
Late Sodium Current of the Heart: Where Do We Stand and Where Are We Going?

 ,
,
Abstract
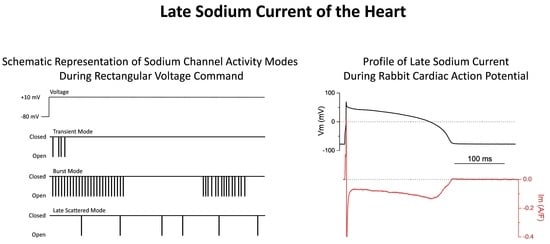
:
{kind=link}
{kind=link}
1. Introduction
2. A Brief History of Cardiac Late Sodium Current Research
3. Cardiac Sodium Channels: Structure and Morphology
3.1. Molecular Identity of Cardiac Sodium Channels
3.2. Morphology of VGSCs
3.3. Localization of Sodium Channels in Cardiac Myocytes and Potential Implications
4. Role of Late Sodium Current in the Homeostasis of Cardiac Cell
4.1. Late Sodium Current and Sodium Homeostasis of Cardiac Cells
4.2. Late Sodium Current and the Cardiac Calcium Homeostasis
5. Electrophysiology of Late Sodium Current
5.1. The Window Mechanism
5.2. Sodium Channel Gating Modes
5.3. Non-Equilibrium Gating
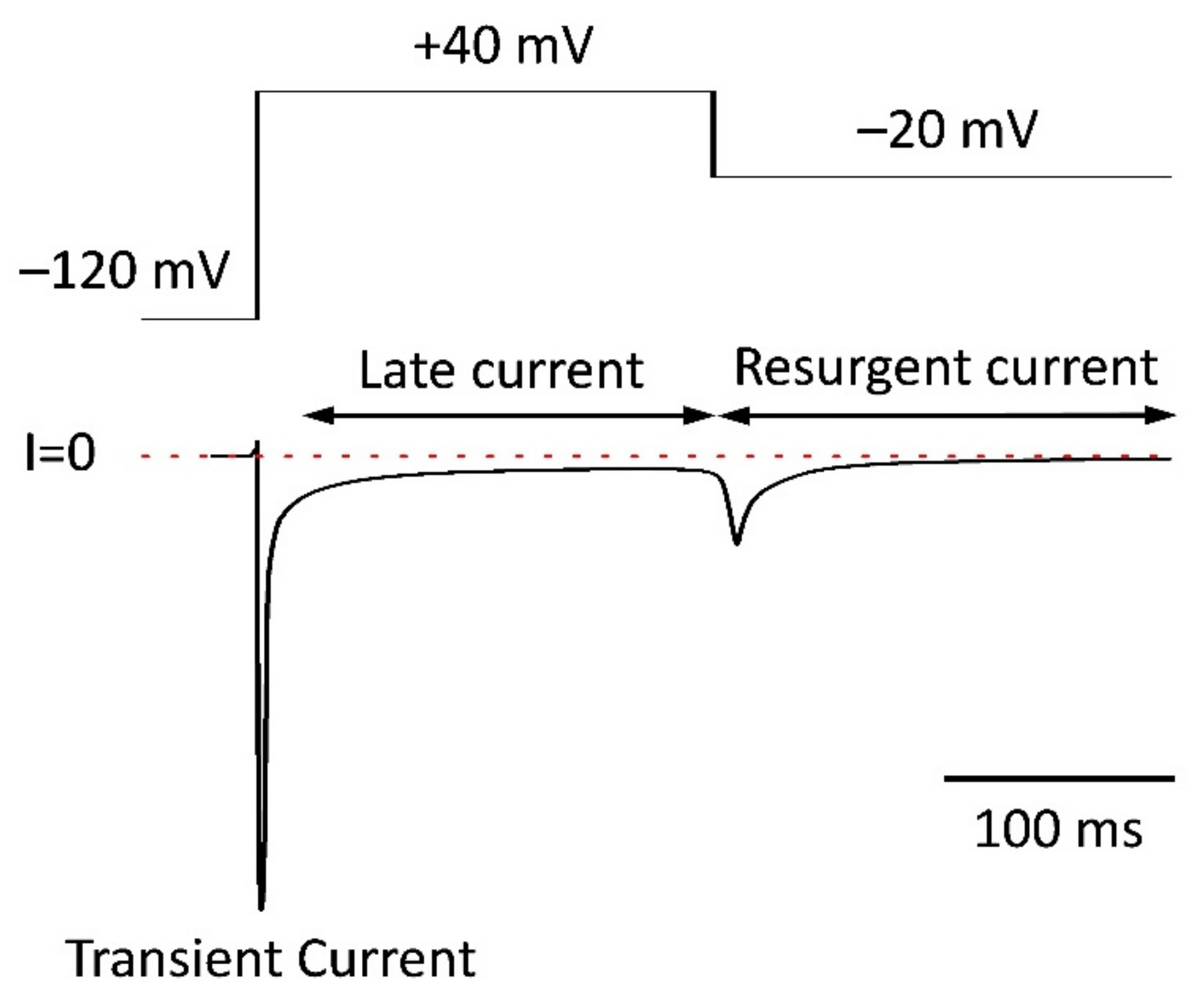
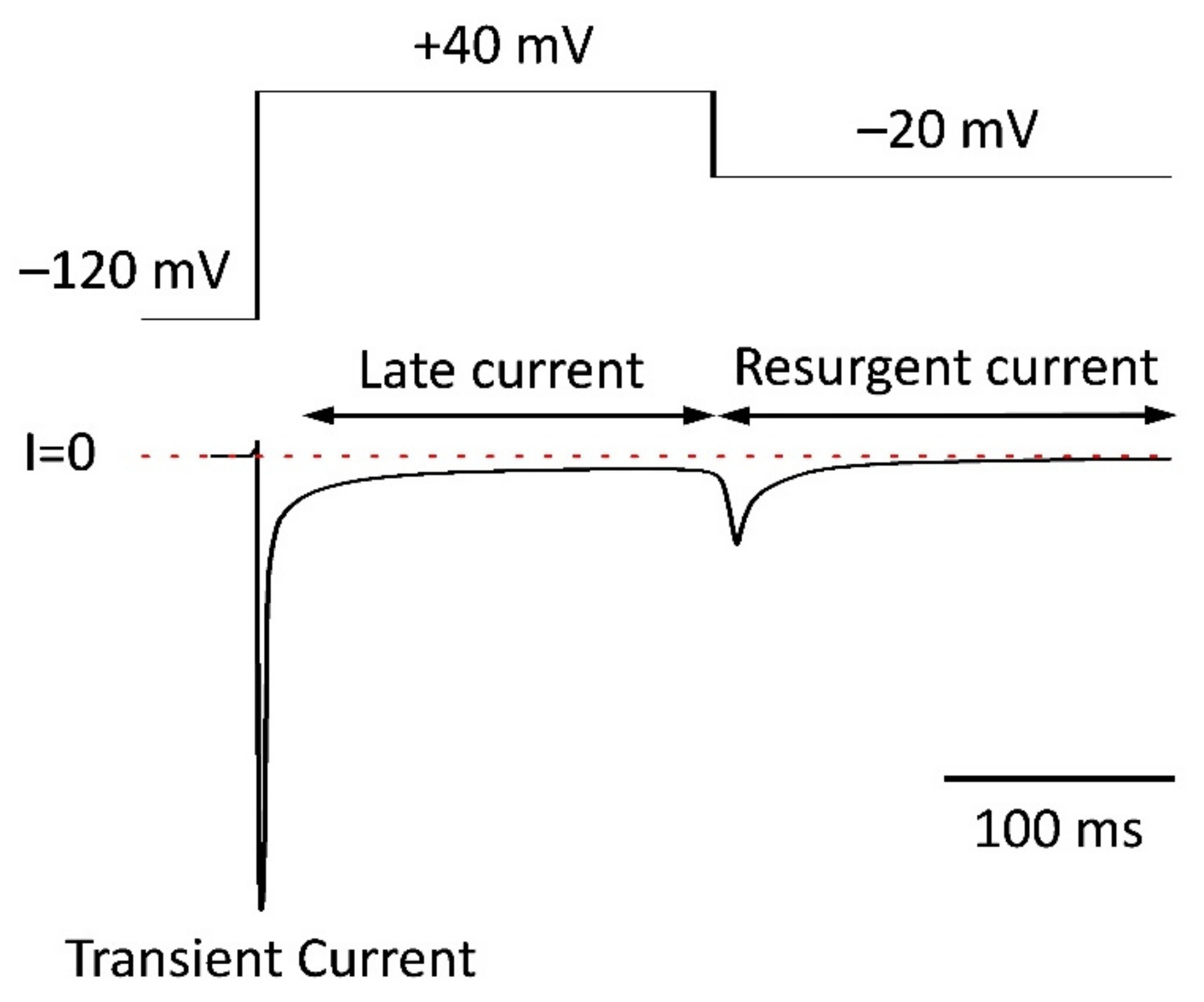
5.4. Resurgent Current
6. The Modulation of Late Sodium Current
6.1. The Calcium—Calmodulin—Calmodulin Kinase Axis
6.1.1. Direct Regulatory Effect of Ca2+ on VGSC
6.1.2. Calmodulin
6.2. Protein Kinases
6.2.1. Calmodulin Kinase
6.2.2. Protein Kinase A (PKA)
6.2.3. Protein Kinase C (PKC)
6.2.4. Serum—And Glucocorticoid-Inducible Kinases (SGKs)
6.3. Metabolic Control
6.4. Mechanical Stress
6.5. Accessory Proteins
7. Pathologic Aspects of Late Sodium Current Function
7.1. Arrhythmias
7.2. Late sodium Current and Dilated Cardiomyopathy
8. Pharmacology of Late Sodium Current
8.1. Eleutheroside B, a New Late Sodium Current Inhibitor
8.2. Old Drugs with New Therapeutic Effects
8.3. ATX-II. New Observations with an Old Tool: Friend or Foe?
9. Summary and Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AP | Action Potential |
| AF | Atrial fibrillation |
| ATX-II | Anemone toxin II |
| CaM | Calmodulin |
| CaMK | Calmodulin Kinase |
| DAD | Delayed Afterdepolarization |
| DCM | Dilated Cardiomyopathy |
| EAD | Early Afterdepolarization |
| NCX | Na+/Ca2+ exchanger |
| NKA | Na+/K+-ATPase |
| SGK | Serum- and Glucocorticoid-Inducible Kinase |
| SSI | Steady State Inactivation |
| TTX | Tetrodotoxin |
| VGSC | Voltage Gated Sodium Channel |
References
- Horvath, B.; Banyasz, T.; Jian, Z.; Hegyi, B.; Kistamas, K.; Nanasi, P.P.; Izu, L.T.; Chen-Izu, Y. Dynamics of the late Na+ current during cardiac action potential and its contribution to afterdepolarizations. J. Mol. Cell. Cardiol. 2013, 64, 59–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegyi, B.; Banyasz, T.; Izu, L.T.; Belardinelli, L.; Bers, D.M.; Chen-Izu, Y. beta-adrenergic regulation of late Na+ current during cardiac action potential is mediated by both PKA and CaMKII. J. Mol. Cell. Cardiol. 2018, 123, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Horvath, B.; Hezso, T.; Szentandrassy, N.; Kistamas, K.; Arpadffy-Lovas, T.; Varga, R.; Gazdag, P.; Veress, R.; Dienes, C.; Baranyai, D.; et al. Late sodium current in human, canine and guinea pig ventricular myocardium. J. Mol. Cell. Cardiol. 2020, 139, 14–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bengel, P.; Ahmad, S.; Tirilomis, P.; Trum, M.; Dybkova, N.; Wagner, S.; Maier, L.S.; Hasenfuß, G.; Sossalla, S. Contribution of the neuronal sodium channel Na(V)1.8 to sodium- and calcium-dependent cellular proarrhythmia. J. Mol. Cell. Cardiol. 2020, 144, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Belardinelli, L. Enhanced basal late sodium current appears to underlie the age-related prolongation of action potential duration in guinea pig ventricular myocytes. J. Appl. Physiol. 2018, 125, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Undrovinas, N.A.; Maltsev, V.; Belardinelli, L.; Sabbah, H.N.; Undrovinas, A. Late sodium current contributes to diastolic cell Ca2+ accumulation in chronic heart failure. J. Physiol. Sci. 2010, 60, 245–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pabel, S.; Ahmad, S.; Tirilomis, P.; Stehle, T.; Mustroph, J.; Knierim, M.; Dybkova, N.; Bengel, P.; Holzamer, A.; Hilker, M.; et al. Inhibition of Na(V)1.8 prevents atrial arrhythmogenesis in human and mice. Basic Res. Cardiol. 2020, 115, 20. [Google Scholar] [CrossRef] [PubMed]
- Wasserstrom, J.A.; Sharma, R.; O’Toole, M.J.; Zheng, J.; Kelly, J.E.; Shryock, J.; Belardinelli, L.; Aistrup, G.L. Ranolazine Antagonizes the Effects of Increased Late Sodium Current on Intracellular Calcium Cycling in Rat Isolated Intact Heart. J. Pharmacol. Exp. Ther. 2009, 331, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Undrovinas, A.I.; Belardinelli, L.; Undrovinas, N.A.; Sabbah, H.N. Ranolazine Improves Abnormal Repolarization and Contraction in Left Ventricular Myocytes of Dogs with Heart Failure by Inhibiting Late Sodium Current. J. Cardiovasc. Electrophysiol. 2006, 17, S169–S177. [Google Scholar] [CrossRef] [Green Version]
- Sossalla, S.; Wagner, S.; Rasenack, E.C.; Ruff, H.; Weber, S.L.; Schöndube, F.A.; Tirilomis, T.; Tenderich, G.; Hasenfuss, G.; Belardinelli, L.; et al. Ranolazine improves diastolic dysfunction in isolated myocardium from failing human hearts—Role of late sodium current and intracellular ion accumulation. J. Mol. Cell. Cardiol. 2008, 45, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Sossalla, S.; Maurer, U.; Schotola, H.; Hartmann, N.; Didie, M.; Zimmermann, W.H.; Jacobshagen, C.; Wagner, S.; Maier, L.S. Diastolic dysfunction and arrhythmias caused by overexpression of CaMKIIdelta(C) can be reversed by inhibition of late Na(+) current. Basic Res. Cardiol. 2011, 106, 263–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoyer, K.; Song, Y.; Wang, D.; Phan, D.; Balschi, J.; Ingwall, J.S.; Belardinelli, L.; Shryock, J.C. Reducing the Late Sodium Current Improves Cardiac Function during Sodium Pump Inhibition by Ouabain. J. Pharmacol. Exp. Ther. 2011, 337, 513–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, D.; Young, L.; Wu, Y.; Belardinelli, L.; Kowey, P.R.; Yan, G.-X. Increased late sodium current in left atrial myocytes of rabbits with left ventricular hypertrophy: Its role in the genesis of atrial arrhythmias. Am. J. Physiol. Circ. Physiol. 2010, 298, H1375–H1381. [Google Scholar] [CrossRef]
- Sossalla, S.; Kallmeyer, B.; Wagner, S.; Mazur, M.; Maurer, U.; Toischer, K.; Schmitto, J.; Seipelt, R.; Schöndube, F.A.; Hasenfuss, G.; et al. Altered Na+Currents in Atrial Fibrillation: Effects of Ranolazine on Arrhythmias and Contractility in Human Atrial Myocardium. J. Am. Coll. Cardiol. 2010, 55, 2330–2342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaza, A.; Belardinelli, L.; Shryock, J.C. Pathophysiology and pharmacology of the cardiac “late sodium current”. Pharmacol. Ther. 2008, 119, 326–339. [Google Scholar] [CrossRef]
- Shryock, J.C.; Song, Y.J.; Rajamani, S.; Antzelevitch, C.; Belardinelli, L. The arrhythmogenic consequences of increasing late I-Na in the cardiomyocyte. Cardiovasc. Res. 2013, 99, 600–611. [Google Scholar] [CrossRef] [Green Version]
- Belardinelli, L.; Giles, W.R.; Rajamani, S.; Karagueuzian, H.S.; Shryock, J.C. Cardiac late Na+ current: Proarrhythmic effects, roles in long QT syndromes, and pathological relationship to CaMKII and oxidative stress. Heart Rhythm. Off. J. Heart Rhythm. Soc. 2015, 12, 440–448. [Google Scholar] [CrossRef]
- Bossu, A.; Houtman, M.J.C.; Meijborg, V.M.F.; Varkevisser, R.; Beekman, H.D.M.; Dunnink, A.; De Bakker, J.M.T.; Mollova, N.; Rajamani, S.; Belardinelli, L.; et al. Selective late sodium current inhibitor GS-458967 suppresses Torsades de Pointes by mostly affecting perpetuation but not initiation of the arrhythmia. J. Cereb. Blood Flow Metab. 2018, 175, 2470–2482. [Google Scholar] [CrossRef] [Green Version]
- Sicouri, S.; Belardinelli, L.; Antzelevitch, C. Antiarrhythmic effects of the highly selective late sodium channel current blocker GS-458967. Heart Rhythm 2013, 10, 1036–1043. [Google Scholar] [CrossRef] [Green Version]
- Belardinelli, L.; Liu, G.; Smith-Maxwell, C.; Wang, W.-Q.; El-Bizri, N.; Hirakawa, R.; Karpinski, S.; Li, C.H.; Hu, L.; Li, X.-J.; et al. A Novel, Potent, and Selective Inhibitor of Cardiac Late Sodium Current Suppresses Experimental Arrhythmias. J. Pharmacol. Exp. Ther. 2012, 344, 23–32. [Google Scholar] [CrossRef]
- Wu, L.; Shryock, J.C.; Song, Y.; Li, Y.; Antzelevitch, C.; Belardinelli, L. Antiarrhythmic Effects of Ranolazine in a Guinea Pig in Vitro Model of Long-QT Syndrome. J. Pharmacol. Exp. Ther. 2004, 310, 599–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhalla, A.K.; Wang, W.Q.; Dow, J.; Shryock, J.C.; Belardinelli, L.; Bhandari, A.; Kloner, R.A. Ranolazine, an antianginal agent, markedly reduces ventricular arrhythmias induced by ischemia and ischemia-reperfusion. Am. J. Physiol.-Heart Circ. Physiol. 2009, 297, H1923–H1929. [Google Scholar] [CrossRef] [Green Version]
- Zaza, A.; Rocchetti, M. The late Na+ current-origin and pathophysiological relevance. Cardiovasc. Drugs Ther. 2013, 27, 61–68. [Google Scholar] [CrossRef] [Green Version]
- Maier, L.S.; Sossalla, S. The late Na current as a therapeutic target: Where are we? J. Mol. Cell. Cardiol. 2013, 61, 44–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kistamás, K.; Hézső, T.; Horváth, B.; Nánási, P.P. Late sodium current and calcium homeostasis in arrhythmogenesis. Channels 2020, 15, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Horváth, B.; Hézső, T.; Kiss, D.; Kistamas, K.; Magyar, J.; Nánási, P.P.; Bányász, T. Late Sodium Current Inhibitors as Potential Antiarrhythmic Agents. Front. Pharmacol. 2020, 11, 413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banyasz, T.; Szentandrassy, N.; Magyar, J.; Szabo, Z.; Nanasi, P.; Chen-Izu, Y.; Izu, L. An Emerging Antiarrhythmic Target: Late Sodium Current. Curr. Pharm. Des. 2014, 21, 1073–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivaud, M.R.; Delmar, M.; Remme, C.A. Heritable arrhythmia syndromes associated with abnormal cardiac sodium channel function: Ionic and non-ionic mechanisms. Cardiovasc. Res. 2020, 116, 1557–1570. [Google Scholar] [CrossRef] [Green Version]
- Jia, S.B.; Lian, J.F.; Guo, D.L.; Xue, X.L.; Patel, C.; Yang, L.; Yuan, Z.Y.; Ma, A.Q.; Yan, G.X. Modulation of the late sodium current by ATX-II and ranolazine affects the reverse use-dependence and proarrhythmic liability of I-Kr blockade. Br. J. Pharmacol. 2011, 164, 308–316. [Google Scholar] [CrossRef] [Green Version]
- Nuyens, D.; Stengl, M.; Dugarmaa, S.; Rossenbacker, T.; Compernolle, V.; Rudy, Y.; Smits, J.F.; Flameng, W.; Clancy, C.E.; Moons, L.; et al. Abrupt rate accelerations or premature beats cause life-threatening arrhythmias in mice with long-QT3 syndrome. Nat. Med. 2001, 7, 1021–1027. [Google Scholar] [CrossRef]
- Stafstrom, C.E. Persistent Sodium Current and Its Role in Epilepsy. Epilepsy Curr. 2007, 7, 15–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wengert, E.R.; Patel, M.K. The Role of the Persistent Sodium Current in Epilepsy. Epilepsy Curr. 2020, 21, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Quignard, J.-F.; Ryckwaert, F.; Albat, B.; Nargeot, J.; Richard, S. A Novel Tetrodotoxin-Sensitive Na sup + Current in Cultured Human Coronary Myocytes. Circ. Res. 1997, 80, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Djamgoz, M.B.; Onkal, R. Persistent Current Blockers of Voltage-Gated Sodium Channels: A Clinical Opportunity for Controlling Metastatic Disease. Recent Patents Anti-Cancer Drug Discov. 2013, 8, 66–84. [Google Scholar] [CrossRef]
- Rizzetto, R.; Rocchetti, M.; Sala, L.; Ronchi, C.; Villa, A.; Ferrandi, M.; Molinari, I.; Bertuzzi, F.; Zaza, A. Late sodium current (INaL) in pancreatic β-cells. Pflug. Arch. Eur. J. Physiol. 2015, 467, 1757–1768. [Google Scholar] [CrossRef]
- Frankenhaeuser, B. Instantaneous potassium currents in myelinated nerve fibres ofXenopus laevis. J. Physiol. 1962, 160, 46–53. [Google Scholar] [CrossRef]
- Frankenhaeuser, B. A quantitative description of potassium currents in myelinated nerve fibres of Xenopus laevis. J. Physiol. 1963, 169, 424–430. [Google Scholar] [CrossRef]
- Frankenhaeuser, B. Potassium permeability in myelinated nerve fibres ofXenopus laevis. J. Physiol. 1962, 160, 54–61. [Google Scholar] [CrossRef]
- Frankenhaeuser, B. Delayed currents in myelinated nerve fibres of Xenopus laevis investigated with voltage clamp technique. J. Physiol. 1962, 160, 40–45. [Google Scholar] [CrossRef] [Green Version]
- Dubois, J.M.; Bergman, C. Late Sodium Current in Node of Ranvier. Pflug. Arch. -Eur. J. Physiol. 1975, 357, 145–148. [Google Scholar] [CrossRef]
- Coraboeuf, E.; Deroubaix, E.; Coulombe, A. Effect of Tetrodotoxin on Action Potentials of the Conductiv System in the Dog Heart. Am. J. Physiol. 1979, 236, H561–H567. [Google Scholar] [PubMed]
- Attwell, D.; Cohen, I.; Eisner, D.; Ohba, M.; Ojeda, C. Steady-state TTX-sensitive (window) sodium current in cardiac Purkinje-fibers. Pflug. Arch.-Eur. J. Physiol. 1979, 379, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Kiyosue, T.; Arita, M. Late sodium current and its contribution to action potential configuration in guinea pig ventricular myocytes. Circ. Res. 1989, 64, 389–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clancy, C.E.; Tateyama, M.; Liu, H.; Wehrens, X.H.; Kass, R.S. Non-equilibrium gating in cardiac Na+ channels: An original mechanism of arrhythmia. Circulation 2003, 107, 2233–2237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, Y.K.; Saint, D.A.; Gage, P.W. Hypoxia increases persistent sodium current in rat ventricular myocytes. J. Physiol. 1996, 497, 337–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Undrovinas, A.I.; Fleidervish, I.A.; Makielski, J.C. Inward sodium current at resting potentials in single cardiac myocytes induced by the ischemic metabolite lysophosphatidylcholine. Circ. Res. 1992, 71, 1231–1241. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Corr, P.B. Palmitoyl carnitine modifies sodium currents and induces transient inward current in ventricular myocytes. Am. J. Physiol. Circ. Physiol. 1994, 266, H1034–H1046. [Google Scholar] [CrossRef]
- Pezhouman, A.; Madahian, S.; Stepanyan, H.; Ghukasyan, H.; Qu, Z.; Belardinelli, L.; Karagueuzian, H.S. Selective inhibition of late sodium current suppresses ventricular tachycardia and fibrillation in intact rat hearts. Heart Rhythm 2013, 11, 492–501. [Google Scholar] [CrossRef] [Green Version]
- Soliman, D.; Wang, L.; Hamming, K.S.C.; Yang, W.; Fatehi, M.; Carter, C.C.; Clanachan, A.S.; Light, P.E. Late Sodium Current Inhibition Alone with Ranolazine Is Sufficient to Reduce Ischemia- and Cardiac Glycoside-Induced Calcium Overload and Contractile Dysfunction Mediated by Reverse-Mode Sodium/Calcium Exchange. J. Pharmacol. Exp. Ther. 2012, 343, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Pignier, C.; Rougier, J.-S.; Vié, B.; Culié, C.; Verscheure, Y.; Vacher, B.; Abriel, H.; Le Grand, B. Selective inhibition of persistent sodium current by F 15845 prevents ischaemia-induced arrhythmias. J. Cereb. Blood Flow Metab. 2010, 161, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Antoons, G.; Oros, A.; Beekman, J.D.; Engelen, M.A.; Houtman, M.J.; Belardinelli, L.; Stengl, M.; Vos, M.A. Late Na+ Current Inhibition by Ranolazine Reduces Torsades de Pointes in the Chronic Atrioventricular Block Dog Model. J. Am. Coll. Cardiol. 2010, 55, 801–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belardinelli, L.; Antzelevitch, C.; Fraser, H. Inhibition of late (sustained/persistent) sodium current: A potential drug target to reduce intracellular sodium-dependent calcium overload and its detrimental effects on cardiomyocyte function. Eur. Heart J. Suppl. 2004, 6, I3–I7. [Google Scholar] [CrossRef]
- Rambarat, C.A.; Elgendy, I.Y.; Handberg, E.M.; Merz, C.N.B.; Wei, J.; Minissian, M.B.; Nelson, M.D.; Thomson, L.E.J.; Berman, D.S.; Shaw, L.J.; et al. Late sodium channel blockade improves angina and myocardial perfusion in patients with severe coronary microvascular dysfunction: Women’s Ischemia Syndrome Evaluation-Coronary Vascular Dysfunction ancillary study. Int. J. Cardiol. 2019, 276, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Ton, A.T.; Nguyen, W.; Sweat, K.; Miron, Y.; Hernandez, E.; Wong, T.; Geft, V.; Macias, A.; Espinoza, A.; Truong, K.; et al. Arrhythmogenic and antiarrhythmic actions of late sustained sodium current in the adult human heart. Sci. Rep. 2021, 11, 12014. [Google Scholar] [CrossRef]
- Dong, C.; Wang, Y.; Ma, A.; Wang, T. Life Cycle of the Cardiac Voltage-Gated Sodium Channel NaV1.5. Front. Physiol. 2020, 11, 9733. [Google Scholar] [CrossRef]
- Catterall, W.A.; Goldin, A.L.; Waxman, S.G. International Union of Pharmacology. XLVII. Nomenclature and Structure-Function Relationships of Voltage-Gated Sodium Channels. Pharmacol. Rev. 2005, 57, 397–409. [Google Scholar] [CrossRef]
- Zimmer, T.; Haufe, V.; Blechschmidt, S. Voltage-gated sodium channels in the mammalian heart. Glob. Cardiol. Sci. Pract. 2014, 2014, 449–463. [Google Scholar] [CrossRef]
- Godazgar, M.; Zhang, Q.; Chibalina, M.V.; Rorsman, P. Biphasic voltage-dependent inactivation of human Na(V)1.3, 1.6 and 1.7 Na+ channels expressed in rodent insulin-secreting cells. J. Physiol.-Lond. 2018, 596, 1601–1626. [Google Scholar] [CrossRef] [Green Version]
- Maltsev, V.A.; Undrovinas, A. Late sodium current in failing heart: Friend or foe? Prog. Biophys. Mol. Biol. 2008, 96, 421–451. [Google Scholar] [CrossRef] [Green Version]
- Pereon, Y.; Lande, G.; Demolombe, S.; Tich, S.N.T.; Sternberg, D.; Le Marec, H.; David, A. Paramyotonia congenita with an SCN4A mutation affecting cardiac repolarization. Neurology 2003, 60, 340–342. [Google Scholar] [CrossRef]
- Biet, M.; Barajas-Martínez, H.; Ton, A.-T.; Delabre, J.-F.; Morin, N.; Dumaine, R. About half of the late sodium current in cardiac myocytes from dog ventricle is due to non-cardiac-type Na+ channels. J. Mol. Cell. Cardiol. 2012, 53, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Atack, T.C.; Stroud, D.M.; Zhang, W.; Hall, L.; Roden, D.M. Blocking Scn10a channels in heart reduces late sodium current and is antiarrhythmic. Circ. Res. 2012, 111, 322–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haufe, V.; Cordeiro, J.M.; Zimmer, T.; Wu, Y.S.; Schiccitano, S.; Benndorf, K.; Dumaine, R. Contribution of neuronal sodium channels to the cardiac fast sodium current INa is greater in dog heart Purkinje fibers than in ventricles. Cardiovasc. Res. 2005, 65, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Maier, S.K.G.; Westenbroek, R.E.; Schenkman, K.A.; Feigl, E.O.; Scheuer, T.; Catterall, W.A. An unexpected role for brain-type sodium channels in coupling of cell surface depolarization to contraction in the heart. Proc. Natl. Acad. Sci. USA 2002, 99, 4073–4078. [Google Scholar] [CrossRef] [Green Version]
- Westenbroek, R.E.; Bischoff, S.; Fu, Y.; Maier, S.K.; Catterall, W.A.; Scheuer, T. Localization of sodium channel subtypes in mouse ventricular myocytes using quantitative immunocytochemistry. J. Mol. Cell. Cardiol. 2013, 64, 69–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haufe, V.; Camacho, J.A.; Dumaine, R.; Günther, B.; Bollensdorff, C.; Von Banchet, G.S.; Benndorf, K.; Zimmer, T. Expression pattern of neuronal and skeletal muscle voltage-gated Na+channels in the developing mouse heart. J. Physiol. 2005, 564, 683–696. [Google Scholar] [CrossRef] [PubMed]
- Blechschmidt, S.; Haufe, V.; Benndorf, K.; Zimmer, T. Voltage-gated Na+ channel transcript patterns in the mammalian heart are species-dependent. Prog. Biophys. Mol. Biol. 2008, 98, 309–318. [Google Scholar] [CrossRef]
- Chambers, J.C.; Zhao, J.; Terracciano, C.M.N.; Bezzina, C.R.; Zhang, W.; Kaba, R.; Navaratnarajah, M.; Lotlikar, A.; Sehmi, J.S.; Kooner, M.K.; et al. Genetic variation in SCN10A influences cardiac conduction. Nat. Genet. 2010, 42, 149–152. [Google Scholar] [CrossRef]
- Verkerk, A.O.; Remme, C.A.; Schumacher, C.A.; Scicluna, B.P.; Wolswinkel, R.; de Jonge, B.; Bezzina, C.R.; Veldkamp, M.W. Functional Nav1.8 channels in intracardiac neurons: The link between SCN10A and cardiac electrophysiology. Circ. Res. 2012, 111, 333–343. [Google Scholar] [CrossRef] [Green Version]
- Jabbari, J.; Olesen, M.S.; Yuan, L.; Nielsen, J.B.; Liang, B.; Macri, V.; Christophersen, I.E.; Nielsen, N.; Sajadieh, A.; Ellinor, P.T.; et al. Common and rare variants in SCN10A modulate the risk of atrial fibrillation. Circ. Cardiovasc. Genet. 2015, 8, 64–73. [Google Scholar] [CrossRef] [Green Version]
- Savio-Galimberti, E.; Weeke, P.; Muhammad, R.; Blair, M.; Ansari, S.; Short, L.; Atack, T.C.; Kor, K.; Vanoye, C.G.; Olesen, M.S.; et al. SCN10A/Nav1.8 modulation of peak and late sodium currents in patients with early onset atrial fibrillation. Cardiovasc. Res. 2014, 104, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Stroud, D.M.; Yang, T.; Bersell, K.; Kryshtal, D.O.; Nagao, S.; Shaffer, C.; Short, L.; Hall, L.; Atack, T.C.; Zhang, W.; et al. Contrasting Nav1.8 Activity in Scn10a−/− Ventricular Myocytes and the Intact Heart. J. Am. Heart Assoc. 2016, 5, e002946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macri, V.; Brody, J.A.; Arking, D.E.; Hucker, W.J.; Yin, X.; Lin, H.; Mills, R.W.; Sinner, M.F.; Lubitz, S.A.; Liu, C.-T.; et al. Common Coding Variants in SCN10A Are Associated with the Nav1.8 Late Current and Cardiac Conduction. Circ. Genom. Precis. Med. 2018, 11, e001663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, L.S.; Sossalla, S.; Schulze-Bahr, E. SCN10A-Dependent Late I(Na) Current: Never Too Late for Cardiac Conduction? Circ. Genom. Precis. Med. 2018, 11, e002167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Kalyanasundaram, A.; Hansen, B.J.; Artiga, E.J.; Sharma, R.; Abudulwahed, S.H.; Helfrich, K.M.; Rozenberg, G.; Wu, P.-J.; Zakharkin, S.; et al. Impaired neuronal sodium channels cause intranodal conduction failure and reentrant arrhythmias in human sinoatrial node. Nat. Commun. 2020, 11, 512. [Google Scholar] [CrossRef]
- Casini, S.; Marchal, G.A.; Kawasaki, M.; Nariswari, F.A.; Portero, V.; van den Berg, N.W.E.; Guan, K.; Driessen, A.H.G.; Veldkamp, M.W.; Mengarelli, I.; et al. Absence of Functional Na(v)1.8 Channels in Non-diseased Atrial and Ventricular Cardiomyocytes. Cardiovasc. Drugs Ther. 2019, 33, 649–660. [Google Scholar] [CrossRef] [Green Version]
- Facer, P.; Punjabi, P.P.; Abrari, A.; Kaba, R.A.; Severs, N.J.; Chambers, J.; Kooner, J.S.; Anand, P. Localisation of SCN10A Gene Product Nav1.8 and Novel Pain-Related Ion Channels in Human Heart. Int. Heart J. 2011, 52, 146–152. [Google Scholar] [CrossRef] [Green Version]
- Xi, Y.; Wu, G.; Yang, L.; Han, K.; Du, Y.; Wang, T.; Lei, X.; Bai, X.; Ma, A. Increased late sodium currents are related to transcription of neuronal isoforms in a pressure-overload model. Eur. J. Heart Fail. 2009, 11, 749–757. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, S.; Tirilomis, P.; Pabel, S.; Dybkova, N.; Hartmann, N.; Molina, C.E.; Tirilomis, T.; Kutschka, I.; Frey, N.; Maier, L.S.; et al. The functional consequences of sodium channel Na(V)1.8 in human left ventricular hypertrophy. Esc. Heart Fail. 2019, 6, 154–163. [Google Scholar] [CrossRef]
- Dybkova, N.; Ahmad, S.; Pabel, S.; Tirilomis, P.; Hartmann, N.; Fischer, T.H.; Bengel, P.; Tirilomis, T.; Ljubojevic, S.; Renner, A.; et al. Differential regulation of sodium channels as a novel proarrhythmic mechanism in the human failing heart. Cardiovasc. Res. 2018, 114, 1728–1737. [Google Scholar] [CrossRef]
- Brackenbury, W.J.; Isom, L.L. Na Channel β Subunits: Overachievers of the Ion Channel Family. Front. Pharmacol. 2011, 2, 53. [Google Scholar] [CrossRef] [Green Version]
- Cusdin, F.S.; Clare, J.J.; Jackson, A.P. Trafficking and Cellular Distribution of Voltage-Gated Sodium Channels. Traffic 2008, 9, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Kazen-Gillespie, K.A.; Ragsdale, D.S.; D’Andrea, M.R.; Mattei, L.N.; Rogers, K.E.; Isom, L.L. Cloning, localization, and functional expression of sodium channel beta1A subunits. J. Biol. Chem. 2000, 275, 1079–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, N.; D’Andrea, M.R.; Lubin, M.-L.; Shafaee, N.; Codd, E.; Correa, A.M. Molecular cloning and functional expression of the human sodium channel beta1B subunit, a novel splicing variant of the beta1 subunit. JBIC J. Biol. Inorg. Chem. 2003, 270, 4762–4770. [Google Scholar]
- Llongueras, J.P.; Das, S.; De Waele, J.; Capulzini, L.; Sorgente, A.; Van Petegem, F.; Bosmans, F. Biophysical Investigation of Sodium Channel Interaction with β-Subunit Variants Associated with Arrhythmias. Bioelectricity 2020, 2, 269–278. [Google Scholar] [CrossRef]
- Isaac, E.; Cooper, S.M.; Jones, S.A.; Loubani, M. Do age-associated changes of voltage-gated sodium channel isoforms expressed in the mammalian heart predispose the elderly to atrial fibrillation? World J. Cardiol. 2020, 12, 123–135. [Google Scholar] [CrossRef]
- Maltsev, V.A.; Kyle, J.W.; Undrovinas, A. Late Na(+) current produced by human cardiac Na(+) channel isoform Na(v)1.5 is modulated by its beta(1) subunit. J. Physiol. Sci. 2009, 59, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Bouza, A.A.; Isom, L.L. Voltage-Gated Sodium Channel β Subunits and Their Related Diseases. Volt.-Gated Sodium Channels 2017, 246, 423–450. [Google Scholar] [CrossRef]
- Angsutararux, P.; Zhu, W.; Voelker, T.L.; Silva, J.R. Molecular Pathology of Sodium Channel Beta-Subunit Variants. Front. Pharmacol. 2021, 12, 3220. [Google Scholar] [CrossRef]
- Grieco, T.M.; Malhotra, J.D.; Chen, C.; Isom, L.L.; Raman, I.M. Open-channel block by the cytoplasmic tail of sodium channel beta4 as a mechanism for resurgent sodium current. Neuron 2005, 45, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Clatot, J.; Hoshi, M.; Wan, X.; Liu, H.; Jain, A.; Shinlapawittayatorn, K.; Marionneau, C.; Ficker, E.; Eckhard, F.; Deschênes, I. Voltage-gated sodium channels assemble and gate as dimers. Nat. Commun. 2017, 8, 2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clatot, J.; Zheng, Y.; Girardeau, A.; Liu, H.; Laurita, K.R.; Marionneau, C.; Deschênes, I. Mutant voltage-gated Na+ channels can exert a dominant negative effect through coupled gating. Am. J. Physiol. Circ. Physiol. 2018, 315, H1250–H1257. [Google Scholar] [CrossRef] [PubMed]
- Rühlmann, A.H.; Körner, J.; Hausmann, R.; Bebrivenski, N.; Neuhof, C.; Detro-Dassen, S.; Hautvast, P.; Benasolo, C.A.; Meents, J.; Machtens, J.-P.; et al. Uncoupling sodium channel dimers restores the phenotype of a pain-linked Nav1.7 channel mutation. Br. J. Pharmacol. 2020, 177, 4481–4496. [Google Scholar] [CrossRef] [PubMed]
- Eshed-Eisenbach, Y.; Peles, E. The clustering of voltage-gated sodium channels in various excitable membranes. Dev. Neurobiol. 2019, 81, 427–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvage, S.C.; Rees, J.S.; McStea, A.; Hirsch, M.; Wang, L.; Tynan, C.J.; Reed, M.W.; Irons, J.R.; Butler, R.; Thompson, A.J.; et al. Supramolecular clustering of the cardiac sodium channel Nav1.5 in HEK293F cells, with and without the auxiliary beta 3-subunit. Faseb J. 2020, 34, 3537–3553. [Google Scholar] [CrossRef]
- Veeraraghavan, R.; Radwański, P.B. Sodium channel clusters: Harmonizing the cardiac conduction orchestra. J. Physiol. 2018, 596, 549–550. [Google Scholar] [CrossRef] [Green Version]
- Hichri, E.; Abriel, H.; Kucera, J.P. Distribution of cardiac sodium channels in clusters potentiates ephaptic interactions in the intercalated disc. J. Physiol. 2018, 596, 563–589. [Google Scholar] [CrossRef]
- Bhargava, A.; Lin, X.; Novak, P.; Mehta, K.; Korchev, Y.; Delmar, M.; Gorelik, J. Super-resolution Scanning Patch Clamp Reveals Clustering of Functional Ion Channels in Adult Ventricular Myocyte. Circ. Res. 2013, 112, 1112–1120. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Liu, N.; Lu, J.; Zhang, J.; Anumonwo, J.M.; Isom, L.; Fishman, G.I.; Delmar, M. Subcellular heterogeneity of sodium current properties in adult cardiac ventricular myocytes. Heart Rhythm 2011, 8, 1923–1930. [Google Scholar] [CrossRef] [Green Version]
- Salvage, S.C.; Huang, C.L.; Jackson, A.P. Cell-Adhesion Properties of β-Subunits in the Regulation of Cardiomyocyte Sodium Channel. Biomolecules 2020, 10, 989. [Google Scholar] [CrossRef]
- Verkerk, A.O.; van Ginneken, A.C.; van Veen, T.A.; Tan, H.L. Effects of heart failure on brain-type Na+ channels in rabbit ventricular myocytes. Europace 2007, 9, 571–577. [Google Scholar] [CrossRef]
- Struckman, H.L.; Baine, S.; Thomas, J.; Mezache, L.; Mykytyn, K.; Györke, S.; Radwański, P.B.; Veeraraghavan, R. Super-Resolution Imaging Using a Novel High-Fidelity Antibody Reveals Close Association of the Neuronal Sodium Channel Na(V)1.6 with Ryanodine Receptors in Cardiac Muscle, Microscopy and microanalysis: The official journal of Microscopy Society of America, Microbeam Analysis Society. Microsc. Soc. Can. 2020, 26, 157–165. [Google Scholar]
- Munger, M.A.; Olğar, Y.; Koleske, M.L.; Struckman, H.L.; Mandrioli, J.; Lou, Q.; Bonila, I.; Kim, K.; Mondragon, R.R.; Priori, S.G.; et al. Tetrodotoxin-Sensitive Neuronal-Type Na + Channels: A Novel and Druggable Target for Prevention of Atrial Fibrillation. J. Am. Heart Assoc. 2020, 9, e015119. [Google Scholar] [CrossRef] [PubMed]
- Jayasinghe, I.; Clowsley, A.H.; De Langen, O.; Sali, S.S.; Crossman, D.; Soeller, C. Shining New Light on the Structural Determinants of Cardiac Couplon Function: Insights from Ten Years of Nanoscale Microscopy. Front. Physiol. 2018, 9, 1472. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Belardinelli, L. Basal late sodium current is a significant contributor to the duration of action potential of guinea pig ventricular myocytes. Physiol. Rep. 2017, 5, e13295. [Google Scholar] [CrossRef]
- Sheu, S.S.; Lederer, W.J. Lidocaine’s negative inotropic and antiarrhythmic actions. Dependence on shortening of action potential duration and reduction of intracellular sodium activity. Circ. Res. 1985, 57, 578–590. [Google Scholar] [CrossRef] [Green Version]
- Makielski, J.C.; Farley, A.L. Na+ Current in Human Ventricle: Implications for Sodium Loading and Homeostasis. J. Cardiovasc. Electrophysiol. 2006, 17, S15–S20. [Google Scholar] [CrossRef]
- Hegyi, B.; Bányász, T.; Shannon, T.R.; Chen-Izu, Y.; Izu, L.T. Electrophysiological Determination of Submembrane Na+ Concentration in Cardiac Myocytes. Biophys. J. 2016, 111, 1304–1315. [Google Scholar] [CrossRef] [Green Version]
- Despa, S. Myocyte [Na+]i Dysregulation in Heart Failure and Diabetic Cardiomyopathy. Front. Physiol. 2018, 9, 1303. [Google Scholar] [CrossRef] [Green Version]
- Despa, S.; Bers, D. Na+ transport in the normal and failing heart—Remember the balance. J. Mol. Cell. Cardiol. 2013, 61, 2–10. [Google Scholar] [CrossRef] [Green Version]
- Schramm, M.; Klieber, H.G.; Daut, J. The energy-expenditure of actomyosin-ATPase, Ca2+-ATPase and Na+/K+-ATPase in guinea-pig cardiac ventricular muscle. J. Physiol.-Lond. 1994, 481, 647–662. [Google Scholar] [CrossRef] [PubMed]
- Brill, D.M.; Wasserstrom, J.A. Intracellular sodium and the positive inotropic effect of veratridine and cardiac glycoside in sheep Purkinje fibers. Circ. Res. 1986, 58, 109–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Brown, J.H. Role of Ca2+/calmodulin-dependent protein kinase II in cardiac hypertrophy and heart failure. Cardiovasc. Res. 2004, 63, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.E. Calmodulin kinase signaling in heart: An intriguing candidate target for therapy of myocardial dysfunction and arrhythmias. Pharmacol. Ther. 2005, 106, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M.; Grandi, E. Calcium/Calmodulin-dependent Kinase II Regulation of Cardiac Ion Channels. J. Cardiovasc. Pharmacol. 2009, 54, 180–187. [Google Scholar] [CrossRef] [Green Version]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef]
- Zhu, W.; Woo, A.Y.; Yang, D.; Cheng, H.; Crow, M.T.; Xiao, R.P. Activation of CaMKIIdeltaC is a common intermediate of diverse death stimuli-induced heart muscle cell apoptosis. J. Biol. Chem. 2007, 282, 10833–10839. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, X.; Kubo, H.; Harris, D.M.; Mills, G.D.; Moyer, J.; Berretta, R.; Potts, S.T.; Marsh, J.D.; Houser, S.R. Ca2+ Influx–Induced Sarcoplasmic Reticulum Ca2+ Overload Causes Mitochondrial-Dependent Apoptosis in Ventricular Myocytes. Circ. Res. 2005, 97, 1009–1017. [Google Scholar] [CrossRef] [Green Version]
- Petroff, M.V.; Salas, M.A.; Said, M.; Valverde, C.A.; Sapia, L.; Portiansky, E.; Hajjar, R.J.; Kranias, E.G.; Mundiña-Weilenmann, C.; Mattiazzi, A. CaMKII inhibition protects against necrosis and apoptosis in irreversible ischemia–reperfusion injury. Cardiovasc. Res. 2007, 73, 689–698. [Google Scholar] [CrossRef]
- Noble, D.; Noble, P.J. Late sodium current in the pathophysiology of cardiovascular disease: Consequences of sodium-calcium overload. Heart 2006, 92, iv1–iv5. [Google Scholar] [CrossRef]
- Zhang, S.; Ma, J.-H.; Zhang, P.-H.; Luo, A.-T.; Ren, Z.-Q.; Kong, L.-H. Sophocarpine Attenuates the Na+-dependent Ca2+ Overload Induced by Anemonia Sulcata Toxin—Increased Late Sodium Current in Rabbit Ventricular Myocytes. J. Cardiovasc. Pharmacol. 2012, 60, 357–366. [Google Scholar] [CrossRef]
- Philippaert, K.; Kalyaanamoorthy, S.; Fatehi, M.; Long, W.; Soni, S.; Byrne, N.J.; Barr, A.; Singh, J.; Wong, J.; Palechuk, T.; et al. Cardiac Late Sodium Channel Current Is a Molecular Target for the Sodium/Glucose Cotransporter 2 Inhibitor Empagliflozin. Circulation 2021, 143, 2188–2204. [Google Scholar] [CrossRef]
- Bengel, P.; Dybkova, N.; Tirilomis, P.; Ahmad, S.; Hartmann, N.; Mohamed, B.A.; Krekeler, M.C.; Maurer, W.; Pabel, S.; Trum, M.; et al. Detrimental proarrhythmogenic interaction of Ca2+/calmodulin-dependent protein kinase II and Na(V)1.8 in heart failure. Nat. Commun. 2021, 12, 6586. [Google Scholar] [CrossRef]
- Hilgemann, D.W. Control of cardiac contraction by sodium: Promises, reckonings, and new beginnings. Cell Calcium 2019, 85, 102129. [Google Scholar] [CrossRef]
- Aksentijevic, D.; O’Brien, B.A.; Eykyn, T.R.; Shattock, M. Is there a causal link between intracellular Na elevation and metabolic remodelling in cardiac hypertrophy? Biochem. Soc. Trans. 2018, 46, 817–827. [Google Scholar] [CrossRef] [Green Version]
- Phuket, T.; Covarrubias, M. Kv4 Channels Underlie the Subthreshold-Operating A-type K+-current in Nociceptive Dorsal Root Ganglion Neurons. Front. Mol. Neurosci. 2009, 2, 3. [Google Scholar] [CrossRef] [Green Version]
- Antoons, G.; Volders, P.G.A.; Stankovicova, T.; Bito, V.; Stengl, M.; Vos, M.A.; Sipido, K.R. Window Ca2+current and its modulation by Ca2+release in hypertrophied cardiac myocytes from dogs with chronic atrioventricular block. J. Physiol. 2007, 579, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Morita, N.; Lee, J.H.; Xie, Y.; Sovari, A.; Qu, Z.; Weiss, J.N.; Karagueuzian, H.S. Suppression of Re-Entrant and Multifocal Ventricular Fibrillation by the Late Sodium Current Blocker Ranolazine. J. Am. Coll. Cardiol. 2011, 57, 366–375. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Sun, H.-Y.; Lau, C.-P.; Li, G.-R. Regulation of voltage-gated cardiac sodium current by epidermal growth factor receptor kinase in guinea pig ventricular myocytes. J. Mol. Cell. Cardiol. 2007, 42, 760–768. [Google Scholar] [CrossRef]
- Wang, D.W.; Yazawa, K.; George, A.L.; Bennett, P.B. Characterization of human cardiac Na+ channel mutations in the congenital long QT syndrome. Proc. Natl. Acad. Sci. USA 1996, 93, 13200–13205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, J.D.; Clancy, C.E. Pathophysiology of the cardiac late Na current and its potential as a drug target. J. Mol. Cell. Cardiol. 2011, 52, 608–619. [Google Scholar] [CrossRef] [Green Version]
- Beyder, A.; Rae, J.L.; Bernard, C.; Strege, P.R.; Sachs, F.; Farrugia, G. Mechanosensitivity of Na(v)1.5, a voltage-sensitive sodium channel. J. Physiol.-Lond. 2010, 588, 4969–4985. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly-Shah, V.; Wingo, T.L.; Weiss, K.L.; Williams, C.K.; Balser, J.R.; Chazin, W.J. Calcium-dependent regulation of the voltage-gated sodium channel hH1: Intrinsic and extrinsic sensors use a common molecular switch. Proc. Natl. Acad. Sci. USA 2006, 103, 3592–3597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wingo, T.L.; Shah, V.N.; Anderson, M.E.; Lybrand, T.P.; Chazin, W.J.; Balser, J.R. An EF-hand in the sodium channel couples intracellular calcium to cardiac excitability. Nat. Struct. Mol. Biol. 2004, 11, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Y.; Liu, N.; Priori, S.G. Sodium channel mutations and arrhythmias. Nat. Rev. Cardiol. 2009, 6, 337–348. [Google Scholar] [CrossRef]
- Rivaud, M.R.; Baartscheer, A.; Verkerk, A.O.; Beekman, L.; Rajamani, S.; Belardinelli, L.; Bezzina, C.R.; Remme, C.A. Enhanced late sodium current underlies pro-arrhythmic intracellular sodium and calcium dysregulation in murine sodium channelopathy. Int. J. Cardiol. 2018, 263, 54–62. [Google Scholar] [CrossRef]
- Rivaud, M.R.; Marchal, G.A.; Wolswinkel, R.; Jansen, J.A.; van der Made, I.; Beekman, L.; Ruiz-Villalba, A.; Baartscheer, A.; Rajamani, S.; Belardinelli, L.; et al. Functional modulation of atrio-ventricular conduction by enhanced late sodium current and calcium-dependent mechanisms in Scn5a(1798insDl+) mice. Europace 2020, 22, 1579–1589. [Google Scholar] [CrossRef]
- Peters, C.H.; Watkins, A.R.; Poirier, O.L.; Ruben, P.C. E1784K, the most common Brugada syndrome and long-QT syndrome type 3 mutant, disrupts sodium channel inactivation through two separate mechanisms. J. Gen. Physiol. 2020, 152, e202012595. [Google Scholar] [CrossRef]
- Mitsuiye, T.; Noma, A. Inactivation of Cardiac Na+ Channel Simply through Open States as Revealed by Single-Channel Analysis in Guinea Pig Ventricular Myocytes. Jpn. J. Physiol. 2002, 52, 457–469. [Google Scholar] [CrossRef] [Green Version]
- Maltsev, V.; Undrovinas, A.I. A multi-modal composition of the late Na+ current in human ventricular cardiomyocytes. Cardiovasc. Res. 2006, 69, 116–127. [Google Scholar] [CrossRef] [Green Version]
- Scanley, B.E.; Hanck, D.A.; Chay, T.; Fozzard, H.A. Kinetic-analysis of single sodium-channels from canine cardiac Purkinje-cells. J. Gen. Physiol. 1990, 95, 411–437. [Google Scholar] [CrossRef] [PubMed]
- Patlak, J.B.; Ortiz, M. Kinetic diversity of Na+ channel bursts in frog skeletal muscle. J. Gen. Physiol. 1989, 94, 279–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patlak, J.B.; Ortiz, M. Two modes of gating during late Na+ channel currents in frog sartorius muscle. J. Gen. Physiol. 1986, 87, 305–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patlak, J.B.; Ortiz, M. Slow currents through single sodium channels of the adult rat heart. J. Gen. Physiol. 1985, 86, 89–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohlhardt, M.; Frobe, U.; Herzig, J.W. Properties of normal and noninactivating single cardic Na+ channels. Proc. R. Soc. Ser. B-Biol. Sci. 1987, 232, 71–93. [Google Scholar]
- Bezzina, C.; Veldkamp, M.W.; Berg, M.V.D.; Postma, A.; Rook, M.B.; Viersma, J.-W.; van Langen, I.M.; Tan-Sindhunata, G.; Bink-Boelkens, M.T.E.; van der Hout, A.H.; et al. A Single Na + Channel Mutation Causing Both Long-QT and Brugada Syndromes. Circ. Res. 1999, 85, 1206–1213. [Google Scholar] [CrossRef] [Green Version]
- Maltsev, V.; Silverman, N.; Sabbah, H.N.; Undrovinas, A.I. Chronic heart failure slows late sodium current in human and canine ventricular myocytes: Implications for repolarization variability. Eur. J. Heart Fail. 2007, 9, 219–227. [Google Scholar] [CrossRef] [Green Version]
- Valdivia, C.R.; Chu, W.W.; Pu, J.; Foell, J.D.; Haworth, R.A.; Wolff, M.R.; Kamp, T.; Makielski, J.C. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J. Mol. Cell. Cardiol. 2005, 38, 475–483. [Google Scholar] [CrossRef]
- Song, Y.; Shryock, J.C.; Belardinelli, L. An increase of late sodium current induces delayed afterdepolarizations and sustained triggered activity in atrial myocytes. Am. J. Physiol. Circ. Physiol. 2008, 294, H2031–H2039. [Google Scholar] [CrossRef] [Green Version]
- Trenor, B.; Cardona, K.; Gómez, J.F.; Rajamani, S.; Jr, J.M.F.; Belardinelli, L.; Saiz, J. Simulation and Mechanistic Investigation of the Arrhythmogenic Role of the Late Sodium Current in Human Heart Failure. PLoS ONE 2012, 7, e32659. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Shryock, J.C.; Song, Y.; Belardinelli, L. An Increase in Late Sodium Current Potentiates the Proarrhythmic Activities of Low-Risk QT-Prolonging Drugs in Female Rabbit Hearts. J. Pharmacol. Exp. Ther. 2005, 316, 718–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belardinelli, L.; Shryock, J.C.; Fraser, H. Inhibition of the late sodium current as a potential cardioprotective principle: Effects of the late sodium current inhibitor ranolazine. Heart 2006, 92, iv6–iv14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magyar, J.; Kiper, C.E.; Dumaine, R.; Burgess, D.E.; Bányász, T.; Satin, J. Divergent action potential morphologies reveal nonequilibrium properties of human cardiac Na channels. Cardiovasc. Res. 2004, 64, 477–487. [Google Scholar] [CrossRef]
- Bant, J.S.; Raman, I.M. Control of transient, resurgent, and persistent current by open-channel block by Na channel β4 in cultured cerebellar granule neurons. Proc. Natl. Acad. Sci. USA 2010, 107, 12357. [Google Scholar] [CrossRef] [Green Version]
- Grieco, T.M.; Raman, I.M. Production of Resurgent Current in NaV1.6-Null Purkinje Neurons by Slowing Sodium Channel Inactivation with β-Pompilidotoxin. J. Neurosci. 2004, 24, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raman, I.M.; Bean, B.P. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J. Neurosci. 1997, 17, 4517–4526. [Google Scholar] [CrossRef] [Green Version]
- Eijkelkamp, N.; Linley, J.; Baker, M.D.; Minett, M.S.; Cregg, R.; Werdehausen, R.; Rugiero, F.; Wood, J.N. Neurological perspectives on voltage-gated sodium channels. Brain 2012, 135, 2585–2612. [Google Scholar] [CrossRef] [PubMed]
- Schiavon, E.; Sacco, T.; Cassulini, R.R.; Gurrola, G.; Tempia, F.; Possani, L.D.; Wanke, E. Resurgent current and voltage sensor trapping enhanced activation by a beta-scorpion toxin solely in Nav1.6 channel. Significance in mice Purkinje neurons. J. Biol. Chem. 2006, 281, 20326–20337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeMarco, K.R.; Clancy, C.E. Cardiac Na Channels: Structure to Function. In Na Channels from Phyla to Function; French, R.J., Noskov, S.Y., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; Volume 78, pp. 287–311. [Google Scholar]
- Maier, L.S. CaMKII regulation of voltage-gated sodium channels and cell excitability. Heart Rhythm. Off. J. Heart Rhythm. Soc. 2011, 8, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Maltsev, V.A.; Reznikov, V.; Undrovinas, N.A.; Sabbah, H.N.; Undrovinas, A. Modulation of late sodium current by Ca2+, calmodulin, and CaMKII in normal and failing dog cardiomyocytes: Similarities and differences. Am. J. Physiol. Circ. Physiol. 2008, 294, H1597–H1608. [Google Scholar] [CrossRef] [Green Version]
- Lewit-Bentley, A.; Réty, S. EF-hand calcium-binding proteins. Curr. Opin. Struct. Biol. 2001, 10, 637–643. [Google Scholar] [CrossRef]
- Van Petegem, F.; Lobo, P.A.; Ahern, C.A. Seeing the Forest through the Trees: Towards a Unified View on Physiological Calcium Regulation of Voltage-Gated Sodium Channels. Biophys. J. 2012, 103, 2243–2251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, H.L.; Kupershmidt, S.; Zhang, R.; Stepanovic, S.; Roden, D.M.; Wilde, A.A.M.; Anderson, M.E.; Balser, J.R. A calcium sensor in the sodium channel modulates cardiac excitability. Nature 2002, 415, 442–447. [Google Scholar] [CrossRef]
- Kim, J.; Ghosh, S.; Liu, H.; Tateyama, M.; Kass, R.S.; Pitt, G.S. Calmodulin mediates Ca2+ sensitivity of sodium channels. J. Biol. Chem. 2004, 279, 45004–45012. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.; DiSilvestre, D.; Tian, Y.; Halperin, V.L.; Tomaselli, G.F. Calcium-Mediated Dual-Mode Regulation of Cardiac Sodium Channel Gating. Circ. Res. 2009, 104, 870–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarhan, M.F.; Tung, C.C.; van Petegem, F.; Ahern, C.A. Crystallographic basis for calcium regulation of sodium channels. Proc. Natl. Acad. Sci. USA 2012, 109, 3558–3563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulbricht, W. Sodium Channel Inactivation: Molecular Determinants and Modulation. Physiol. Rev. 2005, 85, 1271–1301. [Google Scholar] [CrossRef] [Green Version]
- Murray, K.T.; Hu, N.; Daw, J.R.; Shin, H.-G.; Watson, M.T.; Mashburn, A.B.; George, A.L. Functional Effects of Protein Kinase C Activation on the Human Cardiac Na sup + Channel. Circ. Res. 1997, 80, 370–376. [Google Scholar] [CrossRef]
- Scheuer, T. Regulation of sodium channel activity by phosphorylation. Semin. Cell Dev. Biol. 2011, 22, 160–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, Y.; Rogers, J.C.; Tanada, T.N.; Catterall, W.A.; Scheuer, T. Phosphorylation of S1505 in the cardiac Na+ channel inactivation gate is required for modulation by protein kinase C. J. Gen. Physiol. 1996, 108, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Z.; Vermij, S.H.; Sottas, V.; Shestak, A.; Ross-Kaschitza, D.; Zaklyazminskaya, E.V.; Hudmon, A.; Pitt, G.S.; Rougier, J.S.; Abriel, H. Calmodulin binds to the N-terminal domain of the cardiac sodium channel Na(v)1.5. Channels 2020, 14, 268–286. [Google Scholar] [CrossRef] [PubMed]
- Hudmon, A.; Schulman, H. Structure-function of the multifunctional Ca2+/calmodulin-dependent protein kinase II. Biochem. J. 2002, 364, 593–611. [Google Scholar] [CrossRef] [PubMed]
- Marionneau, C.; Lichti, C.F.; Lindenbaum, P.; Charpentier, F.; Nerbonne, J.M.; Townsend, R.R.; Mérot, J. Mass Spectrometry-Based Identification of Native Cardiac Nav1.5 Channel α Subunit Phosphorylation Sites. J. Proteome Res. 2012, 11, 5994–6007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, B.J.; Rogers, J.; Perdichizzi, A.P.; Colvin, A.A.; Catterall, W.A. cAMP-dependent phosphorylation of two sites in the alpha subunit of the cardiac sodium channel. J. Biol. Chem. 1996, 271, 28837–28843. [Google Scholar] [CrossRef] [Green Version]
- Rook, M.B.; Evers, M.M.; Vos, M.A.; Bierhuizen, M.F. Biology of cardiac sodium channel Nav1.5 expression. Cardiovasc. Res. 2012, 93, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Yi, J.; Hu, N.; George, A.L.; Murray, K.T. Activation of Protein Kinase A Modulates Trafficking of the Human Cardiac Sodium Channel in Xenopus Oocytes. Circ. Res. 2000, 87, 33–38. [Google Scholar] [CrossRef] [Green Version]
- Wagner, S.; Dybkova, N.; Rasenack, E.C.L.; Jacobshagen, C.; Fabritz, L.; Kirchhof, P.; Maier, S.K.G.; Zhang, T.; Hasenfuss, G.; Brown, J.H.; et al. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J. Clin. Investig. 2006, 116, 3127–3138. [Google Scholar] [CrossRef]
- Ashpole, N.M.; Herren, A.W.; Ginsburg, K.S.; Brogan, J.D.; Johnson, D.E.; Cummins, T.R.; Bers, D.M.; Hudmon, A. Ca2+/Calmodulin-dependent Protein Kinase II (CaMKII) Regulates Cardiac Sodium Channel NaV1.5 Gating by Multiple Phosphorylation Sites. J. Biol. Chem. 2012, 287, 19856–19869. [Google Scholar] [CrossRef] [Green Version]
- Koval, O.M.; Snyder, J.S.; Wolf, R.M.; Pavlovicz, R.E.; Glynn, P.; Curran, J.; Leymaster, N.D.; Dun, W.; Wright, P.J.; Cardona, N.; et al. Ca2+/calmodulin-dependent protein kinase II-based regulation of voltage-gated Na+ channel in cardiac disease. Circulation 2012, 126, 2084–2094. [Google Scholar] [CrossRef] [Green Version]
- Aiba, T.; Hesketh, G.G.; Liu, T.; Carlisle, R.; Villa-Abrille, M.C.; O’Rourke, B.; Akar, F.G.; Tomaselli, G.F. Na+ channel regulation by Ca2+/calmodulin and Ca2+/calmodulin-dependent protein kinase II in guinea-pig ventricular myocytes. Cardiovasc. Res. 2010, 85, 454–463. [Google Scholar] [CrossRef] [Green Version]
- Fouda, M.A.; Ruben, P.C. Protein Kinases Mediate Anti-Inflammatory Effects of Cannabidiol and Estradiol Against High Glucose in Cardiac Sodium Channels. Front. Pharmacol. 2021, 12, 1004. [Google Scholar] [CrossRef] [PubMed]
- Palaniyandi, S.S.; Sun, L.; Ferreira, J.C.B.; Mochly-Rosen, D. Protein kinase C in heart failure: A therapeutic target? Cardiovasc. Res. 2008, 82, 229–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellor, H.; Parker, P. The extended protein kinase C superfamily. Biochem. J. 1998, 332, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Herren, A.W.; Bers, D.M.; Grandi, E. Post-translational modifications of the cardiac Na channel: Contribution of CaMKII-dependent phosphorylation to acquired arrhythmias. Am. J. Physiol.-Heart Circ. Physiol. 2013, 305, H431–H445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimm, M.; Ling, H.Y.; Brown, J.H. Crossing signals: Relationships between beta-adrenergic stimulation and CaMKII activation. Heart Rhythm. Off. J. Heart Rhythm. Soc. 2011, 8, 1296–1298. [Google Scholar] [CrossRef] [Green Version]
- Tateyama, M.; Rivolta, I.; Clancy, C.E.; Kass, R.S. Modulation of Cardiac Sodium Channel Gating by Protein Kinase A Can Be Altered by Disease-linked Mutation. J. Biol. Chem. 2003, 278, 46718–46726. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Luo, A.; Wu, L.; Wan, W.; Zhang, P.; Ren, Z.; Zhang, S.; Qian, C.; Shryock, J.C.; Belardinelli, L. Calmodulin kinase II and protein kinase C mediate the effect of increased intracellular calcium to augment late sodium current in rabbit ventricular myocytes. Am. J. Physiol. Physiol. 2012, 302, C1141–C1151. [Google Scholar] [CrossRef] [Green Version]
- Qu, Y.; Rogers, J.; Tanada, T.; Scheuer, T.; Catterall, W.A. Modulation of cardiac Na+ channels expressed in a mammalian cell line and in ventricular myocytes by protein kinase C. Proc. Natl. Acad. Sci. USA 1994, 91, 3289–3293. [Google Scholar] [CrossRef] [Green Version]
- Aoyama, T.; Matsui, T.; Novikov, M.; Park, J.; Hemmings, B.; Rosenzweig, A. Serum and Glucocorticoid-Responsive Kinase-1 Regulates Cardiomyocyte Survival and Hypertrophic Response. Circulation 2005, 111, 1652–1659. [Google Scholar] [CrossRef] [Green Version]
- Lang, F.; Shumilina, E. Regulation of ion channels by the serum- and glucocorticoid-inducible kinase SGK1. FASEB J. 2012, 27, 3–12. [Google Scholar] [CrossRef]
- Lang, F.; Böhmer, C.; Palmada, M.; Seebohm, G.; Strutz-Seebohm, N.; Vallon, V. (Patho)physiological Significance of the Serum- and Glucocorticoid-Inducible Kinase Isoforms. Physiol. Rev. 2006, 86, 1151–1178. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Deak, M.; Morrice, N.; Cohen, P. Characterization of the structure and regulation of two novel isoforms of serum- and glucocorticoid-induced protein kinase. Biochem. J. 1999, 344, 189–197. [Google Scholar] [CrossRef]
- Murphy, L.; Renodin, D.; Antzelevitch, C.; Di Diego, J.M.; Cordeiro, J.M. Extracellular proton depression of peak and late Na+ current in the canine left ventricle. Am. J. Physiol. Circ. Physiol. 2011, 301, H936–H944. [Google Scholar] [CrossRef] [Green Version]
- Murphy, L.; Renodin, D.M.; Antzelevitch, C.; Di Diego, J.M.; Cordeiro, J.M. Extracellular Proton Modulation of Peak and Late Sodium Current in the Canine Left Ventricle. Biophys. J. 2011, 100, 574a. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.; Peters, C.H.; Allard, C.R.; Claydon, T.; Ruben, P.C. Proton Sensors in the Pore Domain of the Cardiac Voltage-gated Sodium Channel. J. Biol. Chem. 2013, 288, 4782–4791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Aiba, T.; Rosenberg, M.; Hessler, K.; Xiao, C.; Quintero, P.A.; Ottaviano, F.G.; Knight, A.C.; Graham, E.L.; Boström, P.; et al. Pathological Role of Serum- and Glucocorticoid-Regulated Kinase 1 in Adverse Ventricular Remodeling. Circulation 2012, 126, 2208–2219. [Google Scholar] [CrossRef] [PubMed]
- Boehmer, C.; Wilhelm, V.; Palmada, M.; Wallisch, S.; Henke, G.; Brinkmeier, H.; Cohen, P.; Pieske, B.; Lang, F. Serum and glucocorticoid inducible kinases in the regulation of the cardiac sodium channel SCN5A. Cardiovasc. Res. 2003, 57, 1079–1084. [Google Scholar] [CrossRef] [Green Version]
- Fahmi, A.; Forhead, A.; Fowden, A.; Vandenberg, J.; Forhead, A.; Vandenberg, J. Cortisol influences the ontogeny of both alpha- and beta-subunits of the cardiac sodium channel in fetal sheep. J. Endocrinol. 2004, 180, 449–455. [Google Scholar] [CrossRef]
- Nguyenthi, A.; Ruizceretti, E.; Schanne, O.F. Electrophysiologic effects and electrolite changes in total myocardial ischemia. Can. J. Physiol. Pharmacol. 1981, 59, 876–883. [Google Scholar] [CrossRef]
- Jones, D.; Peters, C.; Tolhurst, S.; Claydon, T.; Ruben, P. Extracellular Proton Modulation of the Cardiac Voltage-Gated Sodium Channel, NaV1.5. Biophys. J. 2011, 101, 2147–2156. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.; Claydon, T.; Ruben, P. Extracellular Protons Inhibit Charge Immobilization in the Cardiac Voltage-Gated Sodium Channel. Biophys. J. 2013, 105, 101–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Q.; Ma, J.H.; Zhang, P.H.; Wan, W.; Kong, L.H.; Wu, L. Persistent sodium current and Na+/H+ exchange contributes to the augmentation of the reverse Na+/Ca2+ exchange during hypoxia or acute ischemia in ventricular myocytes. Pflug. Arch.-Eur. J. Physiol. 2012, 463, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, L.A.; Polak, J. Hypoxia. 4. Hypoxia and ion channel function. Am. J. Physiol.-Cell Physiol. 2011, 300, C951–C967. [Google Scholar] [CrossRef]
- Hammarstrom, A.K.; Gage, P.W. Hypoxia and persistent sodium current. Eur. Biophys. J. 2002, 31, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, E. Cardiac Ionic Currents and Acute Ischemia: From Channels to Arrhythmias. Physiol. Rev. 1999, 79, 917–1017. [Google Scholar] [CrossRef]
- Wang, W.; Ma, J.; Zhang, P.; Luo, A. Redox reaction modulates transient and persistent sodium current during hypoxia in guinea pig ventricular myocytes. Pflugers Arch. 2007, 454, 461–475. [Google Scholar] [CrossRef]
- Plant, L.D.; Xiong, D.; Romero, J.; Dai, H.; Goldstein, S.A. Hypoxia Produces Pro-arrhythmic Late Sodium Current in Cardiac Myocytes by SUMOylation of NaV1.5 Channels. Cell Rep. 2020, 30, 2225–2236. [Google Scholar] [CrossRef] [Green Version]
- Ward, C.A.; Giles, W.R. Ionic mechanism of the effects of hydrogen peroxide in rat ventricular myocytes. J. Physiol. 1997, 500, 631–642. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Shryock, J.C.; Wu, L.; Belardinelli, L. Antagonism by Ranolazine of the Pro-Arrhythmic Effects of Increasing Late INa in Guinea Pig Ventricular Myocytes. J. Cardiovasc. Pharmacol. 2004, 44, 192–199. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Shryock, J.C.; Wagner, S.; Maier, L.S.; Belardinelli, L. Blocking Late Sodium Current Reduces Hydrogen Peroxide-Induced Arrhythmogenic Activity and Contractile Dysfunction. J. Pharmacol. Exp. Ther. 2006, 318, 214–222. [Google Scholar] [CrossRef] [Green Version]
- Pignier, C.; Revenaz, C.; Rauly-Lestienne, I.; Cussac, D.; Delhon, A.; Gardette, J.; Le Grand, B. Direct protective effects of poly-unsaturated fatty acids, DHA and EPA, against activation of cardiac late sodium current. Basic Res. Cardiol. 2007, 102, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Burnashev, N.A.; Undrovinas, A.I.; Fleidervish, I.A.; Makielski, J.C.; Rosenshtraukh, L.V. Modulation of cardiac sodium-channel gating by lysophosphatidylcholine. J. Mol. Cell. Cardiol. 1991, 23, 23–30. [Google Scholar] [CrossRef]
- Ahern, G.P.; Hsu, S.-F.; Klyachko, V.A.; Jackson, M.B. Induction of Persistent Sodium Current by Exogenous and Endogenous Nitric Oxide. J. Biol. Chem. 2000, 275, 28810–28815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.; Valdivia, C.R.; Vaidyanathan, R.; Balijepalli, R.C.; Ackerman, M.J.; Makielski, J.C. Caveolin-3 suppresses late sodium current by inhibiting nNOS-dependent S-nitrosylation of SCN5A. J. Mol. Cell. Cardiol. 2013, 61, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Bussey, C.T.; Erickson, J.R. Physiology and pathology of cardiac CaMKII. Curr. Opin. Physiol. 2018, 1, 52–58. [Google Scholar] [CrossRef]
- Matasic, D.S.; Yoon, J.Y.; McLendon, J.M.; Mehdi, H.; Schmidt, M.S.; Greiner, A.M.; Quinones, P.; Morgan, G.M.; Boudreau, R.L.; Irani, K.; et al. Modulation of the cardiac sodium channel Na(V)1.5 peak and late currents by NAD(+) precursors. J. Mol. Cell. Cardiol. 2020, 141, 70–81. [Google Scholar] [CrossRef]
- Morris, C.E.; Juranka, P.F. Nav Channel Mechanosensitivity: Activation and Inactivation Accelerate Reversibly with Stretch. Biophys. J. 2007, 93, 822–833. [Google Scholar] [CrossRef] [Green Version]
- Beyder, A.; Strege, P.R.; Reyes, S.; Bernard, C.E.; Terzic, A.; Makielski, J.C.; Ackerman, M.J.; Farrugia, G. Ranolazine Decreases Mechanosensitivity of the Voltage-Gated Sodium Ion Channel Na V 1.5. Circulation 2012, 125, 2698–2706. [Google Scholar] [CrossRef] [Green Version]
- Strege, P.; Beyder, A.; Bernard, C.; Crespo-Diaz, R.; Behfar, A.; Terzic, A.; Ackerman, M.; Farrugia, G. Ranolazine inhibits shear sensitivity of endogenous Na+current and spontaneous action potentials in HL-1 cells. Channels 2012, 6, 457–462. [Google Scholar] [CrossRef] [Green Version]
- Nassal, D.; Yu, J.; Min, D.; Lane, C.; Shaheen, R.; Gratz, D.; Hund, T. Regulation of Cardiac Conduction and Arrhythmias by Ankyrin/Spectrin-Based Macromolecular Complexes. J. Cardiovasc. Dev. Dis. 2021, 8, 48. [Google Scholar] [CrossRef]
- Dalton, G.R.; Jones, J.V.; Evans, S.J.; Levi, A.J. Wall stress-induced arrhythmias in the working rat heart as left ventricular hypertrophy regresses during captopril treatment. Cardiovasc. Res. 1997, 33, 561–572. [Google Scholar] [CrossRef] [Green Version]
- Salmon, A.H.; Mays, J.L.; Dalton, G.R.; Jones, J.V.; Levi, A.J. Effect of streptomycin on wall-stress-induced arrhythmias in the working rat heart. Cardiovasc. Res. 1997, 34, 493–503. [Google Scholar] [CrossRef] [Green Version]
- Parker, K.K.; Lavelle, J.A.; Taylor, L.K.; Wang, Z.; Hansen, D.E. Stretch-induced ventricular arrhythmias during acute ischemia and reperfusion. J. Appl. Physiol. 2004, 97, 377–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortada, E.; Serradesanferm, R.; Brugada, R.; Verges, M. The voltage-gated sodium channel beta 2 subunit associates with lipid rafts by S-palmitoylation. J. Cell Sci. 2021, 134, jcs252189. [Google Scholar] [CrossRef]
- Namadurai, S.; Yereddi, N.R.; Cusdin, F.S.; Huang, C.L.-H.; Chirgadze, D.Y.; Jackson, A.P. A new look at sodium channel β subunits. Open Biol. 2015, 5, 140192. [Google Scholar] [CrossRef] [Green Version]
- Nevin, S.T.; Lawrence, N.; Nicke, A.; Lewis, R.J.; Adams, D.J. Functional modulation of the human voltage-gated sodium channel Na(V)1.8 by auxiliary beta subunits. Channels 2021, 15, 79–93. [Google Scholar] [CrossRef]
- Abramochkin, D.V.; Filatova, T.S.; Pustovit, K.B.; Dzhumaniiazova, I.; Karpushev, A.V. Small G—protein RhoA is a potential inhibitor of cardiac fast sodium current. J. Physiol. Biochem. 2020, 77, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Petitprez, S.; Zmoos, A.F.; Ogrodnik, J.; Balse, E.; Raad, N.; El-Haou, S.; Albesa, M.; Bittihn, P.; Luther, S.; Lehnart, S.E.; et al. SAP97 and Dystrophin Macromolecular Complexes Determine Two Pools of Cardiac Sodium Channels Na(v)1.5 in Cardiomyocytes. Circ. Res. 2011, 108, 294–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, B.; Hu, Y.; Wang, Z.; Cheng, C.; Wang, P.; Liang, L.; Xiong, H.; Luo, C.; Xu, C.; Chen, Q.; et al. UBC9 regulates cardiac sodium channel Na(v)1.5 ubiquitination, degradation and sodium current density. J. Mol. Cell. Cardiol. 2019, 129, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; He, Y.; Wang, X.; Ge, J.; Li, H. Ventricular voltage-gated ion channels: Detection, characteristics, mechanisms, and drug safety evaluation. Clin. Transl. Med. 2021, 11, e530. [Google Scholar] [CrossRef]
- Remme, C.A. Cardiac sodium channelopathy associated with SCN5A mutations: Electrophysiological, molecular and genetic aspects. J. Physiol.-Lond. 2013, 591, 4099–4116. [Google Scholar] [CrossRef] [PubMed]
- Lowe, J.S.; Stroud, D.M.; Yang, T.; Hall, L.; Atack, T.C.; Roden, D.M. Increased late sodium current contributes to long QT-related arrhythmia susceptibility in female mice. Cardiovasc. Res. 2012, 95, 300–307. [Google Scholar] [CrossRef] [Green Version]
- Bezzina, C.R.; Remme, C.A. Dilated Cardiomyopathy due to Sodium Channel Dysfunction What Is the Connection? Circ. -Arrhythmia Electrophysiol. 2008, 1, 80–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, B.; El-Sherif, T.; Gidh-Jain, M.; Qin, D.; El-Sherif, N. Alterations of Sodium Channel Kinetics and Gene Expression in the Postinfarction Remodeled Myocardium. J. Cardiovasc. Electrophysiol. 2001, 12, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Zhang, T.; Pereira, L.; Means, C.K.; Cheng, H.; Gu, Y.; Dalton, N.D.; Peterson, K.L.; Chen, J.; Bers, D.; et al. Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J. Clin. Investig. 2009, 119, 1230–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backs, J.; Backs, T.; Neef, S.; Kreusser, M.M.; Lehmann, L.H.; Patrick, D.M.; Grueter, C.E.; Qi, X.; Richardson, J.A.; Hill, J.A.; et al. The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc. Natl. Acad. Sci. USA 2009, 106, 2342–2347. [Google Scholar] [CrossRef] [Green Version]
- Undrovinas, A.I.; Maltsev, V.A.; Sabbah, H.N. Repolarization abnormalities in cardiomyocytes of dogs with chronic heart failure: Role of sustained inward current. Cell. Mol. Life Sci. 1999, 55, 494–505. [Google Scholar] [CrossRef]
- January, C.T.; Riddle, J.M. Early afterdepolarizations: Mechanism of induction and block. A role for L-type Ca2+ current. Circ. Res. 1989, 64, 977–990. [Google Scholar] [CrossRef] [Green Version]
- Marban, E.; Robinson, S.W.; Wier, W.G. Mechanisms of arrhythmogenic delayed and early afterdepolarizations in ferret ventricular muscle. J. Clin. Investig. 1986, 78, 1185–1192. [Google Scholar] [CrossRef]
- Szabo, B.; Sweidan, R.; Rajagopalan, C.V.; Lazzara, R. Role of Na+: Ca2+Exchange Current in Cs+-Induced Early Afterdepolarizations in Purkinje Fibers. J. Cardiovasc. Electrophysiol. 1994, 5, 933–944. [Google Scholar] [CrossRef]
- Szabo, B.; Kovacs, T.; Lazzara, R. Role of Calcium Loading in Early Afterdepolarizations Generated by Cs+in Canine and Guinea Pig Purkinje Fibers. J. Cardiovasc. Electrophysiol. 1995, 6, 796–812. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Kimura, S.; Furukawa, N.; Bassett, A.L.; Myerburg, R.J. Potassium rectifier currents differ in myocytes of endocardial and epicardial origin. Circ. Res. 1992, 70, 91–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.W.; Antzelevitch, C. Characteristics of the delayed rectifier current (I-Kr and I-Ks) in canine ventricular epicardial, midmyocardial, and endocardial myocytes—A weaker I-Ks contributes to the longer action-potential of the M-cell. Circ. Res. 1995, 76, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Wettwer, E.; Amos, G.J.; Posival, H.; Ravens, U. Transient outward current in human ventricular myocytes of supepicardial and subendocardial origin. Circ. Res. 1994, 75, 473–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kass, R.S.; Lederer, W.J.; Tsien, R.W.; Weingart, R. Role of calcium-ions in transient inward currents and after contractions induced by strophantidin in cardiac Purkinje-fibers. J. Physiol. -Lond. 1978, 281, 187–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zygmunt, A.C.; Goodrow, R.J.; Weigel, C.M. I NaCa andI Cl(Ca)contribute to isoproterenol-induced delayed afterdepolarizations in midmyocardial cells. Am. J. Physiol. Circ. Physiol. 1998, 275, H1979–H1992. [Google Scholar] [CrossRef]
- Volders, P.G.; Kulcsár, A.; Vos, M.A.; Sipido, K.R.; Wellens, H.J.; Lazzara, R.; Szabo, B. Similarities between early and delayed afterdepolarizations induced by isoproterenol in canine ventricular myocytes. Cardiovasc. Res. 1997, 34, 348–359. [Google Scholar] [CrossRef] [Green Version]
- Priori, S.G.; Corr, P.B. Mechanisms underlying early and delayed afterdepolarizations induced by catecholamines. Am. J. Physiol. Circ. Physiol. 1990, 258, H1796–H1805. [Google Scholar] [CrossRef]
- January, C.T.; Fozzard, H.A. Delayed afterdepolarizations in heart muscle: Mechanisms and relevance. Pharmacol. Rev. 1988, 40. [Google Scholar]
- Bányász, T.; Fülöp, L.; Magyar, J.; Szentandrássy, N.; Varró, A.; Nánási, P.P. Endocardial versus epicardial differences in L-type calcium current in canine ventricular myocytes studied by action potential voltage clamp. Cardiovasc. Res. 2003, 58, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Nattel, S.; Dobrev, D. The multidimensional role of calcium in atrial fibrillation pathophysiology: Mechanistic insights and therapeutic opportunities. Eur. Heart J. 2012, 33, 1870–1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Wagoner, D.R.; Pond, A.; Lamorgese, M.; Rossie, S.S.; McCarthy, P.M.; Nerbonne, J.M. Atrial L-Type Ca2+ Currents and Human Atrial Fibrillation. Circ. Res. 1999, 85, 428–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biliczki, P.; Virág, L.; Iost, N.; Papp, J.G.; Varró, A. Interaction of different potassium channels in cardiac repolarization in dog ventricular preparations: Role of repolarization reserve. J. Cereb. Blood Flow Metab. 2002, 137, 361–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litovsky, S.H.; Antzelevitch, C. Transient outward current prominent in canine ventricular epicardium but not endocardium. Circ. Res. 1988, 62, 116–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.W.; Gintant, G.A.; Antzelevitch, C. Ionic bases for electrophysiological distinctions among epicardial, mid-myocardial, and endocardial myocytes from the free wall of the canine left-ventricle. Circ. Res. 1993, 72, 671–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szentandrássy, N.; Banyasz, T.; Biro, T.; Szabo, G.; Tóth, B.I.; Magyar, J.; Lazar, J.; Varro, A.; Kovacs, L.; Nánási, P.P. Apico?basal inhomogeneity in distribution of ion channels in canine and human ventricular myocardium. Cardiovasc. Res. 2005, 65, 851–860. [Google Scholar] [CrossRef] [Green Version]
- Bauer, A.; Becker, R.; Karle, C.; Schreiner, K.D.; Senges, J.C.; Voss, F.; Kraft, P.; Kuebler, W.; Schoels, W. Effects of the I-Kr-blocking agent dofetilide and of the I-Ks-blocking agent chromanol 293b on regional disparity of left ventricular repolarization in the intact canine heart. J. Cardiovasc. Pharmacol. 2002, 39, 460–467. [Google Scholar] [CrossRef]
- Cheng, J.H.; Kamiya, K.; Liu, W.R.; Tsuji, Y.; Toyama, J.; Kodama, I. Heterogeneous distribution of the two components of delayed rectifier K+ current: A potential mechanism of the proarrhythmic effects of methanesulfonanilide class III agents. Cardiovasc. Res. 1999, 43, 135–147. [Google Scholar] [CrossRef]
- Kannel, W.; Wolf, P.; Benjamin, E.; Levy, D. Prevalence, incidence, prognosis, and predisposing conditions for atrial fibrillation: Population-based estimates. Am. J. Cardiol. 1998, 82, 2N–9N. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Wolf, P.A.; D’Agostino, R.B.; Silbershatz, H.; Kannel, W.B.; Levy, D. Impact of Atrial Fibrillation on the Risk of Death: The Framingham Heart Study. Circulation 1998, 98, 946–952. [Google Scholar] [CrossRef] [Green Version]
- Blok, M.; Boukens, B.J. Mechanisms of Arrhythmias in the Brugada Syndrome. Int. J. Mol. Sci. 2020, 21, 7051. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Barajas-Martínez, H.; Pfeiffer, R.; Dezi, F.; Pfeiffer, J.; Buch, T.; Betzenhauser, M.J.; Belardinelli, L.; Kahlig, K.M.; Rajamani, S.; et al. Mutations in SCN10A Are Responsible for a Large Fraction of Cases of Brugada Syndrome. J. Am. Coll. Cardiol. 2014, 64, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Cano, J.; Zorio, E.; Mazzanti, A.; Arnau, M.; Trenor, B.; Priori, S.G.; Saiz, J.; Romero, L. Ranolazine as an Alternative Therapy to Flecainide for SCN5A V411M Long QT Syndrome Type 3 Patients. Front. Pharmacol. 2020, 11, 1854. [Google Scholar] [CrossRef] [PubMed]
- Barake, W.; Giudicessi, J.R.; Asirvatham, S.J.; Ackerman, M.J. Purkinje system hyperexcitability and ventricular arrhythmia risk in type 3 long QT syndrome. Heart Rhythm 2020, 17, 1768–1776. [Google Scholar] [CrossRef] [PubMed]
- Giudicessi, J.R.; Wilde, A.A.; Ackerman, M.J. The genetic architecture of long QT syndrome: A critical reappraisal. Trends Cardiovasc. Med. 2018, 28, 453–464. [Google Scholar] [CrossRef] [PubMed]
- McNair, W.P.; Ku, L.; Taylor, M.R.G.; Fain, P.R.; Dao, D.; Wolfel, E.; Mestroni, L.; Familial Cardiomyopathy Registry Research Group. SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation 2004, 110, 2163–2167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, T.M.; Michels, V.V.; Ballew, J.D.; Reyna, S.P.; Karst, M.L.; Herron, K.J.; Horton, S.C.; Rodeheffer, R.J.; Anderson, J.L. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. J. Am. Med. Assoc. 2005, 293, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Groenewegen, W.A.; Wilde, A.A.M. Letter regarding article by McNair et al., “SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia”. Circulation 2005, 112, E9. [Google Scholar] [CrossRef] [Green Version]
- Frustaci, A.; Priori, S.G.; Pieroni, M.; Chimenti, C.; Napolitano, C.; Rivolta, I.; Sanna, T.; Bellocci, F.; Russo, M.A. Cardiac Histological Substrate in Patients with Clinical Phenotype of Brugada Syndrome. Circulation 2005, 112, 3680–3687. [Google Scholar] [CrossRef] [Green Version]
- Ge, J.; Sun, A.; Paajanen, V.; Wang, S.; Su, C.; Yang, Z.; Li, Y.; Wang, S.; Jia, J.; Wang, K.; et al. Molecular and Clinical Characterization of a Novel SCN5A Mutation Associated with Atrioventricular Block and Dilated Cardiomyopathy. Circ. Arrhythmia Electrophysiol. 2008, 1, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Gosselin-Badaroudine, P.; Keller, D.I.; Huang, H.; Pouliot, V.; Chatelier, A.; Osswald, S.; Brink, M.; Chahine, M. A Proton Leak Current through the Cardiac Sodium Channel Is Linked to Mixed Arrhythmia and the Dilated Cardiomyopathy Phenotype. PLoS ONE 2012, 7, e38331. [Google Scholar] [CrossRef]
- Sedaghat-Hamedani, F.; Rebs, S.; El-Battrawy, I.; Chasan, S.; Krause, T.; Haas, J.; Zhong, R.; Liao, Z.; Xu, Q.; Zhou, X.; et al. Identification of SCN5a p.C335R Variant in a Large Family with Dilated Cardiomyopathy and Conduction Disease. Int. J. Mol. Sci. 2021, 22, 12990. [Google Scholar] [CrossRef] [PubMed]
- Asatryan, B. Cardiac Sodium Channel Dysfunction and Dilated Cardiomyopathy: A Contemporary Reappraisal of Pathophysiological Concepts. J. Clin. Med. 2019, 8, 1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calloe, K.; Broendberg, A.K.; Christensen, A.H.; Pedersen, L.N.; Olesen, M.S.; Tejada, M.D.; Friis, S.; Thomsen, M.B.; Bundgaard, H.; Jensen, H.K. Multifocal atrial and ventricular premature contractions with an increased risk of dilated cardiomyopathy caused by a Na(v)1.5 gain-of-function mutation (G213D). Int. J. Cardiol. 2018, 257, 160–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaklyazminskaya, E.; Dzemeshkevich, S. The role of mutations in the SCN5A gene in cardiomyopathies. Biochim. Et Biophys. Acta-Mol. Cell Res. 2016, 1863, 1799–1805. [Google Scholar] [CrossRef] [PubMed]
- Moreau, A.; Gosselin-Badaroudine, P.; Boutjdir, M.; Chahine, M. Mutations in the Voltage Sensors of Domains I and II of Nav1.5 that are Associated with Arrhythmias and Dilated Cardiomyopathy Generate Gating Pore Currents. Front. Pharmacol. 2015, 6, 301. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.-P.; Guo, Z.-F.; Liu, Z.-P.; Song, L.; Zhang, Z.-F.; Jia, Y.-Z.; Cao, Z.-Z.; Ma, J.-H. Eleutheroside B, a selective late sodium current inhibitor, suppresses atrial fibrillation induced by sea anemone toxin II in rabbit hearts. Acta Pharmacol. Sin. 2020, 42, 209–217. [Google Scholar] [CrossRef]
- Munger, M.A.; Mandroli, J.; Kim, K.; Biskupiak, J.; Veeraraghavan, R.; Gyorke, S.; Radwanski, P. Neuronal Na+ Channel Inhibitor Riluzole Prevents Atrial Fibrillation in Humans. Circulation 2019, 140, A13855. [Google Scholar]
- Brand, S.; Seeger, T.; Alzheimer, C. Enhancement of persistent Na+current by sea anemone toxin (ATX II) exerts dual action on hippocampal excitability. Eur. J. Neurosci. 2000, 12, 2387–2396. [Google Scholar] [CrossRef]
- Chahine, M.; Plante, E.; Kallen, R. Sea Anemone Toxin (ATX II) Modulation of Heart and Skeletal Muscle Sodium Channel α-Subunits Expressed in tsA201 Cells. J. Membr. Biol. 1996, 152, 39–48. [Google Scholar] [CrossRef]
- Luo, A.; Ma, J.; Song, Y.; Qian, C.; Wu, Y.; Zhang, P.; Wang, L.; Fu, C.; Cao, Z.; Shryock, J.C. Larger late sodium current density as well as greater sensitivities to ATX II and ranolazine in rabbit left atrial than left ventricular myocytes. Am. J. Physiol. Circ. Physiol. 2014, 306, H455–H461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, J.S.; Redaelli, E.; Zaharenko, A.J.; Cassulini, R.R.; Konno, K.; Pimenta, D.C.; Freitas, J.C.; Clare, J.J.; Wanke, E. Binding specificity of sea anemone toxins to Na(v)1.1-1.6 sodium channels—Unexpected contributions from differences in the IV/S3-S4 outer loop. J. Biol. Chem. 2004, 279, 33323–33335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klinger, A.B.; Eberhardt, M.; Link, A.S.; Namer, B.; Kutsche, L.K.; Schuy, E.T.; Sittl, R.; Hoffmann, T.; Alzheimer, C.; Huth, T.; et al. Sea-Anemone Toxin ATX-II Elicits A-Fiber-Dependent Pain and Enhances Resurgent and Persistent Sodium Currents in Large Sensory Neurons. Mol. Pain 2012, 8, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horváth, B.; Szentandrássy, N.; Almássy, J.; Dienes, C.; Kovács, Z.M.; Nánási, P.P.; Banyasz, T. Late Sodium Current of the Heart: Where Do We Stand and Where Are We Going? Pharmaceuticals 2022, 15, 231. https://doi.org/10.3390/ph15020231
Horváth B, Szentandrássy N, Almássy J, Dienes C, Kovács ZM, Nánási PP, Banyasz T. Late Sodium Current of the Heart: Where Do We Stand and Where Are We Going? Pharmaceuticals. 2022; 15(2):231. https://doi.org/10.3390/ph15020231
Chicago/Turabian StyleHorváth, Balázs, Norbert Szentandrássy, János Almássy, Csaba Dienes, Zsigmond Máté Kovács, Péter P. Nánási, and Tamas Banyasz. 2022. "Late Sodium Current of the Heart: Where Do We Stand and Where Are We Going?" Pharmaceuticals 15, no. 2: 231. https://doi.org/10.3390/ph15020231
APA StyleHorváth, B., Szentandrássy, N., Almássy, J., Dienes, C., Kovács, Z. M., Nánási, P. P., & Banyasz, T. (2022). Late Sodium Current of the Heart: Where Do We Stand and Where Are We Going? Pharmaceuticals, 15(2), 231. https://doi.org/10.3390/ph15020231