
Pyrazole and Triazole Derivatives as Mycobacterium tuberculosis UDP-Galactopyranose Inhibitors


Abstract
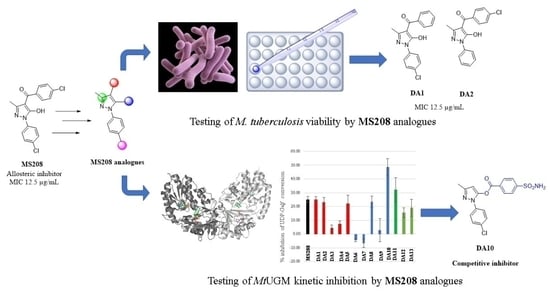
:
1. Introduction
2. Results and Discussions
2.1. Synthesis of the Target Compounds
2.2. Antituberculosis Activity
2.3. MtbUGM Inhibitory Activities
2.3.1. Kinetic Inhibition
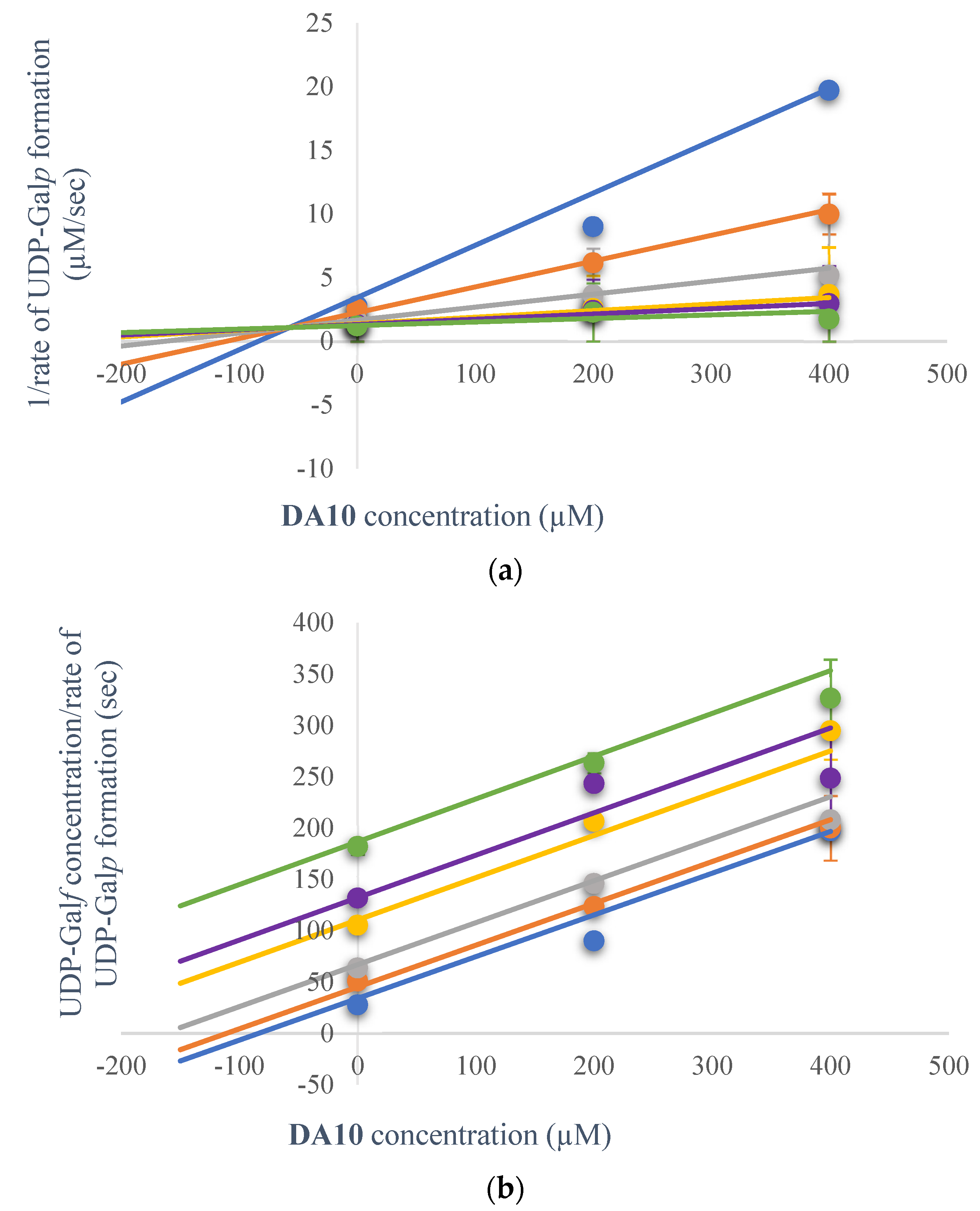
2.3.2. Kinetic Inhibition Mechanism of DA10
3. Materials and Methods
3.1. Chemistry
General Procedure for the Syntheses of Starting Materials
3.2. Expression and Purification of MtbUGM
3.3. Inhibition Assays
3.3.1. Determination of % Inhibition
3.3.2. Determination of Inhibition Model
3.4. Antituberculosis Activity Determination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). Global Tuberculosis Report 2021; World Health Organization (WHO): Geneva, Switzerland, 2021; ISBN 9789240037021. Available online: https://www.who.int/publications/i/item/9789240037021 (accessed on 1 February 2022).
- Pan, F.; Jackson, M.; Ma, Y.; McNeil, M. Cell wall core galactofuran synthesis is essential for growth of mycobacteria. J. Bacteriol. 2001, 183, 3991–3998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peltier, P.; Euzen, R.; Daniellou, R.; Nugier-Chauvin, C.; Ferrières, V. Recent knowledge and innovations related to hexofuranosides: Structure, synthesis and applications. Carbohydr. Res. 2008, 343, 1897–1923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borrelli, S.; Zandberg, W.F.; Mohan, S.; Ko, M.; Martinez-Gutierrez, F.; Partha, S.K.; Sanders, D.A.R.; Av-Gay, Y.; Pinto, B.M. Antimycobacterial activity of UDP-galactopyranose mutase inhibitors. Int. J. Antimicrob. Agents 2010, 36, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Partha, S.K.; Sadeghi-Khomami, A.; Slowski, K.; Kotake, T.; Thomas, N.R.; Jakeman, D.L.; Sanders, D.A.R. Chemoenzymatic Synthesis, Inhibition Studies, and X-ray Crystallographic Analysis of the Phosphono Analog of UDP-Galp as an Inhibitor and Mechanistic Probe for UDP-Galactopyranose Mutase. J. Mol. Biol. 2010, 403, 578–590. [Google Scholar] [CrossRef] [PubMed]
- N’Go, I.; Golten, S.; Ardá, A.; Cañada, J.; Jiménez-Barbero, J.; Linclau, B.; Vincent, S.P. Tetrafluorination of Sugars as Strategy for Enhancing Protein–Carbohydrate Affinity: Application to UDP-Galp Mutase Inhibition. Chem. Eur. J. 2014, 20, 106–112. [Google Scholar] [CrossRef]
- Soltero-Higgin, M.; Carlson, E.E.; Phillips, J.H.; Kiessling, L.L. Identification of Inhibitors for UDP-Galactopyranose Mutase. J. Am. Chem. Soc. 2004, 126, 10532–10533. [Google Scholar] [CrossRef]
- Konyariková, Z.; Savková, K.; Kozmon, S.; Mikušová, K. Biosynthesis of Galactan in Mycobacterium tuberculosis as a Viable TB Drug Target? Antibiotics 2020, 9, 20. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Colombo, C.; Kuttiyatveetil, J.R.A.; Zalatar, N.; van Straaten, K.E.; Mohan, S.; Sanders, D.A.R.; Pinto, B.M. A Second, Druggable Binding Site in UDP-Galactopyranose Mutase from Mycobacterium tuberculosis? ChemBioChem 2016, 17, 2264–2273. [Google Scholar] [CrossRef]
- Manetti, F.; Magnani, M.; Castagnolo, D.; Passalacqua, L.; Botta, M.; Corelli, F.; Saddi, M.; Deidda, D.; De Logu, A. Ligand-based virtual screening, parallel solution-phase and microwave-assisted synthesis as tools to identify and synthesize new inhibitors of mycobacterium tuberculosis. ChemMedChem 2006, 1, 973–989. [Google Scholar] [CrossRef]
- Castagnolo, D.; De Logu, A.; Radi, M.; Bechi, B.; Manetti, F.; Magnani, M.; Supino, S.; Meleddu, R.; Chisu, L.; Botta, M. Synthesis, biological evaluation and SAR study of novel pyrazole analogues as inhibitors of Mycobacterium tuberculosis. Bioorg. Med. Chem. 2008, 16, 8587–8591. [Google Scholar] [CrossRef] [Green Version]
- Castagnolo, D.; Manetti, F.; Radi, M.; Bechi, B.; Pagano, M.; De Logu, A.; Meleddu, R.; Saddi, M.; Botta, M. Synthesis, biological evaluation, and SAR study of novel pyrazole analogues as inhibitors of Mycobacterium tuberculosis: Part 2. Synthesis of rigid pyrazolones. Bioorg. Med. Chem. 2009, 17, 5716–5721. [Google Scholar] [CrossRef] [Green Version]
- Reddyrajula, R.; Dalimba, U.K. Structural modification of zolpidem led to potent antimicrobial activity in imidazo[1,2-a]pyridine/pyrimidine-1,2,3-triazoles. New J. Chem. 2019, 43, 16281–16299. [Google Scholar] [CrossRef]
- Zhang, S.; Xu, Z.; Gao, C.; Ren, Q.-C.; Chang, L.; Lv, Z.-S.; Feng, L.-S. Triazole derivatives and their anti-tubercular activity. Eur. J. Med. Chem. 2017, 138, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.-M.; Wu, J.B.; Lin, H.-C.; Huang, J.-J.; Huang, Y.-Y.; Lin, S.-K.; Lin, T.-P.; Wong, F.F. Dibromination of 5-pyrazolones and 5-hydroxypyrazoles via dibromoisocyanuric acid. J. Heterocycl. Chem. 2010, 47, 1153–1156. [Google Scholar] [CrossRef]
- Tan, J.-N.; Li, M.; Gu, Y. Multicomponent reactions of 1,3-disubstituted 5-pyrazolones and formaldehyde in environmentally benign solvent systems and their variations with more fundamental substrates. Green Chem. 2010, 12, 908–914. [Google Scholar] [CrossRef]
- Metwally, M.; Bondock, S.; El-Desouky, S.; Abdou, M. ChemInform Abstract: Pyrazol-5-ones: Tautomerism, Synthesis and Reactions. Int. J. Mod. Org. Chem. 2013, 1, 19–54. [Google Scholar] [CrossRef]
- Jensen, B.S. The synthesis of 1-phenyl-3-methyl-4-acyl-pyrazolones-5. Acta Chem. Scand. 1959, 13, 1668–1670. [Google Scholar] [CrossRef]
- Maruoka, H.; Yamagata, K.; Okabe, F.; Tomioka, Y. Synthesis of 1-acyl-1,2-dihydro-3H-pyrazol-3-ones VIA lewis acid-mediated rearrangement of 3-acyloxypyrazoles. J. Heterocycl. Chem. 2006, 43, 859–865. [Google Scholar] [CrossRef]
- Kurteva, V.B.; Petrova, M.A. Synthesis of 3-Methyl-4-(4-methylbenzoyl)-1-phenyl-pyrazol-5-one: How To Avoid O-Acylation. J. Chem. Educ. 2015, 92, 382–384. [Google Scholar] [CrossRef]
- García, H.; Primo, J.; Miranda, M.A. The Photo-Fries Rearrangement in the Presence of Potassium Carbonate: A Convenient Synthesis of ortho-Hydroxyacetophenones. Synthesis 1985, 1985, 901–902. [Google Scholar] [CrossRef]
- Salem, O.I.; Schulz, T.; Hartmann, R.W. Synthesis and biological evaluation of 4-(4-(alkyl- and phenylaminocarbonyl)benzoyl)benzoic acid derivatives as non-steroidal inhibitors of steroid 5 alpha-reductase isozymes 1 and 2. Arch. Pharm. Pharm. Med. Chem. 2002, 335, 83–88. [Google Scholar] [CrossRef]
- Bock, V.D.; Hiemstra, H.; van Maarseveen, J.H. CuI-Catalyzed Alkyne–Azide “Click” Cycloadditions from a Mechanistic and Synthetic Perspective. Eur. J. Org. Chem. 2006, 2006, 51–68. [Google Scholar] [CrossRef]
- Dai, Z.-C.; Chen, Y.-F.; Zhang, M.; Li, S.-K.; Yang, T.-T.; Shen, L.; Wang, J.-X.; Qian, S.-S.; Zhu, H.-L.; Ye, Y.-H. Synthesis and antifungal activity of 1,2,3-triazole phenylhydrazone derivatives. Org. Biomol. Chem. 2015, 13, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhuang, X.; Pan, X.; Zhang, Z.; Duan, L.; Liu, Y.; Zhang, L.; Ren, X.; Ding, K. 1-Phenyl-4-benzoyl-1H-1,2,3-triazoles as orally bioavailable transcriptional function suppressors of estrogen-related receptor α. J. Med. Chem. 2013, 56, 4631–4640. [Google Scholar] [CrossRef] [PubMed]
- Pereira, G.R.; Brandão, G.C.; Arantes, L.M.; de Oliveira, H.A.; de Paula, R.C.; do Nascimento, M.F.A.; dos Santos, F.M.; da Rocha, R.K.; Lopes, J.C.D.; de Oliveira, A.B. 7-Chloroquinolinotriazoles: Synthesis by the azide–alkyne cycloaddition click chemistry, antimalarial activity, cytotoxicity and SAR studies. Eur. J. Med. Chem. 2014, 73, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Franzblau, S.G.; Witzig, R.S.; McLaughlin, J.C.; Torres, P.; Madico, G.; Hernandez, A.; Degnan, M.T.; Cook, M.B.; Quenzer, V.K.; Ferguson, R.M.; et al. Rapid, low-technology MIC determination with clinical Mycobacterium tuberculosis isolates by using the microplate Alamar Blue assay. J. Clin. Microbiol. 1998, 36, 362–366. [Google Scholar] [CrossRef] [Green Version]
- Khalifa, R.A.; Nasser, M.S.; Gomaa, A.A.; Osman, N.M.; Salem, H.M. Resazurin Microtiter Assay Plate method for detection of susceptibility of multidrug resistant Mycobacterium tuberculosis to second-line anti-tuberculous drugs. Egypt. J. Chest Dis. Tuberc. 2013, 62, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Franzblau, S.G.; DeGroote, M.A.; Cho, S.H.; Andries, K.; Nuermberger, E.; Orme, I.M.; Mdluli, K.; Angulo-Barturen, I.; Dick, T.; Dartois, V.; et al. Comprehensive analysis of methods used for the evaluation of compounds against Mycobacterium tuberculosis. Tuberculosis 2012, 92, 453–488. [Google Scholar] [CrossRef] [Green Version]
- Nayak, M.; Batchu, H.; Batra, S. Straightforward copper-catalyzed synthesis of pyrrolopyrazoles from halogenated pyrazolecarbaldehydes. Tetrahedron Lett. 2012, 53, 4206–4208. [Google Scholar] [CrossRef]
- Wang, B.-L.; Wu, J.; Liu, Q.-X.; Li, Y.-H.; Song, H.-B.; Li, Z.-M. Synthesis, Structure, and Biological Activities of [5-(Arylthio/sulfinyl/sulfonyl)-3-methyl-1-phenyl-1H-pyrazol-4-yl]-arylmethanones. Phosphorus Sulfur Silicon Relat. Elem. 2015, 190, 66–78. [Google Scholar] [CrossRef]
- Beis, K.; Srikannathasan, V.; Liu, H.; Fullerton, S.W.B.; Bamford, V.A.; Sanders, D.A.R.; Whitfield, C.; McNeil, M.R.; Naismith, J.H. Crystal structures of Mycobacteria tuberculosis and Klebsiella pneumoniae UDP-galactopyranose mutase in the oxidised state and Klebsiella pneumoniae UDP-galactopyranose mutase in the (active) reduced state. J. Mol. Biol. 2005, 348, 971–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Fu, H.; Dieu, M.; Halloum, I.; Kremer, L.; Xia, Y.; Pan, W.; Vincent, S.P. Identification of inhibitors targeting Mycobacterium tuberculosis cell wall biosynthesis via dynamic combinatorial chemistry. Chem. Commun. 2017, 53, 10632–10635. [Google Scholar] [CrossRef] [PubMed]
- Van Straaten, K.E.; Kuttiyatveetil, J.R.A.; Sevrain, C.M.; Villaume, S.A.; Jiménez-Barbero, J.; Linclau, B.; Vincent, S.P.; Sanders, D.A.R. Structural Basis of Ligand Binding to UDP-Galactopyranose Mutase from Mycobacterium tuberculosis Using Substrate and Tetrafluorinated Substrate Analogues. J. Am. Chem. Soc. 2015, 137, 1230–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test Compound | MIC (µg/mL) |
|---|---|
| MS208 | 12.5 |
| DA1 | 12.5 |
| DA2 | 12.5 |
| DA3 | 25 |
| DA4 | >100 |
| DA5 | 25 |
| DA6 | 50 |
| DA7 | >100 |
| DA8 | 50 |
| DA9 | >100 |
| DA10 | >100 |
| DA11 | >100 |
| DA12 | 50 |
| DA13 | >100 |
| INH | 0.25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, D.M.; Chen, J.M.; Sanders, D.A.R. Pyrazole and Triazole Derivatives as Mycobacterium tuberculosis UDP-Galactopyranose Inhibitors. Pharmaceuticals 2022, 15, 197. https://doi.org/10.3390/ph15020197
Ahmed DM, Chen JM, Sanders DAR. Pyrazole and Triazole Derivatives as Mycobacterium tuberculosis UDP-Galactopyranose Inhibitors. Pharmaceuticals. 2022; 15(2):197. https://doi.org/10.3390/ph15020197
Chicago/Turabian StyleAhmed, Dalia M., Jeffrey M. Chen, and David A. R. Sanders. 2022. "Pyrazole and Triazole Derivatives as Mycobacterium tuberculosis UDP-Galactopyranose Inhibitors" Pharmaceuticals 15, no. 2: 197. https://doi.org/10.3390/ph15020197
APA StyleAhmed, D. M., Chen, J. M., & Sanders, D. A. R. (2022). Pyrazole and Triazole Derivatives as Mycobacterium tuberculosis UDP-Galactopyranose Inhibitors. Pharmaceuticals, 15(2), 197. https://doi.org/10.3390/ph15020197