myeloid Metaplasia. PLoS Med. 2006, 3, 1140–1151. [Google Scholar] [CrossRef] [Green Version]
- Pardanani, A.D.; Levine, R.L.; Lasho, T.; Pikman, Y.; Mesa, R.A.; Wadleigh, M.; Steensma, D.P.; Elliott, M.A.; Wolanskyj, A.P.; Hogan, W.J.; et al. MPL515 Mutations in Myeloproliferative and Other Myeloid Disorders: A Study of 1182 Patients. Blood 2006, 108, 3472–3476. [Google Scholar] [CrossRef] [Green Version]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic Mutations of Calreticulin in Myeloproliferative Neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef] [Green Version]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR Mutations in Myeloproliferative Neoplasms with Nonmutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef] [Green Version]
- Elf, S.; Abdelfattah, N.S.; Chen, E.; Perales-Patón, J.; Rosen, E.A.; Ko, A.; Peisker, F.; Florescu, N.; Giannini, S.; Wolach, O.; et al. Mutant Calreticulin Requires Both Its Mutant C-Terminus and the Thrombopoietin Receptor for Oncogenic Transformation. Cancer Discov. 2016, 6, 368–381. [Google Scholar] [CrossRef] [Green Version]
- Chachoua, I.; Pecquet, C.; El-Khoury, M.; Nivarthi, H.; Albu, R.I.; Marty, C.; Gryshkova, V.; Defour, J.P.; Vertenoeil, G.; Ngo, A.; et al. Thrombopoietin Receptor Activation by Myeloproliferative Neoplasm Associated Calreticulin Mutants. Blood 2016, 127, 1325–1335. [Google Scholar] [CrossRef]
- Rumi, E.; Pietra, D.; Ferretti, V.; Klampfl, T.; Harutyunyan, A.S.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Elena, C.; Casetti, I.C.; et al. JAK2 or CALR Mutation Status Defines Subtypes of Essential Thrombocythemia with Substantially Different Clinical Course and Outcomes. Blood 2014, 123, 1544–1551. [Google Scholar] [CrossRef]
- Rampal, R.; Al-Shahrour, F.; Abdel-Wahab, O.; Patel, J.P.; Brunel, J.P.; Mermel, C.H.; Bass, A.J.; Pretz, J.; Ahn, J.; Hricik, T.; et al. Integrated Genomic Analysis Illustrates the Central Role of JAK-STAT Pathway Activation in Myeloproliferative Neoplasm Pathogenesis. Blood 2014, 123, e123–e133. [Google Scholar] [CrossRef]
- Rinaldi, C.R.; Rinaldi, P.; Alagia, A.; Gemei, M.; Esposito, N.; Formiggini, F.; Martinelli, V.; Senyuk, V.; Nucifora, G.; Pane, F. Preferential Nuclear Accumulation of JAK2V617F in CD34+ but Not in Granulocytic, Megakaryocytic, or Erythroid Cells of Patients with Philadelphia-Negative Myeloproliferative Neoplasia. Blood 2010, 116, 6023–6026. [Google Scholar] [CrossRef] [Green Version]
- Dawson, M.A.; Bannister, A.J.; Göttgens, B.; Foster, S.D.; Bartke, T.; Green, A.R.; Kouzarides, T. JAK2 Phosphorylates Histone H3Y41 and Excludes HP1α from Chromatin. Nature 2009, 461, 819–822. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Zhao, X.; Perna, F.; Wang, L.; Koppikar, P.; Abdel-Wahab, O.; Harr, M.W.; Levine, R.L.; Xu, H.; Tefferi, A.; et al. JAK2V617F-Mediated Phosphorylation of PRMT5 Downregulates Its Methyltransferase Activity and Promotes Myeloproliferation. Cancer Cell 2011, 19, 283–294. [Google Scholar] [CrossRef] [Green Version]
- Pastore, F.; Bhagwat, N.; Pastore, A.; Radzisheuskaya, A.; Karzai, A.; Krishnan, A.; Li, B.; Bowman, R.L.; Xiao, W.; Viny, A.D.; et al. PRMT5 Inhibition Modulates E2F1 Methylation and Gene-Regulatory Networks Leading to Therapeutic Efficacy in JAK2V617F-Mutant MPN. Cancer Discov. 2020, 10, 1742–1757. [Google Scholar] [CrossRef]
- Di Battista, V.; Bochicchio, M.T.; Giordano, G.; Napolitano, M.; Lucchesi, A. Review Genetics and Pathogenetic Role of Inflammasomes in Philadelphia Negative Chronic Myeloproliferative Neoplasms: A Narrative Review. Int. J. Mol. Sci. 2021, 22, 561. [Google Scholar] [CrossRef] [PubMed]
- Fleischman, A.G.; Aichberger, K.J.; Luty, S.B.; Bumm, T.G.; Petersen, C.L.; Doratotaj, S.; Vasudevan, K.B.; LaTocha, D.H.; Yang, F.; Press, R.D.; et al. TNFα Facilitates Clonal Expansion of JAK2V617F Positive Cells in Myeloproliferative Neoplasms. Blood 2011, 118, 6392–6398. [Google Scholar] [CrossRef] [PubMed]
- Marty, C.; Lacout, C.; Droin, N.; Le Couédic, J.P.; Ribrag, V.; Solary, E.; Vainchenker, W.; Villeval, J.L.; Plo, I. A Role for Reactive Oxygen Species in JAK2 V617F Myeloproliferative Neoplasm Progression. Leukemia 2013, 27, 2187–2195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasselbalch, H.C. The Role of Cytokines in the Initiation and Progression of Myelofibrosis. Cytokine Growth Factor Rev. 2013, 24, 133–145. [Google Scholar] [CrossRef]
- Găman, M.A.; Cozma, M.A.; Dobrică, E.C.; Crețoiu, S.M.; Găman, A.M.; Diaconu, C.C. Liquid Biopsy and Potential Liquid Biopsy-Based Biomarkers in Philadelphia-Negative Classical Myeloproliferative Neoplasms: A Systematic Review. Life 2021, 11, 677. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- DAMESHEK, W. Some Speculations on the Myeloproliferative Syndromes. Blood 1951, 6, 372–375. [Google Scholar] [CrossRef] [Green Version]
- Godfrey, A.L.; Chen, E.; Pagano, F.; Ortmann, C.A.; Silber, Y.; Bellosillo, B.; Guglielmelli, P.; Harrison, C.N.; Reilly, J.T.; Stegelmann, F.; et al. JAK2V617F Homozygosity Arises Commonly and Recurrently in PV and ET, but PV Is Characterized by Expansion of a Dominant Homozygous Subclone. Blood 2012, 120, 2704–2707. [Google Scholar] [CrossRef]
- Ortmann, C.A.; Kent, D.G.; Nangalia, J.; Silber, Y.; Wedge, D.C.; Grinfeld, J.; Baxter, E.J.; Massie, C.E.; Papaemmanuil, E.; Menon, S.; et al. Effect of Mutation Order on Myeloproliferative Neoplasms. N. Engl. J. Med. 2015, 372, 601–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falanga, A.; Marchetti, M. Thrombosis in Myeloproliferative Neoplasms. Semin. Thromb. Hemost. 2014, 40, 348–358. [Google Scholar] [CrossRef]
- Shao, Y.; Cheng, Z.; Li, X.; Chernaya, V.; Wang, H.; Yang, X.F. Immunosuppressive/Anti-Inflammatory Cytokines Directly and Indirectly Inhibit Endothelial Dysfunction-A Novel Mechanism for Maintaining Vascular Function. J. Hematol. Oncol. 2014, 7, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guy, A.; Gourdou-Latyszenok, V.; Le Lay, N.; Peghaire, C.; Kilani, B.; Dias, J.V.; Duplaa, C.; Renault, M.A.; Denis, C.; Villeval, J.L.; et al. Vascular Endothelial Cell Expression of JAK2 V617F Is Sufficient to Promote a Pro-Thrombotic State Due to Increased P-Selectin Expression. Haematologica 2019, 104, 70–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guglielmelli, P.; Barosi, G.; Specchia, G.; Rambaldi, A.; Lo Coco, F.; Antonioli, E.; Pieri, L.; Pancrazzi, A.; Ponziani, V.; Delaini, F.; et al. Identification of Patients with Poorer Survival in Primary Myelofibrosis Based on the Burden of JAK2V617F Mutated Allele. Blood 2009, 114, 1477–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Guglielmelli, P.; Larson, D.R.; Finke, C.; Wassie, E.A.; Pieri, L.; Gangat, N.; Fjerza, R.; Belachew, A.A.; Lasho, T.L.; et al. Long-Term Survival and Blast Transformation in Molecularly Annotated Essential Thrombocythemia, Polycythemia Vera, and Myelofibrosis. Blood 2014, 124, 2507–2513. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.L.; Abdel-Wahab, O.; Guglielmelli, P.; Patel, J.; Caramazza, D.; Pieri, L.; Finke, C.M.; Kilpivaara, O.; Wadleigh, M.; et al. IDH1 and IDH2 Mutation Studies in 1473 Patients with Chronic-, Fibrotic- or Blast-Phase Essential Thrombocythemia, Polycythemia Vera or Myelofibrosis. Leukemia 2010, 24, 1302–1309. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.J.; Rampal, R.; Manshouri, T.; Patel, J.; Mensah, N.; Kayserian, A.; Hricik, T.; Heguy, A.; Hedvat, C.; Gönen, M.; et al. Genetic Analysis of Patients with Leukemic Transformation of Myeloproliferative Neoplasms Shows Recurrent SRSF2 Mutations That Are Associated with Adverse Outcome. Blood 2012, 119, 4480–4485. [Google Scholar] [CrossRef] [PubMed]
- Rampal, R.; Ahn, J.; Abdel-Wahaba, O.; Nahas, M.; Wang, K.; Lipson, D.; Otto, G.A.; Yelensky, R.; Hricik, T.; McKenney, A.S.; et al. Genomic and Functional Analysis of Leukemic Transformation of Myeloproliferative Neoplasms. Proc. Natl. Acad. Sci. USA 2014, 111, E5401–E5410. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, P.; Karow, A.; Nienhold, R.; Looser, R.; Hao-Shen, H.; Nissen, I.; Girsberger, S.; Lehmann, T.; Passweg, J.; Stern, M.; et al. Clonal Evolution and Clinical Correlates of Somatic Mutations in Myeloproliferative Neoplasms. Blood 2014, 123, 2220–2228. [Google Scholar] [CrossRef] [Green Version]
- Brkic, S.; Meyer, S.C. Challenges and Perspectives for Therapeutic Targeting of Myeloproliferative Neoplasms. Hemasphere 2021, 5, e516. [Google Scholar] [CrossRef]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A Double-Blind, Placebo-Controlled Trial of Ruxolitinib for Myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef] [Green Version]
- Harrison, C.N.; Vannucchi, A.M.; Kiladjian, J.J.; Al-Ali, H.K.; Gisslinger, H.; Knoops, L.; Cervantes, F.; Jones, M.M.; Sun, K.; McQuitty, M.; et al. Long-Term Findings from COMFORT-II, a Phase 3 Study of Ruxolitinib vs Best Available Therapy for Myelofibrosis. Leukemia 2016, 30, 1701–1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannucchi, A.M.; Kiladjian, J.J.; Griesshammer, M.; Masszi, T.; Durrant, S.; Passamonti, F.; Harrison, C.N.; Pane, F.; Zachee, P.; Mesa, R.; et al. Ruxolitinib versus Standard Therapy for the Treatment of Polycythemia Vera. N. Engl. J. Med. 2015, 372, 426–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchioli, R.; Finazzi, G.; Specchia, G.; Cacciola, R.; Cavazzina, R.; Cilloni, D.; De Stefano, V.; Elli, E.; Iurlo, A.; Latagliata, R.; et al. CYTO-PV Collaborative Group. Cardiovascular Events and Intensity of Treatment in Polycythemia Vera. N. Engl. J. Med. 2013, 368, 22–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, C.N.; Mead, A.J.; Panchal, A.; Fox, S.; Yap, C.; Gbandi, E.; Houlton, A.; Alimam, S.; Ewing, J.; Wood, M.; et al. Ruxolitinib vs. Best Available Therapy for et Intolerant or Resistant to Hydroxycarbamide. Blood 2017, 130, 1889–1897. [Google Scholar] [CrossRef] [Green Version]
- Pardanani, A.; Harrison, C.; Cortes, J.E.; Cervantes, F.; Mesa, R.A.; Milligan, D.; Masszi, T.; Mishchenko, E.; Jourdan, E.; Vannucchi, A.M.; et al. Safety and Efficacy of Fedratinib in Patients with Primary or Secondary Myelofibrosis: A Randomized Clinical Trial. JAMA Oncol. 2015, 1, 643–651. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Hoffman, R.; Talpaz, M.; Gerds, A.T.; Stein, B.; Gupta, V.; Szoke, A.; Drummond, M.; Pristupa, A.; Granston, T.; et al. Pacritinib vs Best Available Therapy, Including Ruxolitinib, in Patients with Myelofibrosis: A Randomized Clinical Trial. JAMA Oncol. 2018, 4, 652–659. [Google Scholar] [CrossRef]
- Oh, S.T.; Talpaz, M.; Gerds, A.T.; Gupta, V.; Verstovsek, S.; Mesa, R.; Miller, C.B.; Rivera, C.E.; Fleischman, A.G.; Goel, S.; et al. ACVR1/JAK1/JAK2 Inhibitor Momelotinib Reverses Transfusion Dependency and Suppresses Hepcidin in Myelofibrosis Phase 2 Trial. Blood Adv. 2020, 4, 4282–4291. [Google Scholar] [CrossRef]
- Gupta, V.; Mesa, R.A.; Deininger, M.W.N.; Rivera, C.E.; Sirhan, S.; Brachmann, C.B.; Collins, H.; Kawashima, J.; Xin, Y.; Verstovsek, S. A Phase 1/2, Open-Label Study Evaluating Twice-Daily Administration of Momelotinib in Myelofibrosis. Haematologica 2017, 102, 94–102. [Google Scholar] [CrossRef]
- Zeiser, R.; von Bubnoff, N.; Butler, J.; Mohty, M.; Niederwieser, D.; Or, R.; Szer, J.; Wagner, E.M.; Zuckerman, T.; Mahuzier, B.; et al. Ruxolitinib for Glucocorticoid-Refractory Acute Graft-versus-Host Disease. N. Engl. J. Med. 2020, 382, 1800–1810. [Google Scholar] [CrossRef]
- Zeiser, R.; Polverelli, N.; Ram, R.; Hashmi, S.K.; Chakraverty, R.; Middeke, J.M.; Musso, M.; Giebel, S.; Uzay, A.; Langmuir, P.; et al. Ruxolitinib for Glucocorticoid-Refractory Chronic Graft-versus-Host Disease. N. Engl. J. Med. 2021, 385, 228–238. [Google Scholar] [CrossRef]
- Newberry, K.J.; Patel, K.; Masarova, L.; Luthra, R.; Manshouri, T.; Jabbour, E.; Bose, P.; Daver, N.; Cortes, J.; Kantarjian, H.; et al. Clonal Evolution and Outcomes in Myelofibrosis after Ruxolitinib Discontinuation. Blood 2017, 130, 1125–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiladjian, J.J.; Zachee, P.; Hino, M.; Pane, F.; Masszi, T.; Harrison, C.N.; Mesa, R.; Miller, C.B.; Passamonti, F.; Durrant, S.; et al. Long-Term Efficacy and Safety of Ruxolitinib versus Best Available Therapy in Polycythaemia Vera (RESPONSE): 5-Year Follow up of a Phase 3 Study. Lancet Haematol. 2020, 7, e226–e237. [Google Scholar] [CrossRef]
- Luo, Q.; Xiao, Z.; Peng, L. Effects of Ruxolitinib on Infection in Patients with Myeloproliferative Neoplasm: A Meta-Analysis. Hematology 2021, 26, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Sadjadian, P.; Wille, K.; Griesshammer, M.; Sadjadian, P.; Wille, K.; Griesshammer, M. Ruxolitinib-Associated Infections in Polycythemia Vera: Review of the Literature, Clinical Significance, and Recommendations. Cancers 2020, 12, 3132. [Google Scholar] [CrossRef] [PubMed]
- Khalid, F.; Damlaj, M.; AlZahrani, M.; Abuelgasim, K.A.; Gmati, G.E. Reactivation of Tuberculosis Following Ruxolitinib Therapy for Primary Myelofibrosis: Case Series and Literature Review. Hematol. Oncol. Stem Cell Ther. 2021, 14, 252–256. [Google Scholar] [CrossRef]
- Duan, M.-H.; Cao, X.-X.; Chang, L.; Zhou, D.-B. Risk of Hepatitis B Virus Reactivation Following Ruxolitinib Treatment in Patients with Myeloproliferative Neoplasms. Hematology 2021, 26, 460–464. [Google Scholar] [CrossRef]
- Devos, T.; Selleslag, D.; Granacher, N.; Havelange, V.; Benghiat, F.S. Updated Recommendations on the Use of Ruxolitinib for the Treatment of Myelofibrosis. Hematology 2022, 27, 23–31. [Google Scholar] [CrossRef]
- Barraco, F.; Greil, R.; Herbrecht, R.; Schmidt, B.; Reiter, A.; Willenbacher, W.; Raymakers, R.; Liersch, R.; Wroclawska, M.; Pack, R.; et al. Real-World Non-Interventional Long-Term Post-Authorisation Safety Study of Ruxolitinib in Myelofibrosis. Br. J. Haematol. 2020, 191, 764–774. [Google Scholar] [CrossRef]
- Porpaczy, E.; Tripolt, S.; Hoelbl-Kovacic, A.; Gisslinger, B.; Bago-Horvath, Z.; Casanova-Hevia, E.; Clappier, E.; Decker, T.; Fajmann, S.; Fux, D.A.; et al. Aggressive B-Cell Lymphomas in Patients with Myelofibrosis Receiving JAK1/2 Inhibitor Therapy. Blood 2018, 132, 694–706. [Google Scholar] [CrossRef]
- Sekhri, R.; Sadjadian, P.; Becker, T.; Kolatzki, V.; Huenerbein, K.; Meixner, R.; Marchi, H.; Wallmann, R.; Fuchs, C.; Griesshammer, M.; et al. Ruxolitinib-Treated Polycythemia Vera Patients and Their Risk of Secondary Malignancies. Ann. Hematol. 2021, 100, 2707–2716. [Google Scholar] [CrossRef]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.W.N.; Miller, C.B.; Silver, R.T.; Talpaz, M.; et al. Long-Term Treatment with Ruxolitinib for Patients with Myelofibrosis: 5-Year Update from the Randomized, Double-Blind, Placebo-Controlled, Phase 3 COMFORT-I Trial. J. Hematol. Oncol. 2017, 10, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gisslinger, H.; Schalling, M.; Gisslinger, B.; Skrabs, C.; Müllauer, L.; Kralovics, R. Restoration of Response to Ruxolitinib upon Brief Withdrawal in Two Patients with Myelofibrosis. Am. J. Hematol. 2014, 89, 344–346. [Google Scholar] [CrossRef] [PubMed]
- Koppikar, P.; Bhagwat, N.; Kilpivaara, O.; Manshouri, T.; Adli, M.; Hricik, T.; Liu, F.; Saunders, L.M.; Mullally, A.; Abdel-Wahab, O.; et al. Heterodimeric JAK-STAT Activation as a Mechanism of Persistence to JAK2 Inhibitor Therapy. Nature 2012, 489, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.C. Mechanisms of Resistance to JAK2 Inhibitors in Myeloproliferative Neoplasms. Hematol. Oncol. Clin. N. Am. 2017, 31, 627–642. [Google Scholar] [CrossRef]
- Meyer, S.C.; Keller, M.D.; Chiu, S.; Koppikar, P.; Guryanova, O.A.; Rapaport, F.; Xu, K.; Manova, K.; Pankov, D.; O’Reilly, R.J.; et al. CHZ868, a Type II JAK2 Inhibitor, Reverses Type I JAK Inhibitor Persistence and Demonstrates Efficacy in Myeloproliferative Neoplasms. Cancer Cell 2015, 28, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Li, L.S.; Kopp, N.; Montero, J.; Chapuy, B.; Yoda, A.; Christie, A.L.; Liu, H.; Christodoulou, A.; van Bodegom, D.; et al. Activity of the Type II JAK2 Inhibitor CHZ868 in B Cell Acute Lymphoblastic Leukemia. Cancer Cell 2015, 28, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Hammarén, H.M.; Ungureanu, D.; Grisouard, J.; Skoda, R.C.; Hubbard, S.R.; Silvennoinen, O. ATP Binding to the Pseudokinase Domain of JAK2 Is Critical for Pathogenic Activation. Proc. Natl. Acad. Sci. USA 2015, 112, 4642–4647. [Google Scholar] [CrossRef] [Green Version]
- Harrison, C.; Garcia, J.S.; Mesa, R.; Somervaille, T.; Ritchie, E.K.; Komrokji, R.S.; Pemmaraju, N.; Jamieson, C.; Papadantonakis, N.; Foran, J.M.; et al. MPN-038: Navitoclax in Combination with Ruxolitinib in Patients with Primary or Secondary Myelofibrosis: A Phase 2 Study. Clin. Lymphoma Myeloma Leuk. 2020, 20, S325. [Google Scholar] [CrossRef]
- Waibel, M.; Solomon, V.S.; Knight, D.A.; Ralli, R.A.; Kim, S.K.; Banks, K.M.; Vidacs, E.; Virely, C.; Sia, K.C.S.; Bracken, L.S.; et al. Combined Targeting of JAK2 and Bcl-2/Bcl-XL to Cure Mutant JAK2-Driven Malignancies and Overcome Acquired Resistance to JAK2 Inhibitors. Cell Rep. 2013, 5, 1047–1059. [Google Scholar] [CrossRef] [Green Version]
- Yacoub, A.; Borate, U.; Rampal, R.; Ali, H.; Wang, E.; Gerds, A.; Hobbs, G.; Kremyanskaya, M.; Winton, E.; O’Connell, C.; et al. MPN-127: Add-On Parsaclisib (a PI3K5 Inhibitor) in Patients with Myelofibrosis and Suboptimal Response to Ruxolitinib: Interim Analysis from a Phase 2 Study. Clin. Lymphoma Myeloma Leuk. 2021, 21, S354–S355. [Google Scholar] [CrossRef]
- Stivala, S.; Codilupi, T.; Brkic, S.; Baerenwaldt, A.; Ghosh, N.; Hao-Shen, H.; Dirnhofer, S.; Dettmer, M.S.; Simillion, C.; Kaufmann, B.A.; et al. Targeting Compensatory MEK/ERK Activation Increases JAK Inhibitor Efficacy in Myeloproliferative Neoplasms. J. Clin. Investig. 2019, 129, 1596–1611. [Google Scholar] [CrossRef] [PubMed]
- Brkic, S.; Stivala, S.; Santopolo, A.; Szybinski, J.; Jungius, S.; Passweg, J.R.; Tsakiris, D.; Dirnhofer, S.; Hutter, G.; Leonards, K.; et al. Dual Targeting of JAK2 and ERK Interferes with the Myeloproliferative Neoplasm Clone and Enhances Therapeutic Efficacy. Leukemia 2021, 35, 2875–2884. [Google Scholar] [CrossRef] [PubMed]
- Jayavelu, A.K.; Schnöder, T.M.; Perner, F.; Herzog, C.; Meiler, A.; Krishnamoorthy, G.; Huber, N.; Mohr, J.; Edelmann-Stephan, B.; Austin, R.; et al. Splicing Factor YBX1 Mediates Persistence of JAK2-Mutated Neoplasms. Nature 2020, 588, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-Related Mutations Associated with Clonal Hematopoietic Expansion and Malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef]
- Patel, R.K.; Lea, N.C.; Heneghan, M.A.; Westwood, N.B.; Milojkovic, D.; Thanigaikumar, M.; Yallop, D.; Arya, R.; Pagliuca, A.; Gäken, J.; et al. Prevalence of the Activating JAK2 Tyrosine Kinase Mutation V617F in the Budd-Chiari Syndrome. Gastroenterology 2006, 130, 2031–2038. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Gene/Mutation | Chromosome | Mutational Frequency (%) | ||
|---|---|---|---|---|
| PV | ET | PMF | ||
| Driver mutations in MPN | ||||
| JAK2 V617F (exon 14) | 9p24 | 95 | 50–60 | 50–60 |
| JAK2 exon 12 mutations | 9p24 | 2–3 | - | - |
| CALR | 19p13.2 | <1 | 20–30 | 20–35 |
| MPL | 1p34 | <1 | 1–5 | 5–9 |
| High molecular risk (HMR) mutations in MF | ||||
| ASXL1 | 20q11.1 | 25–35 | ||
| EZH2 | 7q36.1 | 1–10 | ||
| SRSF2 | 17q25.1 | 10–18 (enriched in BP-MPN) | ||
| IDH1/IDH2 | 2q33.3/15q26.1 | 1–6 (enriched in BP-MPN) | ||
| Other mutations enriched in blast phase MPN | ||||
| TP53 | 17p13.1 | 1–5 (enriched in BP-MPN) | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bader, M.S.; Meyer, S.C. JAK2 in Myeloproliferative Neoplasms: Still a Protagonist. Pharmaceuticals 2022, 15, 160. https://doi.org/10.3390/ph15020160
Bader MS, Meyer SC. JAK2 in Myeloproliferative Neoplasms: Still a Protagonist. Pharmaceuticals. 2022; 15(2):160. https://doi.org/10.3390/ph15020160
Chicago/Turabian StyleBader, Michael Stephan, and Sara Christina Meyer. 2022. "JAK2 in Myeloproliferative Neoplasms: Still a Protagonist" Pharmaceuticals 15, no. 2: 160. https://doi.org/10.3390/ph15020160
APA StyleBader, M. S., & Meyer, S. C. (2022). JAK2 in Myeloproliferative Neoplasms: Still a Protagonist. Pharmaceuticals, 15(2), 160. https://doi.org/10.3390/ph15020160