
Design, Synthesis, and Biological Evaluation of a Novel VEGFR-2 Inhibitor Based on a 1,2,5-Oxadiazole-2-Oxide Scaffold with MAPK Signaling Pathway Inhibition

 ,
,  ,
,
Abstract
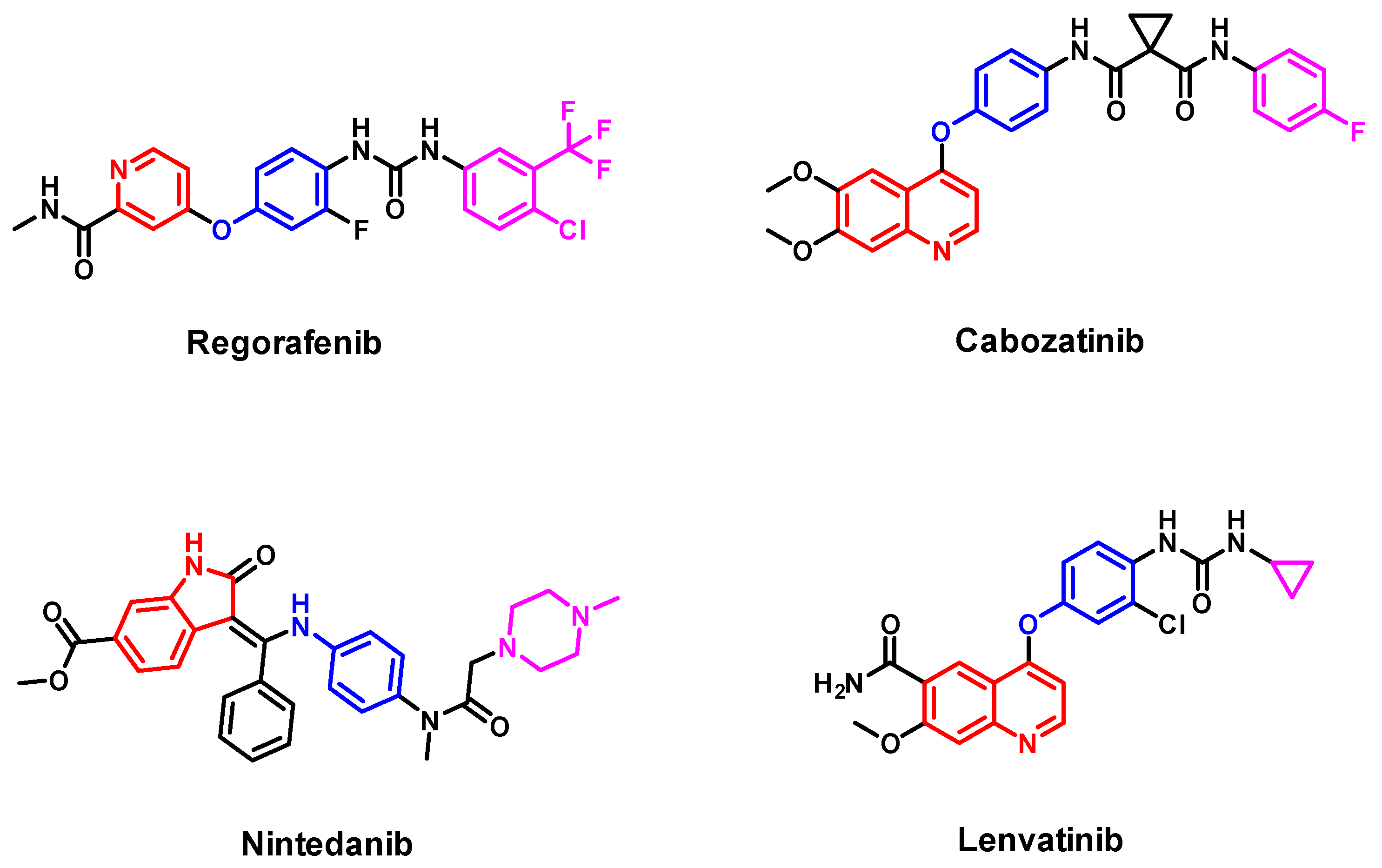
1. Introduction
2. Results and Discussion
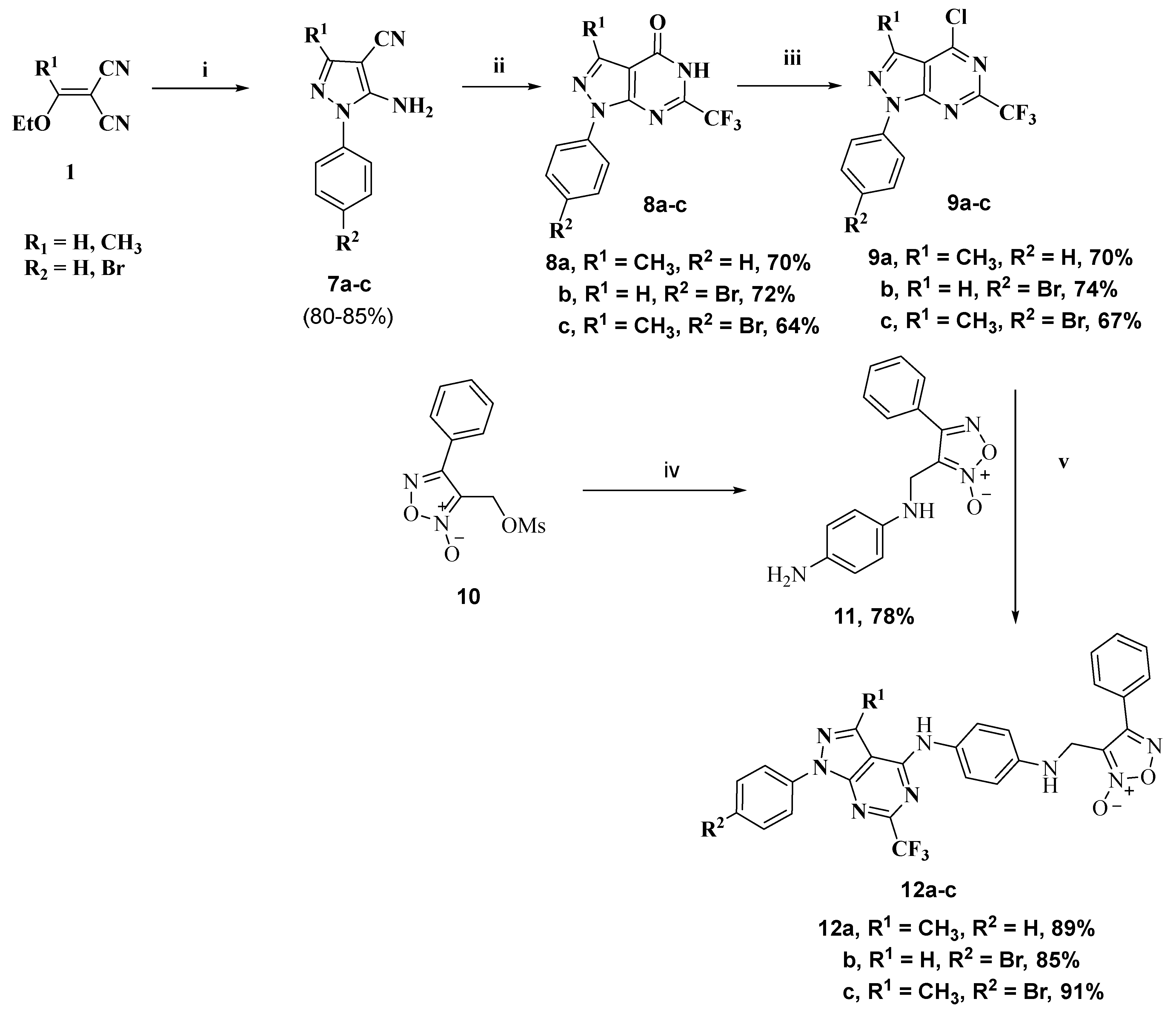
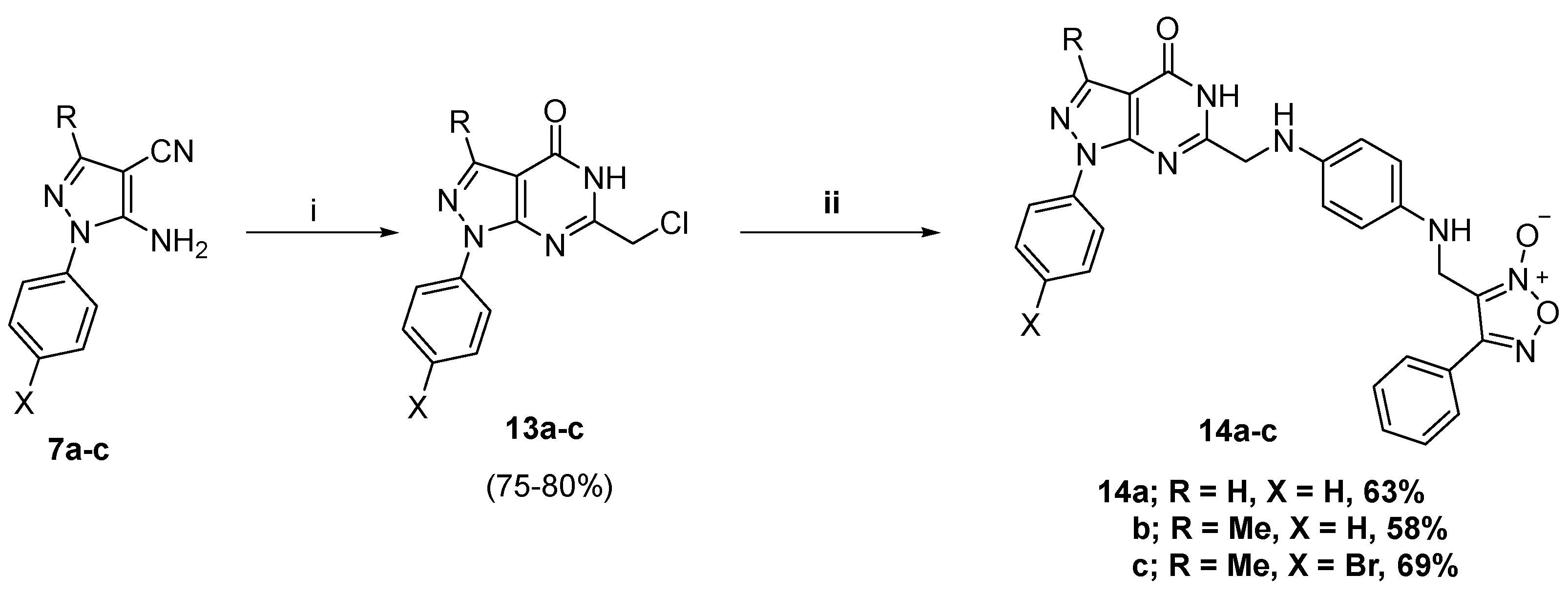
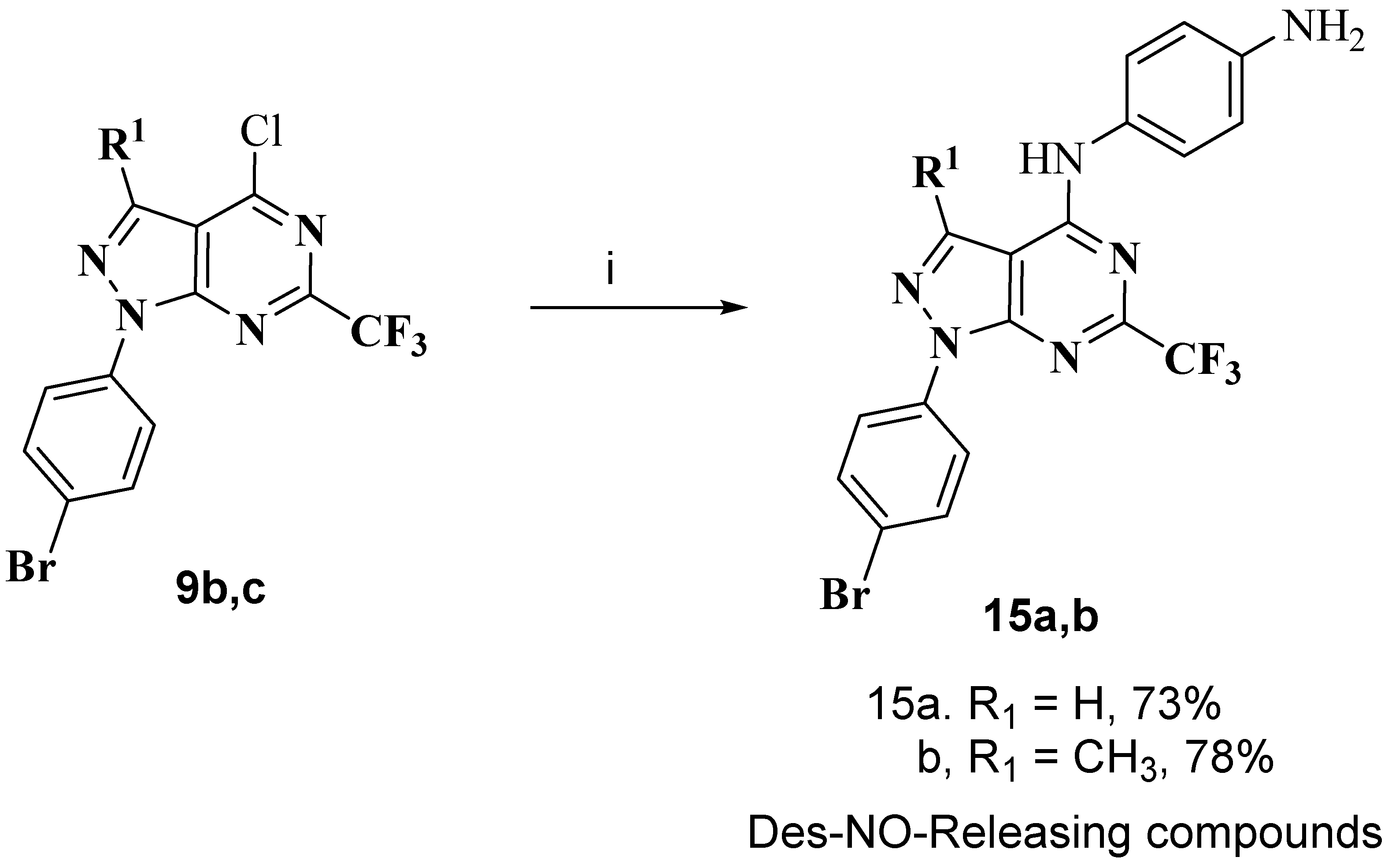
2.1. Chemistry
2.2. Biological Evaluation
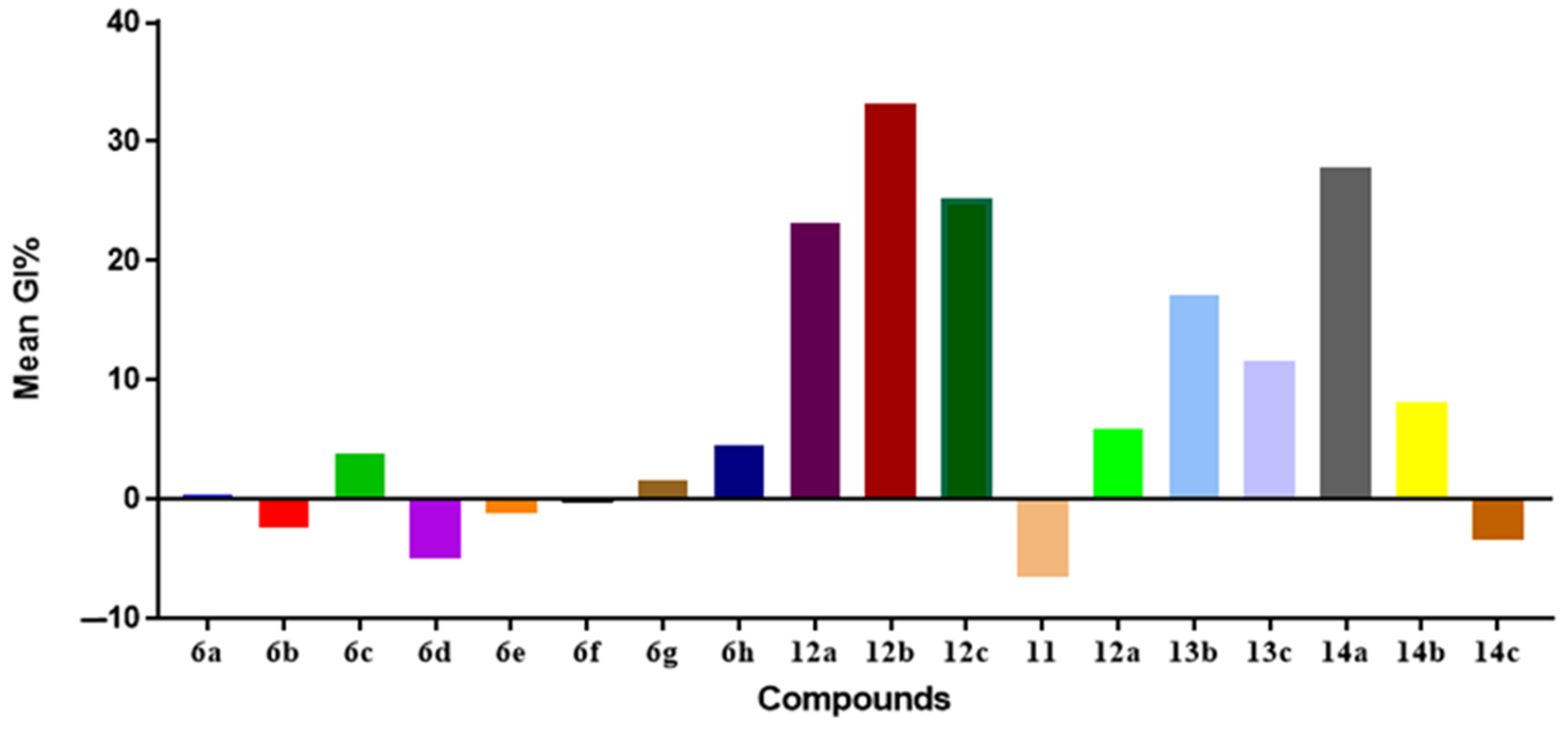
2.2.1. In Vitro Anticancer Activity
NCI-60 Cell Line Screening
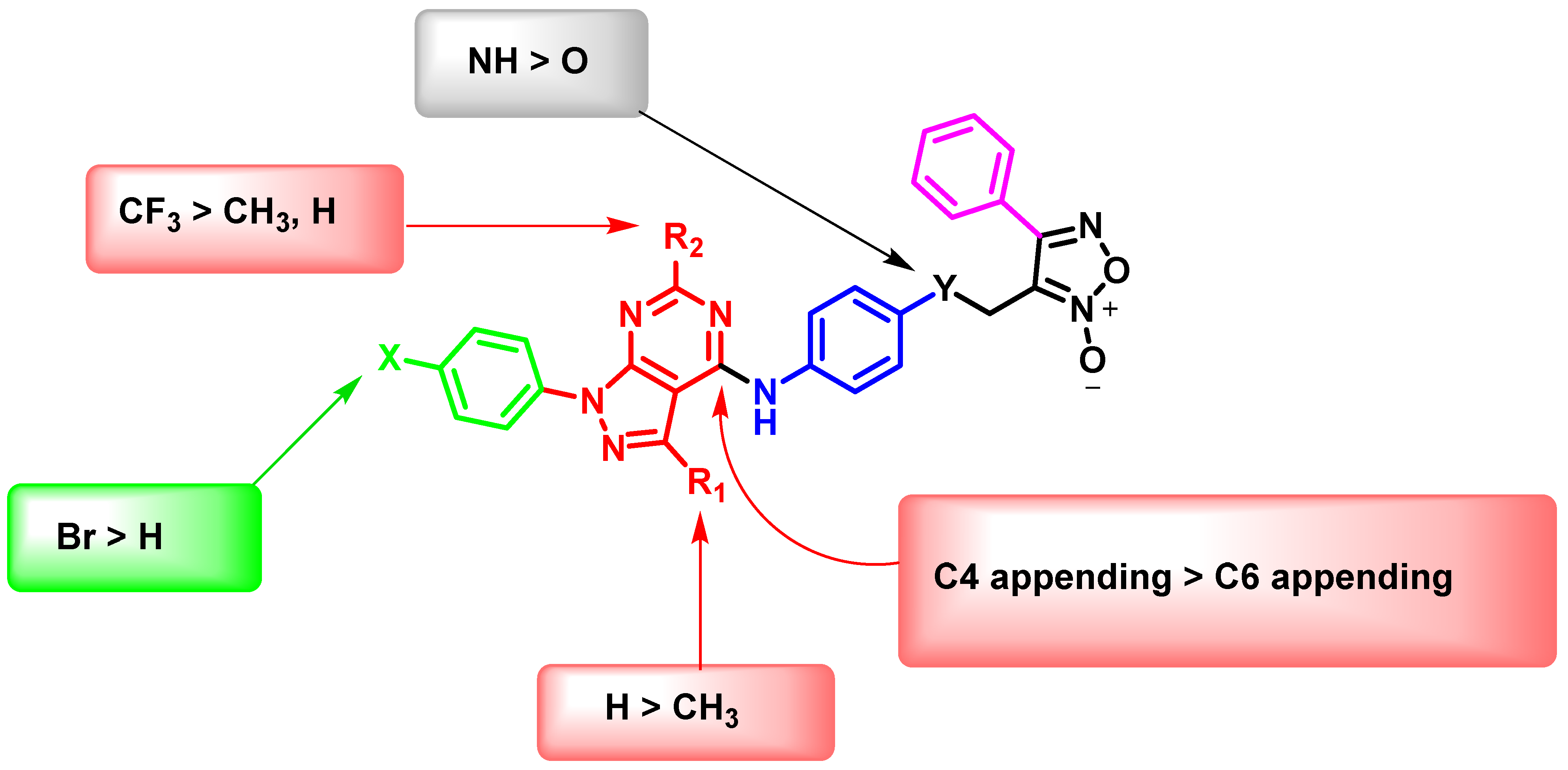
Preliminary SARs Study
In Vitro Anti-Proliferative Activities
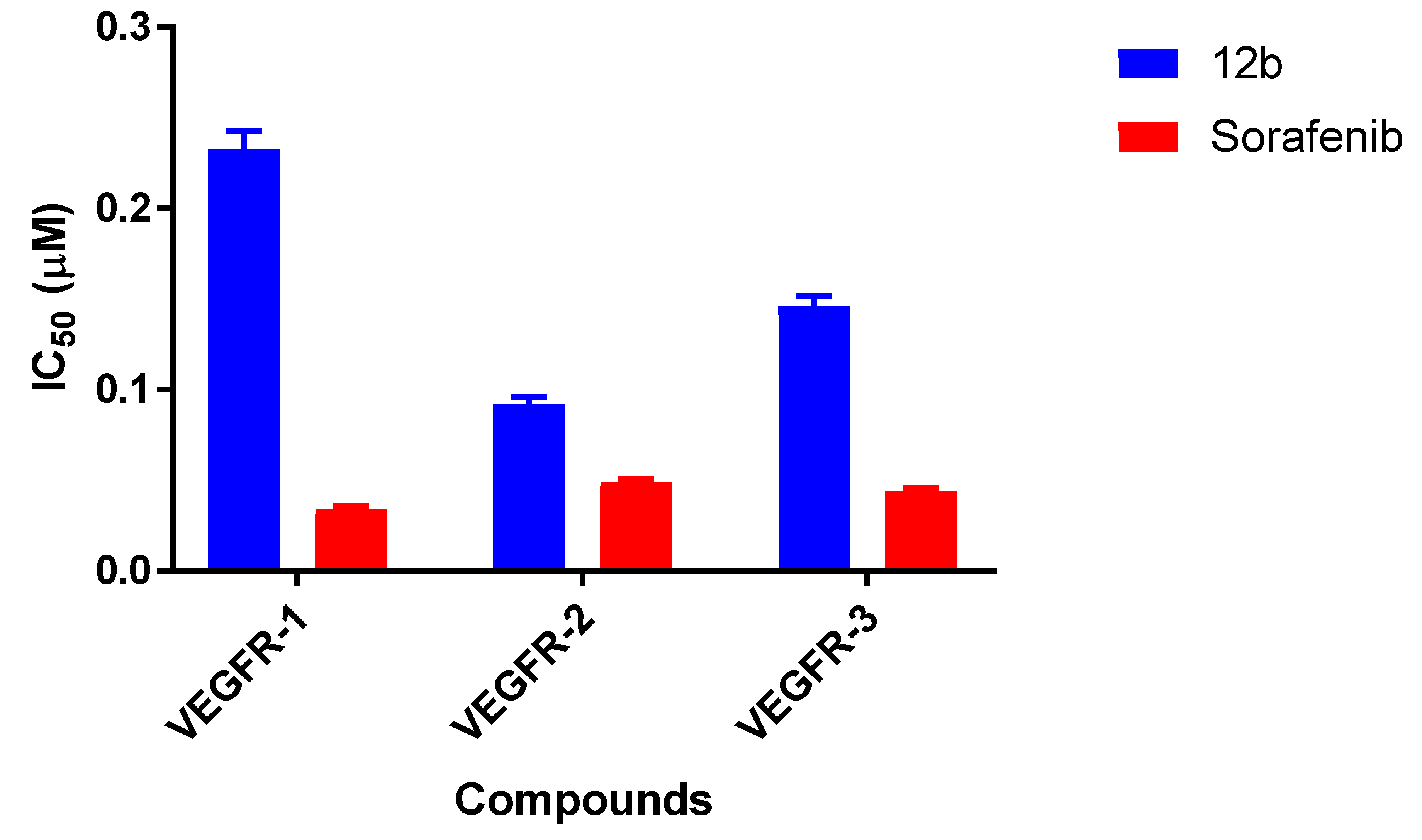
2.2.2. VEGFR Kinase Inhibitory Profile of 12b
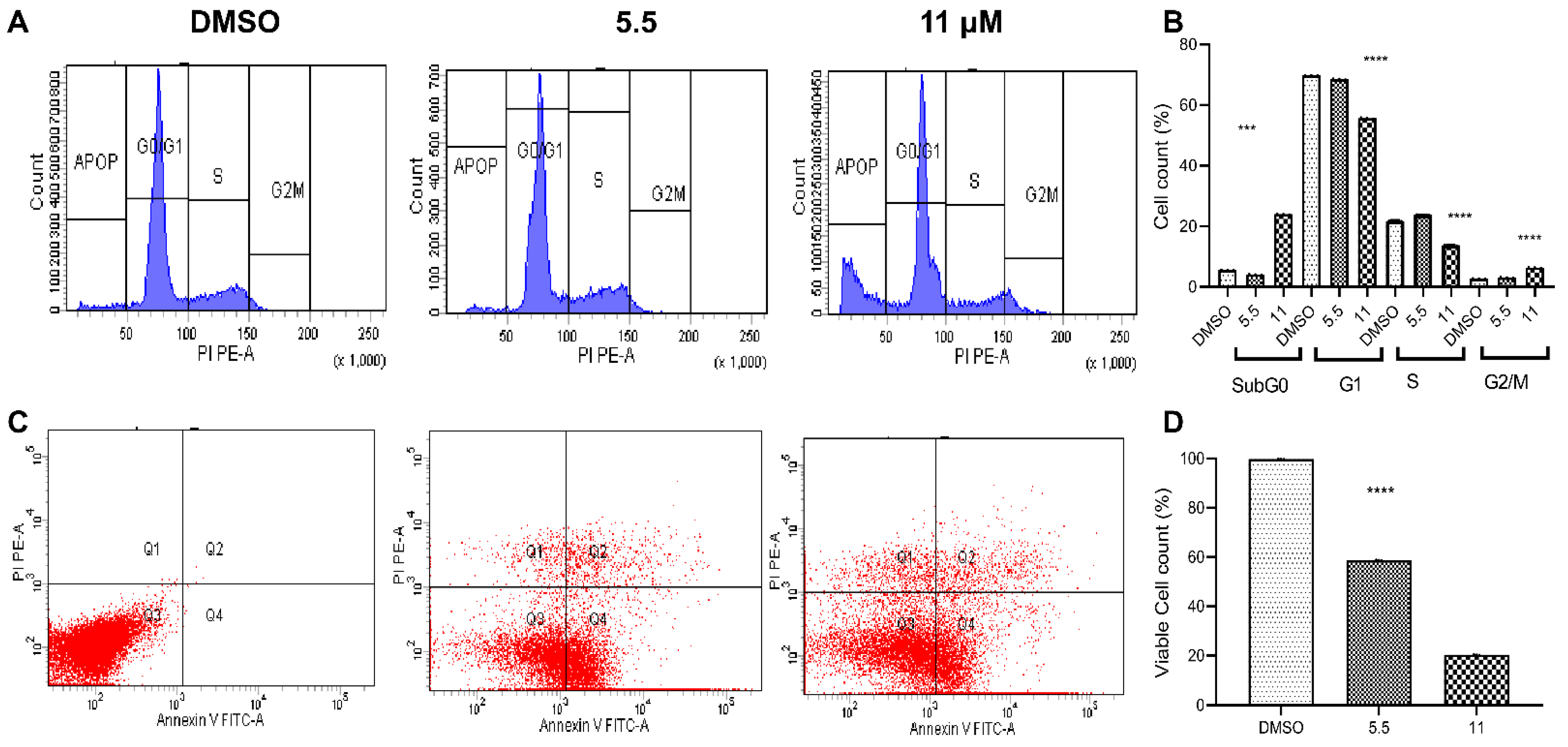
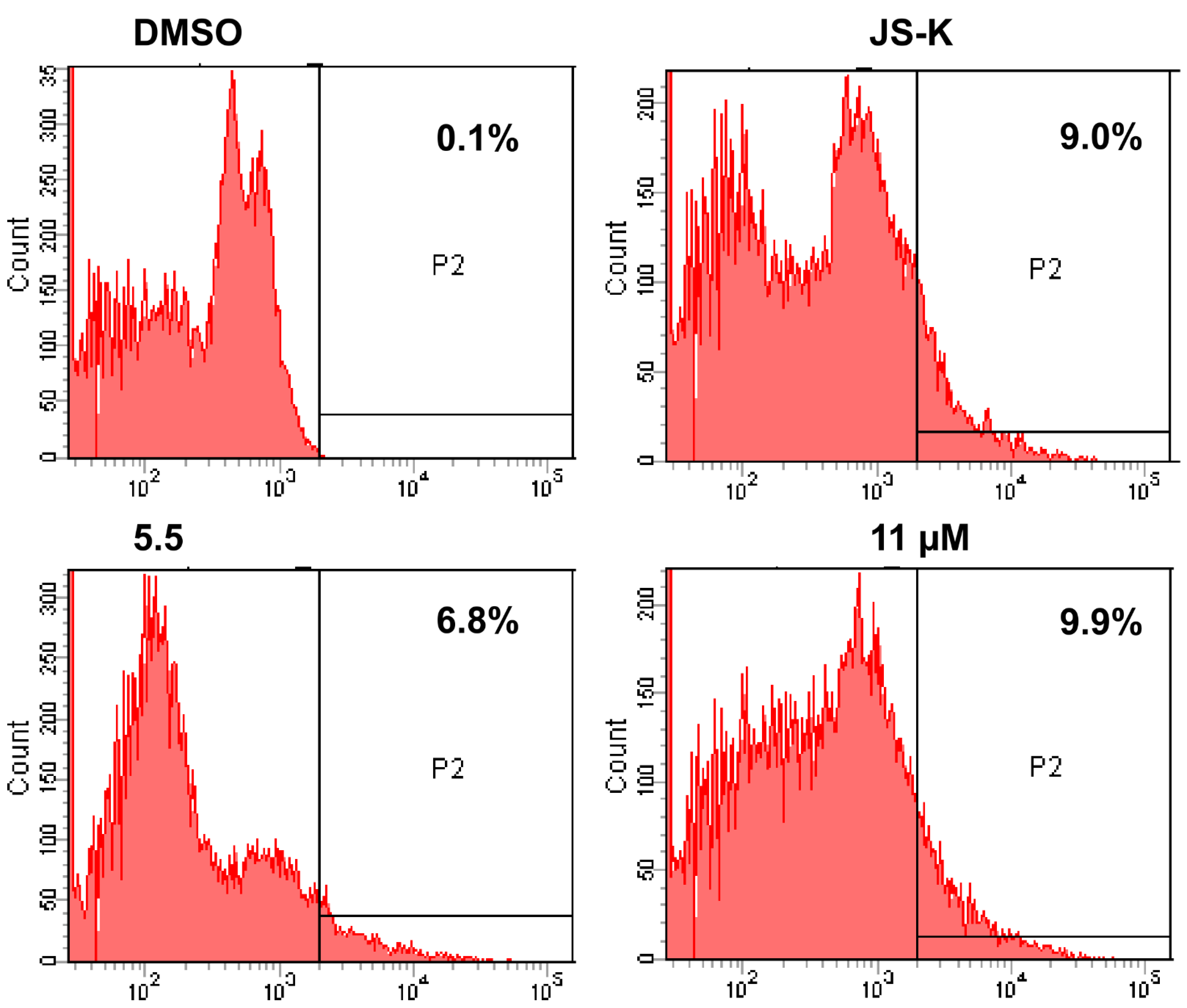
2.2.3. Flow Cytometric Studies
2.2.4. Intracellular Measurement of NO
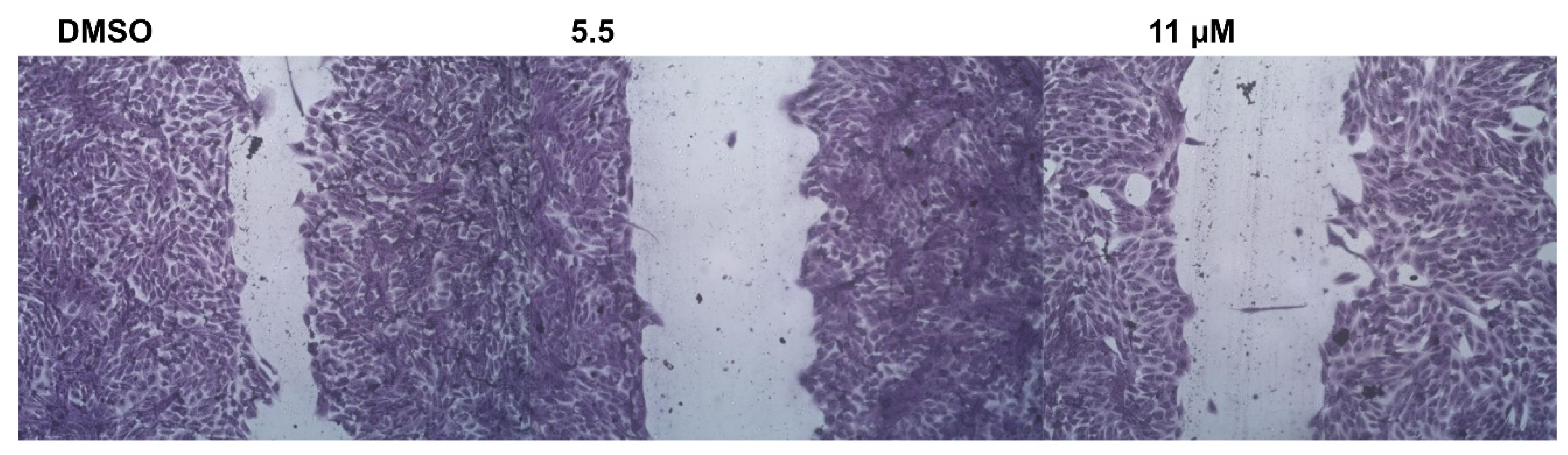
2.2.5. Wound-Healing Assay
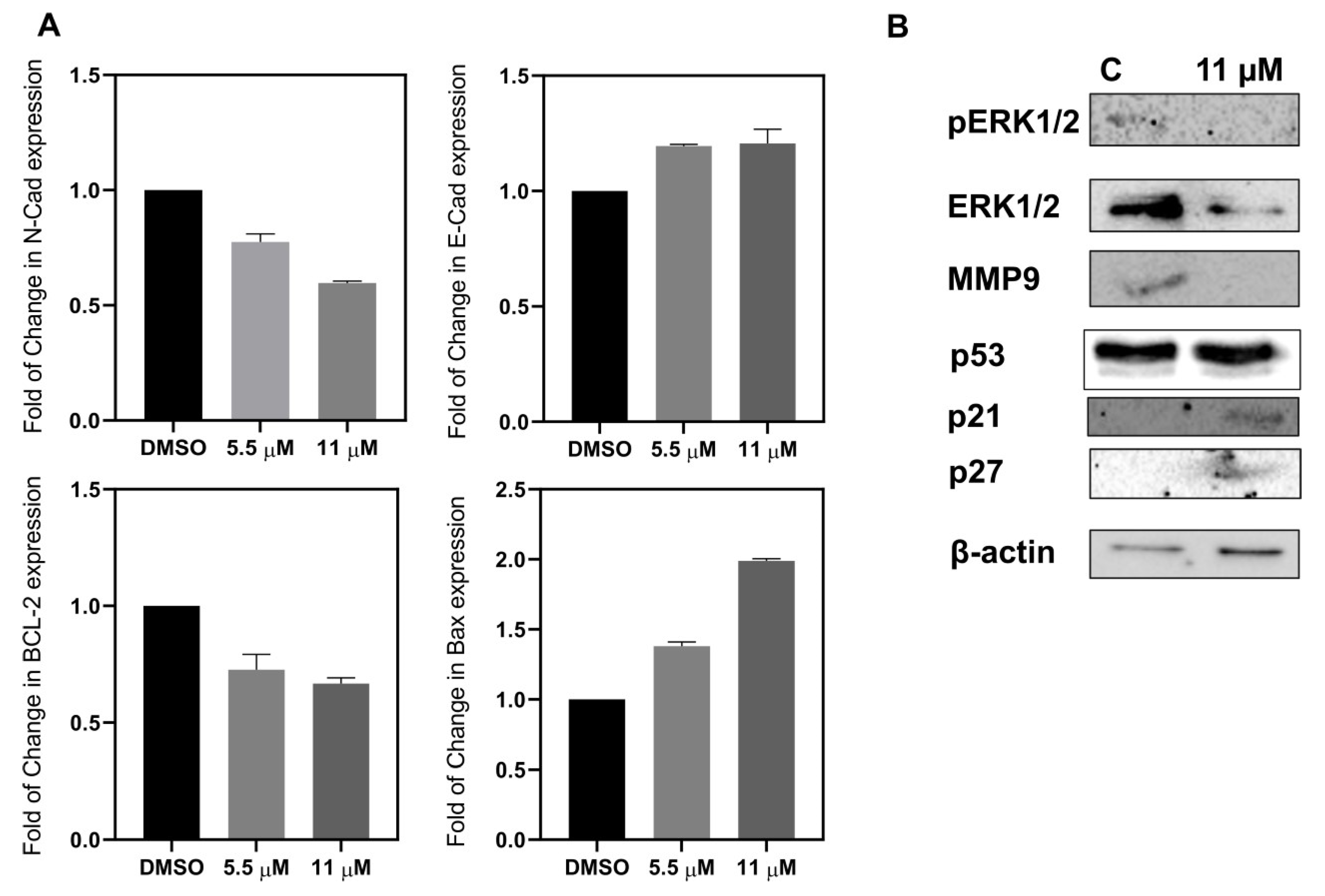
2.2.6. Apoptosis and Metastatic Proteins
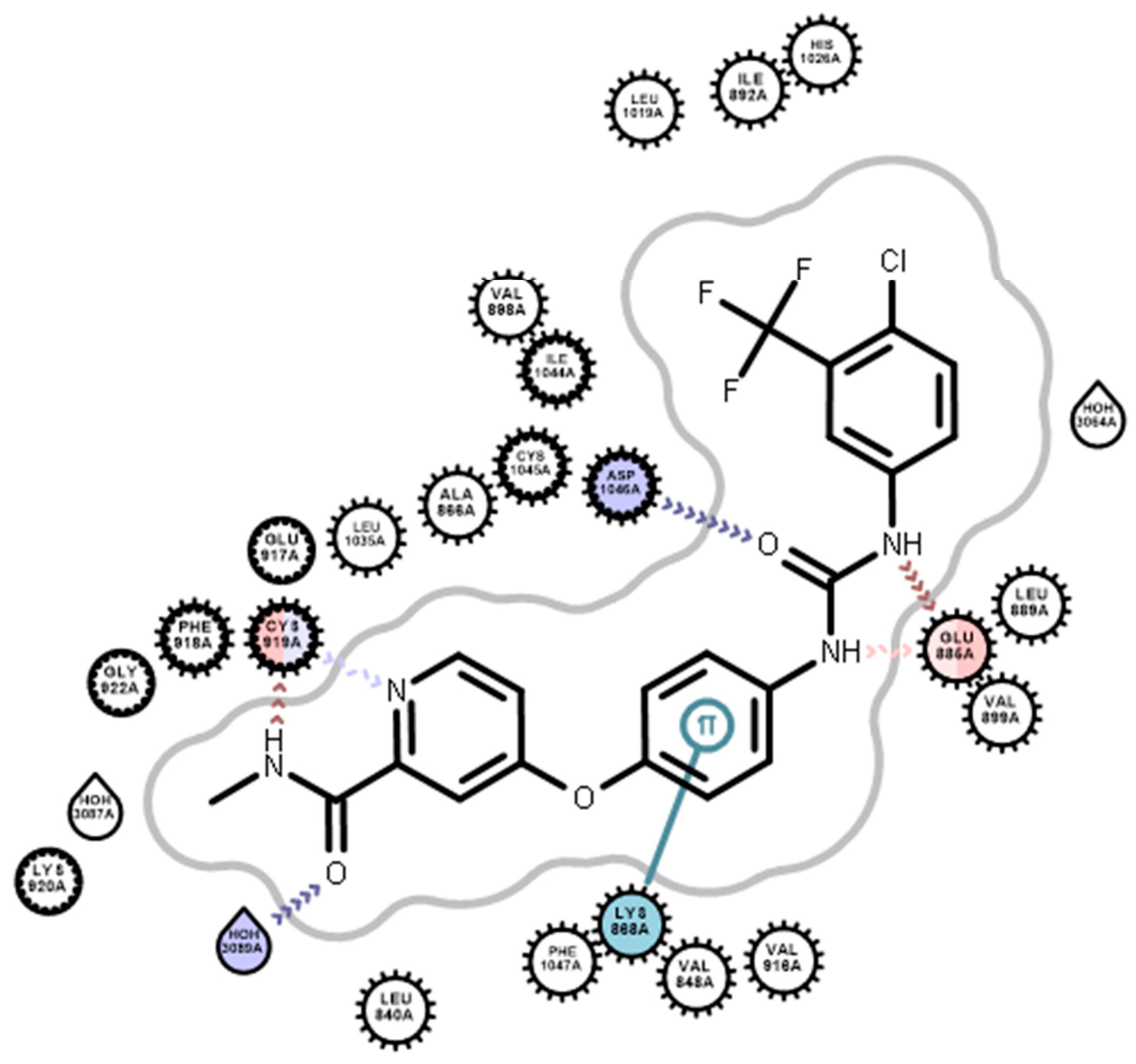
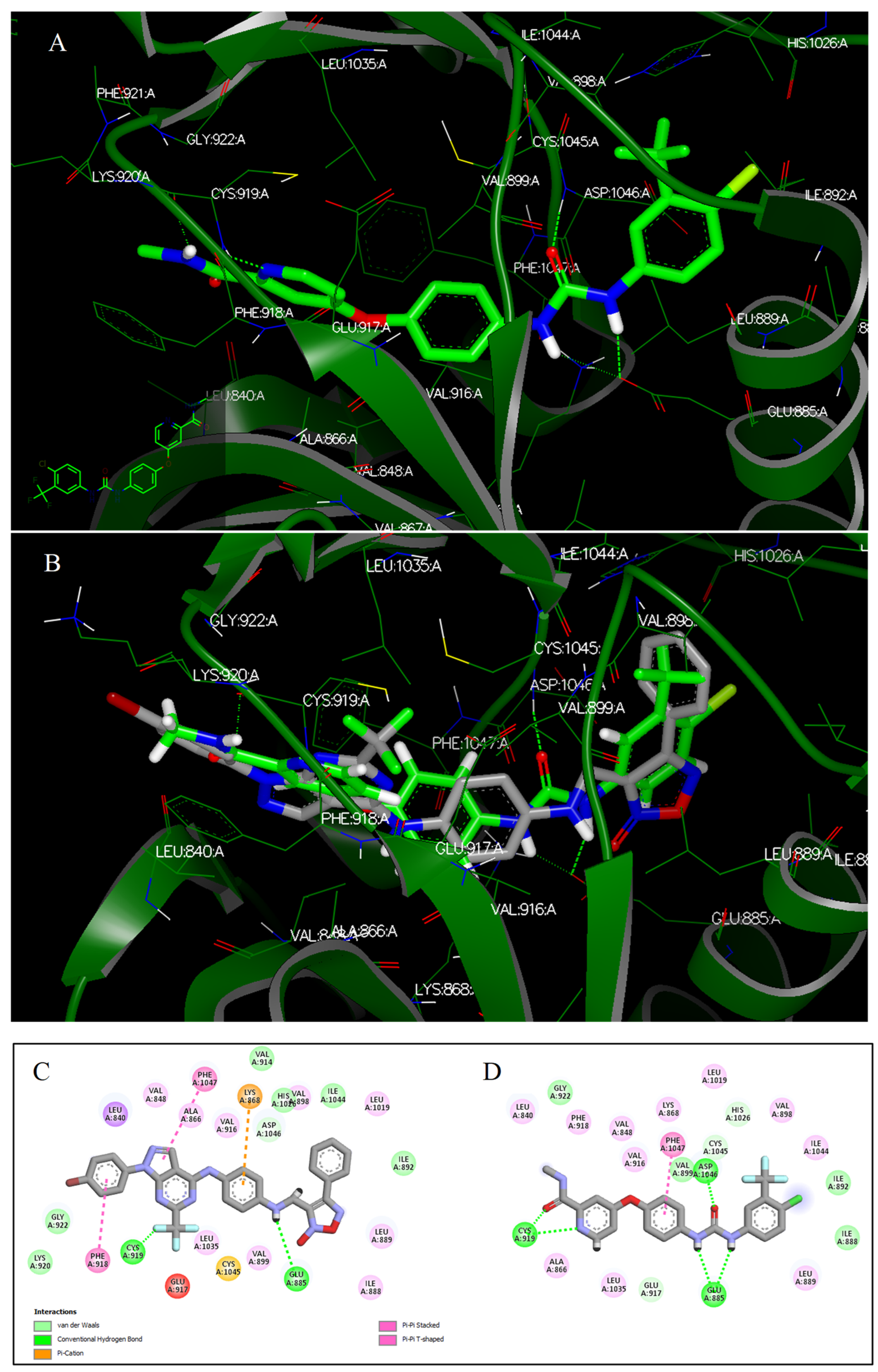
2.3. In Silico Molecular Modeling
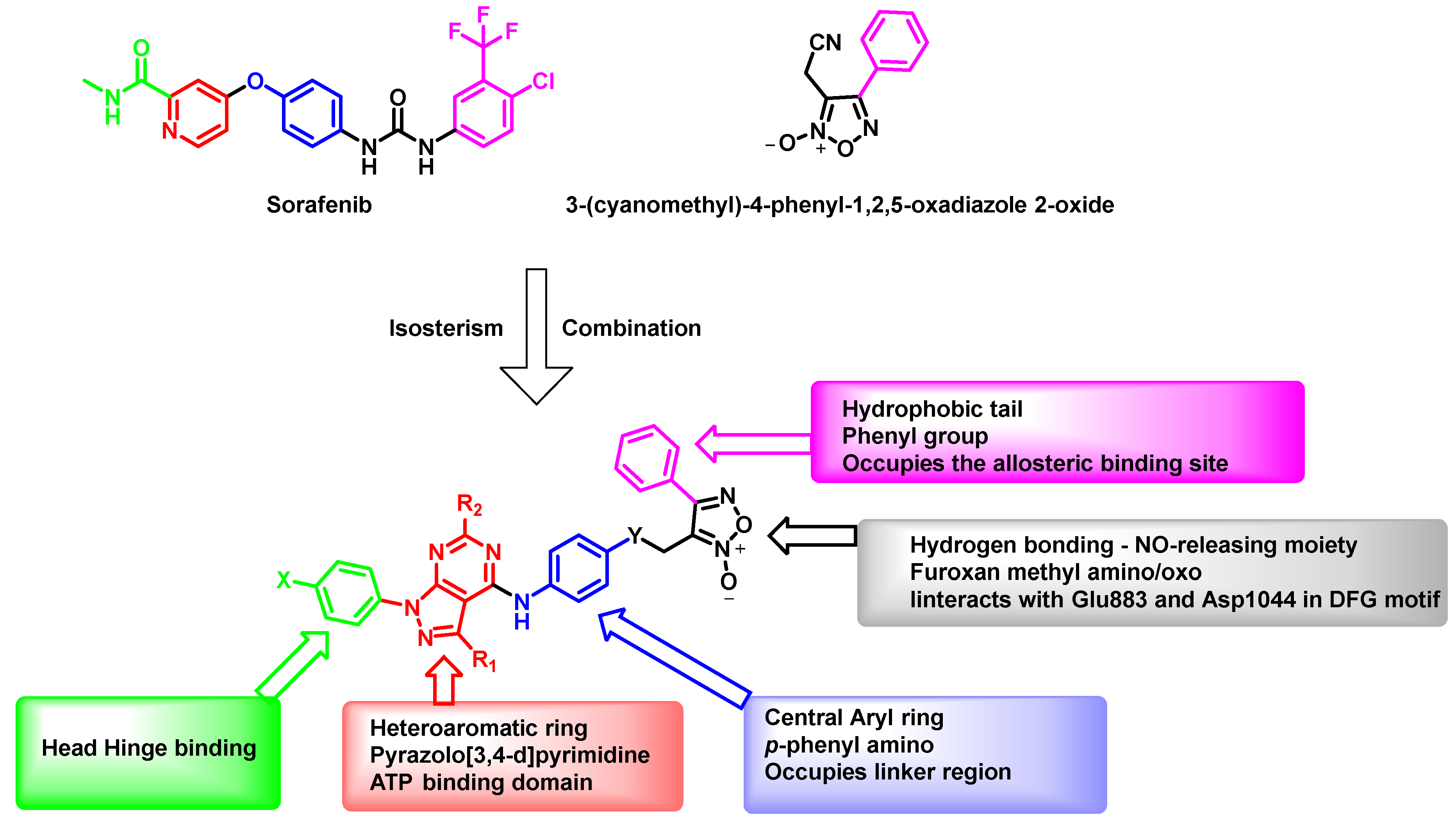
2.3.1. EON Scaffold Hopping
2.3.2. Docking Studies
2.4. ADME/Toxicity Analysis
3. Materials and Methods
3.1. Chemistry
3.1.1. General Procedure for Preparation of Allyloxy Derivatives (3a–d)
N-(4-(allyloxy)phenyl)-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (3a)
N-(4-(allyloxy)phenyl)-6-methyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (3b)
N-(4-(allyloxy)phenyl)-3-methyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (3c)
N-(4-(allyloxy)phenyl)-3,6-dimethyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (3d)
3.1.2. General Procedure for Preparation of Nitrated Allyloxy Derivatives (5a–d)
N-(4-(allyloxy)-3-nitrophenyl)-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (5a)
N-(4-(allyloxy)-3-nitrophenyl)-6-methyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (5b)
N-(4-(allyloxy)-3-nitrophenyl)-3-methyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (5c)
N-(4-(allyloxy)-3-nitrophenyl)-3,6-dimethyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-amine (5d)
3.1.3. General Procedure for Preparation of Target Compounds 6a–h
4-phenyl-3-((4-((1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)amino)phenoxy)methyl)-1,2,5-oxadiazole 2-oxide (6a)
3-((4-((6-methyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)amino)phenoxy)methyl)-4-phenyl-1,2,5-oxadiazole 2-oxide (6b)
3-((4-((3-methyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)amino)phenoxy)methyl)-4-phenyl-1,2,5-oxadiazole 2-oxide (6c)
3-((4-((3,6-dimethyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)amino)phenoxy)methyl)-4-phenyl-1,2,5-oxadiazole 2-oxide (6d)
3-((4-((1-(4-bromophenyl)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)amino)phenoxy)methyl)-4-phenyl-1,2,5-oxadiazole 2-oxide (6e)
3-((4-((1-(4-bromophenyl)-6-methyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)amino)phenoxy)methyl)-4-phenyl-1,2,5-oxadiazole 2-oxide (6f)
3-((4-((1-(4-bromophenyl)-3-methyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)amino)phenoxy)methyl)-4-phenyl-1,2,5-oxadiazole 2-oxide (6g)
3-((4-((1-(4-bromophenyl)-3,6-dimethyl-1H-pyrazolo[3,4-d]pyrimidin-4-yl)amino)phenoxy)methyl)-4-phenyl-1,2,5-oxadiazole 2-oxide (6h)
3.1.4. General Procedure for Preparation of Pyrazolopyrimidinone Derivatives 8b,c
1-(4-bromophenyl)-6-(trifluoromethyl)-1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-one (8b)
1-(4-bromophenyl)-3-methyl-6-(trifluoromethyl)-1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-one (8c)
3.1.5. General Procedure for Preparation of 4-Chloro Derivatives 9a–c
4-chloro-3-methyl-1-phenyl-6-(trifluoromethyl)-1H-pyrazolo[3,4-d]pyrimidine (9a)
1-(4-bromophenyl)-4-chloro-6-(trifluoromethyl)-1H-pyrazolo[3,4-d]pyrimidine (9b)
1-(4-bromophenyl)-4-chloro-3-methyl-6-(trifluoromethyl)-1H-pyrazolo[3,4-d]pyrimidine (9c)
3.1.6. General Procedure for Preparation of Phenylfuroxan Derivative 11
3-(((4-aminophenyl)amino)methyl)-4-phenyl-1,2,5-oxadiazole 2-oxide (11)
3.1.7. General Procedure for Preparation of Fluorinated Target Compounds 12a–c
3-(((4-((3-methyl-1-phenyl-6-(trifluoromethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)amino)phenyl)amino)methyl)-4-phenyl-1,2,5-oxadiazole 2-oxide (12a)
3-(((4-((1-(4-bromophenyl)-6-(trifluoromethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)amino)phenyl)amino)methyl)-4-phenyl-1,2,5-oxadiazole 2-oxide (12b)
3-(((4-((1-(4-bromophenyl)-3-methyl-6-(trifluoromethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)amino)phenyl)amino)methyl)-4-phenyl-1,2,5-oxadiazole 2-oxide (12c)
3.1.8. General Procedure for Preparation of C6 Appending Target Compounds 14a–c
3-(((4-(((4-oxo-1-phenyl-4,5-dihydro-1H-pyrazolo[3,4-d]pyrimidin-6-yl)methyl)amino)phenyl)amino)methyl)-4-phenyl-1,2,5-oxadiazole 2-oxide (14a)
3-(((4-(((3-methyl-4-oxo-1-phenyl-4,5-dihydro-1H-pyrazolo[3,4-d]pyrimidin-6-yl)methyl)amino)phenyl)amino)methyl)-4-phenyl-1,2,5-oxadiazole 2-oxide (14b)
3-(((4-(((1-(4-bromophenyl)-3-methyl-4-oxo-4,5-dihydro-1H-pyrazolo[3,4-d]pyrimidin-6-yl)methyl)amino)phenyl)amino)methyl)-4-phenyl-1,2,5-oxadiazole 2-oxide (14c)
3.1.9. General Procedure for Preparation of Des-NO-Releasing Compounds 15a,b
N1-(1-(4-bromophenyl)-6-(trifluoromethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)benzene-1,4-diamine (15a)
N1-(1-(4-bromophenyl)-3-methyl-6-(trifluoromethyl)-1H-pyrazolo[3,4-d]pyrimidin-4-yl)benzene-1,4-diamine (15b)
3.2. Biological Evaluation
3.2.1. NCI-60 Cell Line Screening
3.2.2. VEGFRx Kinase Assay
3.2.3. Cell Lines and Reagents
The Initial Screening and Cell Viability by MTT Assay
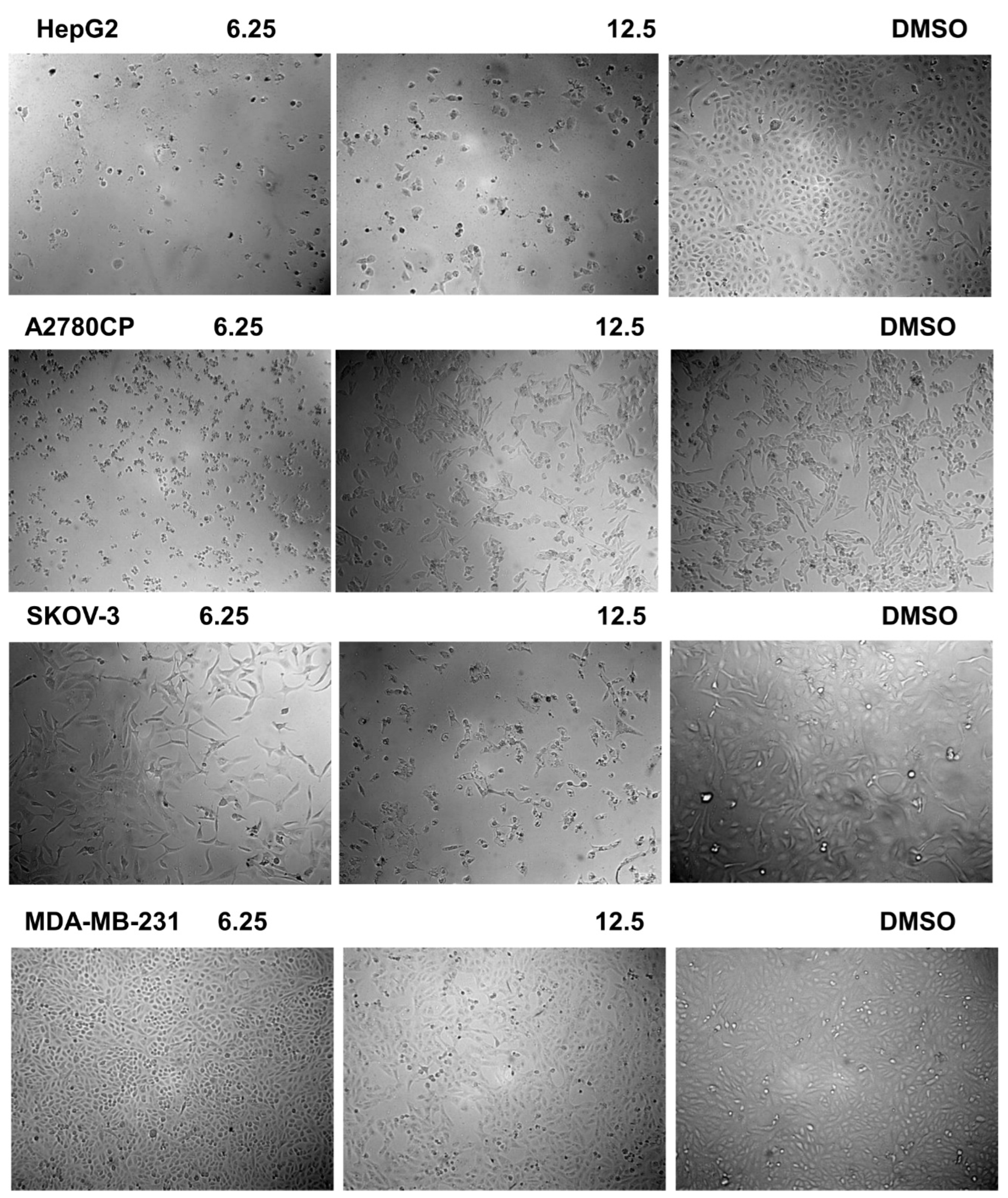
Selectivity of 12b towards Cancer Cells
Cell-Cycle Analysis
Apoptosis Analysis
Detection of Nitric Oxide Level by DAF-FM Diacetate
RT-PCR for Gene Expression
Western Blot Analysis
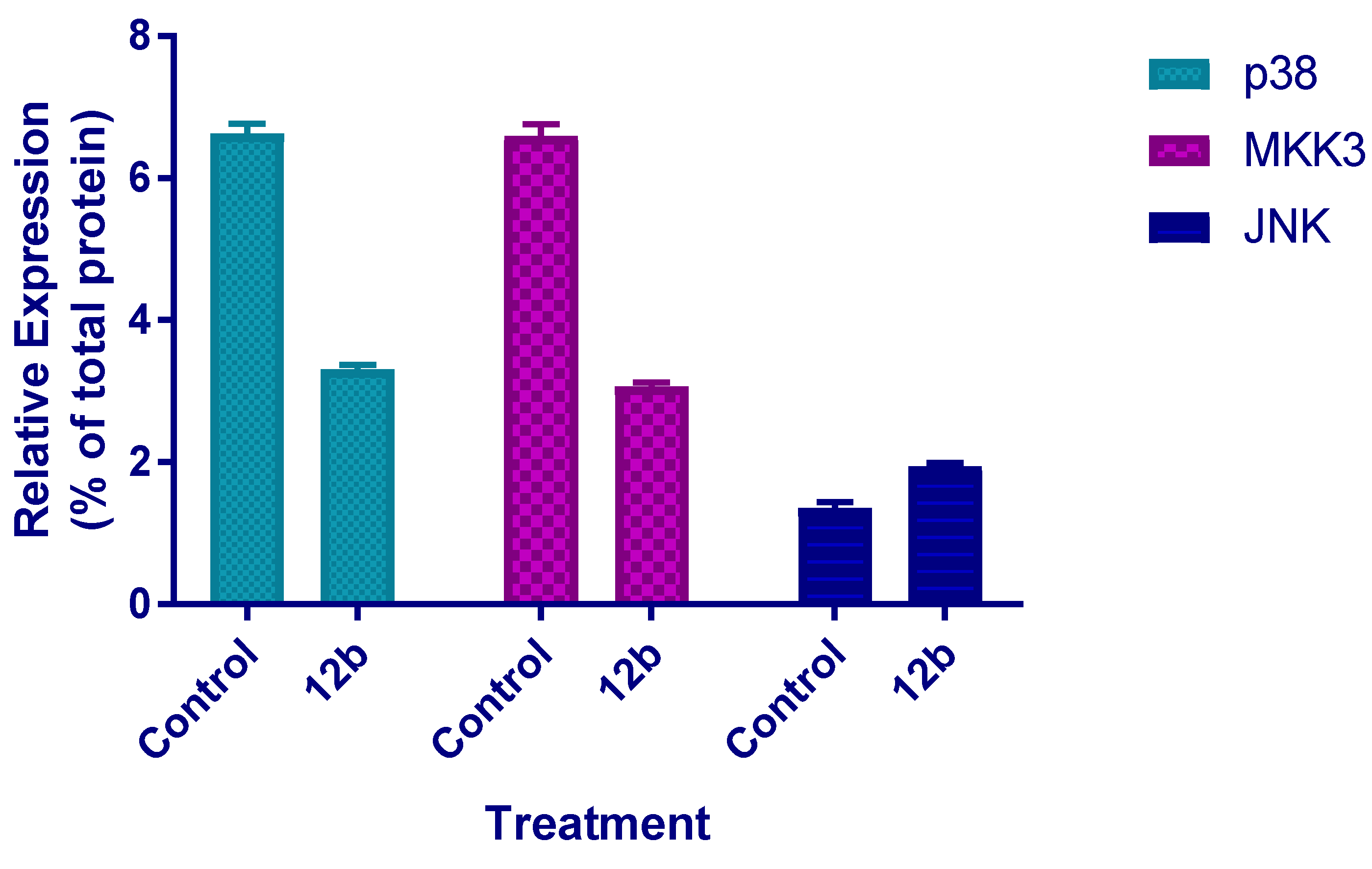
ELISA Assay
3.3. ADME/Toxicity Analysis
3.4. Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO Cancer. Available online: https://www.who.int/health-topics/cancer#tab=tab_1 (accessed on 12 November 2021).
- Fidler-Benaoudia, M.; Bray, F. Chapter 1.3 Transitions in human development and the global cancer burden. In World Cancer Report: Cancer Research for Cancer Prevention; Wild, C.P., Weiderpass, E., Stewart, B.W., Eds.; International Agency for Research on Cancer: Lyon, France, 2020; pp. 34–44. [Google Scholar]
- Goubran, H.A.; Kotb, R.R.; Stakiw, J.; Emara, M.E.; Burnouf, T. Regulation of tumor growth and metastasis: The role of tumor microenvironment. Cancer Growth Metastasis 2014, 7, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.-P.; Liu, K.-L.; Li, X.-Y.; Lu, G.-Q.; Xue, W.-H.; Qian, X.-H.; Mohamed, O.K.; Meng, F.-H. Design, synthesis, and in vitro and in vivo anti-angiogenesis study of a novel vascular endothelial growth factor receptor-2 (VEGFR-2) inhibitor based on 1,2,3-triazole scaffold. Eur. J. Med. Chem. 2021, 211, 113083. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.J.; Kulkarni, V.M. Vascular Endothelial Growth Factor Receptor (VEGFR-2)/KDR Inhibitors: Medicinal Chemistry Perspective. Med. Drug Discov. 2019, 2, 100009. [Google Scholar] [CrossRef]
- Mahnashi, M.H.; Alqahtani, Y.S.; Alyami, B.A.; Alqarni, A.O.; Ullah, F.; Wadood, A.; Sadiq, A.; Shareef, A.; Ayaz, M. Cytotoxicity, anti-angiogenic, anti-tumor and molecular docking studies on phytochemicals isolated from Polygonum hydropiper L. BMC Complementary Med. Ther. 2021, 21, 239. [Google Scholar] [CrossRef]
- NCI Angiogenesis Inhibitors. Available online: https://www.cancer.gov/about-cancer/treatment/types/immunotherapy/angiogenesis-inhibitors-fact-sheet (accessed on 12 November 2021).
- Padro, T.; Bieker, R.; Ruiz, S.; Steins, M.; Retzlaff, S.; Bürger, H.; Büchner, T.; Kessler, T.; Herrera, F.; Kienast, J. Overexpression of vascular endothelial growth factor (VEGF) and its cellular receptor KDR (VEGFR-2) in the bone marrow of patients with acute myeloid leukemia. Leukemia 2002, 16, 1302–1310. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Sullivan, C.A.W.; Zerkowski, M.P.; Molinaro, A.M.; Rimm, D.L.; Camp, R.L.; Chung, G.G. High levels of vascular endothelial growth factor and its receptors (VEGFR-1, VEGFR-2, neuropilin-1) are associated with worse outcome in breast cancer. Hum. Pathol. 2008, 39, 1835–1843. [Google Scholar] [CrossRef] [PubMed]
- Rydén, L.; Linderholm, B.; Nielsen, N.H.; Emdin, S.; Jönsson, P.-E.; Landberg, G. Tumor Specific VEGF-A and VEGFR2/KDR Protein are Co-expressed in Breast Cancer. Breast Cancer Res. Treat. 2003, 82, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Colbert, L.S.; McGlothen, T.Z.; Gonzalez-Perez, R.R. Regulation of angiogenesis in human cancer via vascular endothelial growth factor receptor-2 (VEGFR-2). Tumor Angiogenesis 2012, 27–66. [Google Scholar] [CrossRef]
- Jach, R.; Dulinska-Litewka, J.; Laidler, P.; Szczudrawa, A.; Kopera, A.; Szczudlik, L.; Pawlik, M.; Zajac, K.; Mak, M.; Basta, A. Expression of VEGF, VEGF-C and VEGFR-2 in in situ and invasive SCC of cervix. Front. Biosci. 2010, 2, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Hilbe, W.; Dirnhofer, S.; Oberwasserlechner, F.; Schmid, T.; Gunsilius, E.; Hilbe, G.; Wöll, E.; Kähler, C. CD133 positive endothelial progenitor cells contribute to the tumour vasculature in non-small cell lung cancer. J. Clin. Pathol. 2004, 57, 965–969. [Google Scholar] [CrossRef]
- Donnem, T.; Al-Shibli, K.; Andersen, S.; Al-Saad, S.; Busund, L.-T.; Bremnes, R.M. Combination of low vascular endothelial growth factor A (VEGF-A)/VEGF receptor 2 expression and high lymphocyte infiltration is a strong and independent favorable prognostic factor in patients with nonsmall cell lung cancer. Cancer 2010, 116, 4318–4325. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhang, X.; Tang, Q.; Zhang, F.; Li, Y.; Feng, Z.; Zhu, J. Prognostic significance and potential therapeutic target of VEGFR2 in hepatocellular carcinoma. J. Clin. Pathol. 2011, 64, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Ljungberg, B.J.; Jacobsen, J.; Rudolfsson, S.H.; Lindh, G.; Grankvist, K.; Rasmuson, T. Different vascular endothelial growth factor (VEGF), VEGF-receptor 1 and -2 mRNA expression profiles between clear cell and papillary renal cell carcinoma. BJU Int. 2006, 98, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Fontanella, C.; Ongaro, E.; Bolzonello, S.; Guardascione, M.; Fasola, G.; Aprile, G. Clinical advances in the development of novel VEGFR2 inhibitors. Ann. Transl. Med. 2014, 2, 123. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Mohsen, H.T.; Omar, M.A.; El Kerdawy, A.M.; Mahmoud, A.E.E.; Ali, M.M.; El Diwani, H.I. Novel potent substituted 4-amino-2-thiopyrimidines as dual VEGFR-2 and BRAF kinase inhibitors. Eur. J. Med. Chem. 2019, 179, 707–722. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.S.; Kopel, L.C.; Halaweish, F.T. Structural Optimization and Biological Screening of a Steroidal Scaffold Possessing Cucurbitacin-Like Functionalities as B-Raf Inhibitors. ChemMedChem 2014, 9, 1361–1367. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wan, S.; Li, Z.; Fu, Y.; Wang, G.; Zhang, J.; Wu, X. Design, synthesis, biological evaluation and molecular modeling of novel 1H-pyrazolo[3,4-d]pyrimidine derivatives as BRAFV600E and VEGFR-2 dual inhibitors. Eur. J. Med. Chem. 2018, 155, 210–228. [Google Scholar] [CrossRef] [PubMed]
- Abou-Salim, M.A.; Shaaban, M.A.; Abd El Hameid, M.K.; Elshaier, Y.A.M.M.; Halaweish, F. Design, synthesis and biological study of hybrid drug candidates of nitric oxide releasing cucurbitacin-inspired estrone analogs for treatment of hepatocellular carcinoma. Bioorganic Chem. 2019, 85, 515–533. [Google Scholar] [CrossRef]
- Boiani, M.; Cerecetto, H.; González, M.; Risso, M.; Olea-Azar, C.; Piro, O.E.; Castellano, E.E.; López de Ceráin, A.; Ezpeleta, O.; Monge-Vega, A. 1,2,5-Oxadiazole N-oxide derivatives as potential anti-cancer agents: Synthesis and biological evaluation. Part IV. Eur. J. Med. Chem. 2001, 36, 771–782. [Google Scholar] [CrossRef]
- Damaraju, V.L.; Kuzma, M.; Cass, C.E.; Putman, C.T.; Sawyer, M.B. Multitargeted kinase inhibitors imatinib, sorafenib and sunitinib perturb energy metabolism and cause cytotoxicity to cultured C2C12 skeletal muscle derived myotubes. Biochem. Pharmacol. 2018, 155, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, E.; Troiani, T.; Laus, G.; Pepe, S.; Ciardiello, F. 399 POSTER Synergistic antitumor activity of the combination of the multi-targeted tyrosine kinase inhibitor sorafenib and of EGFR inhibitors in human colon and lung cancer cell lines. Eur. J. Cancer Suppl. 2006, 4, 123. [Google Scholar] [CrossRef]
- Mahdy, H.A.; Ibrahim, M.K.; Metwaly, A.M.; Belal, A.; Mehany, A.B.M.; El-Gamal, K.M.A.; El-Sharkawy, A.; Elhendawy, M.A.; Radwan, M.M.; Elsohly, M.A.; et al. Design, synthesis, molecular modeling, in vivo studies and anticancer evaluation of quinazolin-4(3H)-one derivatives as potential VEGFR-2 inhibitors and apoptosis inducers. Bioorganic Chem. 2020, 94, 103422. [Google Scholar] [CrossRef] [PubMed]
- El-Adl, K.; Sakr, H.M.; Yousef, R.G.; Mehany, A.B.M.; Metwaly, A.M.; Elhendawy, M.A.; Radwan, M.M.; ElSohly, M.A.; Abulkhair, H.S.; Eissa, I.H. Discovery of new quinoxaline-2(1H)-one-based anticancer agents targeting VEGFR-2 as inhibitors: Design, synthesis, and anti-proliferative evaluation. Bioorganic Chem. 2021, 114, 105105. [Google Scholar] [CrossRef]
- Parmar, D.R.; Soni, J.Y.; Guduru, R.; Rayani, R.H.; Kusurkar, R.V.; Vala, A.G.; Talukdar, S.N.; Eissa, I.H.; Metwaly, A.M.; Khalil, A.; et al. Discovery of new anticancer thiourea-azetidine hybrids: Design, synthesis, in vitro antiproliferative, SAR, in silico molecular docking against VEGFR-2, ADMET, toxicity, and DFT studies. Bioorganic Chem. 2021, 115, 105206. [Google Scholar] [CrossRef] [PubMed]
- El-Metwally, S.A.; Abou-El-Regal, M.M.; Eissa, I.H.; Mehany, A.B.M.; Mahdy, H.A.; Elkady, H.; Elwan, A.; Elkaeed, E.B. Discovery of thieno[2,3-d]pyrimidine-based derivatives as potent VEGFR-2 kinase inhibitors and anti-cancer agents. Bioorganic Chem. 2021, 112, 104947. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yuan, X.-H.; Wang, S.-Q.; Zhao, W.; Chen, X.-B.; Yu, B. FDA-approved pyrimidine-fused bicyclic heterocycles for cancer therapy: Synthesis and clinical application. Eur. J. Med. Chem. 2021, 214, 113218. [Google Scholar] [CrossRef] [PubMed]
- Ayati, A.; Moghimi, S.; Toolabi, M.; Foroumadi, A. Pyrimidine-based EGFR TK inhibitors in targeted cancer therapy. Eur. J. Med. Chem. 2021, 221, 113523. [Google Scholar] [CrossRef]
- Alberti, M.J.; Auten, E.P.; Lackey, K.E.; McDonald, O.B.; Wood, E.R.; Preugschat, F.; Cutler, G.J.; Kane-Carson, L.; Liu, W.; Jung, D.K. Discovery and in vitro evaluation of potent kinase inhibitors: Pyrido[1′,2′:1,5]pyrazolo[3,4-d]pyrimidines. Bioorganic Med. Chem. Lett. 2005, 15, 3778–3781. [Google Scholar] [CrossRef]
- Abdelgawad, M.A.; Bakr, R.B.; Alkhoja, O.A.; Mohamed, W.R. Design, synthesis and antitumor activity of novel pyrazolo[3,4-d]pyrimidine derivatives as EGFR-TK inhibitors. Bioorganic Chem. 2016, 66, 88–96. [Google Scholar] [CrossRef]
- Bill Cai, T.; Wang, P.G.; Holder, A.A. NO and NO Donors. In Nitric Oxide Donors; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005; pp. 1–31. [Google Scholar]
- Girotti, A. Modulation of the Anti-Tumor Efficacy of Photodynamic Therapy by Nitric Oxide. Cancers 2016, 8, 96. [Google Scholar] [CrossRef]
- Ridnour, L.A.; Thomas, D.D.; Switzer, C.; Flores-Santana, W.; Isenberg, J.S.; Ambs, S.; Roberts, D.D.; Wink, D.A. Molecular mechanisms for discrete nitric oxide levels in cancer. Nitric Oxide 2008, 19, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Chakrapani, H. Site-directed delivery of nitric oxide to cancers. Nitric Oxide 2014, 43, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Fruttero, R.; Crosetti, M.; Chegaev, K.; Guglielmo, S.; Gasco, A.; Berardi, F.; Niso, M.; Perrone, R.; Panaro, M.A.; Colabufo, N.A. Phenylsulfonylfuroxans as Modulators of Multidrug-Resistance-Associated Protein-1 and P-Glycoprotein. J. Med. Chem. 2010, 53, 5467–5475. [Google Scholar] [CrossRef] [PubMed]
- De Ridder, M.; Verellen, D.; Verovski, V.; Storme, G. Hypoxic tumor cell radiosensitization through nitric oxide. Nitric Oxide 2008, 19, 164–169. [Google Scholar] [CrossRef]
- Bonavida, B.; Baritaki, S. Inhibition of Epithelial-to-Mesenchymal Transition (EMT) in Cancer by Nitric Oxide: Pivotal Roles of Nitrosylation of NF-κB, YY1 and Snail. Immunopathol. Dis. Ther. 2012, 3, 125–133. [Google Scholar] [CrossRef]
- Kashfi, K.; Duvalsaint, P.L. Chapter 4—Nitric Oxide Donors and Therapeutic Applications in Cancer A2—Seabra, Amedea Barozzi. In Nitric Oxide Donors, 1st ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 75–119. [Google Scholar]
- Turnbull, C.M.; Cena, C.; Fruttero, R.; Gasco, A.; Rossi, A.G.; Megson, I.L. Mechanism of action of novel NO-releasing furoxan derivatives of aspirin in human platelets. Br. J. Pharmacol. 2006, 148, 517–526. [Google Scholar] [CrossRef]
- Elshaier, Y.A.; Shaaban, M.A.; Abd El Hamid, M.K.; Abdelrahman, M.H.; Abou-Salim, M.A.; Elgazwi, S.M.; Halaweish, F. Design and synthesis of pyrazolo [3,4-d] pyrimidines: Nitric oxide releasing compounds targeting hepatocellular carcinoma. Bioorganic Med. Chem. 2017, 25, 2956–2970. [Google Scholar] [CrossRef]
- Pinto, T.A.; Hrdina, R.; Kirsch, G.; Campos, A.M.; Rodrigues, L.M.; Esteves, A.P. Synthesis of esters derived from 2,3,4-tri-O-benzyl-alpha-D-methylglucoside. Arkivoc 2012, 6, 185–193. [Google Scholar] [CrossRef]
- Pinto, T.A.D.; Silva, M.; Cunha, S.; Oliveira-Campos, A.M.F.; Rodrigues, L.M.; Hrdina, R.; Esteves, A.P. Synthesis of esters derived from 2,3 4,6-tetra-O-acetyl-1-[4-(2-hydroxyethyl)-1H-1,2,3-triazol-1-yl]-β-D-glucopyranose. Eur. J. Chem. 2013, 4, 64–69. [Google Scholar] [CrossRef][Green Version]
- Ahmed, M.M.; Khan, M.A.; Rainsford, K.D. Synthesis of thiophene and NO-curcuminoids for antiinflammatory and anti-cancer activities. Molecules 2013, 18, 1483–1501. [Google Scholar] [CrossRef]
- Hopf, H.; Mourad, A.F.; Jones, P.G. A surprising new route to 4-nitro-3-phenylisoxazole. Beilstein J. Org. Chem. 2010, 6, 68. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lin, Q.; Zhong, P. A Facile One-pot Synthesis of 1-Arylpyrazolo [3,4-d] pyrimidin-4-ones. Molecules 2010, 15, 3079–3086. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Robins, R.K. Potential Purine Antagonists. VI. Synthesis of 1-Alkyl-and 1-Aryl-4-substituted Pyrazolo [3,4-d] pyrimidines. J. Org. Chem. 1956, 21, 1240–1256. [Google Scholar] [CrossRef]
- El-Mekabaty, A. Synthesis and Antioxidant Activity of Some New Heterocycles Incorporating the Pyrazolo-[3,4-D]Pyrimidin-4-One Moiety. Chem. Heterocycl. Compd. 2015, 50, 1698–1706. [Google Scholar] [CrossRef]
- Shaaban, M.A.; Elshaier, Y.A.M.M.; Hammad, A.H.; Farag, N.A.; Hassan Haredy, H.; AbdEl-Ghany, A.A.; Mohamed, K.O. Design and synthesis of pyrazolo[3,4-d]pyrimidinone derivatives: Discovery of selective phosphodiesterase-5 inhibitors. Bioorganic Med. Chem. Lett. 2020, 30, 127337. [Google Scholar] [CrossRef]
- Elbadawi, M.M.; Eldehna, W.M.; Wang, W.; Agama, K.K.; Pommier, Y.; Abe, M. Discovery of 4-alkoxy-2-aryl-6,7-dimethoxyquinolines as a new class of topoisomerase I inhibitors endowed with potent in vitro anticancer activity. Eur. J. Med. Chem. 2021, 215, 113261. [Google Scholar] [CrossRef]
- Aucejo, F.; Kim, R.; Zein, N.; Quintini, C.; Uso, T.D.; Lopez, R.; Eghtesad, B.; Fung, J.; Miller, C.; Yerian, L. Vascular endothelial growth factor receptor 2 expression in non-tumorous cirrhotic liver is higher when hepatoma is beyond Milan criteria. Liver Transplant. Off. Publ. Am. Assoc. Study Liver Dis. Int. Liver Transplant. Soc. 2009, 15, 169–176. [Google Scholar] [CrossRef]
- Liu, L.; Qin, S.; Zheng, Y.; Han, L.; Zhang, M.; Luo, N.; Liu, Z.; Gu, N.; Gu, X.; Yin, X. Molecular targeting of VEGF/VEGFR signaling by the anti-VEGF monoclonal antibody BD0801 inhibits the growth and induces apoptosis of human hepatocellular carcinoma cells in vitro and in vivo. Cancer Biol. Ther. 2017, 18, 166–176. [Google Scholar] [CrossRef]
- Guo, S.; Colbert, L.S.; Fuller, M.; Zhang, Y.; Gonzalez-Perez, R.R. Vascular endothelial growth factor receptor-2 in breast cancer. Biochim. Biophys. Acta 2010, 1806, 108–121. [Google Scholar] [CrossRef]
- Adham, S.A.; Sher, I.; Coomber, B.L. Molecular blockade of VEGFR2 in human epithelial ovarian carcinoma cells. Lab. Investig. 2010, 90, 709–723. [Google Scholar] [CrossRef]
- He, Z.; Li, B.; Rankin, G.O.; Rojanasakul, Y.; Chen, Y.C. Selecting bioactive phenolic compounds as potential agents to inhibit proliferation and VEGF expression in human ovarian cancer cells. Oncol. Lett. 2015, 9, 1444–1450. [Google Scholar] [CrossRef] [PubMed]
- Kojima, H.; Sakurai, K.; Kikuchi, K.; Kawahara, S.; Kirino, Y.; Nagoshi, H.; Hirata, Y.; Nagano, T. Development of a fluorescent indicator for nitric oxide based on the fluorescein chromophore. Chem. Pharm. Bull. 1998, 46, 373–375. [Google Scholar] [CrossRef] [PubMed]
- Greco, A.; De Virgilio, A.; Rizzo, M.I.; Pandolfi, F.; Rosati, D.; De Vincentiis, M. The prognostic role of E-cadherin and β-catenin overexpression in laryngeal squamous cell carcinoma. Laryngoscope 2016, 126, E148–E155. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed]
- ABCAM JNK 1/2. Available online: https://www.abcam.com/jnk-12-pt183y185--total-elisa-kit-ab176662.html (accessed on 1 February 2022).
- ABCAM p38. Available online: https://www.abcam.com/p38-mapk-alpha-elisa-kit-ab221012.html (accessed on 1 February 2022).
- RayBiotech MKK3. Available online: https://www.biocompare.com/25138-Assay-Kit/9505514-Human-Mouse-Rat-Phospho-MKK3-S189-and-Total-MKK3-ELISA/?pda=9956%7C9505514_0_1%7C%7C3%7CMKK3&dfp=true (accessed on 2 February 2022).
- OpenEye Lead Optimization EON. Available online: https://www.eyesopen.com/eon (accessed on 10 January 2021).
- OpenEye EON, Version 2.3.5; OpenEye Scientific Software: Santa Fe, NM, USA. Available online: http://www.eyesopen.com (accessed on 20 July 2021).
- OpenEye EON. Available online: https://docs.eyesopen.com/applications/eon/index.html (accessed on 10 January 2021).
- OpenEye VIDA, Version 5.0.1.0; OpenEye Scientific Software: Santa Fe, NM, USA. Available online: https://docs.eyesopen.com/applications/vida/index.html (accessed on 8 December 2021).
- Ramu-Phone, R. Comparative molecular docking and simulation analysis of molnupiravir and remdesivir with SARS-CoV-2 RNA dependent RNA polymerase (RdRp). Bioinformation 2021, 17, 932–939. [Google Scholar] [CrossRef]
- BIOVIA Free Download: BIOVIA Discovery Studio Visualizer. Available online: https://discover.3ds.com/discovery-studio-visualizer-download (accessed on 2 December 2021).
- Group, C.B.D.D. ADMETLAB2.0. Available online: https://admetmesh.scbdd.com/ (accessed on 6 November 2021).
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, w5–w14. [Google Scholar] [CrossRef]
- Ishii, H.K.M.; Koseki, J.; Asai, A.; Meguro, K.; Akai, S.; Kawano, Y.; Nunomura, K.; Morie, T. Novel Drug Targeted On Epigenetics Containing pyrazolo[3,4-d]pyrimidine Derivative, and Use Thereof. Japan Patent No. WO 2020218470 A1, Application No. PCT/JP2020/017607; Osaka University, Osaka, Japan, 29 October 2020. pp. 1–84. [Google Scholar]
- Lamie, P.F. RETRACTED: Design, synthesis, structure–activity relationship and kinase inhibitory activity of substituted 3-methyl-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4-ones. Bioorganic Med. Chem. Lett. 2016, 26, 3093–3097. [Google Scholar] [CrossRef]
- NCI NCI-60 Screening Methodology. Available online: https://dtp.cancer.gov/discovery_development/nci-60/methodology.htm (accessed on 19 November 2021).
- Sharma, K.; Suresh, P.; Mullangi, R.; Srinivas, N. Quantitation of VEGFR2 (vascular endothelial growth factor receptor) inhibitors–review of assay methodologies and perspectives. Biomed. Chromatogr. 2015, 29, 803–834. [Google Scholar] [CrossRef]
- Bioscience VEGFR2 (KDR) Kinase Assay Kit. Available online: https://bpsbioscience.com/vegfr2-kdr-kinase-assay-kit-40325 (accessed on 5 December 2021).
- Bioscience FLT1 Kinase Assay Kit. Available online: https://bpsbioscience.com/flt1-kinase-assay-kit-78019 (accessed on 5 December 2021).
- Bioscience FLT3 Kinase Assay Kit. Available online: https://bpsbioscience.com/flt3-kinase-assay-kit-79797 (accessed on 5 December 2021).
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New Colorimetric Cytotoxicity Assay for Anticancer-Drug Screening. JNCI J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef]
- Plumb, J.A. Cell sensitivity assays: The MTT assay. In Cancer Cell Culture; Springer: Berlin/Heidelberg, Germany, 2004; pp. 165–169. [Google Scholar]
- Bézivin, C.; Tomasi, S.; Lohézic-Le Dévéhat, F.; Boustie, J.J.P. Cytotoxic activity of some lichen extracts on murine and human cancer cell lines. Phytomedicine 2003, 10, 499–503. [Google Scholar] [CrossRef]
- Prayong, P.; Barusrux, S.; Weerapreeyakul, N. Cytotoxic activity screening of some indigenous Thai plants. Fitoterapia 2008, 79, 598–601. [Google Scholar] [CrossRef] [PubMed]
- El-Senduny, F.F.; Zidane, M.M.; Youssef, M.M.; Badria, F. An Approach to Treatment of Liver Cancer by Novel Glycyrrhizin Derivative. Anti-Cancer Agents Med. Chem. 2019, 19, 1863–1873. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.W.; Dolbeare, F.; Pallavicini, M.G.; Beisker, W.; Waldman, F. Cell Cycle Analysis Using Flow Cytometry. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1986, 49, 237–255. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, M.; Miller, B.; Newmark, J.; Al-Kofahi, Y.; Holden, E. Chapter 7—Laser Scanning Cytometry and Its Applications: A Pioneering Technology in the Field of Quantitative Imaging Cytometry. In Methods in Cell Biology; Darzynkiewicz, Z., Holden, E., Orfao, A., Telford, W., Wlodkowic, D., Eds.; Academic Press: Cambridge, MA, USA, 2011; Volume 102, pp. 159–205. [Google Scholar]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Liu, M.-M.; Chen, X.-Y.; Huang, Y.-Q.; Feng, P.; Guo, Y.-L.; Yang, G.; Chen, Y. Hybrids of Phenylsulfonylfuroxan and Coumarin as Potent Antitumor Agents. J. Med. Chem. 2014, 57, 9343–9356. [Google Scholar] [CrossRef]
- Li, X.; Wang, X.; Xu, C.; Huang, J.; Wang, C.; Wang, X.; He, L.; Ling, Y. Synthesis and biological evaluation of nitric oxide-releasing hybrids from gemcitabine and phenylsulfonyl furoxans as anti-tumor agents. MedChemComm 2015, 6, 1130–1136. [Google Scholar] [CrossRef]
- Maciag, A.E.; Holland, R.J.; Robert Cheng, Y.S.; Rodriguez, L.G.; Saavedra, J.E.; Anderson, L.M.; Keefer, L.K. Nitric oxide-releasing prodrug triggers cancer cell death through deregulation of cellular redox balance. Redox Biol. 2013, 1, 115–124. [Google Scholar] [CrossRef]
- Abdel-Wahab, B.A.; Ali, F.E.M.; Alkahtani, S.A.; Alshabi, A.M.; Mahnashi, M.H.; Hassanein, E.H.M. Hepatoprotective effect of rebamipide against methotrexate-induced hepatic intoxication: Role of Nrf2/GSK-3β, NF-κβ-p65/JAK1/STAT3, and PUMA/Bax/BCL-2 signaling pathways. Immunopharmacol. Immunotoxicol. 2020, 42, 493–503. [Google Scholar] [CrossRef]
- Noolvi, M.N.; Patel, H.M. A comparative QSAR analysis and molecular docking studies of quinazoline derivatives as tyrosine kinase (EGFR) inhibitors: A rational approach to anticancer drug design. J. Saudi Chem. Soc. 2013, 17, 361–379. [Google Scholar] [CrossRef]
- Yang, Y.-A.; Tang, W.-J.; Zhang, X.; Yuan, J.-W.; Liu, X.-H.; Zhu, H.-L. Synthesis, molecular docking and biological evaluation of glycyrrhizin analogs as anticancer agents targeting EGFR. Molecules 2014, 19, 6368–6381. [Google Scholar] [CrossRef]
- Rajendra Prasad, V.V.S.; Deepak Reddy, G.; Kathmann, I.; Amareswararao, M.; Peters, G.J. Nitric oxide releasing acridone carboxamide derivatives as reverters of doxorubicin resistance in MCF7/Dx cancer cells. Bioorganic Chem. 2016, 64, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Mahnashi, M.; Elgazwi, S.M.; Ahmed, M.S.; Halaweish, F.T. Cucurbitacins inspired organic synthesis: Potential dual inhibitors targeting EGFR—MAPK pathway. Eur. J. Med. Chem. 2019, 173, 294–304. [Google Scholar] [CrossRef] [PubMed]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
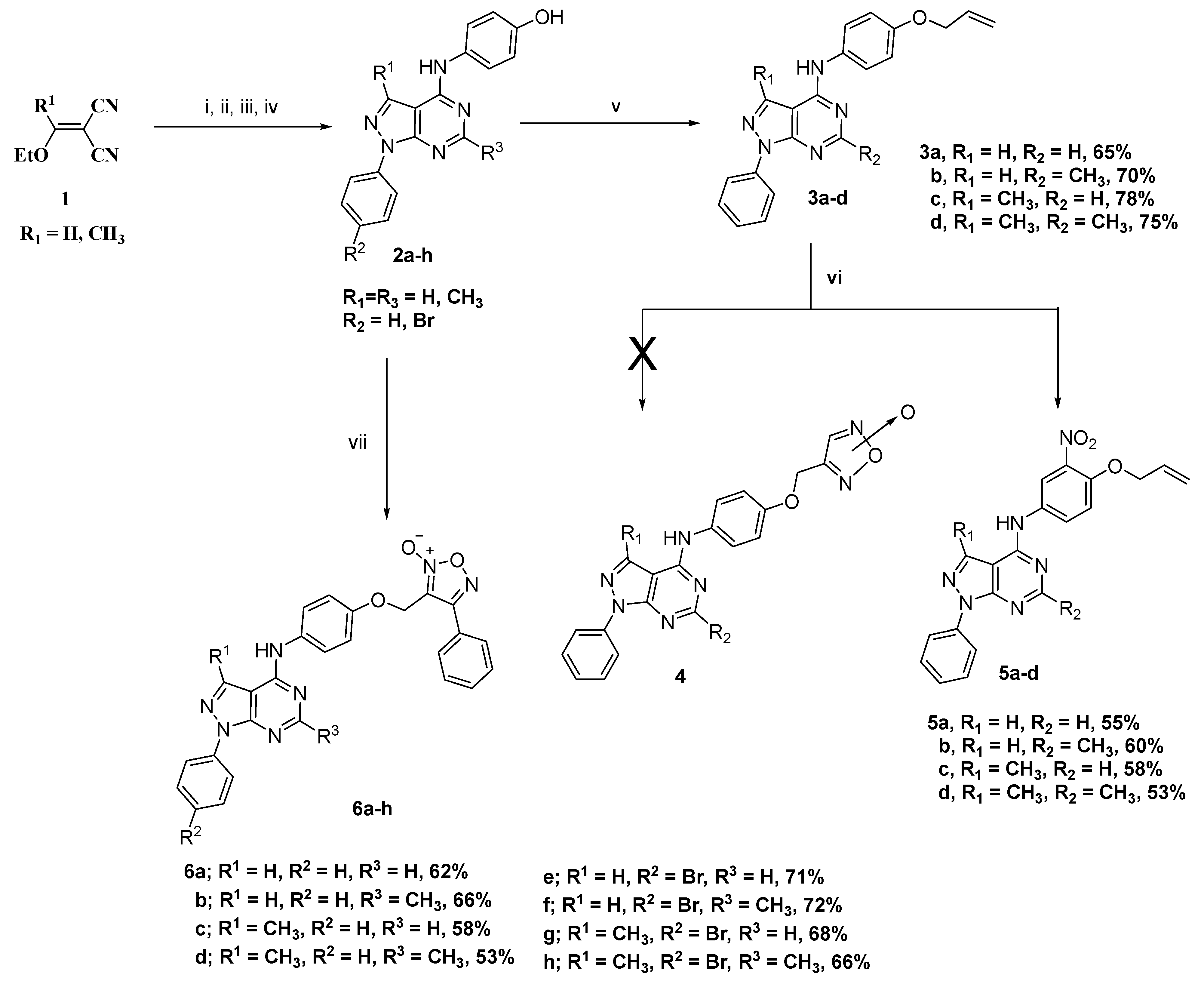{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer | Cell Line | IC50 (µM) | |
|---|---|---|---|
| 12b | Sorafenib | ||
| Liver | HepG2 | 11.5 ± 0.5 | 14 ± 0.4 |
| Ovarian | SKOV-3 | 18.6 ± 0.2 | 16.9 ± 0.2 |
| A2780 | 15 ± 0.15 | 10.5 ± 0.5 | |
| A2780CP | 11.6 ± 0.1 | 15.2 ± 0.1 | |
| Breast | MDA-MB-231 | 13 ± 0.2 | 16.6 ± 0.3 |
| MCF-7 | 22.3 ± 0.3 | 5.1 ± 0.4 | |
| Colon | HT-29 | 15 ± 0.24 | 11 ± 0.2 |
| CPD Name | EON ET coul a | EON ET pb b | EON Shape Tanimoto c | EON ET combo d | EON Rank |
|---|---|---|---|---|---|
| Sorafenib | 1 | 1 | 1 | 2 | 1 |
| 14a | 0.153 | 0.126 | 0.106 | 0.232 | 2 |
| 14b | 0.155 | 0.127 | 0.102 | 0.229 | 3 |
| 14c | 0.153 | 0.126 | 0.103 | 0.228 | 4 |
| 6d | 0.067 | 0.057 | 0.086 | 0.143 | 5 |
| 12a | 0.023 | 0.032 | 0.107 | 0.139 | 6 |
| 6h | 0.056 | 0.049 | 0.089 | 0.138 | 7 |
| 12b | 0.012 | 0.019 | 0.117 | 0.136 | 8 |
| 6b | 0.055 | 0.048 | 0.088 | 0.135 | 9 |
| 12c | 0.016 | 0.022 | 0.112 | 0.135 | 10 |
| 6f | 0.045 | 0.039 | 0.091 | 0.13 | 11 |
| 6a | 0.02 | 0.032 | 0.078 | 0.11 | 12 |
| 6c | 0.072 | 0.069 | 0.04 | 0.109 | 13 |
| 6g | 0.065 | 0.06 | 0.044 | 0.104 | 14 |
| 6e | 0.013 | 0.027 | 0.076 | 0.104 | 15 |
| Compound | FRED Chemgauss4 Score |
|---|---|
| 6e | −12.8024 |
| 6a | −11.6995 |
| 6b | −11.5059 |
| 6g | −11.4424 |
| 12b | −11.3747 |
| 6c | −11.3160 |
| 6h | −11.1254 |
| 6f | −11.0437 |
| 14c | −10.9153 |
| 12a | −10.7872 |
| 6d | −10.3713 |
| 14a | −10.1469 |
| 14b | −9.9950 |
| 12c | −9.8026 |
| Sorafenib | −18.5752 |
| Gene | Primer Sequence 5′-3′ |
|---|---|
| Bax | GTT TCA TCC AGG ATC GAG CAG |
| CAT CTT CTT CCA GAT GGT GA | |
| BCL-2 | CAG AGA CAG CCA GGA GAA ATC A |
| TCG CCC TGT GGA TGA CTG A | |
| E-cadherin | GAG TGC CAA CTG GAC CA T TCA GTA |
| AGT CAC CCA CCT CTA AGG CCA TC | |
| N-cadherin | GAG ATC CTA CTG GAC GGT TCG |
| TCT TGG CGA ATG ATC TTA GGA | |
| GAPDH | ACC ACA GTC CAT GCC ATC AC |
| TCC ACC ACC CTG TTG CTG TA |
| Antibody | Dilution | Cat. No. |
|---|---|---|
| β-actin | 1:5000 | 4970 |
| P27 | 1:4000 | 2552 |
| P21 | 1:2000 | 2947 |
| pERK | 1:2000 | 4370 |
| ERK | 1:2000 | 4695 |
| MMP9 | 1:2000 | 13667 |
| P53 | 1:5000 | 9282 |
| Anti-Rabbit IgG-HRP- linked | 1:5000 | 7074 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahnashi, M.H.; El-Senduny, F.F.; Alshahrani, M.A.; Abou-Salim, M.A. Design, Synthesis, and Biological Evaluation of a Novel VEGFR-2 Inhibitor Based on a 1,2,5-Oxadiazole-2-Oxide Scaffold with MAPK Signaling Pathway Inhibition. Pharmaceuticals 2022, 15, 246. https://doi.org/10.3390/ph15020246
Mahnashi MH, El-Senduny FF, Alshahrani MA, Abou-Salim MA. Design, Synthesis, and Biological Evaluation of a Novel VEGFR-2 Inhibitor Based on a 1,2,5-Oxadiazole-2-Oxide Scaffold with MAPK Signaling Pathway Inhibition. Pharmaceuticals. 2022; 15(2):246. https://doi.org/10.3390/ph15020246
Chicago/Turabian StyleMahnashi, Mater H., Fardous F. El-Senduny, Mohammed Abdulrahman Alshahrani, and Mahrous A. Abou-Salim. 2022. "Design, Synthesis, and Biological Evaluation of a Novel VEGFR-2 Inhibitor Based on a 1,2,5-Oxadiazole-2-Oxide Scaffold with MAPK Signaling Pathway Inhibition" Pharmaceuticals 15, no. 2: 246. https://doi.org/10.3390/ph15020246
APA StyleMahnashi, M. H., El-Senduny, F. F., Alshahrani, M. A., & Abou-Salim, M. A. (2022). Design, Synthesis, and Biological Evaluation of a Novel VEGFR-2 Inhibitor Based on a 1,2,5-Oxadiazole-2-Oxide Scaffold with MAPK Signaling Pathway Inhibition. Pharmaceuticals, 15(2), 246. https://doi.org/10.3390/ph15020246