
Novel Small-Molecule Inhibitors of the SARS-CoV-2 Spike Protein Binding to Neuropilin 1



Abstract
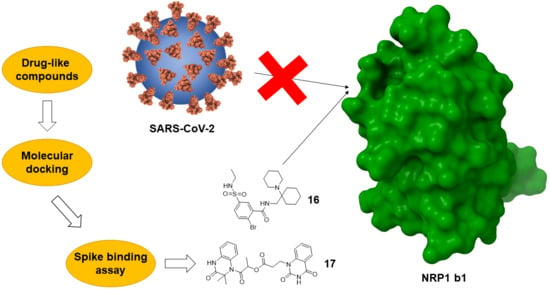
:
1. Introduction
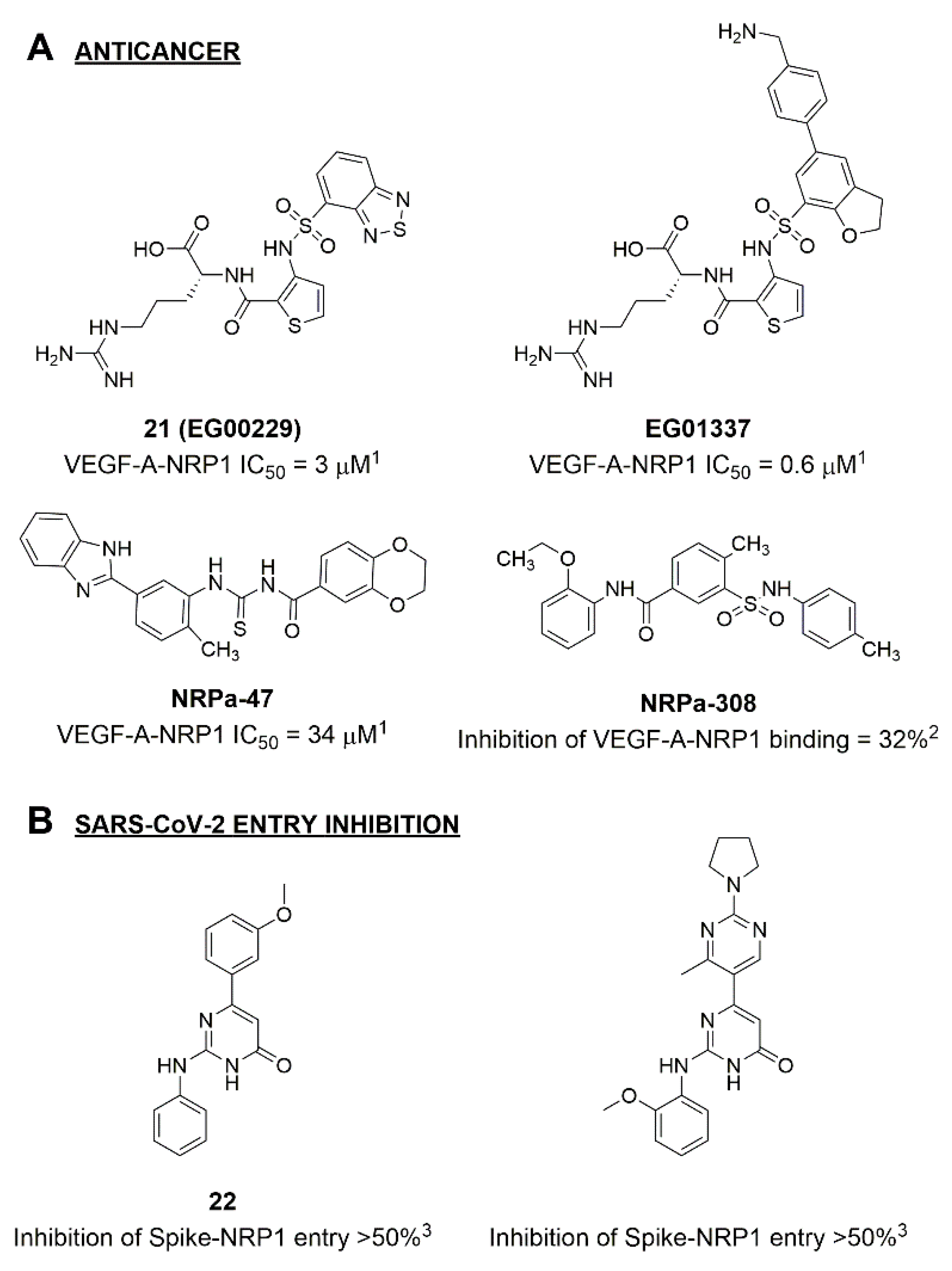
2. Results and Discussion
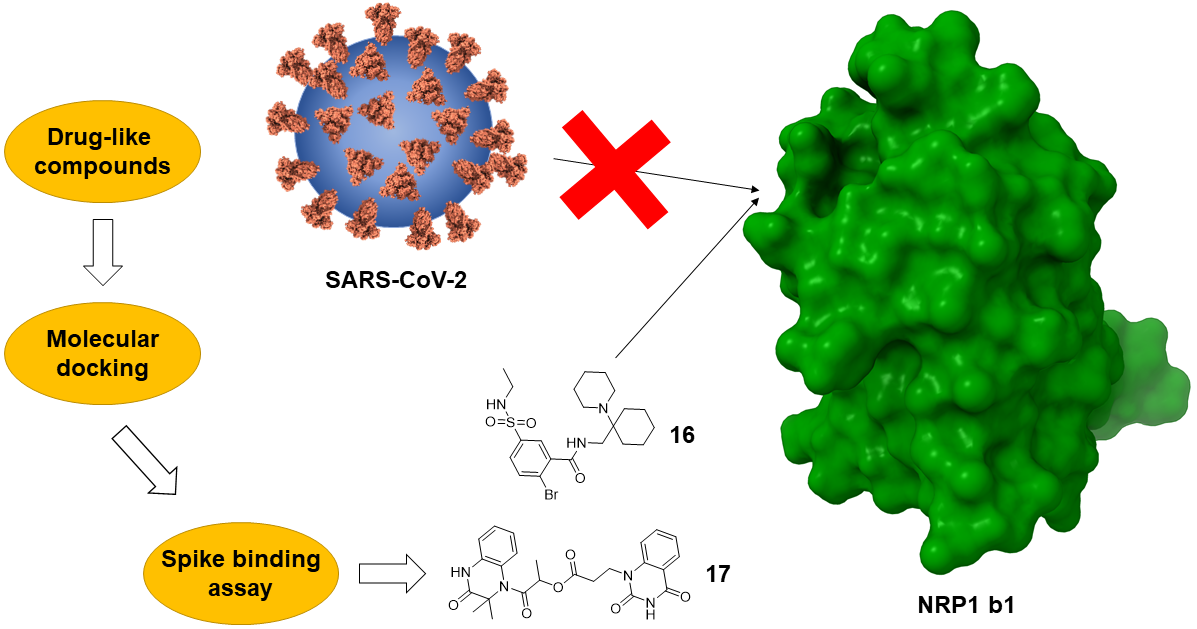
2.1. Molecular Docking and Selection of Top Compounds
2.2. In Vitro Evaluation of the Spike Binding Inhibition to NRP1
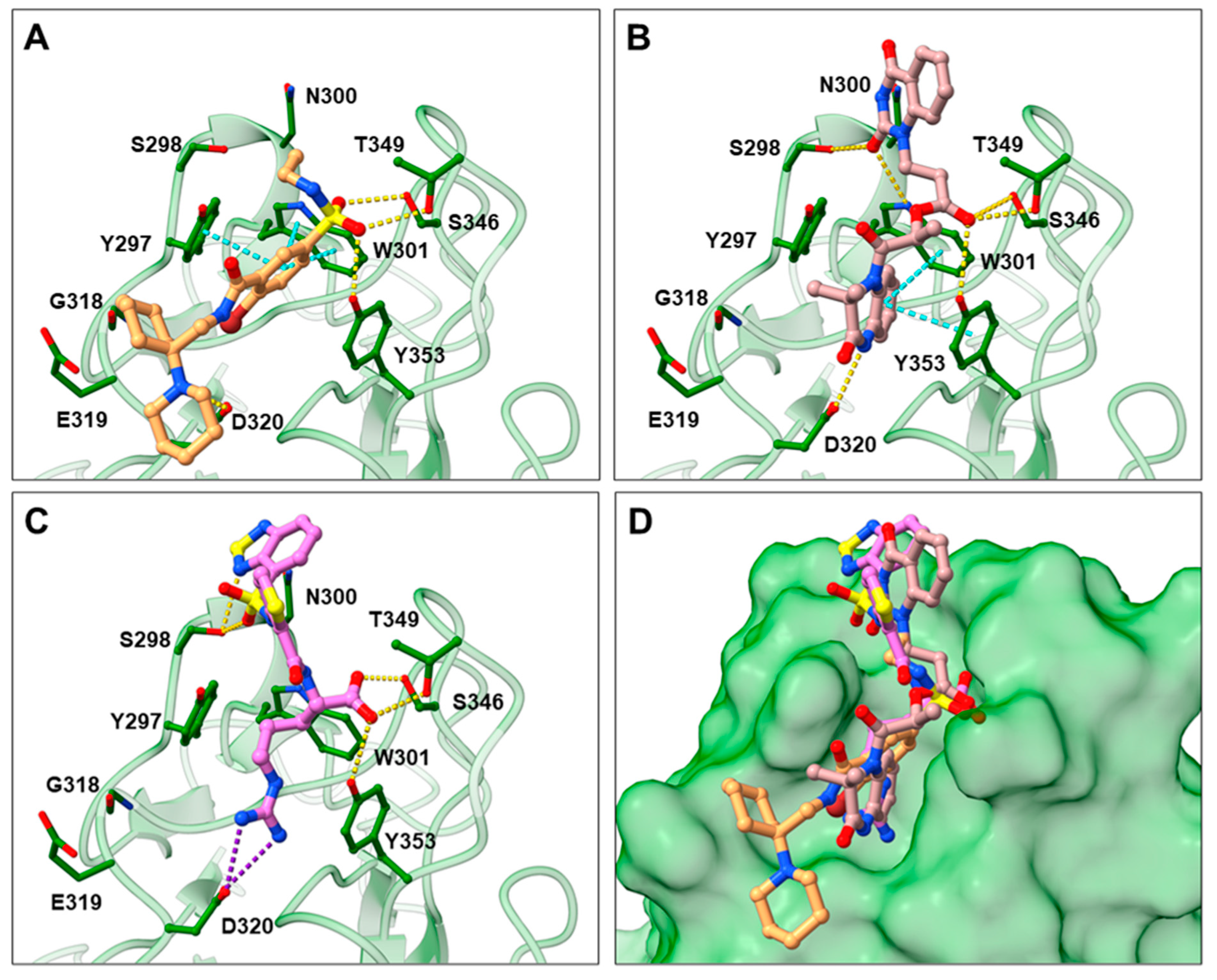
2.3. Prediction of Compounds’ 16 and 17 Binding to NRP1
3. Materials and Methods
3.1. Molecular Docking Calculations
3.2. Inhibition of SARS-CoV-2 Spike Binding to NRP1
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Available online: https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---5-october-2021 (accessed on 6 October 2021).
- Gupta, A.; Madhavan, M.V.; Sehgal, K.; Nair, N.; Mahajan, S.; Sehrawat, T.S.; Bikdeli, B.; Ahluwalia, N.; Ausiello, J.C.; Wan, E.Y.; et al. Extrapulmonary manifestations of COVID-19. Nat. Med. 2020, 26, 1017–1032. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Leon, S.; Wegman-Ostrosky, T.; Perelman, C.; Sepulveda, R.; Rebolledo, P.A.; Cuapio, A.; Villapol, S. More than 50 Long-term effects of COVID-19: A systematic review and meta-analysis. medRxiv 2021. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 Spike glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef]
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.-E.; Kavanagh Williamson, M.; Antón-Plágaro, C.; Shoemark, D.K.; Simón-Gracia, L.; Bauer, M.; Holland, R.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 2020, 370, 861–865. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef]
- Pellet-Many, C.; Frankel, P.; Jia, H.; Zachary, I. Neuropilins: Structure, function and role in disease. Biochem. J. 2008, 411, 211–226. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.; Bag, A.K.; Singh, R.K.; Talmadge, J.E.; Batra, S.K.; Datta, K. Multifaceted role of neuropilins in the immune system: Potential targets for immunotherapy. Front. Immunol. 2017, 8, 1228. [Google Scholar] [CrossRef] [Green Version]
- Gudowska-Sawczuk, M.; Mroczko, B. The Role of Neuropilin-1 (NRP-1) in SARS-CoV-2 Infection. J. Clin. Med. 2021, 10, 2772. [Google Scholar] [CrossRef]
- Heidari, F.; Karimi, E.; Firouzifar, M.; Khamushian, P.; Ansari, R.; Mohammadi Ardehali, M. Anosmia as a prominent symptom of COVID-19 infection. Rhinology 2020, 58, 302–303. [Google Scholar] [CrossRef]
- Mayi, B.S.; Leibowitz, J.A.; Woods, A.T.; Ammon, K.A.; Liu, A.E.; Raja, A. The role of Neuropilin-1 in COVID-19. PLoS Pathog. 2021, 17, e1009153. [Google Scholar] [CrossRef] [PubMed]
- Berlit, P.; Bösel, J.; Gahn, G.; Isenmann, S.; Meuth, S.G.; Nolte, C.H.; Pawlitzki, M.; Rosenow, F.; Schoser, B.; Thomalla, G.; et al. Neurological manifestations of COVID-19—Guideline of the German society of neurology. Neurol. Res. Pract. 2020, 2, 51. [Google Scholar] [CrossRef] [PubMed]
- Machhi, J.; Herskovitz, J.; Senan, A.M.; Dutta, D.; Nath, B.; Oleynikov, M.D.; Blomberg, W.R.; Meigs, D.D.; Hasan, M.; Patel, M.; et al. The natural history, pathobiology, and clinical manifestations of SARS-CoV-2 infections. J. Neuroimmune Pharmacol. 2020, 15, 359–386. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Bai, Y.; Zhu, Q.; Hu, B.; Xu, Y. Targeting VEGF–neuropilin interactions: A promising antitumor strategy. Drug Discov. 2019, 24, 656–664. [Google Scholar] [CrossRef]
- Niland, S.; Eble, J.A. Neuropilin: Handyman and power broker in the tumor microenvironment. In Tumor Microenvironment. Advances in Experimental Medicine and Biology, 1st ed.; Birbrair, A., Ed.; Springer: Cham, Switzerland, 2020; Volume 1223, pp. 31–67. [Google Scholar] [CrossRef]
- Parker, M.W.; Xu, P.; Li, X.; Vander Kooi, C.W. Structural basis for selective vascular endothelial growth factor-A (VEGF-A) binding to neuropilin-1. JBC 2012, 287, 11082–11089. [Google Scholar] [CrossRef] [Green Version]
- Osman, E.E.A.; Rehemtulla, A.; Neamati, N. Why all the fury over furin? J. Med. Chem. 2021; in press. [Google Scholar] [CrossRef]
- Teesalu, T.; Sugahara, K.N.; Kotamraju, V.R.; Ruoslahti, E. C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc. Natl. Acad. Sci. USA 2009, 106, 16157–16162. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.B.; Zhang, H.; Zhang, J.P.; Li, Y.; Zhao, B.; Feng, G.K.; Du, Y.; Xiong, D.; Zhong, Q.; Liu, W.-L.; et al. Neuropilin 1 is an entry factor that promotes EBV infection of nasopharyngeal epithelial cells. Nat. Commun. 2015, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Lambert, S.; Bouttier, M.; Vassy, R.; Seigneuret, M.; Petrow-Sadowski, C.; Janvier, S.; Heveker, N.; Ruscetti, F.W.; Perret, G.; Jones, K.S.; et al. HTLV-1 uses HSPG and neuropilin-1 for entry by molecular mimicry of VEGF165. Blood 2009, 113, 5176–5185. [Google Scholar] [CrossRef] [Green Version]
- Moutal, A.; Martin, L.F.; Boinon, L.; Gomez, K.; Ran, D.; Zhou, Y.; Stratton, H.J.; Cai, S.; Luo, S.; Gonzalez, K.B.; et al. SARS-CoV-2 spike protein co-opts VEGF-A/Neuropilin-1 receptor signaling to induce analgesia. Pain 2021, 162, 243–252. [Google Scholar] [CrossRef]
- Showers, W.M.; Leach, S.M.; Kechris, K.; Strong, M. Longitudinal analysis of SARS-CoV-2 spike and RNA-dependent RNA polymerase protein sequences reveals the emergence and geographic distribution of diverse mutations. Infect. Genet. Evol. 2022, 97, 105153. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.; Basiouni, S.; Parvin, R.; Hafez, H.M.; Shehata, A.A. Evolutionary insights into the furin cleavage sites of SARS-CoV-2 variants from humans and animals. Arch. Virol. 2021, 166, 2541–2549. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Li, X.; Gao, X.; Dong, Q. Natural polymorphisms are present in the furin cleavage site of the SARS-CoV-2 spike glycoprotein. Front. Genet. 2020, 11, 783. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Bagherzadeh, A.; Hartzoulakis, B.; Jarvis, A.; Löhr, M.; Shaikh, S.; Aqil, R.; Cheng, L.; Tickner, M.; Esposito, D.; et al. Characterization of a bicyclic peptide neuropilin-1 (NP-1) antagonist (EG3287) reveals importance of vascular endothelial growth factor exon 8 for NP-1 binding and role of NP-1 in KDR signaling. JBC 2006, 281, 13493–13502. [Google Scholar] [CrossRef] [Green Version]
- Jia, H.; Aqil, R.; Cheng, L.; Chapman, C.; Shaikh, S.; Jarvis, A.; Chan, A.W.E.; Hartzoulakis, B.; Evans, I.M.; Frolov, A.; et al. N-terminal modification of VEGF-A C terminus-derived peptides delineates structural features involved in neuropilin-1 binding and functional activity. ChemBioChem 2014, 15, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Starzec, A.; Vassy, R.; Martin, A.; Lecouvey, M.; Di Benedetto, M.; Crépin, M.; Perret, G.Y. Antiangiogenic and antitumor activities of peptide inhibiting the vascular endothelial growth factor binding to neuropilin-1. Life Sci. 2006, 79, 2370–2381. [Google Scholar] [CrossRef] [PubMed]
- Tymecka, D.; Puszko, A.K.; Lipiński, P.F.; Fedorczyk, B.; Wilenska, B.; Sura, K.; Perret, G.; Misicka, A. Branched pentapeptides as potent inhibitors of the vascular endothelial growth factor 165 binding to Neuropilin-1: Design, synthesis and biological activity. Eur. J. Med. Chem. 2018, 158, 453–462. [Google Scholar] [CrossRef]
- Grabowska, K.; Puszko, A.K.; Lipiński, P.F.; Laskowska, A.K.; Wileńska, B.; Witkowska, E.; Misicka, A. Design, synthesis and in vitro biological evaluation of a small cyclic peptide as inhibitor of vascular endothelial growth factor binding to neuropilin-1. Bioorg. Med. Chem. Lett. 2016, 26, 2843–2846. [Google Scholar] [CrossRef]
- Grabowska, K.; Puszko, A.K.; Lipiński, P.F.; Laskowska, A.K.; Wileńska, B.; Witkowska, E.; Perret, G.Y.; Misicka, A. Structure-activity relationship study of a small cyclic peptide Hc [Lys-Pro-Glu]-Arg-OH: A potent inhibitor of Vascular Endothelial Growth Factor interaction with Neuropilin-1. Bioorg. Med. Chem. 2017, 25, 597–602. [Google Scholar] [CrossRef]
- Tymecka, D.; Lipiński, P.F.; Fedorczyk, B.; Puszko, A.K.; Wileńska, B.; Perret, G.Y.; Misicka, A. Structure-activity relationship study of tetrapeptide inhibitors of the Vascular Endothelial Growth Factor A binding to Neuropilin-1. Peptides 2017, 94, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Fedorczyk, B.; Lipiński, P.F.; Tymecka, D.; Puszko, A.K.; Wilenska, B.; Perret, G.Y.; Misicka, A. Conformational latitude–activity relationship of KPPR tetrapeptide analogues toward their ability to inhibit binding of vascular endothelial growth factor 165 to neuropilin-1. J. Pept. Sci. 2017, 23, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Mo, Z.; Yu, F.; Han, S.; Yang, S.; Wu, L.; Li, P.; Jiao, S. New peptide MY1340 revert the inhibition effect of VEGF on dendritic cells differentiation and maturation via blocking VEGF-NRP-1 axis and inhibit tumor growth in vivo. Int. Immunopharmacol. 2018, 60, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, A.; Allerston, C.K.; Jia, H.; Herzog, B.; Garza-Garcia, A.; Winfield, N.; Ellard, K.; Aqil, R.; Lynch, R.; Chapman, C.; et al. Small molecule inhibitors of the neuropilin-1 vascular endothelial growth factor A (VEGF-A) interaction. J. Med. Chem. 2010, 53, 2215–2226. [Google Scholar] [CrossRef] [PubMed]
- Novoa, A.; Pellegrini-Moïse, N.; Bechet, D.; Barberi-Heyob, M.; Chapleur, Y. Sugar-based peptidomimetics as potential inhibitors of the vascular endothelium growth factor binding to neuropilin-1. Bioorg. Med. Chem. 2010, 18, 3285–3298. [Google Scholar] [CrossRef]
- Richard, M.; Chateau, A.; Jelsch, C.; Didierjean, C.; Manival, X.; Charron, C.; Bernardi, M.; Murielc, B.-H.; Yvesa, C.; Cédricc, B.; et al. Carbohydrate-based peptidomimetics targeting neuropilin-1: Synthesis, molecular docking study and in vitro biological activities. Bioorg. Med. Chem. 2016, 24, 5315–5325. [Google Scholar] [CrossRef]
- Powell, J.; Mota, F.; Steadman, D.; Soudy, C.; Miyauchi, J.T.; Crosby, S.; Jarvis, A.; Reisinger, T.; Winfield, N.; Evans, G.; et al. Small molecule neuropilin-1 antagonists combine antiangiogenic and antitumor activity with immune modulation through reduction of transforming growth factor beta (TGFβ) production in regulatory T-cells. J. Med. Chem. 2018, 61, 4135–4154. [Google Scholar] [CrossRef]
- Borriello, L.; Montès, M.; Lepelletier, Y.; Leforban, B.; Liu, W.Q.; Demange, L.; Delhomme, B.; Pavoni, S.; Jarray, R.; Boucher, J.L.; et al. Structure-based discovery of a small non-peptidic Neuropilins antagonist exerting in vitro and in vivo anti-tumor activity on breast cancer model. Cancer Lett. 2014, 349, 120–127. [Google Scholar] [CrossRef]
- Starzec, A.; Miteva, M.A.; Ladam, P.; Villoutreix, B.O.; Perret, G.Y. Discovery of novel inhibitors of vascular endothelial growth factor-A–Neuropilin-1 interaction by structure-based virtual screening. Bioorg. Med. Chem. 2014, 22, 4042–4048. [Google Scholar] [CrossRef]
- Liu, W.Q.; Lepelletier, Y.; Montès, M.; Borriello, L.; Jarray, R.; Grépin, R.; Leforban, B.; Loukaci, A.; Benhida, R.; Hermine, O.; et al. NRPa-308, a new neuropilin-1 antagonist, exerts in vitro anti-angiogenic and anti-proliferative effects and in vivo anti-cancer effects in a mouse xenograft model. Cancer Lett. 2018, 414, 88–98. [Google Scholar] [CrossRef]
- Liu, W.Q.; Megale, V.; Borriello, L.; Leforban, B.; Montès, M.; Goldwaser, E.; Gresh, N.; Piquemal, J.-P.; Hadj-Slimane, R.; Hermine, O.; et al. Synthesis and structure–activity relationship of non-peptidic antagonists of neuropilin-1 receptor. Bioorg. Med. Chem. Lett. 2014, 24, 4254–4259. [Google Scholar] [CrossRef]
- Brachet, E.; Dumond, A.; Liu, W.Q.; Fabre, M.; Selkti, M.; Raynaud, F.; Hermine, O.; Benhida, R.; Belmont, P.; Garbay, C.; et al. Synthesis, 3D-structure and stability analyses of NRPa-308, a new promising anti-cancer agent. Bioorg. Med. Chem. Lett. 2019, 29, 126710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, K.; Li, Y.; Bai, Y.; Jiang, T.; Sun, H.; Zhu, Q.; Xu, Y. Discovery of novel nonpeptide small-molecule NRP1 antagonists: Virtual screening, molecular simulation and structural modification. Bioorg. Med. Chem. 2020, 28, 115183. [Google Scholar] [CrossRef] [PubMed]
- Perez-Miller, S.; Patek, M.; Moutal, A.; Duran, P.; Cabel, C.R.; Thorne, C.A.; Campos, S.K.; Khanna, R. Novel compounds targeting neuropilin receptor 1 with potential to interfere with SARS-CoV-2 virus entry. ACS Chem. Neurosci. 2021, 12, 1299–1312. [Google Scholar] [CrossRef] [PubMed]
- Klaewkla, M.; Charoenwongpaiboon, T.; Mahalapbutr, P. Molecular basis of the new COVID-19 target neuropilin-1 in complex with SARS-CoV-2 S1 C-end rule peptide and small-molecule antagonists. J. Mol. Liq. 2021, 335, 116537. [Google Scholar] [CrossRef]
- ZINC. Available online: https://zinc15.docking.org/ (accessed on 6 October 2021).
- Shoichet, B.K. Interpreting steep dose-response curves in early inhibitor discovery. J. Med. Chem. 2006, 49, 7274–7277. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Saubern, S.; Guha, R.; Baell, J.B. KNIME workflow to assess PAINS filters in SMARTS format. Comparison of RDKit and Indigo cheminformatics libraries. Mol. Inform. 2011, 30, 847–850. [Google Scholar] [CrossRef]
- Li, Y.; Han, L.; Liu, Z.; Wang, R. Comparative assessment of scoring functions on an updated benchmark: 2. Evaluation methods and general results. J. Chem. Inf. Model. 2014, 54, 1717–1736. [Google Scholar] [CrossRef]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Thiel, K.; Wiswedel, B. KNIME-the Konstanz information miner: Version 2.0 and beyond. SIGKDD Explor. 2009, 11, 26–31. [Google Scholar] [CrossRef] [Green Version]
- Irwin, J.J.; Duan, D.; Torosyan, H.; Doak, A.K.; Ziebart, K.T.; Sterling, T.; Tumanian, G.; Shoichet, B.K. An aggregation advisor for ligand discovery. J. Med. Chem. 2015, 58, 7076–7087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylo, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins Struct. Funct. Bioinf. 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Structure | ChemPLP Score 1 | Inhibition of Spike-NRP1 Binding [%] 2 |
|---|---|---|---|
| 1 |  | 75.5 | 45.43 ± 1.58 |
| 2 |  | 74.5 | 34.81 ± 1.09 |
| 3 |  | 74.0 | 16.35 ± 1.95 |
| 4 |  | 76.0 | 25.38 ± 0.82 |
| 5 |  | 75.4 | 15.67 ± 1.02 |
| 6 |  | 75.0 | 35.62 ± 1.43 |
| 7 |  | 77.3 | 9.37 ± 0.59 |
| 8 |  | 77.1 | 13.00 ± 6.39 |
| 9 |  | 75.7 | 35.25 ± 4.35 |
| 10 |  | 73.1 | 41.52 ± 0.57 |
| 11 |  | 73.8 | 32.16 ± 1.28 |
| 12 |  | 80.7 | 38.71 ± 1.68 |
| 13 |  | 74.3 | 31.86 ± 2.79 |
| 14 |  | 77.3 | 19.46 ± 2.37 |
| 15 |  | 76.7 | 25.48 ± 5.11 |
| 16 |  | 74.6 | 61.44 ± 2.48 |
| 17 |  | 77.1 | 63.58 ± 1.27 |
| 18 |  | 73.0 | 16.88 ± 0.31 |
| 19 |  | 72.7 | 9.73 ± 0.04 |
| 20 |  | 72.1 | 17.56 ± 1.65 |
| 21 EG00229 [33] |  | 50.57 ± 2.26 | |
| 22 [43] |  | 28.71 ± 0.80 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolarič, A.; Jukič, M.; Bren, U. Novel Small-Molecule Inhibitors of the SARS-CoV-2 Spike Protein Binding to Neuropilin 1. Pharmaceuticals 2022, 15, 165. https://doi.org/10.3390/ph15020165
Kolarič A, Jukič M, Bren U. Novel Small-Molecule Inhibitors of the SARS-CoV-2 Spike Protein Binding to Neuropilin 1. Pharmaceuticals. 2022; 15(2):165. https://doi.org/10.3390/ph15020165
Chicago/Turabian StyleKolarič, Anja, Marko Jukič, and Urban Bren. 2022. "Novel Small-Molecule Inhibitors of the SARS-CoV-2 Spike Protein Binding to Neuropilin 1" Pharmaceuticals 15, no. 2: 165. https://doi.org/10.3390/ph15020165
APA StyleKolarič, A., Jukič, M., & Bren, U. (2022). Novel Small-Molecule Inhibitors of the SARS-CoV-2 Spike Protein Binding to Neuropilin 1. Pharmaceuticals, 15(2), 165. https://doi.org/10.3390/ph15020165