3.1. Chemistry
3.1.1. General
All reagents and solvents were purchased from commercial suppliers and used without further purification. Anhydrous tetrahydrofuran (THF) was obtained from an SG water solvent purification system (Pure Process Technology). Anhydrous dimethyl sulfoxide (DMSO),
N,
N-dimethylacetamide (DMA), MeCN, and pyridine were purchased from commercially suppliers and stored under argon. Reactions requiring anhydrous conditions were carried out under an inert atmosphere (nitrogen or argon) and using oven-dried glassware (152 °C). Syringes used to transfer anhydrous solvents or reagents were purged with argon prior to use. Other solvents were analytical or HPLC grade and were used as received. NMR spectra were acquired on a 600 MHz Bruker Avance III HD (600 MHz for
1H and 151 MHz for
13C), a 400 MHz Bruker Avance II (400 MHz for
1H, 376 MHz for
19F, and 101 MHz for
13C), and a 400 MHz Bruker Avance UltraShield (400 MHz for
1H, 376 MHz for
19F, and 101 MHz for
13C) using chloroform
-d, MeOD, or DMSO
-d6 as a deuterated solvent, and with the residual solvent as the internal reference. For all NMR experiments, the deuterated solvent signal was used as the internal lock. Coupling constants (
J values) are given in Hertz (Hz). Multiplicities of
1H NMR signals are reported as follows: s, singlet; d, doublet; dd, doublet of doublets; ddd, doublet of doublets of doublets; dt, doublet of triplets; t, triplet; q, quartet; m, multiplet; br, broad signal. NMR spectra of all compounds were reprocessed in MestReNova software (version 12.0.22023) from the original FID files. Yields refer to isolated compounds estimated to be >90% pure as determined by
1H NMR (25 °C) and analytical HPLC (Please refer to the
Supplementary Materials). Analytical HPLC method: Thermo Fisher UltiMate 3000 with a C-18 column (Luna 5 μm C18(2) 100 Å, 150 mm × 4.6 mm). Eluents: A, H
2O with 0.1% TFA; B, MeCN with 0.1% TFA. Gradient from 100% A to 100% B for 12 min, back to 100% A for 3 min, flow rate 2 mL/min. Detection by UV absorption at λ = 254 nm on a UVD 170U detector. Thin-layer chromatography (TLC) was carried out on silica gel 60 F
254 plates from Merck (Germany). Visualization was accomplished by UV lamp (254 nm). Preparative HPLC was carried out on an UltiMate HPLC system (Thermo Scientific) consisting of an LPG-3200BX pump (10 mL/min), a Rheodyne 9725i injector, a 10 mL loop, a MWD-3000SD detector (254 nm), and an AFC-3000SD automated fraction collector, using a Gemini-NX C18 column (21.2 × 250 mm, 5 µm, 110Å) (Phenomenex) equipped with a guard. Purifications were performed using linear gradients of 0.1% TFA in MilliQ-H
2O (A) and 0.1% TFA, 10% MilliQ-H
2O in MeCN (B). Data were acquired and processed using Chromeleon software v. 6.80. Semipreparative HPLC was performed on the same system using a Luna 5µ C18 column (250 × 10 mm) with a flow rate of 3 mL/min. Automated flash column chromatography was performed on a CombiFlash NextGen 300+ system supplied by TeleDyne ISCO, equipped with RediSep silica-packed columns. Detection of the compounds was carried out by means of a UV–vis variable wavelength detector operating at 200 to 800 nm and by an evaporative light-scattering detector (ELSD). Solvent systems for separation were particular for each compound, but consisted of various mixtures of heptane, EtOAc, CH
2Cl
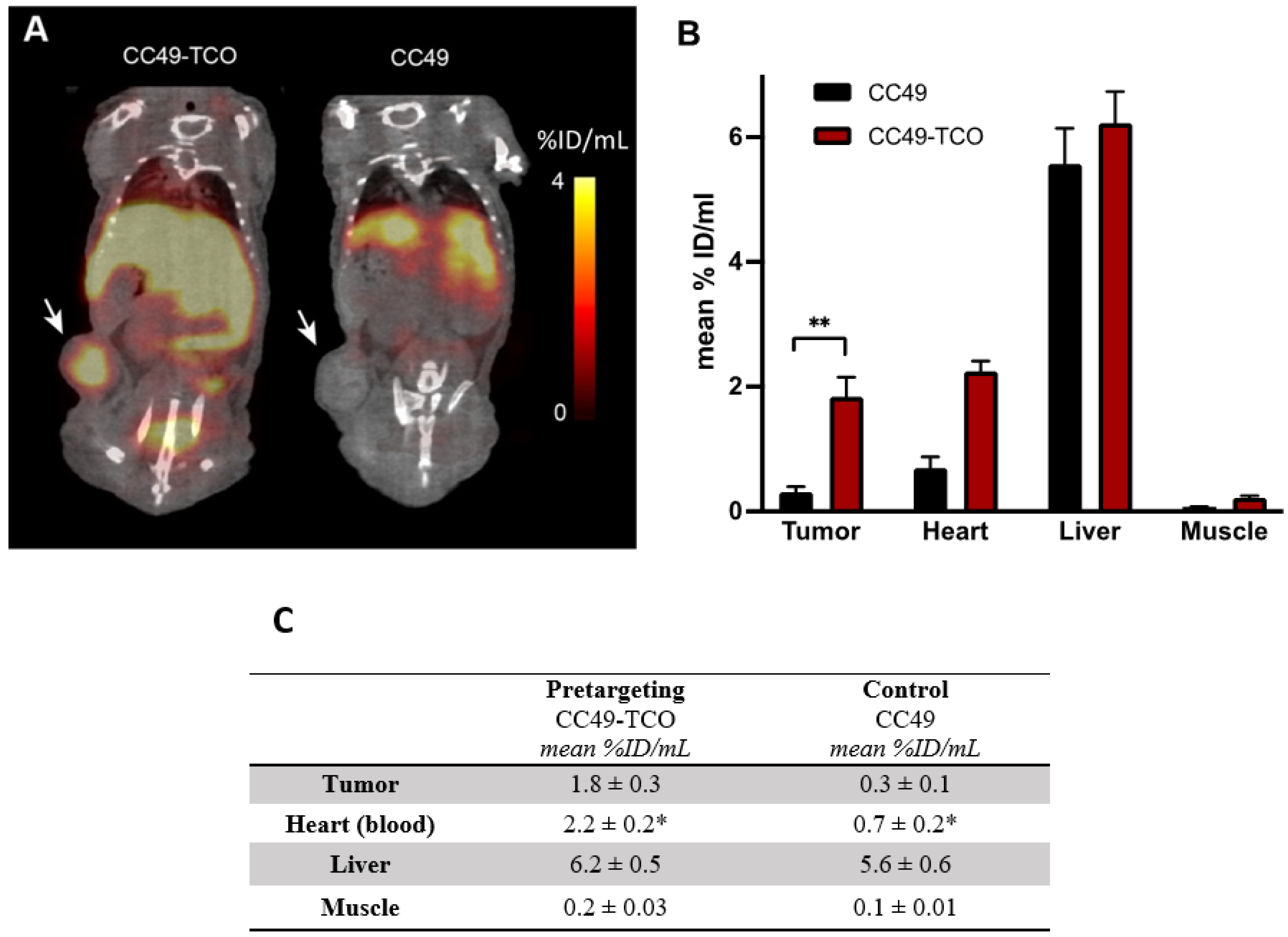
2, and MeOH. Microwave-assisted synthesis was carried out in a Biotage Initiator apparatus operating in single mode; the microwave cavity produced controlled irradiation at 2.45 GHz (Biotage AB, Uppsala, Sweden). The reactions were run in sealed vessels. These experiments were performed by employing magnetic stirring and a fixed hold time using variable power to reach (during 1–2 min) and then maintain the desired temperature in the vessel for the programmed time period. The temperature was monitored by an IR sensor focused on a point on the reactor vial glass. The IR sensor was calibrated to the internal solution’s reaction temperature by the manufacturer. Mass spectra analysis was completed using MS-Acquity-A: Waters Acquity UPLC with QDa-detector. CC49-TCO was kindly provided by Tagworks Pharmaceuticals, and it was obtained as previously described [
14].
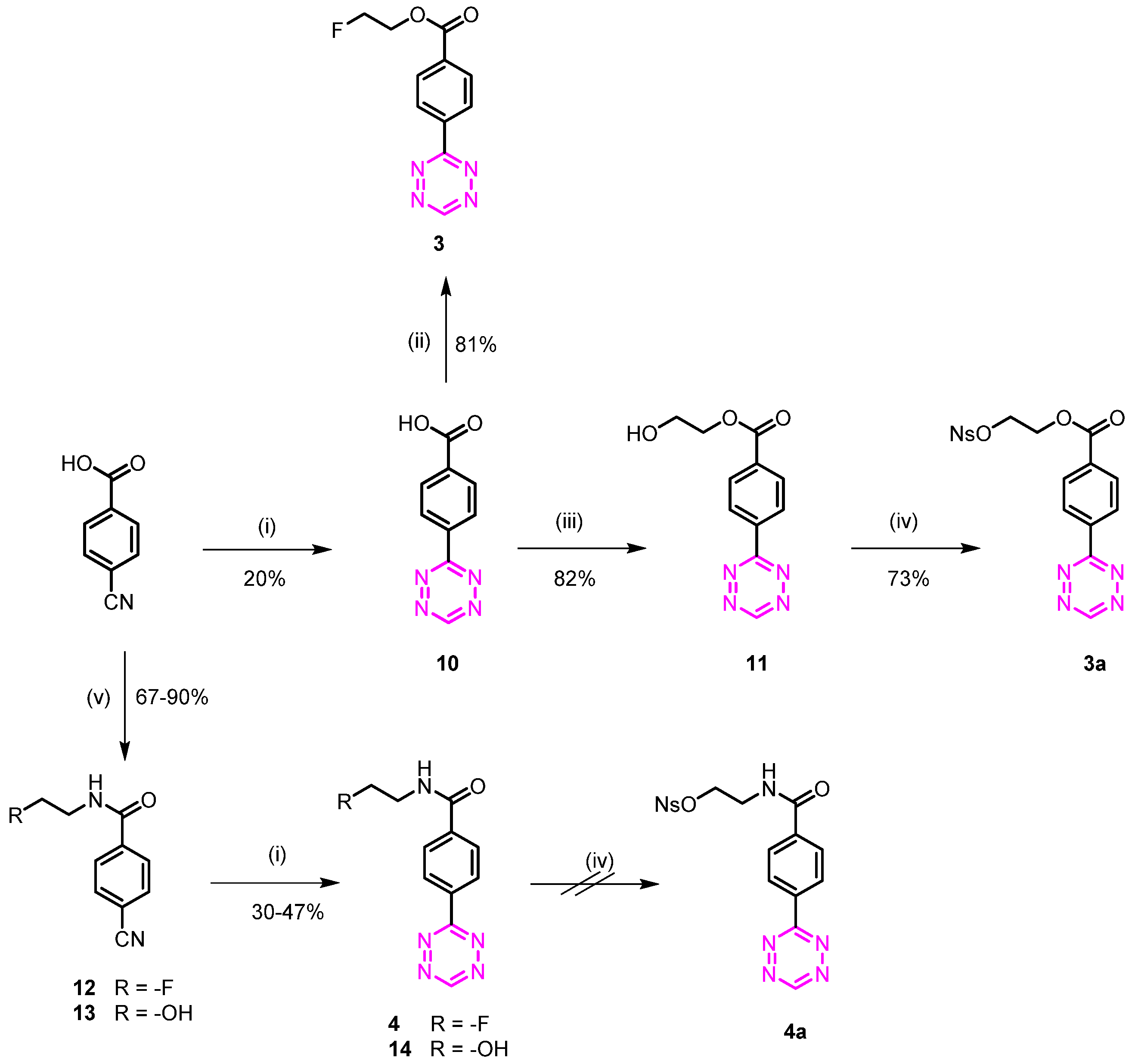
3.1.2. 2-Fluoroethyl 4-(1,2,4,5-tetrazin-3-yl)benzoate (3)
4-(1,2,4,5-Tetrazin-3-yl)benzoic acid (10)
The compound was synthesized as previously reported [
42]. 4-Cyanobenzoic acid (0.3 g, 2.00 mmol), CH
2Cl
2 (0.12 mL, 2.00 mmol), sulfur (0.12 g, 0.5 mmol), and ethanol (4.0 mL) were mixed together in a microwave reaction vial. Hydrazine monohydrate (0.82 mL, 16.00 mmol) was added dropwise with stirring. The vessel was sealed, and the reaction mixture was heated to 50 °C for 24 h. The reaction was diluted with 3 mL of CH
2Cl
2, and sodium nitrite (1.44 g, 20.00 mmol) in 20 mL of H
2O was added dropwise to the mixture under cooling. Excess acetic acid (7 mL) was then added slowly, during which the solution turned bright red in color. The reaction mixture was extracted with CH
2CL
2 (3 × 20 mL). The organic phase was dried over MgSO
4 and concentrated under reduced pressure. The resulting residue was purified using flash chromatography (CH
2Cl
2/MeOH 98/2) to give 0.08 g (20%) of the desired product as a pink solid. Rf = 0.31 (CH
2Cl
2/MeOH 95/5);
1H NMR (400 MHz, DMSO) δ 13.32 (s, 1H), 10.66 (s, 1H), 8.62 (d,
J = 8.5 Hz, 2H), 8.22 (d,
J = 8.5 Hz, 2H);
13C NMR (101 MHz, DMSO) δ 166.67, 165.08, 158.24, 135.70, 134.32, 130.20, 127.97.
2-Fluoroethyl 4-(1,2,4,5-tetrazin-3-yl)benzoate (3)
Compound 10 (0.04 g, 0.20 mmol) and 1-fluoro-2-iodoethane (0.05 g, 0.30 mmol) were dissolved in 2 mL dry DMF, and DIPEA (0.10 mL, 0.60 mmol) was added. The reaction was left at 70 °C overnight. The reaction was diluted with CH2Cl2 (15 mL) and washed with a saturated aqueous solution of NH4Cl (10 mL) and H2O (2 × 10 mL). The organic phase was dried over MgSO4 and concentrated under reduced pressure. Purification by flash chromatography (85/15 heptane/EtOAc) afforded 0.04 g (81%) of 3 as a red solid. Rf = 0.23 (heptane/EtOAc 80/20); 1H NMR (600 MHz, CDCl3) δ 10.21 (s, 1H), 8.65 (d, J = 8.6 Hz, 2H), 8.23 (d, J = 8.5 Hz, 2H), 4.89–4.72 (m, 1H), 4.69–4.64 (m, 1H), 4.63–4.57 (m, 1H), 4.57–4.50 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 165.92, 165.50, 157.99, 135.75, 133.63, 130.58, 128.27, 81.26 (d, J = 171.1 Hz), 64.30 (d, J = 20.1 Hz).
3.1.3. 2-(((4-Nitrophenyl)sulfonyl)oxy)ethyl 4-(1,2,4,5-tetrazin-3-yl)benzoate (3a)
2-Hydroxyethyl 4-(1,2,4,5-tetrazin-3-yl)benzoate (11)
Compound 10 (0.03 g, 0.15 mmol) and 2-bromoethanol (0.02 mL, 0.22 mmol) were dissolved in 3 mL dry DMF, DIPEA (0.08 mL, 0.44 mmol) was dissolved in 1 mL DMF and added dropwise, and the reaction was left at 70 °C. After the reaction was completed, it was cooled down, diluted with water (30 mL) and extracted with CH2Cl2 (3 × 20 mL). The crude was purified by flash chromatography (60/40 heptane/EtOAc) to give 0.03 g (82%) of the desired product as a red solid. Rf = 0.24 (heptane/EtOAc 70/30); 1H NMR (600 MHz, CDCl3) δ 10.28 (s, 1H), 8.72 (d, J = 8.1 Hz, 2H), 8.29 (d, J = 8.1 Hz, 2H), 4.54 (t, J = 4.6 Hz, 2H), 4.02 (t, J = 4.7 Hz, 2H), 2.56–1.97 (m, 1H), 13C NMR (151 MHz, CDCl3) δ 166.04, 165.93, 158.00, 135.68, 133.87, 130.55, 128.28, 67.10, 61.31.
2-(((4-Nitrophenyl)sulfonyl)oxy)ethyl 4-(1,2,4,5-tetrazin-3-yl)benzoate (3a)
To a solution of compound 11 (0.03 g, 0.12 mmol) and DIPEA (0.04 mL, 0.24 mmol) in CH2Cl2 (10 mL) were added nosyl chloride (0.041 g, 0.18 mmol) and DMAP (0.001 g, 0.01 mmol). The reaction was stirred at room temperature for 1 h. The solvent was removed under reduced pressure. Purification by flash chromatography (80/20 heptane/EtOAc) afforded 0.035 (67%) of 3a as a red solid. Rf = 0.38 (heptane/EtOAc 70/30); 1H NMR (600 MHz, CDCl3) δ 10.31 (s, 1H), 8.71 (d, J = 8.5 Hz, 2H), 8.31 (d, J = 8.8 Hz, 2H), 8.16 (d, J = 8.5 Hz, 2H), 8.14–8.09 (m, 2H), 4.64–4.58 (m, 2H), 4.58–4.53 (m, 2H); 13C NMR (151 MHz, CDCl3) δ 165.77, 165.07, 158.06, 150.73, 141.65, 136.14, 132.92, 130.48, 129.17, 128.27, 124.50, 68.72, 62.28.
3.1.4. N-(2-Fluoroethyl)-4-(1,2,4,5-tetrazin-3-yl)benzamide (4)
4-Cyano-N-(2-fluoroethyl)benzamide (12)
To a solution of 4-cyanobenzoic acid (0.73 g, 5.00 mmol) in CH3CN (20 mL) was added CDI (1.21 g, 7.50 mmol). The mixture was stirred at room temperature for 45 min, before addition of 2-fluoroethylamine hydrochloride (0.55 g, 5.50 mmol) and Et3N (2.09 mL, 15.00 mmol). The reaction mixture was stirred for 2 h. Water (30 mL) was added, and the mixture was extracted with EtOAc (3 × 20 mL). The organic phase was dried over MgSO4 and concentrated under reduced pressure to give 0.65 g (68%) of the desire compound as a white solid. Rf = 0.26 (heptane/EtOAc 60/40); 1H NMR (400 MHz, DMSO) δ 8.96 (s, 1H), 8.02 (d, J = 8.7 Hz, 2H), 7.97 (d, J = 8.6 Hz, 2H), 4.61 (t, J = 5.1 Hz, 1H), 4.50 (t, J = 5.1 Hz, 1H), 3.62 (q, J = 5.2 Hz, 1H), 3.56 (q, J = 5.2 Hz, 1H); 13C NMR (101 MHz, DMSO) δ 165.61, 138.57, 132.92, 128.55, 114.17, 82.49 (d, J = 165.8 Hz), 40.57 (d, J = 20.9 Hz).
N-(2-Fluoroethyl)-4-(1,2,4,5-tetrazin-3-yl)benzamide (4)
The compound was obtained from 12 (0.60 g, 3.12 mmol) as described for compound 10. Purification by flash chromatography (60/40 heptane/EtOAc) afforded 0.36 g (47%) of 4 as a red solid. Rf = 0.38 (heptane/EtOAc 50/50); 1H NMR (400 MHz, DMSO) δ 10.65 (s, 1H), 8.95 (t, J = 5.5 Hz, 1H), 8.60 (d, J = 8.6 Hz, 2H), 8.15 (d, J = 8.6 Hz, 2H), 4.65 (t, J = 5.1 Hz, 1H), 4.53 (t, J = 5.1 Hz, 1H), 3.66 (q, J = 5.3 Hz, 1H), 3.59 (q, J = 5.2 Hz, 1H); 13C NMR (101 MHz, DMSO) δ 166.24, 165.59, 158.70, 138.21, 134.81, 128.75, 128.23, 82.57 (d, J = 165.8 Hz), 40.56 (d, J = 16.2 Hz).
3.1.5. 2-(4-(1,2,4,5-Tetrazin-3-yl)benzamido)ethyl 4-nitrobenzenesulfonate (4a)
4-Cyano-N-(2-hydroxyethyl)benzamide (13)
The compound was obtained from 4-cyanobenzoic acid (1.17 g, 8.00 mmol) and ethanolamine (4.82 mL, 80.00 mmol) as described for compound 12. Purification by flash chromatography (CH2Cl2/MeOH 95/5) afforded 1.37 g (90%) of the desired compound as a colorless oil. Rf = 0.22 (CH2Cl2/MeOH 95/5); 1H NMR (400 MHz, DMSO) δ 8.70 (t, J = 5.6 Hz, 1H), 8.01 (d, J = 8.6 Hz, 2H), 7.96 (d, J = 8.6 Hz, 2H), 4.75 (t, J = 5.6 Hz, 1H), 3.53 (q, J = 5.9 Hz, 2H), 3.39–3.32 (m, 2H); 13C NMR (101 MHz, DMSO) δ 165.38, 139.03, 132.83, 128.54, 118.83, 113.94, 60.01, 42.82.
N-(2-Hydroxyethyl)-4-(1,2,4,5-tetrazin-3-yl)benzamide (14)
The compound was obtained from 13 (1.00 g, 5.25 mmol) as described for compound 10. Purification by flash chromatography (CH2Cl2/MeOH 97/3) afforded 0.39 g (30%) of the desired compound as a colorless oil. Rf = 0.31 (CH2Cl2/MeOH 95/5); 1H NMR (600 MHz, DMSO) δ 10.64 (s, 1H), 8.68 (t, J = 5.6 Hz, 1H), 8.61–8.56 (m, 2H), 8.16–8.10 (m, 2H), 4.75 (t, J = 5.6 Hz, 1H), 3.56 (q, J = 6.1 Hz, 2H), 3.38 (q, J = 6.0 Hz, 2H); 13C NMR (151 MHz, DMSO) δ 166.00, 165.61, 158.71, 158.68, 138.66, 128.72, 128.16, 60.13, 42.80.
2-(4-(1,2,4,5-Tetrazin-3-yl)phenyl)-4,5-dihydrooxazole
The compound was obtained from 14 (0.05 g, 0.21 mmol) as described for compound 3a. An intramolecular substitution occurred during the reaction, giving this side product. Purification by flash chromatography (60/40 heptane/EtOAc) afforded 0.35 g (75%) of this side product as a red solid. Rf = 0.41 (heptane/EtOAc 50/50); 1H NMR (600 MHz, CDCl3) δ 10.18 (s, 1H), 9.09–8.44 (m, 2H), 8.11 (d, J = 8.1 Hz, 2H), 4.43 (t, J = 9.5 Hz, 2H), 4.06 (t, J = 9.5 Hz, 2H); 13C NMR (151 MHz, CDCl3) δ 166.07, 163.83, 157.88, 133.94, 132.05, 129.06, 128.22, 67.91, 55.17.
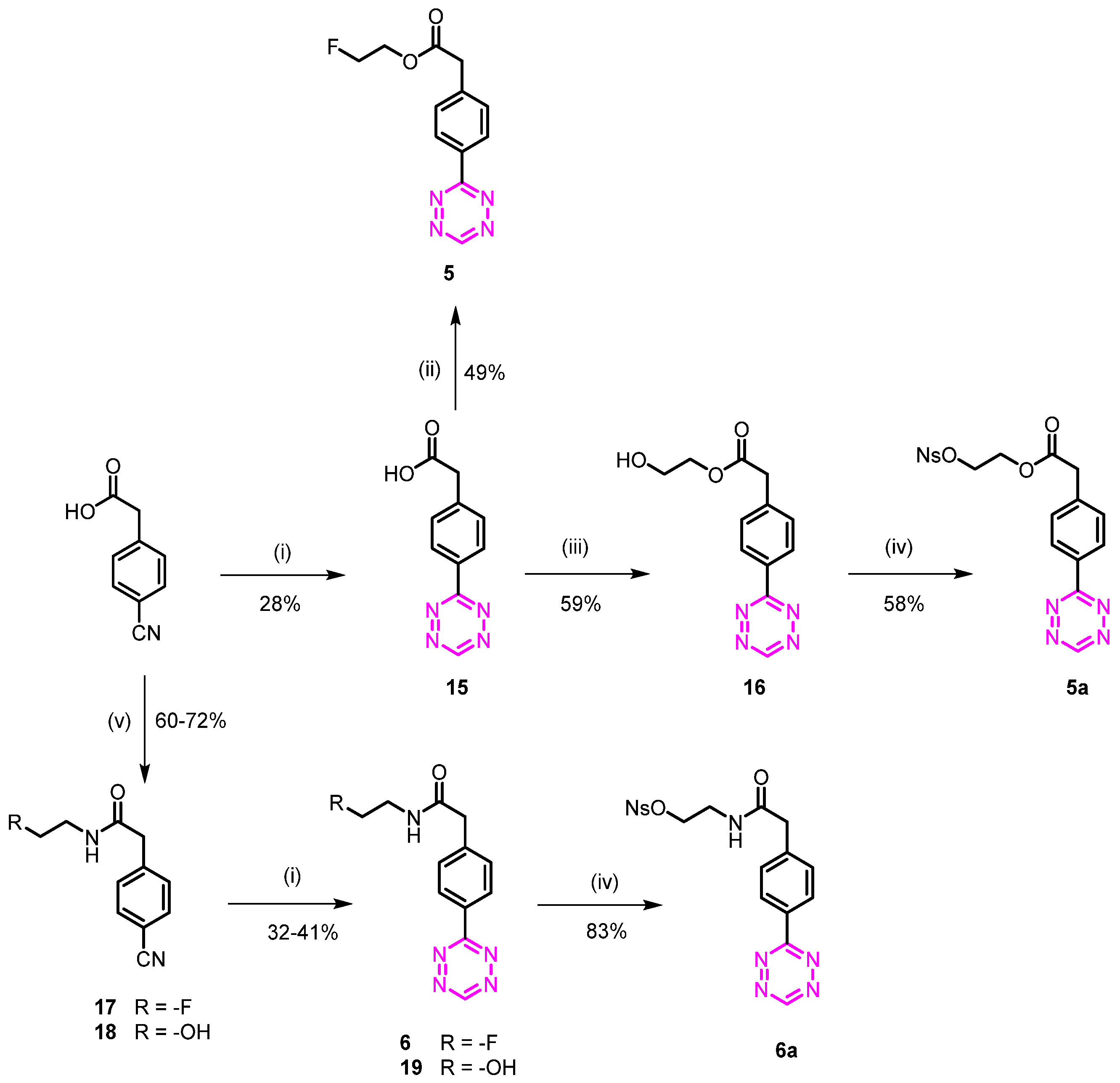
3.1.6. 2-Fluoroethyl 2-(4-(1,2,4,5-tetrazin-3-yl)phenyl)acetate (5)
2-(4-(1,2,4,5-Tetrazin-3-yl)phenyl)acetic acid (15)
The compound was obtained from 2-(4-cyanophenyl)acetic acid (0.97 g, 6.00 mmol) as described for compound 10. Purification by flash chromatography (98/2 CH2Cl2/MeOH) to afford 0.36 g (28%) of the desired compound as a pink solid. Rf = 0.19 (95/5 CH2Cl2/MeOH); 1H NMR (400 MHz, MeOD) δ 10.33 (s, 1H), 8.56 (d, J = 8.4 Hz, 2H), 7.59 (d, J = 8.3 Hz, 2H), 3.78 (s, 2H); 13C NMR (101 MHz, MeOD) δ 173.27, 166.24, 157.83, 140.15, 130.69, 130.13, 127.78, 40.39.
2-Fluoroethyl 2-(4-(1,2,4,5-tetrazin-3-yl)phenyl)acetate (5)
The compound was obtained from 15 (0.07 g, 0.32 mmol) as described for compound 3. Purification by flash chromatography (80/20 heptane/EtOAc) afforded 0.042 g (49%) of 5 as a red solid. Rf = 0.20 (80/20 heptane/EtOAc); 1H NMR (400 MHz, CDCl3) δ 10.14 (s, 1H), 8.53 (d, J = 8.4 Hz, 2H), 7.47 (d, J = 8.1 Hz, 2H), 4.63–4.57 (m, 1H), 4.51–4.46 (m, 1H), 4.37–4.32 (m, 1H), 4.30–4.25 (m, 1H), 3.74 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 170.58, 166.25, 157.79, 139.06, 130.62, 130.38, 128.56, 81.14 (d, J = 170.9 Hz), 63.96 (d, J = 20.2 Hz), 41.01.
3.1.7. 2-(((4-Nitrophenyl)sulfonyl)oxy)ethyl 2-(4-(1,2,4,5-tetrazin-3-yl)phenyl)acetate (5a)
2-Hydroxyethyl 2-(4-(1,2,4,5-tetrazin-3-yl)phenyl)acetate (16)
The compound was obtained from 15 (0.07 g, 0.32 mmol) as described for compound 11. Purification by flash chromatography (80/20 heptane/EtOAc) afforded 0.05 g (59%) of the desired product as a red solid. Rf = 0.17 (60/40 heptane/EtOAc); 1H NMR (400 MHz, CDCl3) δ 10.14 (s, 1H), 8.51 (d, J = 8.4 Hz, 2H), 7.46 (d, J = 8.3 Hz, 2H), 4.24–4.17 (m, 2H), 3.81–3.74 (m, 2H), 3.73 (s, 2H), 1.91 (s, 1H); 13C NMR (101 MHz, CDCl3) δ 171.09, 166.22, 157.78, 139.23, 130.58, 130.36, 128.55, 66.68, 61.06, 41.13.
2-(((4-Nitrophenyl)sulfonyl)oxy)ethyl 2-(4-(1,2,4,5-tetrazin-3-yl)phenyl)acetate (5a)
The compound was obtained from 16 (0.50 g, 0.19 mmol) as described for compound 3a. Purification by flash chromatography (75/25 heptane/EtOAc) afforded 0.05 g (58%) of 5a as a red solid. Rf = 0.27 (heptane/EtOAc 60/40); 1H NMR (400 MHz, CDCl3) δ 10.16 (s, 1H), 8.51 (d, J = 8.2 Hz, 2H), 8.30 (d, J = 8.8 Hz, 2H), 8.02 (d, J = 8.8 Hz, 2H), 7.43 (d, J = 8.2 Hz, 2H), 4.30 (s, 4H), 3.69 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 170.30, 157.84, 150.86, 141.58, 138.66, 130.76, 130.35, 129.20, 128.56, 124.52, 68.35, 62.00, 40.86.
3.1.8. 2-(4-(1,2,4,5-Tetrazin-3-yl)phenyl)-N-(2-fluoroethyl)acetamide (6)
2-(4-Cyanophenyl)-N-(2-fluoroethyl)acetamide (17)
The compound was obtained from 2-(4-cyanophenyl)acetic acid (0.5 g, 3.10 mmol) as described for compound
12 [
17]. Concentration under reduced pressure afforded 0.46 g (72%) of the desired product as a white solid. Rf: 0.35 (heptane/EtOAc 30/70);
1H NMR (400 MHz, CDCl
3) δ 7.57 (d,
J = 8.3 Hz, 2H), 7.34 (d,
J = 8.2 Hz, 2H), 5.88 (s, 1H), 4.47 (t,
J = 4.8 Hz, 1H), 4.35 (t,
J = 4.8 Hz, 1H), 3.59–3.49 (m, 3H), 3.46 (dt,
J = 5.8, 4.7 Hz, 1H);
13C NMR (101 MHz, CDCl
3) δ 169.58, 140.02, 132.56, 130.10, 118.58, 111.32, 82.48 (d,
J = 166.7 Hz), 43.36, 40.23 (d,
J = 19.5 Hz).
2-(4-(1,2,4,5-Tetrazin-3-yl)phenyl)-N-(2-fluoroethyl)acetamide (6)
The compound was obtained from
17 (0.40 g, 1.94 mmol) as described for compound
3 [17]. Purification by flash chromatography (30/70 heptane/EtOAc) afforded 0.21 g (41%) of
6 as a red solid. Rf = 0.22 (heptane/EtOAc 40/60);
1H NMR (400 MHz, DMSO) δ 10.58 (s, 1H), 8.48–8.40 (m, 3H), 7.57 (d,
J = 8.2 Hz, 2H), 4.51 (t,
J = 5.0 Hz, 1H), 4.39 (t,
J = 5.0 Hz, 1H), 3.62 (s, 2H), 3.43 (q,
J = 5.3 Hz, 1H), 3.36 (q,
J = 5.2 Hz, 1H);
13C NMR (101 MHz, DMSO) δ 170.27, 165.92, 158.54, 142.02, 130.61, 130.53, 128.15, 82.90 (d,
J = 165.0 Hz), 42.56, 40.56 (d,
J = 15.8 Hz).
3.1.9. 2-(2-(4-(1,2,4,5-Tetrazin-3-yl)phenyl)acetamido)ethyl 4-nitrobenzenesulfonate (6a)
2-(4-Cyanophenyl)-N-(2-hydroxyethyl)acetamide (18)
The compound was obtained from 2-(4-cyanophenyl)acetic acid (2.1 g, 13.03 mmol) as described for compound 13. Recrystallization from EtOAc afforded 1.60 g (60%) of the desired product as a white solid. Rf = 0.15 (heptane/EtOAc 20/80); 1H NMR (400 MHz, DMSO) δ 8.15 (s, 1H), 7.76 (d, J = 8.2 Hz, 2H), 7.46 (d, J = 8.0 Hz, 2H), 4.68 (t, J = 5.4 Hz, 1H), 3.54 (s, 2H), 3.40 (q, J = 5.8 Hz, 2H), 3.12 (q, J = 5.9 Hz, 2H); 1H NMR (400 MHz, DMSO) δ 8.15 (s, 1H), 7.76 (d, J = 8.2 Hz, 2H), 7.46 (d, J = 8.0 Hz, 2H), 4.68 (t, J = 5.4 Hz, 1H), 3.54 (s, 2H), 3.40 (q, J = 5.8 Hz, 2H), 3.12 (q, J = 5.9 Hz, 2H).
2-(4-(1,2,4,5-Tetrazin-3-yl)phenyl)-N-(2-hydroxyethyl)acetamide (19)
The compound was obtained from 18 (0.82 g, 4.00 mmol) as described for compound 3. Purification by flash chromatography (98/2 CH2Cl2/MeOH) and recrystallization from EtOAc afforded 0.33 g (32%) of the desired product as a pink solid. Rf = 0.31 (CH2Cl2/MeOH 95/5); 1H NMR (400 MHz, DMSO) δ 10.31 (s, 1H), 8.54 (d, J = 8.2 Hz, 2H), 7.58 (d, J = 8.1 Hz, 2H), 3.67 (s, 2H), 3.62 (t, J = 5.8 Hz, 2H), 3.36–3.32 (m, 2H); 13C NMR (101 MHz, DMSO) δ 171.23, 165.43, 157.02, 140.25, 129.84, 129.01, 127.04, 59.30, 41.46, 40.99.
2-(2-(4-(1,2,4,5-Tetrazin-3-yl)phenyl)acetamido)ethyl 4-nitrobenzenesulfonate (6a)
The compound was obtained from 19 (0.06 g, 0.23 mmol) as described for compound 3a. Purification by flash chromatography (99/1 CH2Cl2/MeOH) afforded 0.08 g (83%) of 3a as a red solid. Rf = 0.31 (heptane/EtOAc 60/40); 1H NMR (400 MHz, DMSO) δ 10.51 (s, 1H), 8.46 (d, J = 8.9 Hz, 2H), 8.39 (d, J = 8.6 Hz, 2H), 8.29 (d, J = 8.9 Hz, 2H), 7.69 (d, J = 8.6 Hz, 2H), 5.83 (s, 1H), 4.29 (t, J = 7.0 Hz, 2H), 4.10 (t, J = 7.0 Hz, 2H), 3.31 (s, 2H); 13C NMR (101 MHz, DMSO) δ 165.77, 158.22, 151.32, 149.36, 141.57, 140.68, 129.72, 128.22, 128.11, 128.07, 125.43, 86.21, 67.35, 47.47.
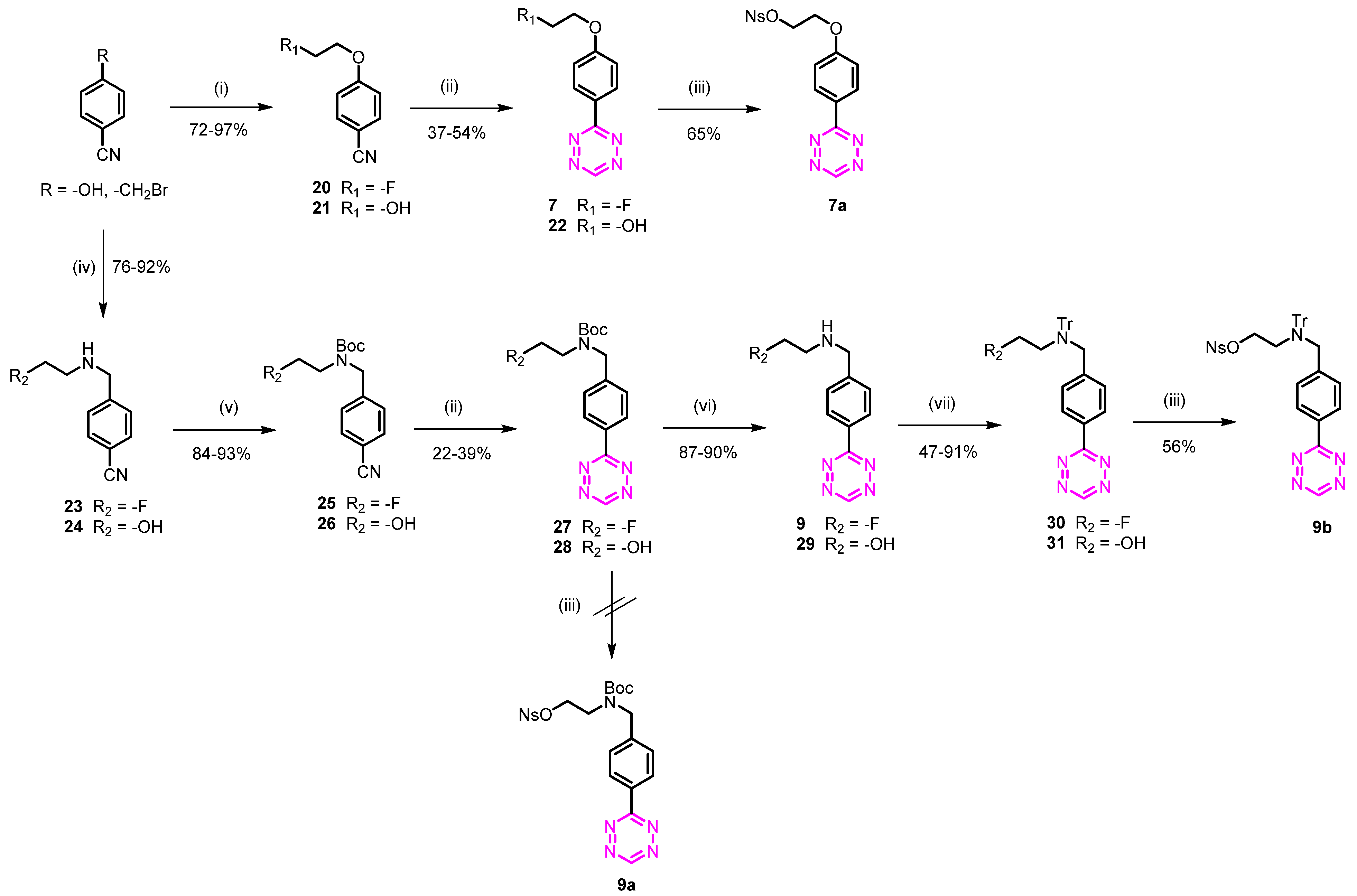
3.1.10. 3-(4-(2-Fluoroethoxy)phenyl)-1,2,4,5-tetrazine (7)
4-(2-Fluoroethoxy)benzonitrile (20)
To a solution of 4-hydroxybenzonitrile (0.6 g, 5.00 mmol) and K2CO3 (1.38 g, 10.00 mmol) in CH3CN (20 mL) was added 1-fluoro-2-iodoethane (1.04 g, 6.00 mmol). The reaction was refluxed for 12 h and then concentrated under reduced pressure. The resulting mixture was diluted with water (50 mL), extracted with EtOAc (3 × 50 mL), washed with brine (50 mL), dried over MgSO4, filtered, and concentrated under reduced pressure. Purification by flash chromatography (heptane/EtOAc 80/20) afforded 0.8 g (97%) of the desired compound as a yellow oil. Rf = 0.37 (heptane/EtOAc 70/30); 1H NMR (400 MHz, CDCl3) δ 7.62 (d, J = 8.9 Hz, 1H), 7.01 (d, J = 8.9 Hz, 1H), 5.05–4.79 (m, 1H), 4.77–4.71 (m, 1H), 4.36–4.29 (m, 1H), 4.29–4.22 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 161.69, 134.06, 119.03, 115.33, 104.59, 81.48 (d, J = 171.7 Hz), 67.37 (d, J = 20.5 Hz).
3-(4-(2-Fluoroethoxy)phenyl)-1,2,4,5-tetrazine (7)
The compound was obtained from 20 (0.73 g, 4.42 mmol) as described for compound 3. Purification by flash chromatography (heptane/EtOAc 90/10) afforded 0.36 g (37%) of the desired compound as a red oil. Rf = 0.33 (heptane/EtOAc 80/20); 1H NMR (400 MHz, CDCl3) δ 10.07 (s, 1H), 8.53 (d, J = 8.9 Hz, 2H), 7.06 (d, J = 8.9 Hz, 2H), 4.89–4.78 (m, 1H), 4.74–4.62 (m, 1H), 4.43–4.29 (m, 1H), 4.27–4.16 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 166.06, 162.56, 157.40, 130.26, 124.56, 115.38, 81.63 (d, J = 171.5 Hz), 67.29 (d, J = 20.6 Hz).
3.1.11. 2-(4-(1,2,4,5-Tetrazin-3-yl)phenoxy)ethyl 4-nitrobenzenesulfonate (7a)
4-(2-Hydroxyethoxy)benzonitrile (21)
To a solution of 4-hydroxybenzonitrile (1.42 g, 12.00 mmol) and K2CO3 (8.29 g, 60.00 mmol) in CH3CN (20 mL) was added 2-bromoethanol (2.55 mL, 36.00 mmol). The reaction was refluxed for 12 h and then concentrated under reduced pressure. The resulting mixture was diluted with water (50 mL), extracted with EtOAc (3 × 50 mL), washed with brine (50 mL), dried over MgSO4, filtered, and concentrated under reduced pressure. Purification by flash chromatography (heptane/EtOAc 40/60) afforded 1.41 g (72%) of the desired compound as a yellow oil. Rf = 0.18 (heptane/EtOAc 50/50); 1H NMR (400 MHz, DMSO) δ 7.76 (d, J = 8.8 Hz, 2H), 7.11 (d, J = 8.8 Hz, 2H), 4.92 (t, J = 5.5 Hz, 1H), 4.09 (t, J = 5.0 Hz, 2H), 3.73 (q, J = 5.0 Hz, 2H), 13C NMR (101 MHz, DMSO) δ 162.70, 134.63, 119.64, 116.05, 103.15, 70.56, 59.79.
2-(4-(1,2,4,5-Tetrazin-3-yl)phenoxy)ethan-1-ol (22)
The compound was obtained from 21 (0.98 g, 6.00 mmol) as described for compound 3. Purification by flash chromatography (heptane/EtOAc 50/50) afforded 0.71 g (54%) of the desired product as a red solid. Rf = 0.27 (heptane/EtOAc 50/50); 1H NMR (400 MHz, DMSO) δ 10.50 (s, 1H), 8.47 (d, J = 9.0 Hz, 2H), 7.23 (d, J = 9.0 Hz, 2H), 4.94 (t, J = 5.5 Hz, 1H), 4.15 (t, J = 4.9 Hz, 2H), 3.78 (q, J = 5.2 Hz, 2H); 13C NMR (101 MHz, DMSO) δ 165.65, 163.10, 158.16, 130.10, 124.35, 115.95, 70.44, 59.92.
2-(4-(1,2,4,5-Tetrazin-3-yl)phenoxy)ethyl 4-nitrobenzenesulfonate (7a)
Compound 21 (0.10 g, 0.46 mmol) was mixed with nitrobenzenesulfonyl chloride (0.15 g, 0.69 mmol) and dissolved in 4 mL dry CH2Cl2 under argon. A mixture of DIPEA (0.33 mL, 1.83 mmol) and DMAP (5 mg, 0.04 mmol) in 1 mL dry CH2Cl2 was added at 0 °C under argon. The reaction was slowly heated to room temperature and left for 1 h. The reaction was diluted with 10 mL CH2Cl2 and washed with 20 mL NH4Cl (sat.) and H2O (2 × 20 mL). The organic phase was dried over N2SO4 and concentrated under reduced pressure. Purification by flash chromatography (heptane/EtOAc 60/40) afforded 0.12 g (65%) of the desired product as a red solid. Rf = 0.41 (heptane/EtOAc 50/50); 1H NMR (400 MHz, DMSO) δ 10.52 (s, 1H), 8.97–8.31 (m, 5H), 8.33–7.87 (m, 2H), 7.11 (d, J = 8.9 Hz, 2H), 4.97–4.50 (m, 2H), 4.48–4.06 (m, 2H); 13C NMR (101 MHz, DMSO) δ 165.57, 161.88, 158.28, 154.85, 147.71, 130.25, 127.38, 125.26, 123.79, 116.05, 75.01, 67.33.
3.1.12. N-(4-(1,2,4,5-Tetrazin-3-yl)benzyl)-2-fluoroethan-1-amine (9)
4-(((2-Fluoroethyl)amino)methyl)benzonitrile (23)
To a solution of 4-(bromomethyl)benzonitrile (0.78 g, 4.00 mmol) in CH3CN (40 mL) was added K2CO3 (0.33 g, 24.0 mmol) and 2-fluoroethylamine hydrochloride (0.16 g, 16.0 mmol). The mixture was stirred at room temperature overnight. The solvent was removed under reduced pressure, and the residue was diluted with water (20 mL) and extracted with EtOAc. The combined organic layer was washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography using EtOAc (heptane/EtOAc 50/50) in heptane to afford 0.54 g (76%) of 23 as a colorless oil. Rf = 0.24 (heptane/EtOAc 40/60). 1H NMR (400 MHz, CDCl3) δ 7.55 (d, J = 8.2 Hz, 2H), 7.40 (d, J = 8.0 Hz, 2H), 4.63–4.48 (m, 1H), 4.47–4.37 (m, 1H), 3.84 (s, 2H), 2.93–2.84 (m, 1H), 2.84–2.72 (m, 1H), 1.65 (s, 1H). 13C NMR (101 MHz, CDCl3) δ 145.6, 132.3, 128.6, 118.9, 110.9, 83.5 (d, J = 165.5 Hz), 53.1, 49.1 (d, J = 19.7 Hz)
Tert-butyl 4-cyanobenzyl(2-fluoroethyl)carbamate (25)
To a solution of 23 (540 mg, 3.03 mmol) and Et3N (1.27 mL, 9.09 mmol) in CH2Cl2 (40 mL) was added Boc2O (790 mg, 3.63 mmol), and the mixture was stirred at room temperature for 12 h. The solution was washed with water and saturated K2CO3 solution, dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by flash column chromatography using (heptane/EtOAc 70/30) to afford 0.710 g (84%) of the desired product as a colorless oil (mixture of rotamers). Rf = 0.42 (heptane/EtOAc 80/20). 1H NMR (400 MHz, CDCl3) δ 7.55 (d, J = 7.8 Hz, 2H), 7.27 (d, J = 7.8 Hz, 2H), 4.79–4.10 (m, 4H), 3.62–3.28 (m, 2H), 1.96–1.05 (m, 9H). 13C NMR (101 MHz, CDCl3) δ 155.4, 144.2, 143.8, 132.4, 128.1, 127.5, 118.7, 111.1, 83.2 (d, J = 168.2 Hz), 82.7 (d, J = 170.5 Hz), 52.1, 51.2, 47.7, 28.3.
Tert-butyl 4-(1,2,4,5-tetrazin-3-yl)benzyl(2-fluoroethyl)carbamate (27)
The compound was obtained from 25 (0.68 g, 2.44 mmol) as described for compound 3. Purification by flash chromatography (heptane/EtOAc 80/20) afforded 0.18 g (22%) of the desired product as a red solid (mixture of rotamers). Rf = 0.21 (heptane/EtOAc 80/20); 1H NMR (400 MHz, CDCl3) δ 10.23 (s, 1H), 8.62 (d, J = 7.8 Hz, 2H), 7.49 (d, J = 7.8 Hz, 2H), 4.76–4.42 (m, 4H), 3.83–3.38 (m, 2H), 1.64–1.38 (m, 9H); 13C NMR (101 MHz, CDCl3) δ 166.32, 157.77, 155.51, 144.25, 132.37, 130.58, 128.55, 127.86, 83.46, 83.17 (d, J = 165.4 Hz), 82.67 (d, J = 170.4 Hz), 80.63, 52.07, 51.07, 47.49, 28.36.
N-(4-(1,2,4,5-tetrazin-3-yl)benzyl)-2-fluoroethan-1-amine (9)
To a solution of 27 (0.10 mg, 0.30 mmol) in dioxane (10 mL) was added a solution of HCl in dioxane (4.0 M, 3.0 mL). The mixture was stirred at room temperature for 2 h and then concentrated under reduced pressure. The obtained solid was washed with Et2O to afford 0.07 g (86%) of 9 as hydrochloride salt. 1H NMR (400 MHz, DMSO-d6) δ 10.64 (s, 1H), 9.87 (s, 2H), 8.55 (d, J = 8.3 Hz, 2H), 7.89 (d, J = 8.3 Hz, 2H), 4.89 (t, J = 4.6 Hz, 1H), 4.77 (t, J = 4.6 Hz, 1H), 4.35 (s, 2H), 3.38 (t, J = 4.7 Hz, 1H), 3.31 (t, J = 4.7 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 165.7, 158.7, 137.0, 132.8, 131.6, 128.4, 80.0 (d, J = 165.3 Hz), 50.1, 47.1 (d, J = 20.0 Hz).
3.1.13. 2-((4-(1,2,4,5-Tetrazin-3-yl)benzyl)(tert-butoxycarbonyl)amino)ethyl 4-nitro Benzenesulfonate (9a)
4-(((2-Hydroxyethyl)amino)methyl)benzonitrile (24)
The compound was obtained from 4-(bromomethyl)benzonitrile (2.00 g, 10.20 mmol) and ethanolamine (12.30 mL, 200.00 mmol) as described for compound 23. Concentration under reduced pressure afforded 1.65 g (92%) of the desired compound as a white solid. Rf = 0.15 (90/10 CH2Cl2/MeOH); 1H NMR (400 MHz, CDCl3) δ 7.62–7.52 (m, 2H), 7.46–7.37 (m, 2H), 3.88–3.77 (m, 2H), 3.69–3.59 (m, 2H), 2.79–2.70 (m, 2H), 2.58–2.23 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 145.76, 132.22, 128.67, 118.88, 110.71, 60.91, 53.07, 50.78.
Tert-butyl (4-cyanobenzyl)(2-hydroxyethyl)carbamate (26)
The compound was obtained from 24 (1.62 g, 9.19 mmol) as described for compound 25. Purification by flash chromatography (heptane/EtOAc 60/40) afforded 2.36 g (93%) of the desired product as a colorless oil (rotamers mixture). Rf = 0.4 (heptane/EtOAc 50/50); 1H NMR (600 MHz, CDCl3) δ 7.63 (d, J = 7.9 Hz, 2H), 4.55–4.52 (m, 2H), 3.74 (s, 2H), 3.51–2.94 (m, 2H), 2.69 (s, 1H), 1.76–0.53 (m, 9H); 13C NMR (151 MHz, CDCl3) δ 156.91, 144.11, 132.43, 128.02, 127.50, 118.72, 111.19, 81.02, 62.17, 61.46, 52.10, 51.17, 50.37, 49.51, 28.32.
Tert-butyl (4-(1,2,4,5-tetrazin-3-yl)benzyl)(2-hydroxyethyl)carbamate (28)
The compound was obtained from 26 (2.32 g, 8.32 mmol) as described for compound 3. Purification by flash chromatography (80/20 heptane/EtOAc) to give 1.07 g (39%) of the desired product as a red oil (rotamers mixture). Rf. = 0.21 (heptane/EtOAc 70/30); 1H NMR (400 MHz, CDCl3) δ 10.17 (s, 1H), 8.53 (d, J = 8.0 Hz, 2H), 7.42 (d, J = 8.1 Hz, 2H), 4.57 (s, 2H), 3.71 (d, J = 5.8 Hz, 2H), 3.46–3.41 (m, 2H), 3.23 (q, J = 5.4 Hz, 1H), 1.39 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 166.23, 157.75, 156.89, 144.17, 132.36, 130.55, 128.52, 127.89, 80.75, 62.36, 61.76, 51.98, 51.12, 50.13, 49.44, 28.34.
3-(4-(1,2,4,5-Tetrazin-3-yl)benzyl)oxazolidin-2-one
The compound was obtained from 28 (0.06 g, 0.18 mmol) as described for compound 3a. An intramolecular substitution occurred during the reaction giving this side product. Purification by flash chromatography (50/50 heptane/EtOAc) afforded 0.035 (67%) of the side product as a red solid. Rf = 0.21 (heptane/EtOAc 50/50); 1H NMR (400 MHz, CDCl3) δ 10.16 (s, 1H), 8.55 (d, J = 8.3 Hz, 2H), 7.47 (d, J = 8.3 Hz, 2H), 4.46–3.83 (m, 2H), 3.45 (dd, J = 8.7, 7.3 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 166.16, 158.59, 157.88, 141.30, 131.38, 128.96, 128.83, 61.87, 48.23, 44.21.
3.1.14. N-(4-(1,2,4,5-Tetrazin-3-yl)benzyl)-2-fluoro-N-tritylethan-1-amine (30)
Compound 9 (0.05 g, 0.18 mmol) was mixed with trityl chloride (0.05 g, 0.20 mmol) and dissolved in 4 mL dry CH2Cl2 under argon. A mixture of Et3N (0.08 mL, 0.55 mmol) in 1 mL dry CH2Cl2 was added at 0 °C under argon. The reaction was slowly heated to room temperature and left for 1 h. The reaction was diluted with 10 mL CH2Cl2 and washed with NH4Cl (sat.) 20 mL and H2O (2 × 20 mL). The organic phase was dried over N2SO4 and concentrated under reduced pressure. Purification by flash chromatography (heptane/EtOAc 80/20) afforded 0.08 g (78%) as a red solid. Rf = 0.41 (heptane/EtOAc 80/20); 1H NMR (600 MHz, CDCl3) δ 10.13 (s, 1H), 8.57 (d, J = 8.4 Hz, 2H), 7.77 (d, J = 8.4 Hz, 2H), 7.74–7.55 (m, 6H), 7.32–7.23 (m, 6H), 7.16–7.10 (m, 3H), 3.99 (t, J = 6.0 Hz, 1H), 3.91 (t, J = 6.1 Hz, 1H), 3.69 (s, 2H), 2.92–2.55 (m, 2H); 13C NMR (151 MHz, CDCl3) δ 166.46, 157.73, 147.36, 143.10, 130.32, 129.32, 128.56, 128.43, 127.93, 127.87, 126.46, 81.94 (d, J = 168.6 Hz), 79.00, 57.49, 54.66 (d, J = 22.8 Hz).
3.1.15. 2-((4-(1,2,4,5-Tetrazin-3-yl)benzyl)(trityl)amino)ethyl 4-nitrobenzenesulfonate (9b)
2-((4-(1,2,4,5-Tetrazin-3-yl)benzyl)amino)ethan-1-ol (29)
To a solution of 28 (0.99 g, 2.98 mmol) in dioxane (10 mL) was added a solution of HCl in dioxane (4.0 M, 7.7 mL). The mixture was stirred at room temperature for 2 h and then concentrated under reduced pressure. The obtained solid was washed with Et2O to afford 0.72 g (90%) of 29 as hydrochloride salt. 1H NMR (400 MHz, DMSO) δ 10.64 (s, 1H), 9.50 (s, 3H), 8.96–8.31 (m, 2H), 8.09–7.60 (m, 2H), 4.32 (t, J = 5.7 Hz, 2H), 3.73 (t, J = 5.4 Hz, 2H), 3.01 (q, J = 5.6 Hz, 2H); 13C NMR (101 MHz, DMSO) δ 165.74, 158.72, 137.22, 132.77, 131.62, 128.37, 66.82, 56.83, 49.93, 49.16.
2-((4-(1,2,4,5-Tetrazin-3-yl)benzyl)(trityl)amino)ethan-1-ol (31)
The compound was obtained from 29 (0.05 g, 0.19 mmol) as described for compound 30. Purification by flash chromatography (60/40 heptane/EtOAc) to give 0.04 g (48%) of the desired product as a red solid. Rf = 0.53 (60/40 heptane/EtOAc); 1H NMR (600 MHz, CDCl3) δ 10.22 (s, 1H), 8.65 (d, J = 8.2 Hz, 2H), 7.86 (d, J = 8.1 Hz, 2H), 7.69 (d, J = 7.9 Hz, 6H), 7.33 (t, J = 7.7 Hz, 6H), 7.22 (t, J = 7.3 Hz, 3H), 3.76 (s, 2H), 3.29 (t, J = 6.8 Hz, 2H), 2.56 (d, J = 7.0 Hz, 2H), 1.63 (s, 1H); 13C NMR (101 MHz, CDCl3) δ 166.40, 157.73, 147.86, 143.28, 130.35, 129.37, 128.55, 128.49, 126.38, 79.05, 61.76, 57.79, 57.12.
2-((4-(1,2,4,5-Tetrazin-3-yl)benzyl)(trityl)amino)ethyl 4-nitrobenzenesulfonate (9b)
The compound was obtained from 31 (0.04 g, 0.08 mmol) as described for compound 3a. Purification by flash chromatography (90/10 heptane/EtOAc) to give 0.04 g (48%) of the desired product as a red solid. Rf = 0.25 (80/20 heptane/EtOAc); 1H NMR (400 MHz, CDCl3) δ 10.19 (s, 1H), 8.46 (d, J = 8.3 Hz, 2H), 8.08 (d, J = 8.8 Hz, 2H), 7.69 (d, J = 8.8 Hz, 2H), 7.64 (d, J = 8.2 Hz, 2H), 7.61–7.43 (m, 6H), 7.23 (t, J = 7.6 Hz, 6H), 7.16–7.09 (m, 3H), 4.32–3.39 (m, 4H), 2.57 (t, J = 7.2 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 166.20, 157.92, 150.45, 146.36, 142.60, 141.64, 130.79, 129.11, 128.98, 128.91, 128.50, 127.98, 126.69, 124.19, 79.05, 57.38.
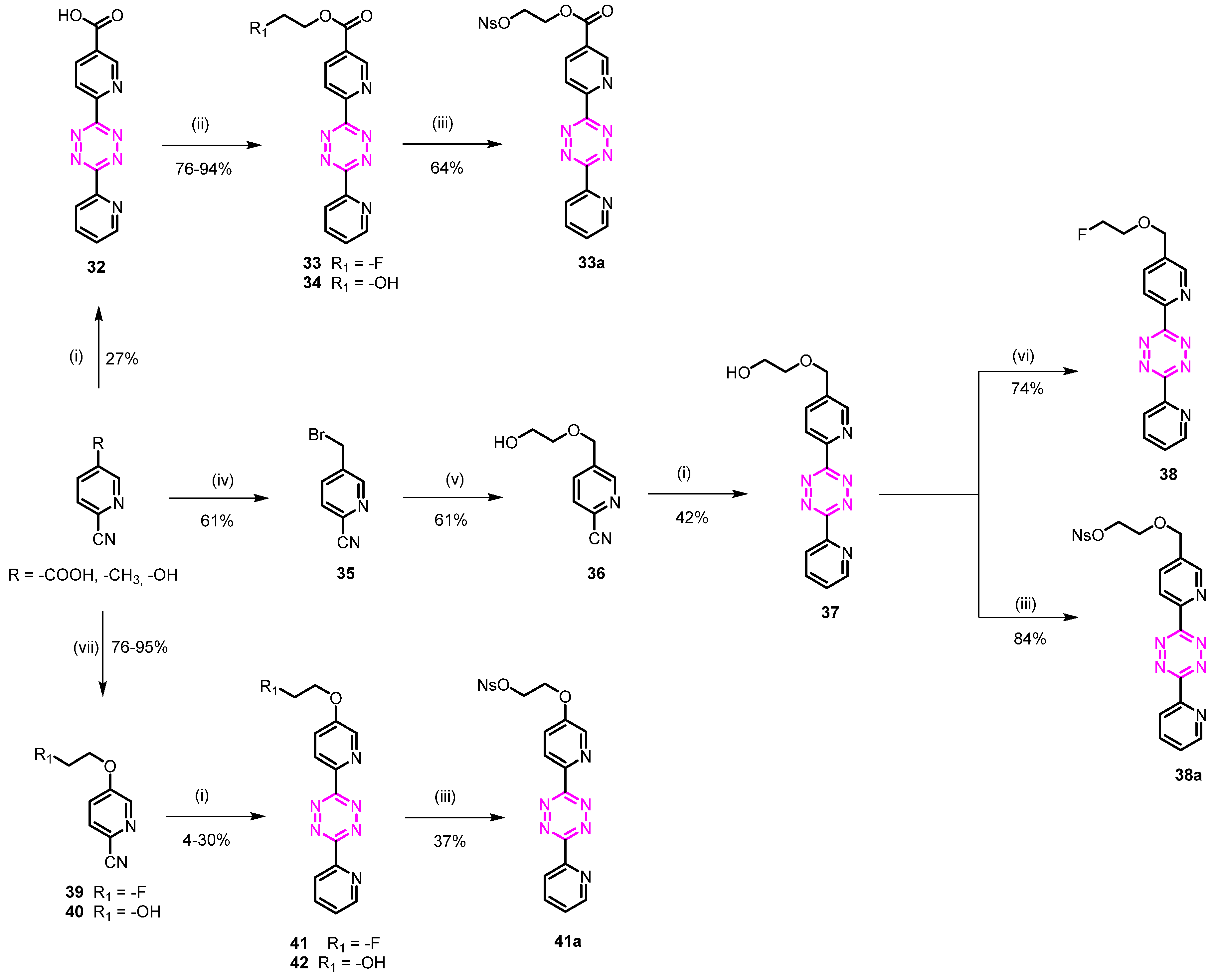
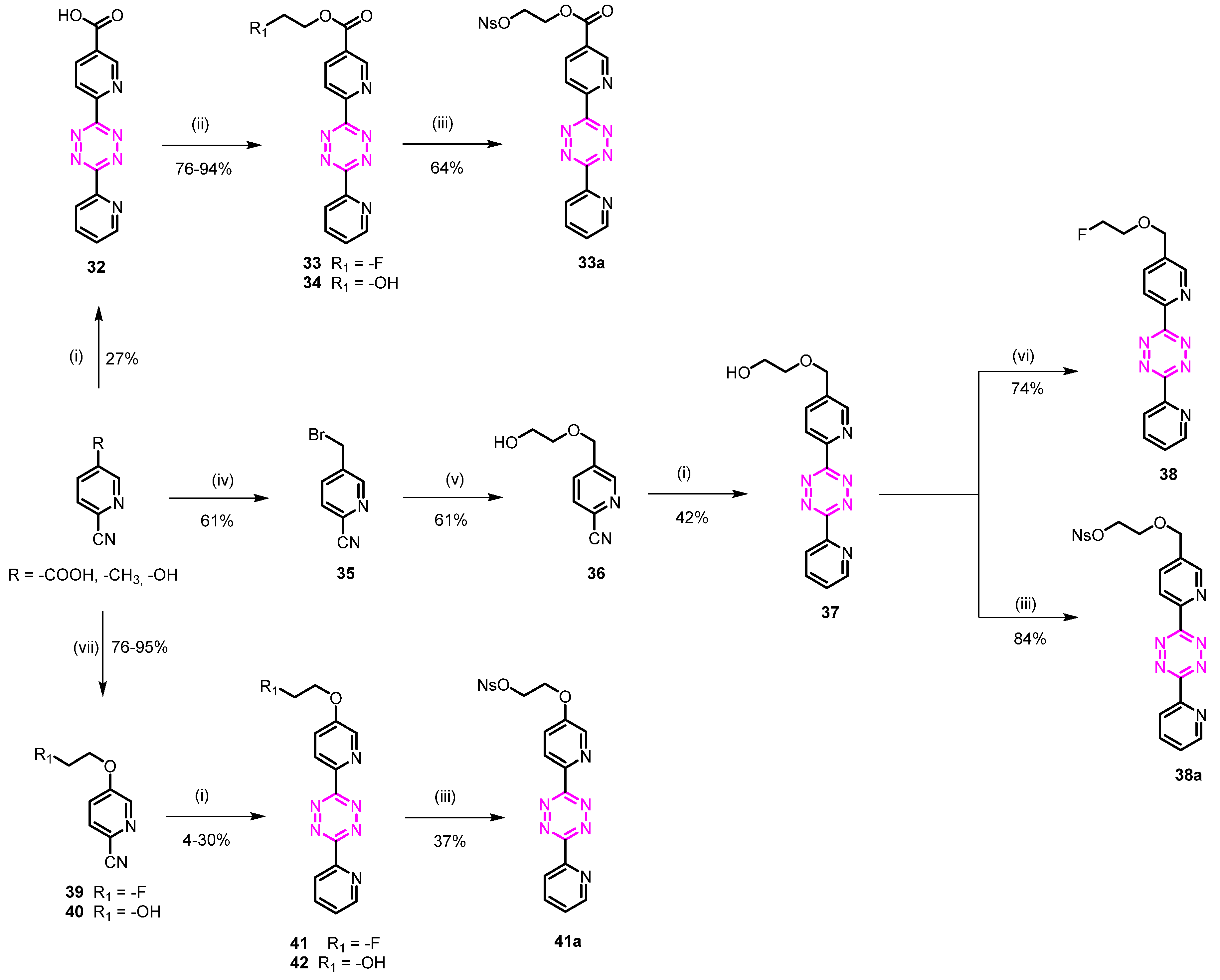
3.1.16. 2-Fluoroethyl 6-(4-(pyridin-2-yl)phenyl)nicotinate (33)
6-(6-(Pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)nicotinic acid (32)
6-Cyanonicotinic acid (1.0 g, 6.75 mmol), 2-cyanopyridine (5.6 g, 45.01 mmol), and sulfur (0.43 g, 1.69 mmol) were suspended in EtOH (10 mL), followed by the addition of hydrazine hydrate (4.93 mL, 101.26 mmol). The reaction was heated to 90 °C for 2 h. The mixture was cooled to room temperature, and the formed precipitate was removed by filtration. Water (20 mL) and a solution of NaNO2 (9.31 g, 135.32 mmol) in 50 mL water were added, and the mixture was cautiously acidified to pH 2–3 by addition of AcOH. The mixture was extracted with CH2Cl2, and the combined organic layer was washed with water and brine, dried over MgSO4, and concentrated. The residue was purified by flash column chromatography (CH2Cl2/MeOH 95/5 + 0.1% AcOH) to afford 0.52 g (27%) of the desired compound as a pink solid. Rf = 0.21 (CH2Cl2/MeOH 95/5 + 0.1% AcOH); 1H NMR (400 MHz, DMSO) δ 9.37 (d, J = 2.1 Hz, 1H), 8.96 (q, J = 1.6 Hz, 1H), 8.71 (d, J = 8.1 Hz, 1H), 8.64 (d, J = 7.9 Hz, 1H), 8.58 (dd, J = 8.1, 2.1 Hz, 1H), 8.18 (td, J = 7.8, 1.8 Hz, 1H), 7.95–7.67 (m, 1H); 13C NMR (101 MHz, DMSO) δ 165.74, 163.21, 162.97, 153.13, 151.01, 150.70, 149.96, 138.66, 137.86, 128.69, 126.81, 124.53, 124.
2-Fluoroethyl 6-(6-(pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)nicotinate (33)
The compound was obtained from 6-(6-(pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)nicotinic acid (0.05 g, 0.18 mmol) as described for compound 3 to give 0.55 g (95%) of the desired product as a pink solid. Rf = 0.41 (heptane/EtOAc 20/80); 1H NMR (400 MHz, CDCl3) δ 9.56 (dd, J = 2.2, 0.9 Hz, 1H), 9.00 (ddd, J = 4.8, 1.8, 0.9 Hz, 1H), 8.85 (dd, J = 8.2, 0.9 Hz, 1H), 8.77 (dt, J = 7.9, 1.1 Hz, 1H), 8.02 (td, J = 7.8, 1.8 Hz, 1H), 7.60 (ddd, J = 7.7, 4.7, 1.2 Hz, 1H), 4.88–4.79 (m, 1H), 4.72 (td, J = 5.9, 3.6 Hz, 2H), 4.64 (dd, J = 5.0, 3.0 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 164.34, 163.84, 163.50, 151.97, 149.86, 137.55, 127.81, 124.78, 123.95, 81.04 (d, J = 171.5 Hz), 64.62 (d, J = 20.3 Hz).
3.1.17. 2-(((4-Nitrophenyl)sulfonyl)oxy)ethyl 6-(6-(pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)nicotinate (33a)
2-Hydroxyethyl 6-(6-(pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)nicotinate (34)
The compound was obtained from 32 (0.04 g, 0.14 mmol) as described for compound 11. The compound was used as a crude due to low solubility (0.035 g, 76%). Rf = 0.31 (CH2Cl2/MeOH 90/10); 1H NMR (400 MHz, CDCl3) δ 9.47 (dd, J = 2.1, 0.9 Hz, 1H), 9.01–8.90 (m, 1H), 8.86–8.74 (m, 1H), 8.70 (dd, J = 7.9, 1.1 Hz, 1H), 8.55 (dd, J = 8.2, 2.1 Hz, 1H), 7.96 (ddt, J = 10.4, 7.7, 2.2 Hz, 1H), 7.62–7.48 (m, 1H), 4.76–4.45 (m, 2H), 4.26–3.68 (m, 2H), 3.24 (s, 1H).
2-(((4-Nitrophenyl)sulfonyl)oxy)ethyl 6-(6-(pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)nicotinate (33a)
The compound was obtained from 34 (0.035 g, 0.11 mmol) as reported for compound 3a. Purification by flash chromatography (CH2Cl2/MeOH 95/5) afforded 0.035 g (64%) of the desired compound as a red solid. Rf = 0.35 (CH2Cl2/MeOH 95/5); 1H NMR (400 MHz, DMSO) δ 9.25 (d, J = 2.2 Hz, 1H), 8.76 (d, J = 8.4 Hz, 2H), 8.66 (d, J = 7.8 Hz, 1H), 8.50 (dd, J = 8.2, 2.2 Hz, 1H), 8.40 (d, J = 8.9 Hz, 2H), 8.24 (d, J = 8.9 Hz, 2H), 8.22–8.17 (m, 1H), 7.87–7.68 (m, 1H), 4.63 (s, 4H); 13C NMR (101 MHz, DMSO) δ 164.1, 163.71, 163.51, 154.88, 154.14, 151.23, 150.39, 141.06, 139.06, 138.39, 129.87, 127.37, 125.42, 125.08, 124.59, 123.78, 70.42, 63.28.
3.1.18. 5-((2-Fluoroethoxy)methyl)-2-(4-(pyridin-2-yl)phenyl)pyridine (38)
5-(Bromomethyl)picolinonitrile (35)
To a solution of 5-methylpicolinonitrile (7.00 g, 59.25 mmol) and N-bromo succinimide (13.71 g, 77.03 mmol) in CH3CN (100 mL) was added AIBN (3.89 g, 23.70 mmol). The resulting solution was refluxed for 12 h. The reaction was cooled down, and EtOAc (200 mL) was added. The organic layer was washed with water (2 × 100 mL) and brine (2 × 100 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. Purification by flash chromatography (n-heptane/EtOAc 90/10) afforded 7.1 g (61%) of 35 as a white solid. Rf = 0.29 (n-heptane/EtOAc 80/20); 1H NMR (400 MHz, CDCl3) δ 8.72 (d, J = 2.2 Hz, 1H), 7.87 (dd, J = 8.1, 2.3 Hz, 1H), 7.69 (dd, J = 8.1, 0.9 Hz, 1H), 4.49 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 151.19, 137.50, 137.45, 133.40, 128.36, 116.84, 27.87.
5-((2-Hydroxyethoxy)methyl)picolinonitrile (36)
NaH (60% weight, 0.22 g, 5.58 mmol) was suspended in dry THF (10 mL), and ethylene glycol (2.83 mL, 50.75 mmol) was added dropwise under argon at 0 °C. The solution was left for 30 min at 0 °C before dropwise addition of 5-(bromomethyl)picolinonitrile (1.00 g, 5.87 mmol) in dry THF (10 mL) under argon at 0 °C. The reaction was stirred to room temperature for 10 min and refluxed for 3 h. The reaction was cooled to room temperature and quenched by adding EtOAc (40 mL), and the crude was washed with NH4Cl (sat, 50 mL × 2) and water (50 mL). The organic phase was dried over Na2SO4 and concentrated under reduced pressure. Purification by flash chromatography (DCM/MeOH 98/2) afforded 0.55 g (61%) of the desired product as a yellow oil. Rf = 0.31 (heptane/EtOAc 30/70); 1H NMR (400 MHz, CDCl3) δ 8.60 (d, J = 2.2 Hz, 1H), 7.82 (dd, J = 8.1, 2.2 Hz, 1H), 7.95–7.32 (m, 1H), 4.61 (s, 2H), 3.74 (dd, J = 5.4, 3.8 Hz, 2H), 3.61 (dd, J = 5.4, 3.8 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 149.94, 138.24, 135.89, 132.41, 128.30, 117.20, 72.47, 69.77, 61.51.
2-((6-(6-(Pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)pyridin-3-yl)methoxy)ethan-1-ol (37)
Compound 36 (0.55 g, 3.08 mmol), 2-cyanopyridine (1.6 g, 15.43 mmol), and sulfur (0.2 g, 0.77 mmol) were suspended in EtOH (5 mL), followed by the addition of hydrazine hydrate (2.26 mL, 43.3 mmol). The reaction was heated to 90 °C for 2 h. The mixture was cooled to room temperature, and the formed precipitate was removed by filtration. Water (20 mL) and a solution of NaNO2 (4.26 g, 61.73 mmol) in 30 mL water were added, and the mixture was cautiously acidified to pH 2 by addition of AcOH. The mixture was extracted with CH2Cl2, and the combined organic layer was washed with water and brine, dried over MgSO4, and concentrated. The residue was purified by flash column chromatography (CH2Cl2/MeOH 95/5) to afford 0.4 g (42%) of the desired compound as a pink solid. Rf = 0.21 (CH2Cl2/MeOH 95/5); 1H NMR (400 MHz, CDCl3) δ 9.03–8.94 (m, 1H), 8.90 (d, J = 2.1 Hz, 1H), 8.79–8.65 (m, 2H), 8.00 (ddd, J = 7.9, 5.5, 2.1 Hz, 3H), 7.57 (ddd, J = 7.6, 4.7, 1.2 Hz, 1H), 4.74 (s, 2H), 3.88–3.78 (m, 2H), 3.70 (dd, J = 5.3, 3.9 Hz, 2H), 2.76 (s, 1H); 13C NMR (101 MHz, CDCl3) δ 163.79, 163.69, 150.99, 150.03, 149.30, 137.50, 137.21, 136.47, 126.59, 124.50, 124.25, 72.24, 70.26, 61.80.
3-(5-((2-Fluoroethoxy)methyl)pyridin-2-yl)-6-(pyridin-2-yl)-1,2,4,5-tetrazine (38)
The compound was obtained from 2-((6-(6-(pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)pyridin-3-yl)methoxy)ethan-1-ol (0.1 g, 0.33 mmol) dissolved in 5 mL dry THF and PBSF (0.20 mg, 0.66 mmol), and DIPEA (0.34 mL, 1.93 mmol) was added. Et3N.3HF (0.11 mL, 0.66 mmol) was dissolved in 2 mL THF and added dropwise. The reaction was left at room temperature for 12 h. The reaction was diluted with 20 mL CH2Cl2 and washed with NH4Cl (sat). The aqueous phase was extracted with CH2Cl2 (2 × 10 mL), and the combined organic layers were dried over MgSO4 and concentrated under reduced pressure. Purification by flash chromatography (CH2Cl2/MeOH 95/5) afforded 0.075 g (75%) of the desired product as a pink solid. Rf = 0.45 (CH2Cl2/MeOH 95/5); 1H NMR (400 MHz, CDCl3) δ 9.05–8.80 (m, 2H), 8.78–8.58 (m, 2H), 8.21–7.86 (m, 2H), 7.51 (ddd, J = 7.6, 4.7, 1.2 Hz, 1H), 4.71 (s, 2H), 4.65–4.58 (m, 1H), 4.57–4.42 (m, 1H), 3.97–3.78 (m, 1H), 3.77–3.68 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 163.82, 163.73, 150.98, 150.03, 149.97, 149.36, 137.50, 137.03, 136.43, 126.60, 124.48, 124.26, 82.98 (d, J = 169.5 Hz), 70.35, 70.00 (d, J = 19.6 Hz).
3.1.19. 2-((6-(4-(Pyridin-2-yl)phenyl)pyridin-3-yl)methoxy)ethyl 4-methylbenzene sulfonate (38a)
The compound was obtained from 37 ((0.05 g, 0.17 mmol) as reported for compound 3a. Purification by flash chromatography (CH2Cl2/MeOH 98/2) afforded 0.07 g (84%) of the desired compound as a red oil. Rf = 0.48 (CH2Cl2/MeOH 90/10); 1H NMR (600 MHz, CDCl3) δ 9.02 (dt, J = 4.5, 1.4 Hz, 1H), 8.86 (d, J = 2.1 Hz, 1H), 8.80–8.63 (m, 2H), 8.41–8.33 (m, 1H), 8.17–8.11 (m, 1H), 8.04 (td, J = 7.7, 1.8 Hz, 1H), 7.93 (dd, J = 8.0, 2.2 Hz, 1H), 7.61 (ddd, J = 7.6, 4.7, 1.1 Hz, 1H), 4.69 (s, 2H), 4.44–4.38 (m, 2H), 3.90–3.82 (m, 2H); 13C NMR (151 MHz, CDCl3) δ 163.90, 163.68, 151.09, 150.76, 150.11, 150.00, 149.76, 141.81, 137.50, 136.40, 136.21, 129.25, 126.62, 124.58, 124.41, 124.22, 70.44, 70.15, 68.20.
3.1.20. 5-(2-Fluoroethoxy)-2-(4-(pyridin-2-yl)phenyl)pyridine (41)
5-(2-Fluoroethoxy)picolinonitrile (39)
The compound was obtained from 5-hydroxypicolinonitrile (0.5 g, 4.16 mmol) as described for compound 20 to give 0.56 g (81%) of the desired product as a pink solid. Rf = 0.34 (heptane/EtOAc 70/30); 1H NMR (400 MHz, CDCl3) δ 8.34 (d, J = 2.9 Hz, 1H), 7.59 (d, J = 8.6 Hz, 1H), 7.27–7.20 (m, 1H), 4.83–4.76 (m, 1H), 4.71–4.64 (m, 1H), 4.35–4.28 (m, 1H), 4.28–4.21 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 155.04, 138.50, 127.74, 123.96, 118.83, 115.55, 79.47 (d, J = 172.4 Hz), 66.05 (d, J = 20.4 Hz).
3-(5-(2-Fluoroethoxy)pyridin-2-yl)-6-(pyridin-2-yl)-1,2,4,5-tetrazine (41)
The compound was obtained from 39 (0.55 g, 3.31 mmol) as described for compound 37, followed by purification by preparative HPLC to give 0.04 g (4%) of the desired product as a pink solid. Rf = 0.32 (CH2Cl2/MeOH 97/3); 1H NMR (400 MHz, CDCl3) δ 8.94 (dt, J = 4.7, 1.4 Hz, 1H), 8.70 (d, J = 8.5 Hz, 2H), 8.65 (d, J = 2.9 Hz, 1H), 8.00 (td, J = 7.8, 1.7 Hz, 1H), 7.57 (ddd, J = 7.7, 4.8, 1.2 Hz, 1H), 7.46 (dd, J = 8.8, 2.9 Hz, 1H), 4.88–4.81 (m, 1H), 4.76–4.69 (m, 1H), 4.44–4.38 (m, 1H), 4.38–4.25 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 163.26, 163.21, 157.38, 150.46, 149.69, 142.33, 139.43, 138.19, 126.84, 125.92 124.58, 121.99, 81.39 (d, J = 172.7 Hz), 67.99 (d, J = 20.4 Hz); HPLC-MS [M+H]+ m/z calc. for [C14H12FN6O]+: 299.10; found: 299.12.
3.1.21. 2-((6-(4-(Pyridin-2-yl)phenyl)pyridin-3-yl)oxy)ethyl 4-nitrobenzenesulfonate (41a)
5-(2-Hydroxyethoxy)picolinonitrile (40)
The compound was obtained from 5-hydroxypicolinonitrile (1.4 g, 11.65 mmol) as described for compound 21. Purification by flash chromatography (n-heptane/EtOAc 40/60) afforded 1.45 g (76%) of the desired product as a white solid. Rf = 0.21 (heptane/EtOAc 40/60); 1H NMR (400 MHz, CDCl3) δ 8.32 (d, J = 2.9 Hz, 1H), 7.59 (d, J = 8.6 Hz, 1H), 7.42–6.71 (m, 1H), 4.66–4.05 (m, 2H), 3.97 (dd, J = 5.1, 3.9 Hz, 2H), 2.27 (s, 1H); 13C NMR (101 MHz, CDCl3) δ 157.27, 140.42, 129.65, 125.41, 120.58, 117.39, 70.21, 60.86.
2-((6-(6-(Pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)pyridin-3-yl)oxy)ethan-1-ol (42)
The compound was obtained from 40 (0.5 g, 3.04 mmol) as described for compound 37. Purification by flash chromatography (CH2Cl2/MeOH 97/3) afforded 0.27 g (30%) of the desired product as a pink solid. Rf = 0.25 (CH2Cl2/MeOH 95/5); 1H NMR (600 MHz, DMSO) δ 8.93 (ddd, J = 4.7, 1.8, 0.9 Hz, 1H), 8.65 (dd, J = 2.9, 0.6 Hz, 1H), 8.62–8.49 (m, 2H), 8.16 (td, J = 7.7, 1.8 Hz, 1H), 7.82–7.56 (m, 2H), 5.00 (s, 1H), 4.50–3.97 (m, 2H), 3.81 (td, J = 5.4, 4.3 Hz, 2H); 13C NMR (151 MHz, DMSO) δ 163.49, 163.30, 157.81, 151.05, 150.71, 142.37, 139.97, 138.25, 127.00, 126.05, 124.59, 121.95, 70.96, 59.88.
2-((6-(4-(Pyridin-2-yl)phenyl)pyridin-3-yl)oxy)ethyl 4-nitrobenzenesulfonate (41a)
The compound was obtained from 42 (0.05 g, 0.17 mmol) as reported for compound 3a. Purification by flash chromatography (CH2Cl2/MeOH 98/2) afforded 0.03 g (37%) of the desired compound as a red solid (mixture of conformers 70/30). Rf = 0.61 (CH2Cl2/MeOH 90/10); 1H NMR (600 MHz, DMSO) δ 8.94 (ddd, J = 4.7, 1.8, 0.9 Hz, 1H), 8.70 (d, J = 2.9 Hz, 0.3H), 8.65 (d, J = 8.5 Hz, 0.3H), 8.60 (d, J = 7.8 Hz, 1H), 8.56 (d, J = 8.8 Hz, 0.7H), 8.51 (d, J = 2.9 Hz, 0.7H), 8.47–8.43 (m, 1.4H), 8.27–8.22 (m, 1.4H), 8.21–8.18 (m, 0.6H), 8.16 (td, J = 7.8, 1.8 Hz, 1H), 7.90–7.81 (m, 0.6H), 7.78 (dd, J = 8.8, 2.9 Hz, 0.3H), 7.73 (ddd, J = 7.6, 4.7, 1.2 Hz, 1H), 7.64 (dd, J = 8.8, 3.0 Hz, 0.7H), 4.81–4.71 (m, 0.6H), 4.68–4.59 (m, 1.4H), 4.57–4.54 (m, 0.6H), 4.51–4.43 (m, 1.4H); 13C NMR (151 MHz, DMSO) δ 163.57, 163.53, 163.26, 163.21, 156.74, 156.60, 154.89, 151.16, 151.08, 150.66, 147.70, 143.22, 142.95, 140.95, 139.86, 139.77, 138.28, 129.93, 127.38, 127.08, 127.05, 126.08, 125.89, 125.43, 124.63, 123.78, 122.36, 122.12, 74.80, 70.58, 67.80, 66.45.
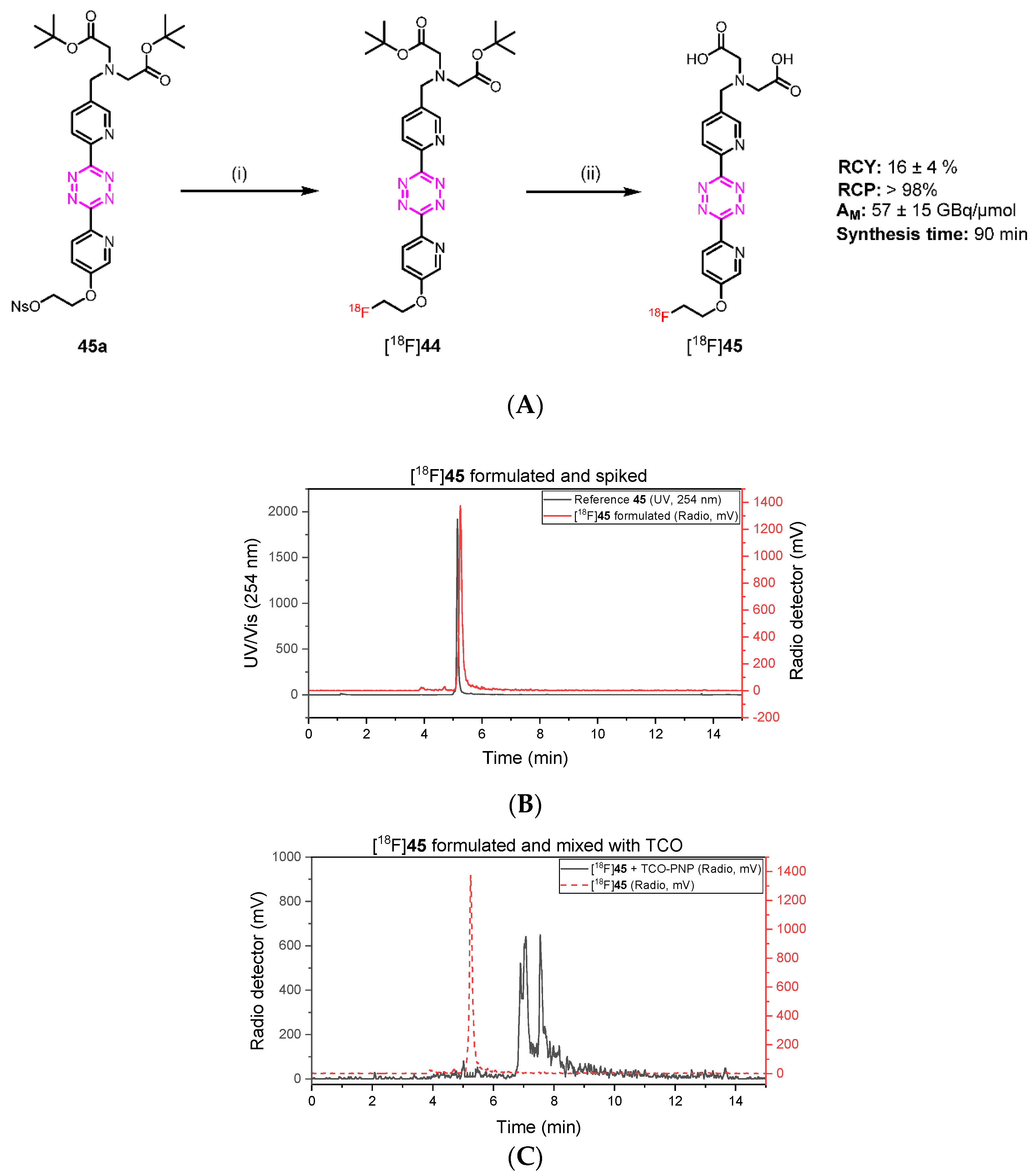
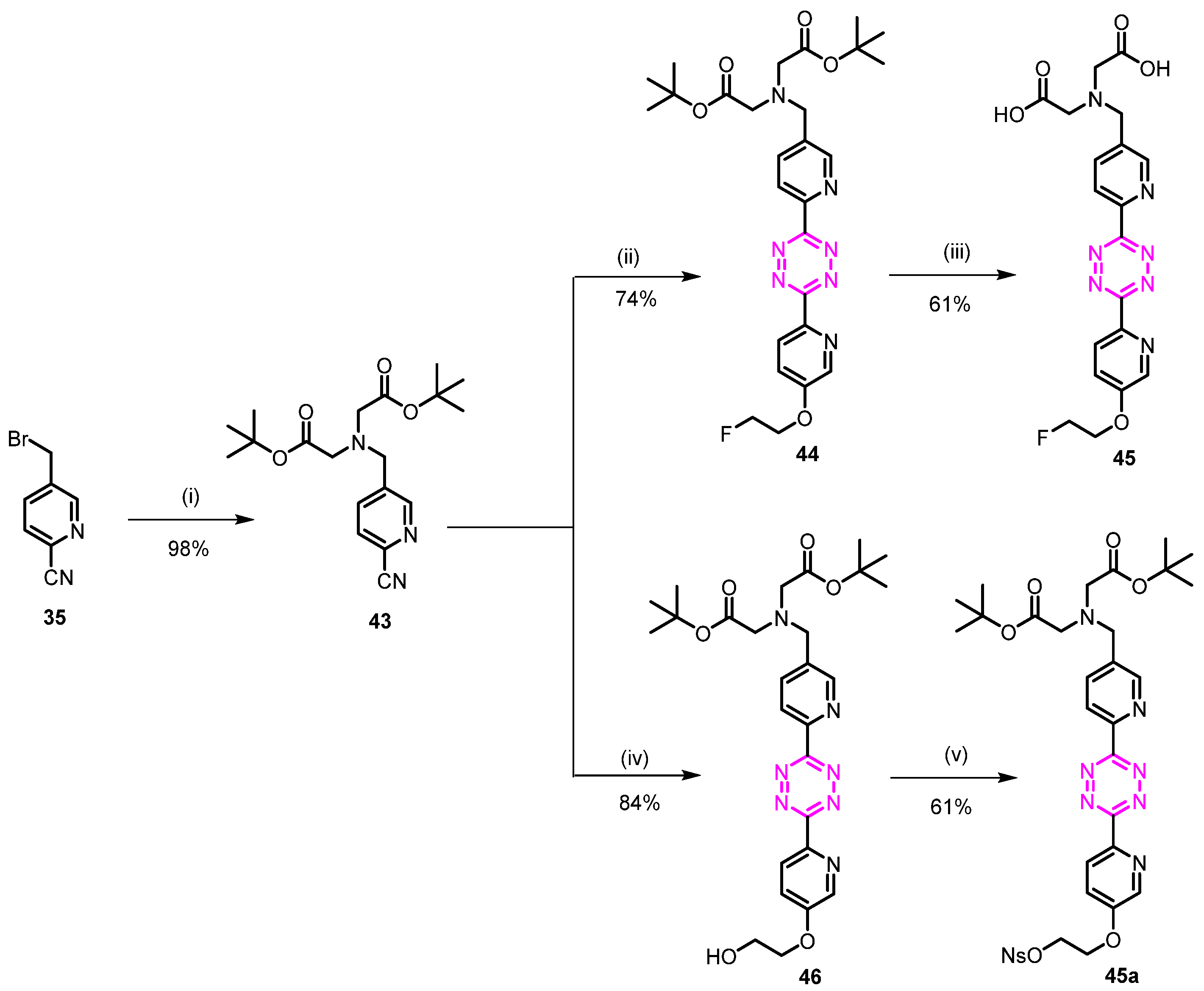
3.1.22. 2,2′-(((6-(6-(5-(2-Fluoroethoxy)pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)pyridin-3-yl)methyl)azanediyl)diacetic Acid (45)
Di-tert-butyl 2,2′-(((6-cyanopyridin-3-yl)methyl)azanediyl)diacetate (43)
To a solution of 5-(bromomethyl)picolinonitrile (1.5 g, 7.61 mmol) in CH3CN (50 mL) was added K2CO3 (3.15 g, 22.84 mmol) and di-tert-butyl iminodiacetate (1.96 g, 7.99 mmol). The reaction mixture was stirred at room temperature overnight, and then the solvent was concentrated under reduced pressure. The resulting mixture was diluted with water (100 mL), extracted with EtOAc (3 × 40 mL), washed with brine (50 mL), dried over MgSO4, filtered, and concentrated under reduced pressure. Purification by flash chromatography (n-heptane/EtOAc 90/10) afforded 2.7 g (98%) of the desired compound as a white solid. Rf = 0.33 (n-heptane/EtOAc 80/20); 1H NMR (400 MHz, CDCl3) δ 8.61 (dd, J = 2.1, 0.9 Hz, 1H), 7.96 (dd, J = 8.0, 2.1 Hz, 1H), 7.60 (dd, J = 7.9, 0.9 Hz, 1H), 3.93 (s, 2H), 3.33 (s, 4H), 1.40 (s, 18H); 13C NMR (101 MHz, CDCl3) δ 170.09, 151.41, 138.96, 137.51, 132.77, 128.27, 117.35, 81.52, 55.41, 54.57, 28.16.
Di-tert-butyl 2,2′-(((6-(6-(5-(2-fluoroethoxy)pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)pyridin-3-yl) methyl)-azanediyl)diacetate (44)
Compound 43 (0.2 g, 0.55 mmol), compound 39 (0.46 g, 2.77 mmol), and sulfur (0.36 g, 0.14 mmol) were suspended in EtOH (5 mL), followed by the addition of hydrazine hydrate (0.40 mL, 8.31 mmol). The reaction was heated to 90 °C for 2 h. The mixture was cooled to room temperature and the formed precipitate was removed by filtration. Water (20 mL) and a solution of NaNO2 (0.76 g, 11.07 mmol) in 10 mL water were added, and the mixture was cautiously acidified to pH 2–3 by addition of AcOH. The mixture was extracted with CH2Cl2, and the combined organic layer was washed with water and brine, dried over MgSO4, and concentrated. The residue was purified by flash column chromatography (CH2Cl2/MeOH 95/5) and crystallized from MeOH to afford 0.07 g (23%) of the desired compound as a pink solid. Rf = 0.36 (CH2Cl2/MeOH 95/5); 1H NMR (600 MHz, DMSO) δ 8.86 (d, J = 2.1 Hz, 1H), 8.68 (dd, J = 7.9, 3.1 Hz, 1H), 8.62 (d, J = 8.8 Hz, 1H), 8.58 (d, J = 8.0 Hz, 1H), 8.11 (dd, J = 8.1, 2.1 Hz, 1H), 7.76 (dd, J = 8.8, 3.0 Hz, 1H), 4.97–4.86 (m, 1H), 4.81 (dd, J = 4.8, 2.8 Hz, 1H), 4.54 (dd, J = 4.8, 2.7 Hz, 1H), 4.51–4.43 (m, 1H), 4.01 (s, 2H), 3.45 (s, 4H), 1.43 (s, 18H); 13C NMR (151 MHz, DMSO) δ 170.35, 163.47, 163.24, 157.21, 151.20, 149.63, 142.89, 139.86, 138.19, 138.11, 126.00, 124.30, 82.43 (d, J = 166.8 Hz), 80.97, 68.39 (d, J = 18.9 Hz), 55.57, 54.87, 28.29.
2,2′-(((6-(6-(5-(2-Fluoroethoxy)pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)pyridin-3-yl)methyl)azane-diyl)diacetic acid (45)
To a solution of 44 (0.006 g, 0.83 mmol) in 5 mL of CH2Cl2 was added 2 mL of TFA. The mixture was stirred at room temperature for 4 h. The solvent was then removed under reduced pressure. Purification by preparative HPLC afforded 0.35 g (58%) of the desired compound (TFA salt) as a red solid. 1H NMR (600 MHz, DMSO) δ 8.89 (d, J = 2.1 Hz, 1H), 8.68 (d, J = 2.9 Hz, 1H), 8.62 (d, J = 8.8 Hz, 1H), 8.57 (dd, J = 8.0, 0.8 Hz, 1H), 8.14 (dd, J = 8.1, 2.1 Hz, 1H), 7.76 (dd, J = 8.8, 3.0 Hz, 1H), 4.91–4.86 (m, 1H), 4.83–4.79 (m, 1H), 4.57–4.52 (m, 1H), 4.52–4.47 (m, 1H), 4.06 (s, 2H), 3.53 (s, 4H); 13C NMR (151 MHz, DMSO) δ 172.62, 163.47, 163.23, 157.22, 151.33, 149.58, 142.89, 139.86, 138.28, 126.00, 124.27, 122.16, 82.43 (d, J = 166.8 Hz), 68.39 (d, J = 18.8 Hz), 55.00, 54.49; HPLC-MS [M+H]+ m/z calc. for [C19H19FN7O5]+: 444.14; found: 444.13.
3.1.23. Di-tert-butyl 2,2′-(((6-(6-(5-(2-(((4-nitrophenyl)sulfonyl)oxy)ethoxy)pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)pyridin-3-yl)methyl)azanediyl)diacetate (45a)
Di-tert-butyl 2,2′-(((6-(6-(5-(2-hydroxyethoxy)pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)pyridin-3-yl)methyl)azanediyl)diacetate (46)
Compound 43 (1.05 g, 2.92 mmol), compound 40 (0.12 g, 0.73 mmol), and sulfur (0.05 g, 0.18 mmol) were suspended in EtOH (3 mL), followed by the addition of hydrazine hydrate (0.53 mL, 10.96 mmol). The reaction was heated to 90 °C for 2 h. The mixture was cooled to room temperature and the formed precipitate was removed by filtration. Water (10 mL) and a solution of NaNO2 (1.0 g, 14.62 mmol) in 10 mL water were added, and the mixture was cautiously acidified to pH 2 by addition of AcOH. The mixture was extracted with CH2Cl2, and the combined organic layer was washed with water and brine, dried over MgSO4, and concentrated. The residue was purified by flash column chromatography (CH2Cl2/MeOH 95/5) to give 0.11 g (27%) of the desired compound as a pink solid. Rf = 0.33 (CH2Cl2/MeOH 95/5); 1H NMR (400 MHz, MeOD) δ 8.88 (d, J = 2.0 Hz, 1H), 8.81–8.69 (m, 2H), 8.56 (d, J = 2.9 Hz, 1H), 8.22 (dd, J = 8.1, 2.1 Hz, 1H), 7.71 (dd, J = 8.9, 2.9 Hz, 1H), 4.45–4.25 (m, 2H), 4.10 (s, 2H), 4.04–3.93 (m, 2H), 3.50 (s, 4H), 3.33 (t, J = 1.6 Hz, 1H), 1.50 (s, 18H); 13C NMR (101 MHz, MeOD) δ 170.55, 163.15, 163.04, 158.24, 150.67, 148.89, 141.66, 139.29, 138.58, 138.49, 125.57, 123.75, 121.49, 81.08, 70.31, 59.99, 55.12, 54.74, 27.04.
Di-tert-butyl 2,2′-(((6-(6-(5-(2-(((4-nitrophenyl)sulfonyl)oxy)ethoxy)pyridin-2-yl)-1,2,4,5-tetrazine-3-yl)pyridin-3-yl)methyl)azanediyl)diacetate (45a)
The compound was obtained from 46 (0.05 g, 0.09 mmol) as reported for compound 3a. Purification by flash chromatography (heptane/EtOAc 40/60) afforded 0.045 g (67%) of the desired compound as a red solid. Rf = 0.26 (heptane/EtOAc 30/70); 1H NMR (600 MHz, CDCl3) δ 8.82 (d, J = 2.1 Hz, 1H), 8.70–8.59 (m, 2H), 8.48 (d, J = 2.9 Hz, 1H), 8.35 (d, J = 8.8 Hz, 2H), 8.15–7.98 (m, 3H), 7.32 (d, J = 2.9 Hz, 1H), 4.76–4.47 (m, 2H), 4.41–4.31 (m, 2H), 4.01 (s, 2H), 3.40 (s, 4H), 1.41 (s, 18H); 13C NMR (151 MHz, CDCl3) δ 169.22, 162.64, 162.15, 155.32, 150.32, 149.95, 148.26, 142.30, 140.49, 138.34, 137.23, 137.03, 128.33, 124.54, 123.55, 123.24, 120.47, 80.37, 67.51, 65.00, 54.31, 53.61, 27.18; HPLC-MS [M+H]+ m/z calc. for [C33H39N8O10S]+: 739.25; found: 739.26.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

