
A Convenient Route to New (Radio)Fluorinated and (Radio)Iodinated Cyclic Tyrosine Analogs

 , ,
, ,  ,
,
Abstract
:1. Introduction
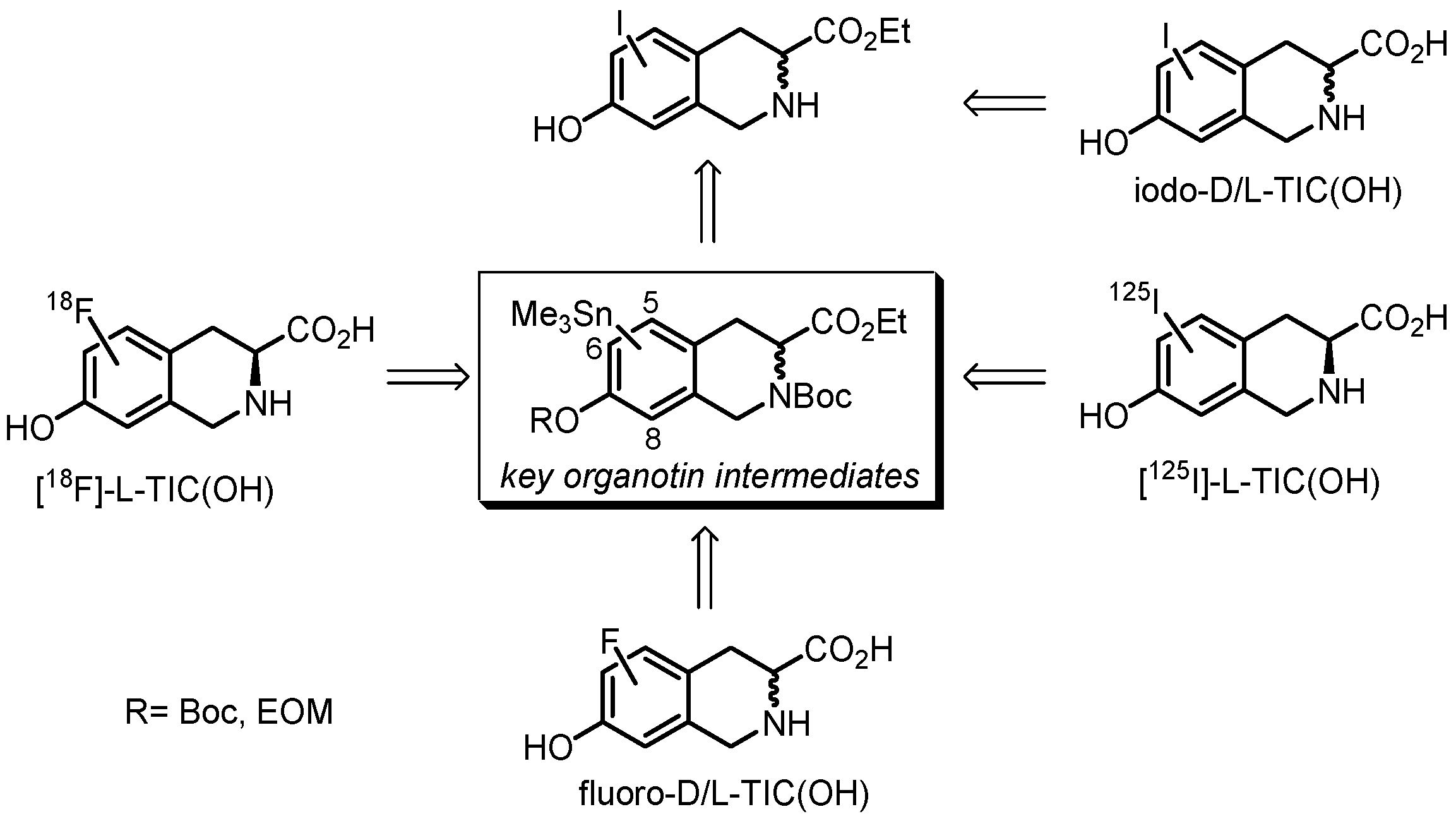
2. Results and Discussion
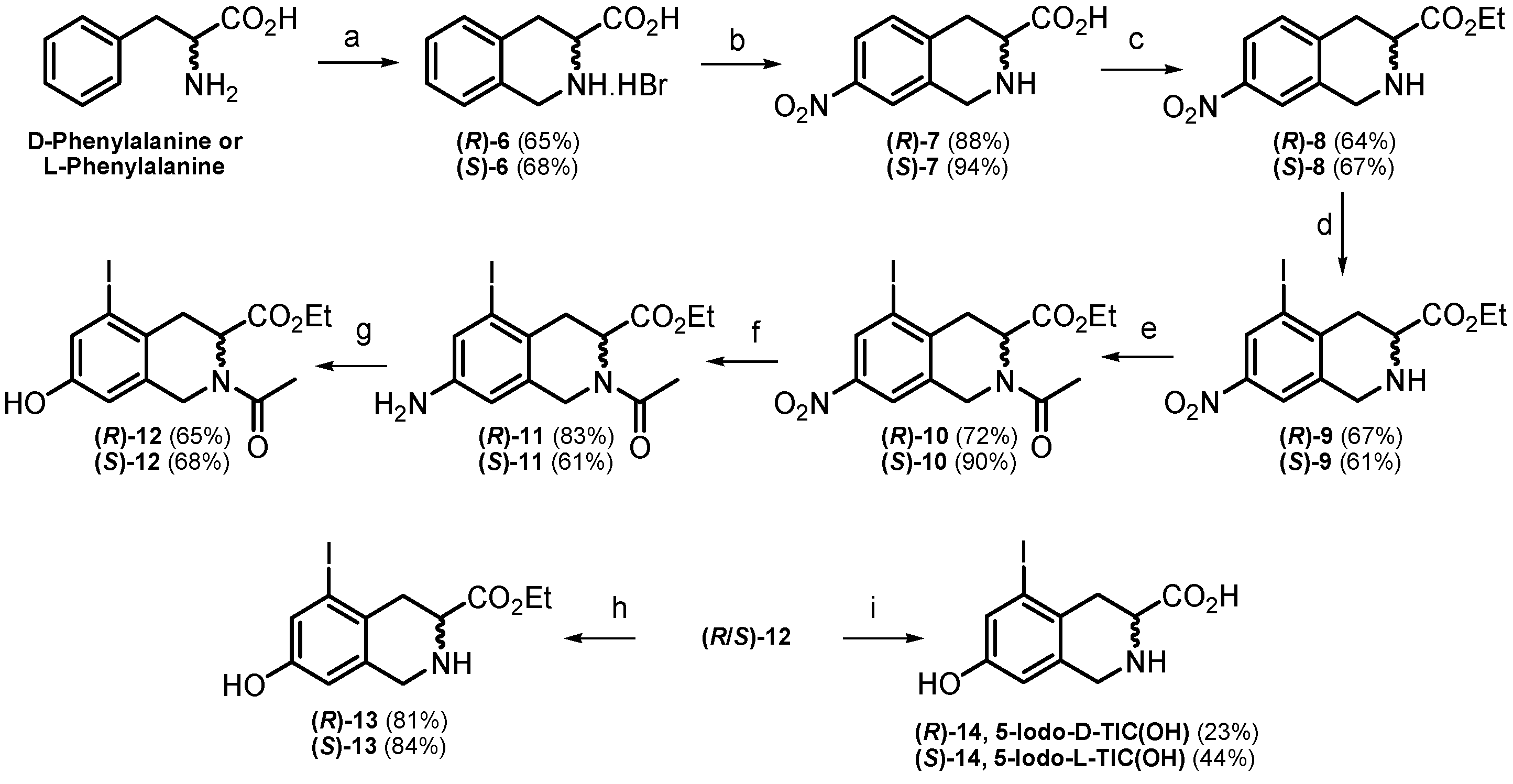
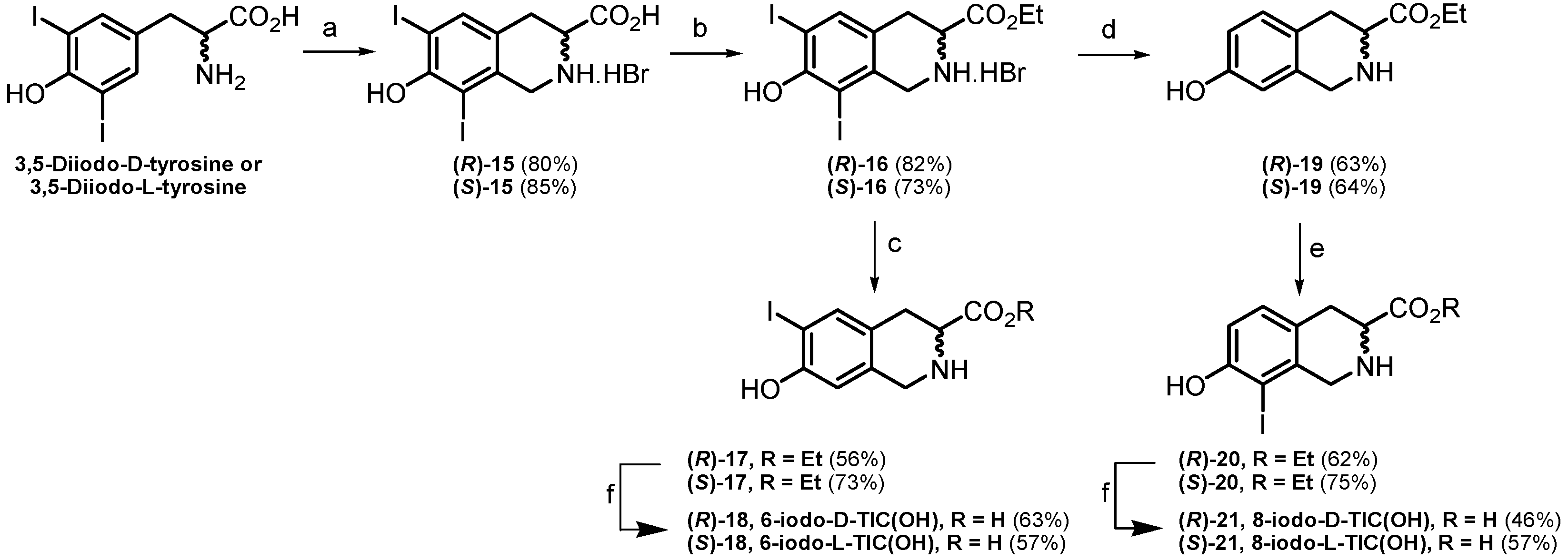
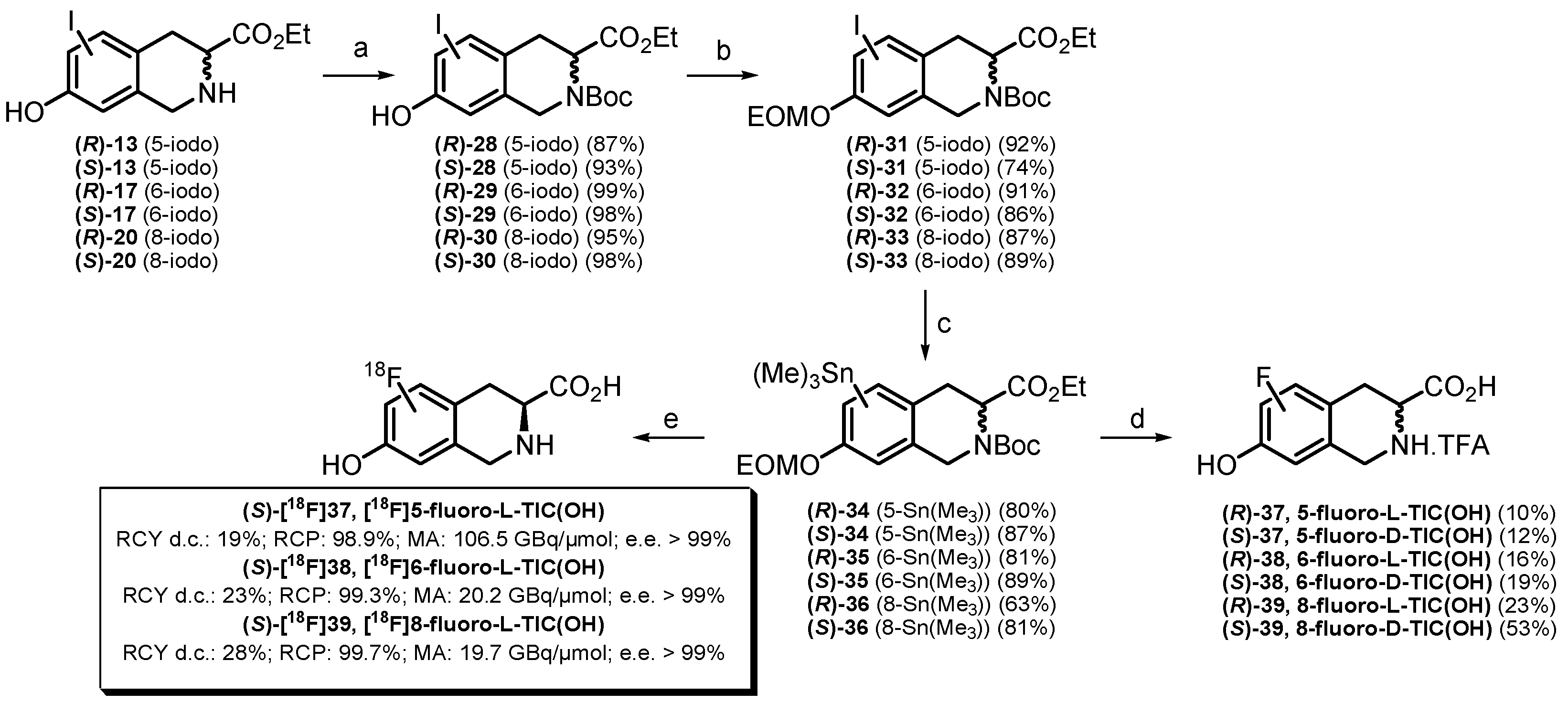
2.1. Chemistry
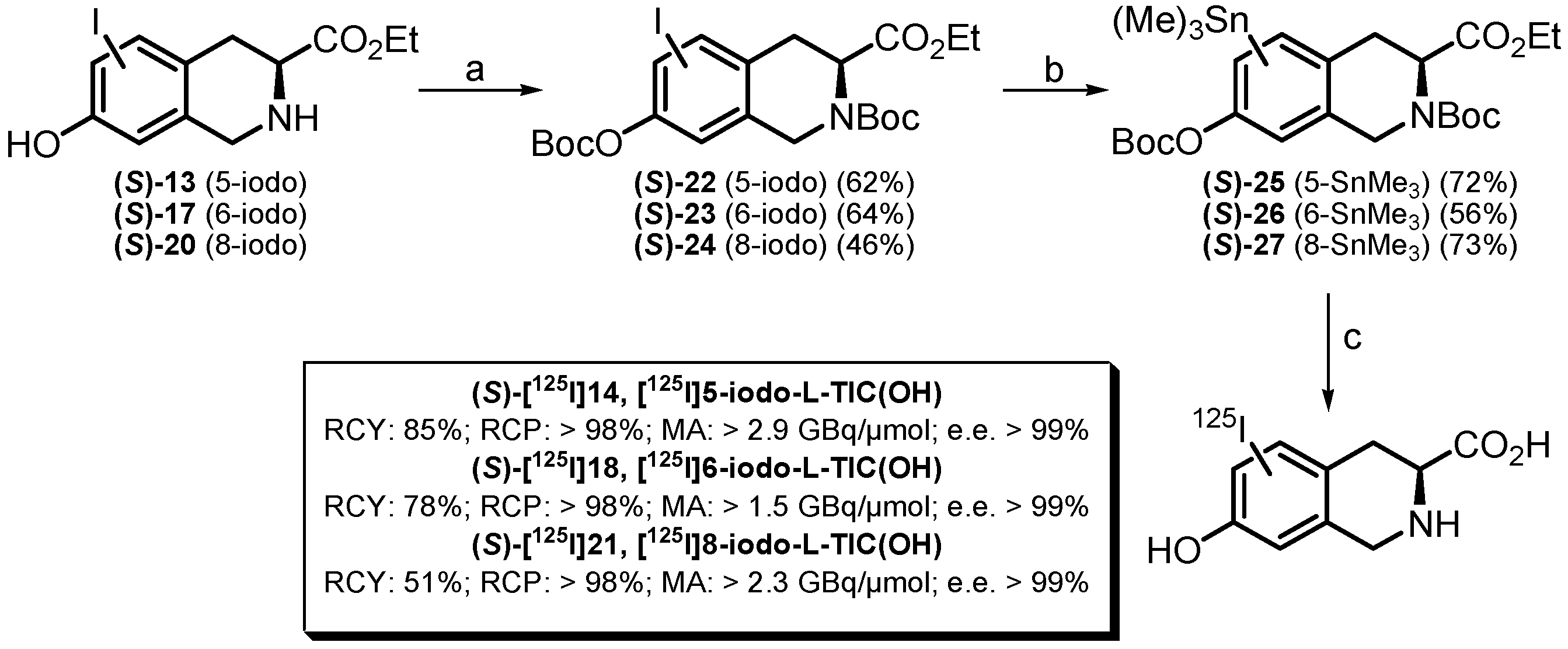
2.2. Radiochemistry
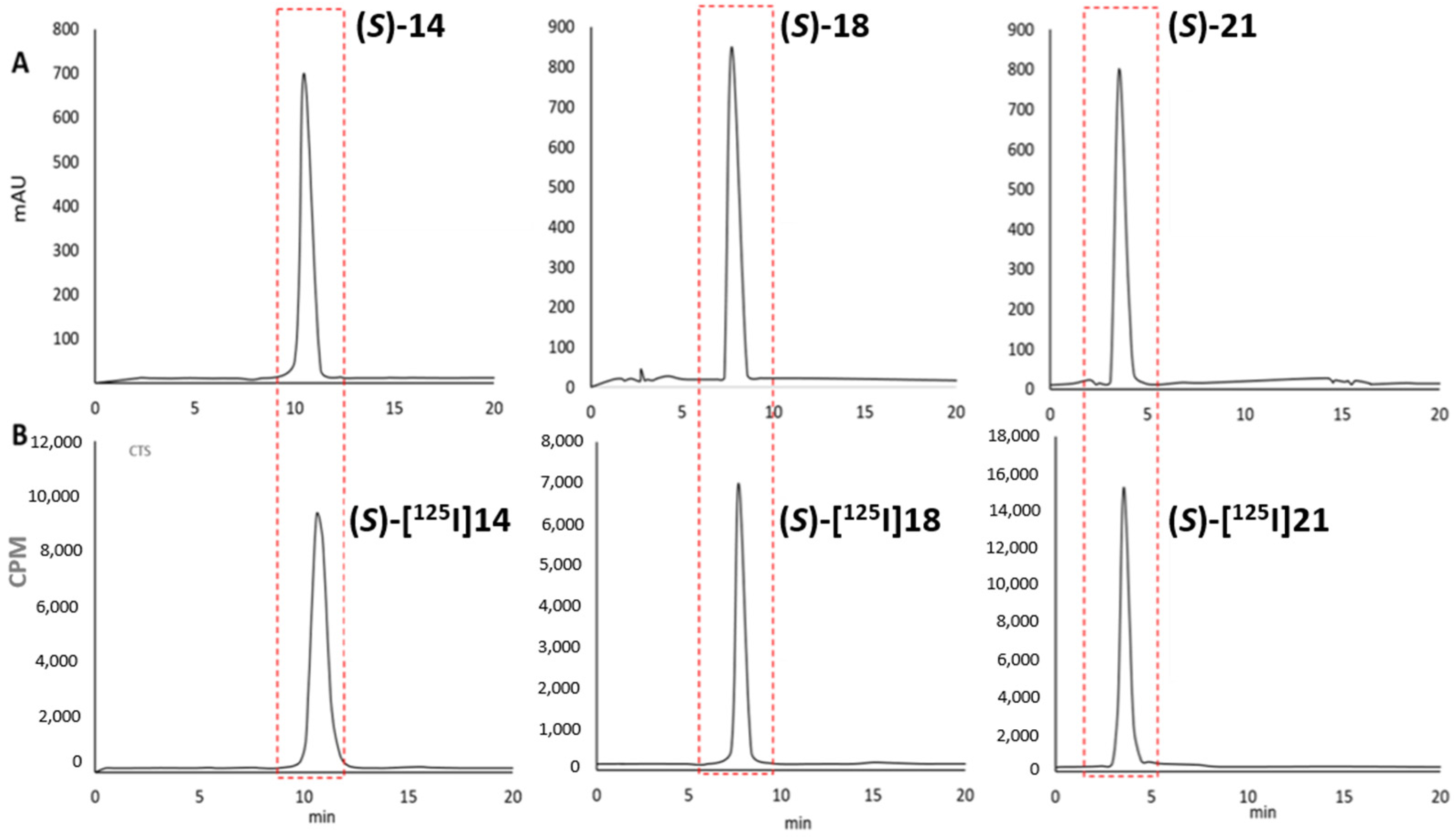
2.2.1. Synthesis of [125I]iodo-L-TIC(OH) Compounds
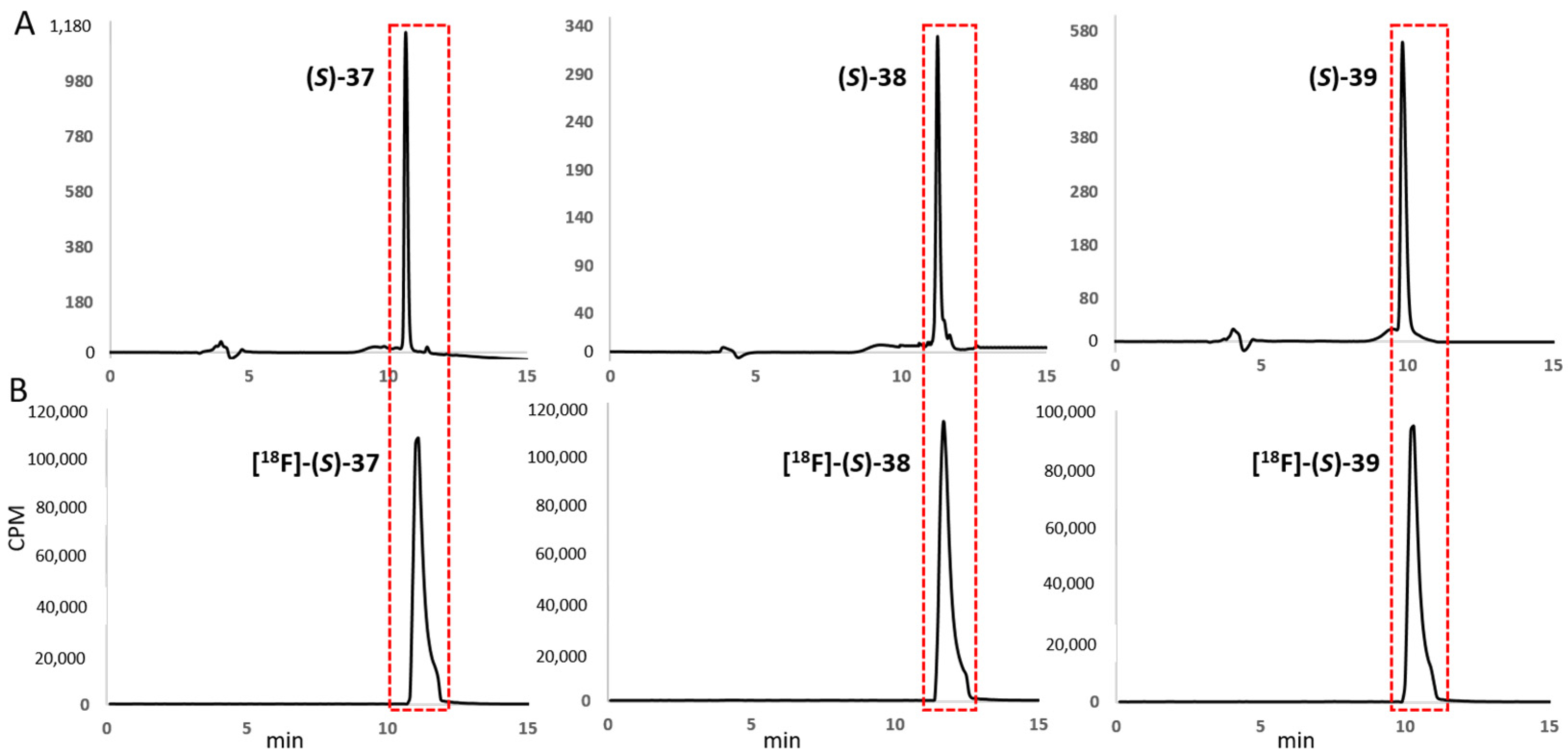
2.2.2. Synthesis of [19F]fluoro-D/L-TIC(OH) References and [18F]fluoro-L-TIC(OH) Radiotracers
3. Materials and Methods
3.1. Radiochemistry with Iodine-125
3.1.1. General Information
3.1.2. General Method for the Syntheses of (S)-[125I]14 ([125I]5-iodo-L-TIC(OH)), (S)-[125I]18 ([125I]6-iodo-L-TIC(OH)) and (S)-[125I]21 ([125I]8-iodo-L-TIC(OH))
3.2. Radiochemistry with Fluorine-18
3.2.1. General Information
3.2.2. Preparation of Anhydrous [18F]NaF in DMA
3.2.3. General Method for the Syntheses of (S)-[18F]37 ([18F]5-fluoro-L-TIC(OH)), (S)-[18F]38 ([18F]6-fluoro-L-TIC(OH)) and (S)-[18F]39 ([18F]8-fluoro-L-TIC(OH))
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taylor, P.M. Role of Amino Acid Transporters in Amino Acid Sensing. Am. J. Clin. Nutr. 2014, 99, 223S–230S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannai, S.; Christensen, H.N.; Vadgama, J.V.; Ellory, J.C.; Englesberg, E.; Guidotti, G.G.; Gazzola, G.C.; Kilberg, M.S.; Lajtha, A.; Sacktor, B. Amino Acid Transport Systems. Nature 1984, 311, 308. [Google Scholar] [PubMed] [Green Version]
- Christensen, H.N. Organic Ion Transport during Seven Decades. The Amino Acids. Biochim. Biophys. Acta 1984, 779, 255–269. [Google Scholar] [CrossRef] [Green Version]
- Oxender, D.L.; Christensen, H.N. Distinct Mediating Systems for The Transport of Neutral Amino Acids By The Ehrlich Cell. J. Biol. Chem. 1963, 238, 3686–3699. [Google Scholar] [CrossRef]
- Christensen, H.N. Distinguishing Amino Acid Transport Systems of a given Cell or Tissue. Meth. Enzymol. 1989, 173, 576–616. [Google Scholar]
- McGivan, J.D. Mammalian Amino Acid Transporters and Their Regulation: Introduction. Biochem. Soc. Trans. 1996, 24, 837–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdulhussein, A.A.; Wallace, H.M. Polyamines and Membrane Transporters. Amino Acids 2014, 46, 655–660. [Google Scholar] [CrossRef]
- Hediger, M.A.; Romero, M.F.; Peng, J.B.; Rolfs, A.; Takanaga, H.; Bruford, E.A. The ABCs of Solute Carriers: Physiological, Pathological and Therapeutic Implications of Human Membrane Transport Proteins, Introduction. Pflug. Arch. 2004, 447, 465–468. [Google Scholar] [CrossRef] [PubMed]
- César-Razquin, A.; Snijder, B.; Frappier-Brinton, T.; Isserlin, R.; Gyimesi, G.; Bai, X.; Reithmeier, R.A.; Hepworth, D.; Hediger, M.A.; Edwards, A.M.; et al. A Call for Systematic Research on Solute Carriers. Cell 2015, 162, 478–487. [Google Scholar] [CrossRef] [Green Version]
- Kandasamy, P.; Gyimesi, G.; Kanai, Y.; Hediger, M.A. Amino Acid Transporters Revisited: New Views in Health and Disease. Trends Biochem. Sci. 2018, 43, 752–789. [Google Scholar] [CrossRef]
- Bröer, S.; Palacín, M. The Role of Amino Acid Transporters in Inherited and Acquired Diseases. Biochem. J. 2011, 436, 193–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhutia, Y.D.; Babu, E.; Ramachandran, S.; Ganapathy, V. Amino Acid Transporters in Cancer and Their Relevance to “Glutamine Addiction”: Novel Targets for the Design of a New Class of Anticancer Drugs. Cancer Res. 2015, 75, 1782–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukey, M.J.; Katt, W.P.; Cerione, R.A. Targeting Amino Acid Metabolism for Cancer Therapy. Drug Discov. Today 2017, 22, 796–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; McConathy, J. Radiolabeled Amino Acids for Oncologic Imaging. J. Nucl. Med. 2013, 54, 1007–1010. [Google Scholar] [CrossRef] [Green Version]
- McConathy, J.; Yu, W.; Jarkas, N.; Seo, W.; Schuster, D.M.; Goodman, M.M. Radiohalogenated Nonnatural Amino Acids as PET and SPECT Tumor Imaging Agents. Med. Res. Rev. 2012, 32, 868–905. [Google Scholar] [CrossRef] [PubMed]
- Häfliger, P.; Charles, R.P. The L-Type Amino Acid Transporter LAT1-An Emerging Target in Cancer. Int. J. Mol. Sci. 2019, 20, 2428. [Google Scholar] [CrossRef] [Green Version]
- Salisbury, T.B.; Arthur, S. The Regulation and Function of the L-Type Amino Acid Transporter 1 (LAT1) in Cancer. Int. J. Mol. Sci. 2018, 19, 2373. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Ecker, G.F. Insights into the Structure, Function, and Ligand Discovery of the Large Neutral Amino Acid Transporter 1, LAT1. Int. J. Mol. Sci. 2018, 19, 1278. [Google Scholar] [CrossRef] [Green Version]
- Filss, C.P.; Cicone, F.; Shah, N.J.; Galldiks, N.; Langen, K.J. Amino Acid PET and MR Perfusion Imaging in Brain Tumours. Clin. Transl. Imaging 2017, 5, 209–223. [Google Scholar] [CrossRef] [Green Version]
- Galldiks, N.; Law, I.; Pope, W.B.; Arbizu, J.; Langen, K.J. The Use of Amino Acid PET and Conventional MRI for Monitoring of Brain Tumor Therapy. Neuroimage Clin. 2017, 13, 386–394. [Google Scholar] [CrossRef] [Green Version]
- Langen, K.J.; Stoffels, G.; Filss, C.; Heinzel, A.; Stegmayr, C.; Lohmann, P.; Willuweit, A.; Neumaier, B.; Mottaghy, F.M.; Galldiks, N. Imaging of Amino Acid Transport in Brain Tumours: Positron Emission Tomography with O-(2-[18F]Fluoroethyl)-L-Tyrosine (FET). Methods 2017, 130, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Langen, K.J.; Pauleit, D.; Coenen, H.H. 3-[(123)I]Iodo-Alpha-Methyl-L-Tyrosine: Uptake Mechanisms and Clinical Applications. Nucl. Med. Biol. 2002, 29, 625–631. [Google Scholar] [CrossRef]
- Giovacchini, G.; Riondato, M.; Giovannini, E.; Ciarmiello, A. Diagnostic Applications of Nuclear Medicine: Brain Tumors. In Nuclear Oncology; Strauss, H.W., Mariani, G., Volterrani, D., Larson, S.M., Eds.; Springer International Publishing: Cham, Germany, 2017; pp. 467–505. [Google Scholar]
- Krummeich, C.; Holschbach, M.; Stöcklin, G. Direct n.c.a. Electrophilic Radioiodination of Tyrosine Analogues; Their in Vivo Stability and Brain-Uptake in Mice. Appl. Radiat. Isot. 1994, 45, 929–935. [Google Scholar] [CrossRef]
- Hamacher, K.; Coenen, H.H. Efficient Routine Production of the 18F-Labelled Amino Acid O-2-18F Fluoroethyl-L-Tyrosine. Appl. Radiat. Isot. 2002, 57, 853–856. [Google Scholar] [CrossRef]
- Lu, M.Y.; Liu, Y.L.; Chang, H.H.; Jou, S.T.; Yang, Y.L.; Lin, K.H.; Lin, D.T.; Lee, Y.L.; Lee, H.; Wu, P.Y.; et al. Characterization of Neuroblastic Tumors Using 18F-FDOPA PET. J. Nucl. Med. 2013, 54, 42–49. [Google Scholar] [CrossRef] [Green Version]
- Balogova, S.; Talbot, J.N.; Nataf, V.; Michaud, L.; Huchet, V.; Kerrou, K.; Montravers, F. 18F-Fluorodihydroxyphenylalanine vs Other Radiopharmaceuticals for Imaging Neuroendocrine Tumours According to Their Type. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 943–966. [Google Scholar] [CrossRef] [Green Version]
- Taïeb, D.; Imperiale, A.; Pacak, K. [18]F-DOPA: The Versatile Radiopharmaceutical. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 1187–1189. [Google Scholar] [CrossRef]
- Campbell, M.G.; Ritter, T. Modern Carbon–Fluorine Bond Forming Reactions for Aryl Fluoride Synthesis. Chem. Rev. 2015, 115, 612–633. [Google Scholar] [CrossRef]
- Xu, P.; Zhao, D.; Berger, F.; Hamad, A.; Rickmeier, J.; Petzold, R.; Kondratiuk, M.; Bohdan, K.; Ritter, T. Site-selective late-stage aromatic [18F]fluorination via aryl sulfonium Salts. Angew. Chem. Int. Ed. 2020, 59, 1956–1960. [Google Scholar] [CrossRef] [Green Version]
- Preshlock, S.; Tredwell, M.; Gouverneur, V. 18F-Labeling of Arenes and Heteroarenes for Applications in Positron Emission Tomography. Chem. Rev. 2016, 116, 719–766. [Google Scholar] [CrossRef]
- Deng, X.; Rong, J.; Wang, L.; Vasdev, N.; Zhang, L.; Josephson, L.; Liang, S.H. Chemistry for Positron Emission Tomography: Recent Advances in 11C-, 18F-, 13N-, and 15O-Labeling Reactions. Angew. Chem. Int. Ed. 2019, 58, 2580–2605. [Google Scholar] [CrossRef]
- Maisonial-Besset, A.; Serre, A.; Ouadi, A.; Schmitt, S.; Canitrot, D.; Léal, F.; Miot-Noirault, E.; Brasse, D.; Marchand, P.; Chezal, J.M. Base/Cryptand/Metal-Free Automated Nucleophilic Radiofluorination of [18F]FDOPA from Iodonium Salts: Importance of Hydrogen Carbonate Counterion. Eur. J. Org. Chem. 2018, 2018, 7058–7065. [Google Scholar] [CrossRef]
- Ichiishi, N.; Brooks, A.F.; Topczewski, J.J.; Rodnick, M.E.; Sanford, M.S.; Scott, P.J.H. Copper-Catalyzed [18F]Fluorination of (Mesityl)(Aryl)Iodonium Salts. Org. Lett. 2014, 16, 3224–3227. [Google Scholar] [CrossRef] [Green Version]
- Makaravage, K.J.; Brooks, A.F.; Mossine, A.V.; Sanford, M.S.; Scott, P.J.H. Copper-Mediated Radiofluorination of Arylstannanes with [18F]KF. Org. Lett. 2016, 18, 5440–5443. [Google Scholar] [CrossRef]
- Lee, E.; Hooker, J.M.; Ritter, T. Nickel-Mediated Oxidative Fluorination for PET with Aqueous [18F] Fluoride. J. Am. Chem. Soc. 2012, 134, 17456–17458. [Google Scholar] [CrossRef] [Green Version]
- Preshlock, S.; Calderwood, S.; Verhoog, S.; Tredwell, M.; Huiban, M.; Hienzsch, A.; Gruber, S.; Wilson, T.C.; Taylor, N.J.; Cailly, T.; et al. Enhanced Copper-Mediated 18F-Fluorination of Aryl Boronic Esters Provides Eight Radiotracers for PET Applications. Chem. Commun. 2016, 52, 8361–8364. [Google Scholar] [CrossRef]
- Kuik, W.J.; Kema, I.P.; Brouwers, A.H.; Zijlma, R.; Neumann, K.D.; Dierckx, R.A.J.O.; DiMagno, S.G.; Elsinga, P.H. In Vivo Biodistribution of No-Carrier-Added 6-18F-Fluoro-3,4-Dihydroxy-L-Phenylalanine (18F-DOPA), Produced by a New Nucleophilic Substitution Approach, Compared with Carrier-Added 18F-DOPA, Prepared by Conventional Electrophilic Substitution. J. Nucl. Med. 2015, 56, 106–112. [Google Scholar] [CrossRef] [Green Version]
- Zarrad, F.; Zlatopolskiy, B.D.; Krapf, P.; Zischler, J.; Neumaier, B. A Practical Method for the Preparation of 18F-Labeled Aromatic Amino Acids from Nucleophilic [18F]Fluoride and Stannyl Precursors for Electrophilic Radiohalogenation. Molecules 2017, 22, 2231. [Google Scholar] [CrossRef] [Green Version]
- Zischler, J.; Kolks, N.; Modemann, D.; Neumaier, B.; Zlatopolskiy, B.D. Alcohol-Enhanced Cu-Mediated Radiofluorination. Chem. Eur. J. 2017, 23, 3251–3256. [Google Scholar] [CrossRef]
- Mossine, A.V.; Tanzey, S.S.; Brooks, A.F.; Makaravage, K.J.; Ichiishi, N.; Miller, J.M.; Henderson, B.D.; Skaddan, M.B.; Sanford, M.S.; Scott, P.J.H. One-Pot Synthesis of High Molar Activity 6-[18F]Fluoro-L-DOPA by Cu-Mediated Fluorination of a BPin Precursor. Org. Biomol. Chem. 2019, 17, 8701–8705. [Google Scholar] [CrossRef]
- Thomas, J.B.; Atkinson, R.N.; Rothman, R.B.; Fix, S.E.; Mascarella, S.W.; Vinson, N.A.; Xu, H.; Dersch, C.M.; Lu, Y.F.; Cantrell, B.E.; et al. Identification of the First Trans-(3R,4R)-Dimethyl-4-(3-Hydroxyphenyl)Piperidine Derivative To Possess Highly Potent and Selective Opioid κ Receptor Antagonist Activity. J. Med. Chem. 2001, 44, 2687–2690. [Google Scholar] [CrossRef] [PubMed]
- Carroll, F.I.; Dolle, R.E. The Discovery and Development of the N-Substituted Trans-3,4-Dimethyl-4-(3′-Hydroxyphenyl)Piperidine Class of Pure Opioid Receptor Antagonists. ChemMedChem 2014, 9, 1638–1654. [Google Scholar] [CrossRef] [PubMed]
- Buda, J.J.; Carroll, F.I.; Kosten, T.R.; Swearingen, D.; Walters, B.B. A Double-Blind, Placebo-Controlled Trial to Evaluate the Safety, Tolerability, and Pharmacokinetics of Single, Escalating Oral Doses of JDTic. Neuropsychopharmacology 2015, 40, 2059–2065. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Fang, H.; Feng, J.; Jia, Y.; Wang, X.; Xu, W. Discovery of a Tetrahydroisoquinoline-Based Hydroxamic Acid Derivative (ZYJ-34c) as Histone Deacetylase Inhibitor with Potent Oral Antitumor Activities. J. Med. Chem. 2011, 54, 5532–5539. [Google Scholar] [CrossRef]
- Maltais, R.; Poirier, D. Compounds for the Treatment of Hormone-Dependent Diseases. WO2015024111A1, 26 February 2015. [Google Scholar]
- Otake, K.; Azukizawa, S.; Fukui, M.; Kunishiro, K.; Kamemoto, H.; Kanda, M.; Miike, T.; Kasai, M.; Shirahase, H. Novel (S)-1,2,3,4-Tetrahydroisoquinoline-3-Carboxylic Acids: Peroxisome Proliferator-Activated Receptor γ Selective Agonists with Protein-Tyrosine Phosphatase 1B Inhibition. Bioorg. Med. Chem. 2012, 20, 1060–1075. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.X.; Njoroge, F.G.; Pichardo, J.; Prongay, A.; Butkiewicz, N.; Yao, N.; Madison, V.; Girijavallabhan, V. Potent 7-Hydroxy-1,2,3,4-Tetrahydroisoquinoline-3-Carboxylic Acid-Based Macrocyclic Inhibitors of Hepatitis C Virus NS3 Protease. J. Med. Chem. 2006, 49, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Donella-Deana, A.; Ruzza, P.; Cesaro, L.; Brunati, A.M.; Calderan, A.; Borin, G.; Pinna, L.A. Specific Monitoring of Syk Protein Kinase Activity by Peptide Substrates Including Constrained Analogs of Tyrosine. FEBS Lett. 2002, 523, 48–52. [Google Scholar] [CrossRef] [Green Version]
- Turner, A.H.; Lebhar, M.S.; Proctor, A.; Wang, Q.; Lawrence, D.S.; Allbritton, N.L. Rational Design of a Dephosphorylation-Resistant Reporter Enables Single-Cell Measurement of Tyrosine Kinase Activity. ACS Chem. Biol. 2016, 11, 355–362. [Google Scholar] [CrossRef]
- Samnick, S.; Schaefer, A.; Siebert, S.; Richter, S.; Vollmar, B.; Kirsch, C.M. Preparation and Investigation of Tumor Affinity, Uptake Kinetic and Transport Mechanism of Iodine-123-Labelled Amino Acid Derivatives in Human Pancreatic Carcinoma and Glioblastoma Cells. Nucl. Med. Biol. 2001, 28, 13–23. [Google Scholar] [CrossRef]
- Samnick, S.; Fozing, T.; Kirsch, C.M. Preparation and Tumor Affinity Testing of the Radioiodinated Tetrahydroisoquinoline Derivative [123I]TIC(OH) for Targeting Prostate Cancer. Appl. Radiat. Isot. 2006, 64, 563–569. [Google Scholar] [CrossRef]
- Samnick, S.; Nestle, U.; Wagner, M.; Fozing, T.; Schaefer, A.; Menger, M.D.; Kirsch, C.M. Validation of 8-[123I]Iodo-L-1,2,3,4-Tetrahydro-7-Hydroxyisoquinoline-3-Carboxylic Acid as an Imaging Agent for Prostate Cancer in Experimental Models of Human Prostate Cancer. Nucl. Med. Biol. 2007, 34, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Chelopo, M.P.; Pawar, S.A.; Sokhela, M.K.; Govender, T.; Kruger, H.G.; Maguire, G.E.M. Anticancer Activity of Ruthenium(II) Arene Complexes Bearing 1,2,3,4-Tetrahydroisoquinoline Amino Alcohol Ligands. Eur. J. Med. Chem. 2013, 66, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Flynn, D.L.; Petillo, P.A. Enzyme Modulators and Treatments. WO2006071940A2, 6 July 2006. [Google Scholar]
- Harling, J.D.; Thompson, M. Novel Compounds. U.S. Patent 2004142964A1, 22 July 2004. [Google Scholar]
- Ohta, M.; Takahashi, K.; Kasai, M.; Shoji, Y.; Kunishiro, K.; Miike, T.; Kanda, M.; Mukai, C.; Shirahase, H. Novel Tetrahydroisoquinoline Derivatives with Inhibitory Activities against Acyl-CoA: Cholesterol Acyltransferase and Lipid Peroxidation. Chem. Pharm. Bull. 2010, 58, 1066–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, M.L.; Kellar, K.J.; Levin, E.D.; Paige, M.A.; Rezvani, A.H.; Xiao, Y.; Yenugonda, V.M. Phenyl-Substituted Nicotinic Ligands, and Methods of Use Thereof. WO2013071067A1, 16 May 2013. [Google Scholar]
- Bohnert, G.; Chen, S.; Jiang, J.; Xia, Z. Compounds for Inflammation and Immune-Related Uses. WO2012151355A1, 8 November 2012. [Google Scholar]
- Moreno-Mañas, M.; Trepat, E.; Sebastián, R.M.; Vallribera, A. Asymmetric Synthesis of Quaternary α-Amino Acids Using d-Ribonolactone Acetonide as Chiral Auxiliary. Tetrahedron Asymmetry 1999, 10, 4211–4224. [Google Scholar] [CrossRef]
- Berrang, B.D.; Lewin, A.H.; Carroll, F.I. Preparation of Carbon-14 Labeled (3R)-7-Hydroxy-N-(1S)-1-{[(3R, 4R)-4-(3-Hydroxyphenyl)-3,4-Dimethyl-1-Piperidinyl]Methyl}-2-Methylpropyl-1,2,3,4-Tetrahydroisoquinolinecarboxamide (JDTic). J. Label. Compd. Radiopharm. 2008, 51, 440–443. [Google Scholar] [CrossRef]
- Beshore, D.C.; Dinsmore, C.J. Preparation of Ethyl 5-Iodo-1H-Indole-2-Carboxylate. Synth. Commun. 2003, 33, 2423–2427. [Google Scholar] [CrossRef]
- Zhang, Y.; Feng, J.; Liu, C.; Zhang, L.; Jiao, J.; Fang, H.; Su, L.; Zhang, X.; Zhang, J.; Li, M.; et al. Design, Synthesis and Preliminary Activity Assay of 1,2,3,4-Tetrahydroisoquinoline-3-Carboxylic Acid Derivatives as Novel Histone Deacetylases (HDACs) Inhibitors. Bioorg. Med. Chem. 2010, 18, 1761–1772. [Google Scholar] [CrossRef]
- Caldwell, R.D.; Guckian, K.M.; Kumaravel, G.; Lee, W.C.; Lin, E.Y.S.; Liu, X.; Ma, B.; Scott, D.M.; Shi, Z.; Taveras, A.G.; et al. Heterobicyclic Sphingosine 1-Phosphate Analogs. WO2010051031A1, 6 May 2010. [Google Scholar]
- Mentzel, U.V.; Tanner, D.; Tønder, J.E. Comparative Study of the Kumada, Negishi, Stille, and Suzuki−Miyaura Reactions in the Synthesis of the Indole Alkaloids Hippadine and Pratosine. J. Org. Chem. 2006, 71, 5807–5810. [Google Scholar] [CrossRef]
- Furuya, T.; Strom, A.E.; Ritter, T. Silver-Mediated Fluorination of Functionalized Aryl Stannanes. J. Am. Chem. Soc. 2009, 131, 1662–1663. [Google Scholar] [CrossRef]
- Tang, P.; Furuya, T.; Ritter, T. Silver-Catalyzed Late-Stage Fluorination. J. Am. Chem. Soc. 2010, 132, 12150–12154. [Google Scholar] [CrossRef] [Green Version]
- Bowden, G.D.; Pichler, B.J.; Maurer, A. A Design of Experiments (DoE) Approach Accelerates the Optimization of Copper-Mediated 18F-Fluorination Reactions of Arylstannanes. Sci. Rep. 2019, 9, 11370. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | (S)-35 (µmol) | Cu Complex | Eq. Cu Complex 1 | Eq. of Pyrizdine 1 | RCY 2 (%) |
|---|---|---|---|---|---|
| 1 | 10 | Cu(OTf)2 | 2 | 15 | 5 |
| 2 | 10 | Cu(OTf)2 | 4 | 15 | 28 |
| 3 | 20 | Cu(OTf)2 | 2 | 2.5 | 7 |
| 4 | 20 | Cu(OTf)2 | 2 | 4 | 30 |
| 5 | 20 | Cu(OTf)2 | 2 | 5 | 31 |
| 6 | 20 | Cu(OTf)2 | 2 | 7.5 | 24 |
| 7 | 20 | Cu(OTf)2 | 2 | 15 | 11 |
| 8 | 20 | Cu(OTf)2 | 2 | 25 | 9 |
| 9 | 20 | Cu(OTf)2 | 4 | 5 | 15 |
| 10 | 20 | Cu(OTf)2 | 4 | 7.5 | 18 |
| 11 | 20 | Cu(OTf)2 | 4 | 15 | 12 |
| 12 | 20 | Cu(OTf)2 | 4 | 25 | 5 |
| 13 | 30 | Cu(OTf)2 | 2 | 5 | 27 |
| 14 | 30 | Cu(OTf)2 | 4 | 15 | 12 |
| 15 | 20 | Cu(OTf)2(py)4 | 1 | 0 | 37 |
| 16 | 20 | Cu(OTf)2(py)4 | 1.5 | 0 | 54 |
| 17 | 20 | Cu(OTf)2(py)4 | 2 | 0 | 41 |
| 18 | 20 | Cu(OTf)2(py)4 | 2.5 | 0 | 36 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chao, M.N.; Chezal, J.-M.; Debiton, E.; Canitrot, D.; Witkowski, T.; Levesque, S.; Degoul, F.; Tarrit, S.; Wenzel, B.; Miot-Noirault, E.; et al. A Convenient Route to New (Radio)Fluorinated and (Radio)Iodinated Cyclic Tyrosine Analogs. Pharmaceuticals 2022, 15, 162. https://doi.org/10.3390/ph15020162
Chao MN, Chezal J-M, Debiton E, Canitrot D, Witkowski T, Levesque S, Degoul F, Tarrit S, Wenzel B, Miot-Noirault E, et al. A Convenient Route to New (Radio)Fluorinated and (Radio)Iodinated Cyclic Tyrosine Analogs. Pharmaceuticals. 2022; 15(2):162. https://doi.org/10.3390/ph15020162
Chicago/Turabian StyleChao, Maria Noelia, Jean-Michel Chezal, Eric Debiton, Damien Canitrot, Tiffany Witkowski, Sophie Levesque, Françoise Degoul, Sébastien Tarrit, Barbara Wenzel, Elisabeth Miot-Noirault, and et al. 2022. "A Convenient Route to New (Radio)Fluorinated and (Radio)Iodinated Cyclic Tyrosine Analogs" Pharmaceuticals 15, no. 2: 162. https://doi.org/10.3390/ph15020162
APA StyleChao, M. N., Chezal, J. -M., Debiton, E., Canitrot, D., Witkowski, T., Levesque, S., Degoul, F., Tarrit, S., Wenzel, B., Miot-Noirault, E., Serre, A., & Maisonial-Besset, A. (2022). A Convenient Route to New (Radio)Fluorinated and (Radio)Iodinated Cyclic Tyrosine Analogs. Pharmaceuticals, 15(2), 162. https://doi.org/10.3390/ph15020162