
Imidazole-Based pH-Sensitive Convertible Liposomes for Anticancer Drug Delivery


Abstract
:1. Introduction
2. Results
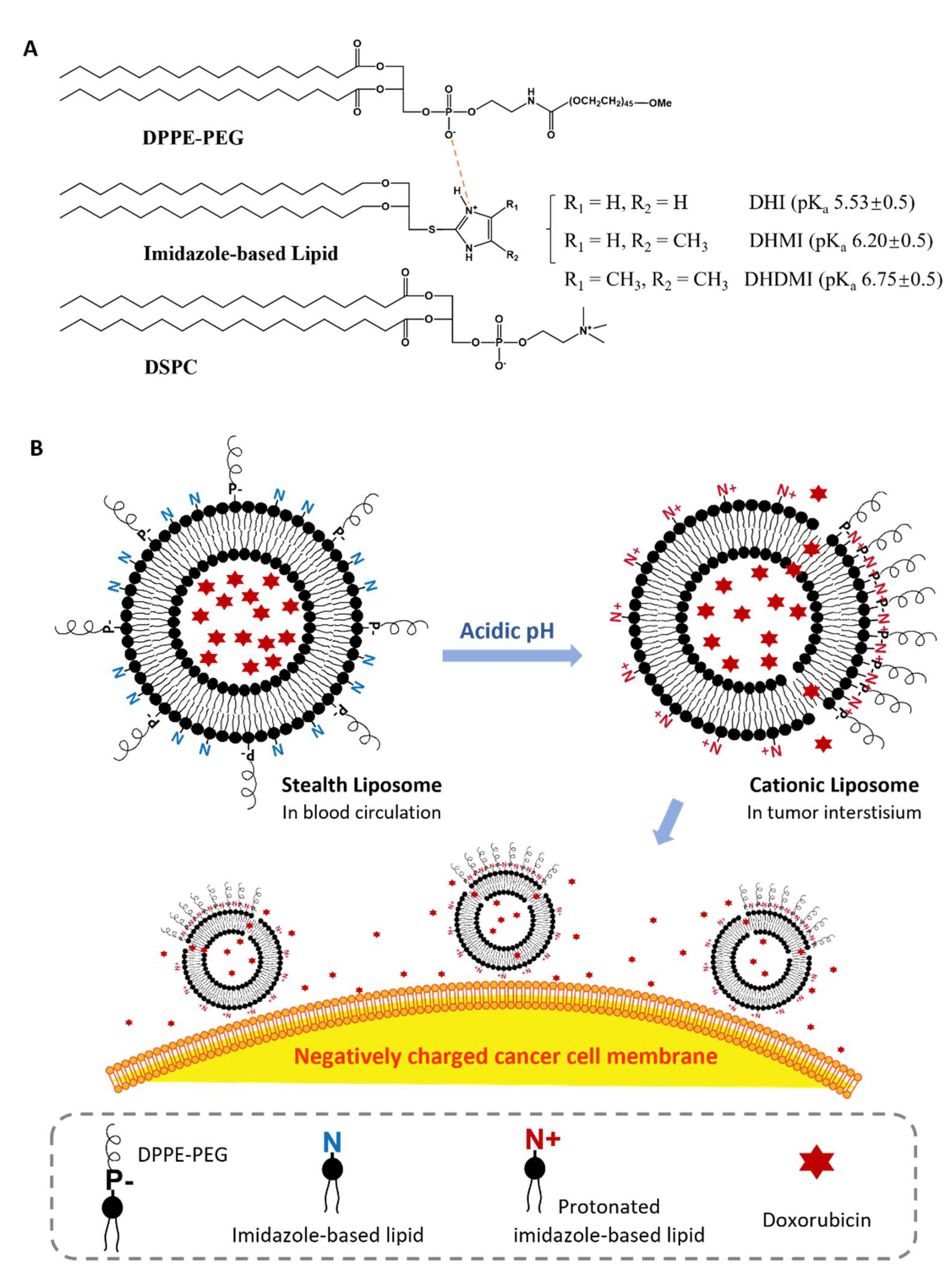
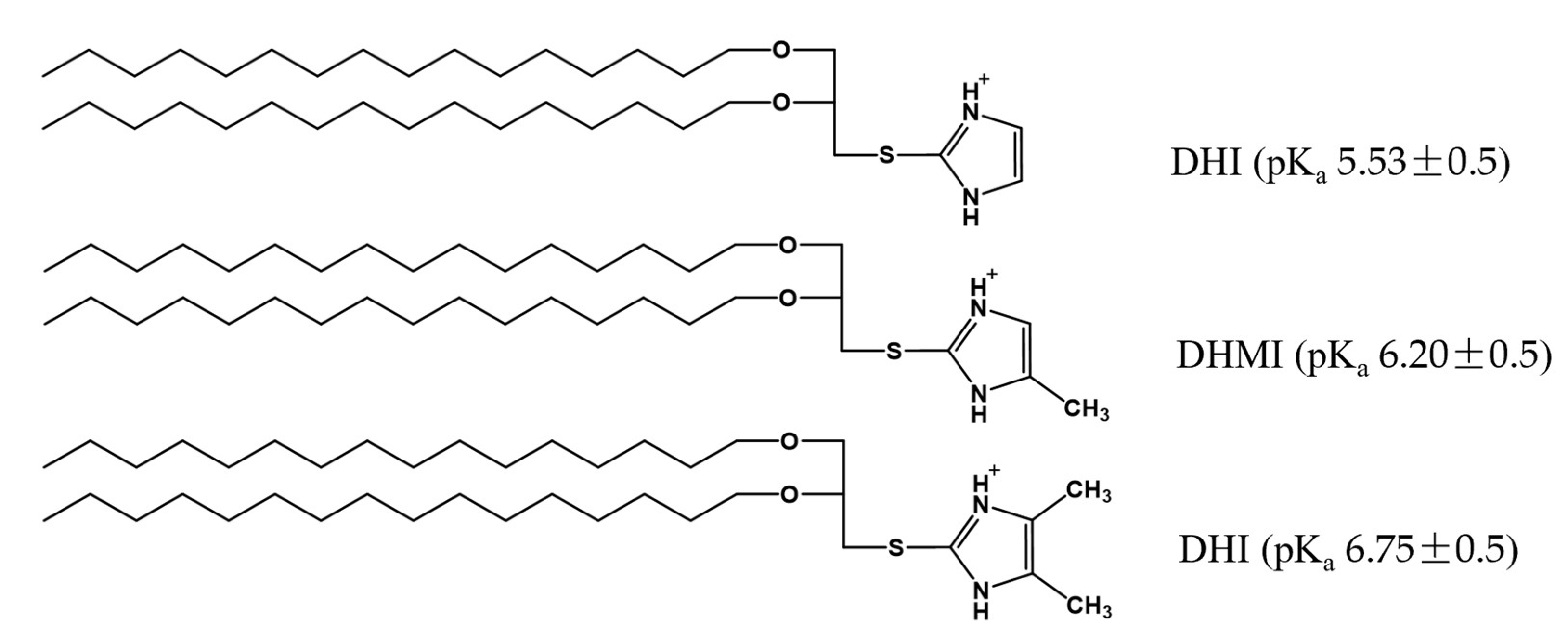
2.1. Imidazole-Based pH-Sensitive Lipids
2.2. Composition of Imidazole-Based Convertible Liposomes (ICL)
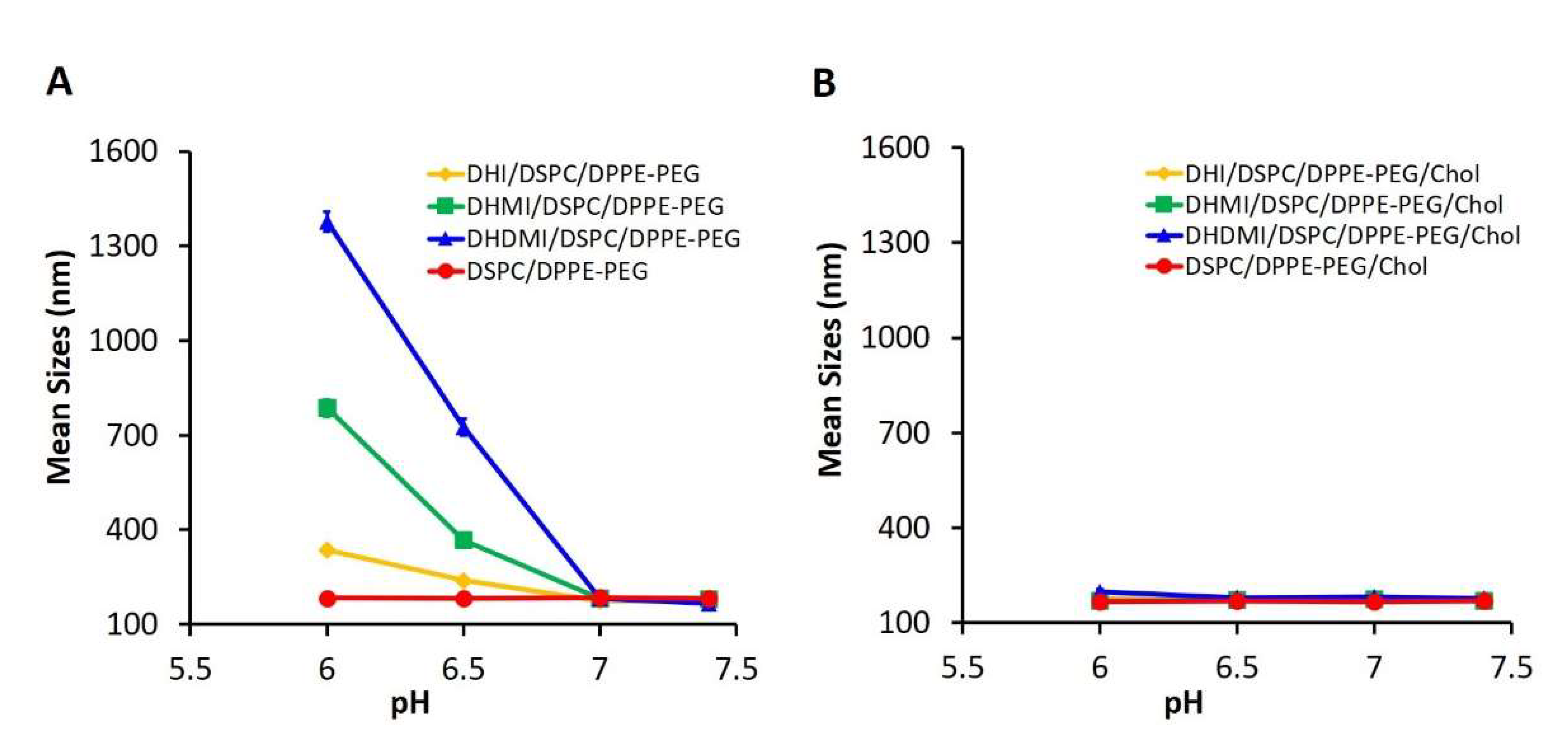
2.3. Physicochemical Characteristics of ICL
2.4. pH-Triggered Acquisition of Positive Charges on ICL Surface
2.5. pH-Triggered Interaction between ICL and Model Liposome
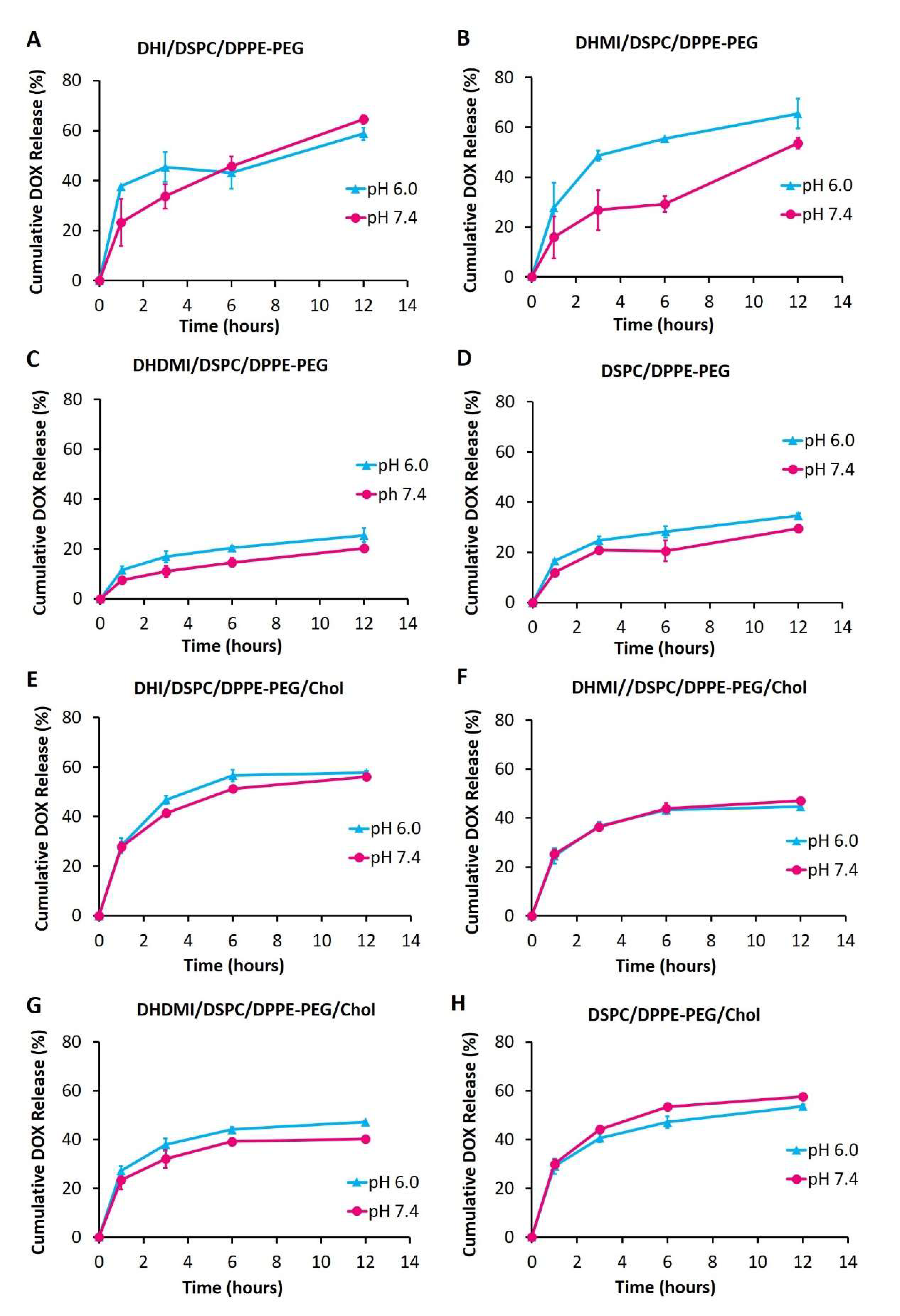
2.6. pH-Triggered Drug Release from ICL
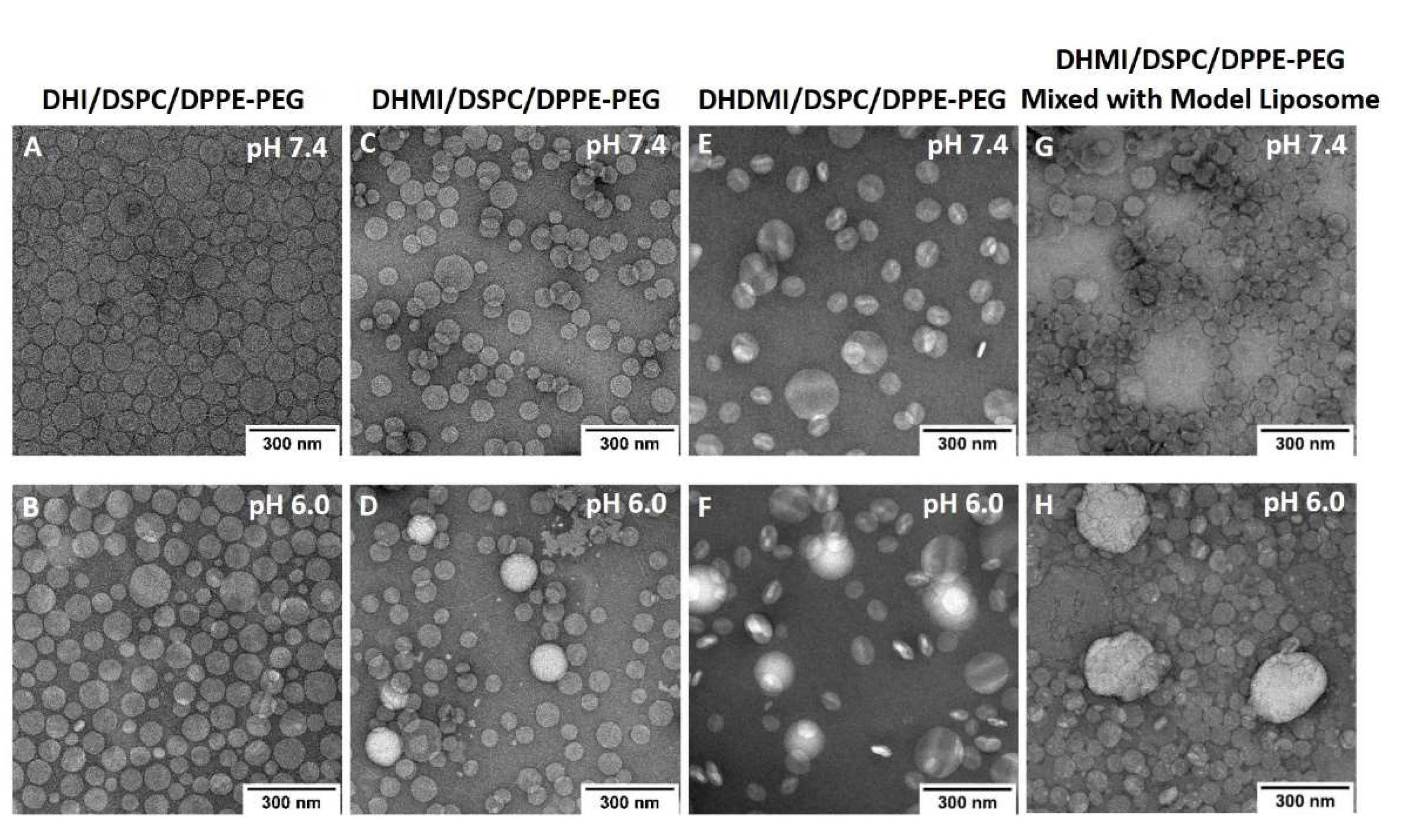
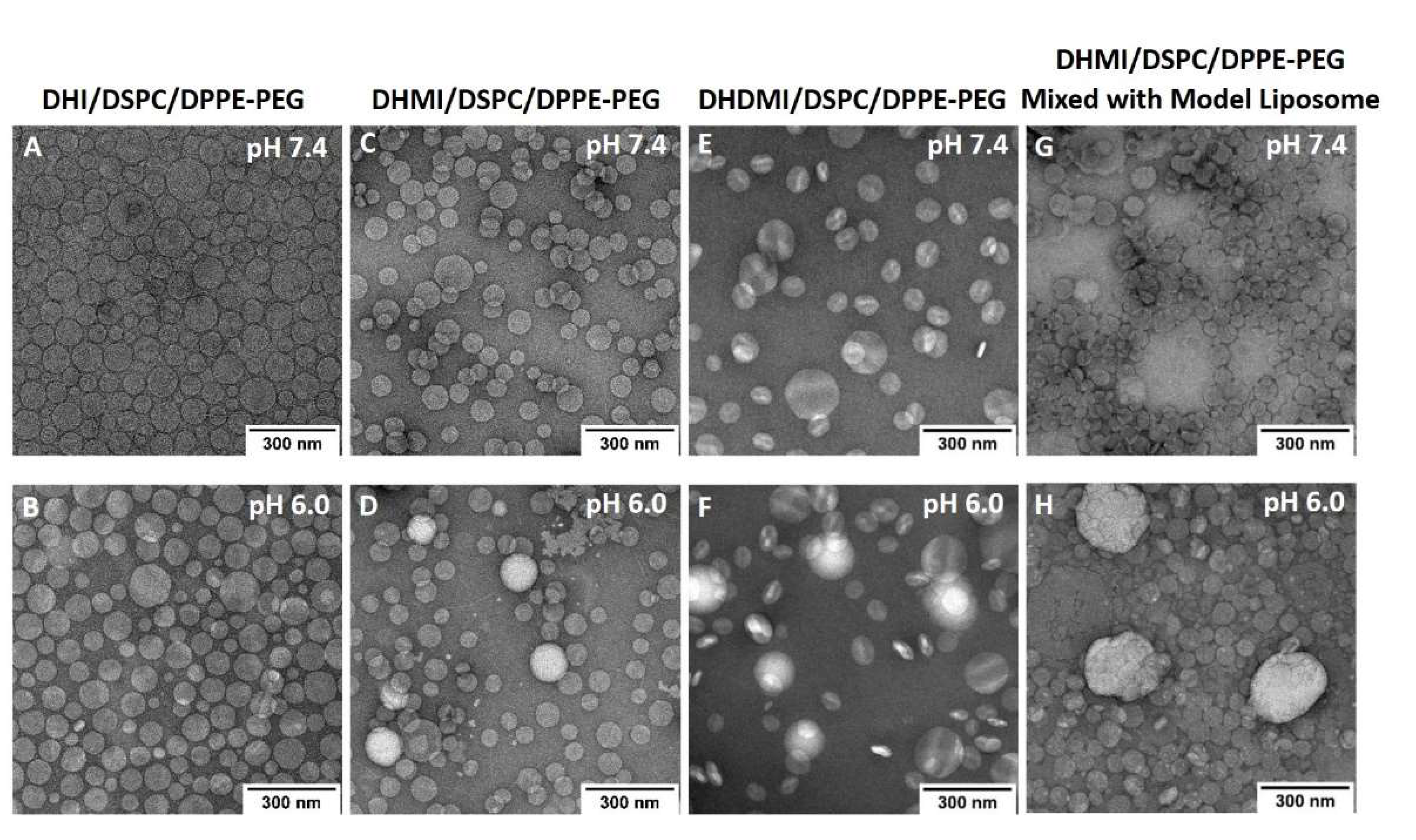
2.7. Morphological Studies on ICL under TEM
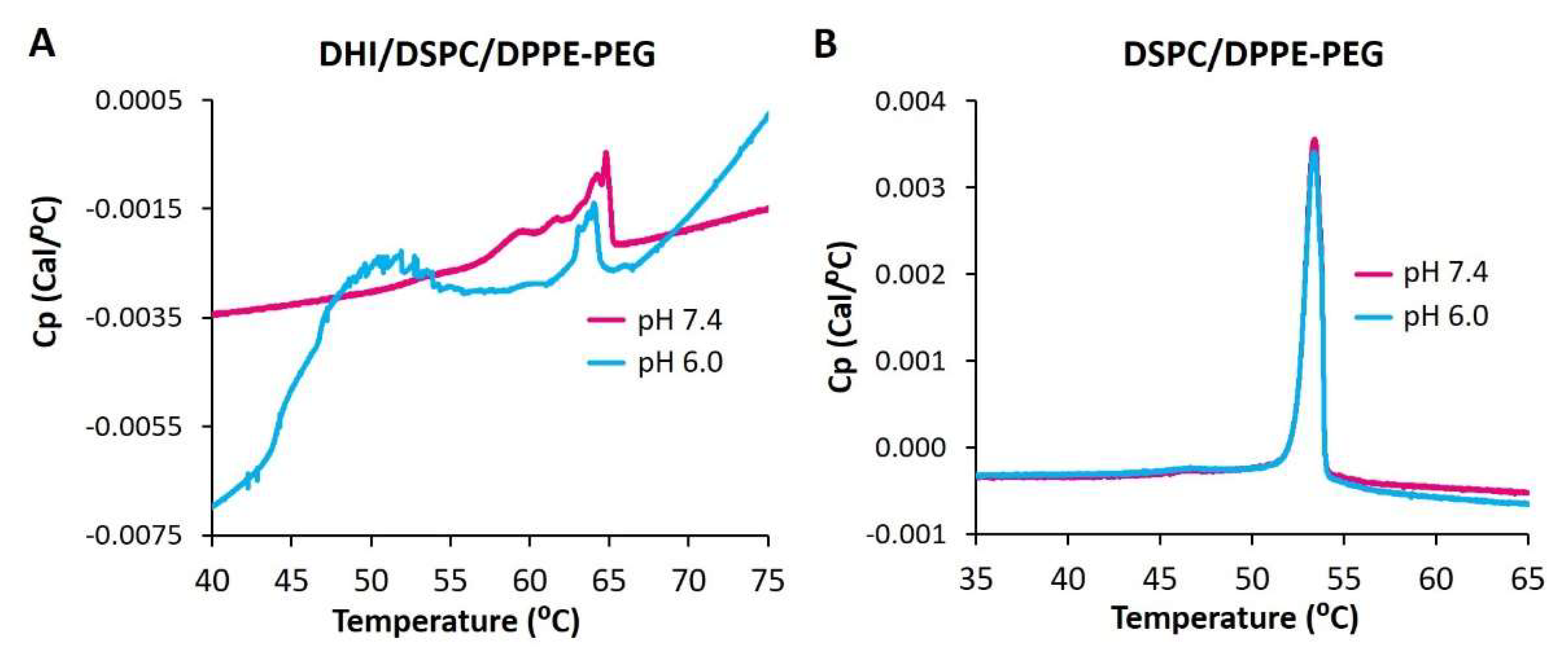
2.8. Differential Scanning Calorimetry of ICL
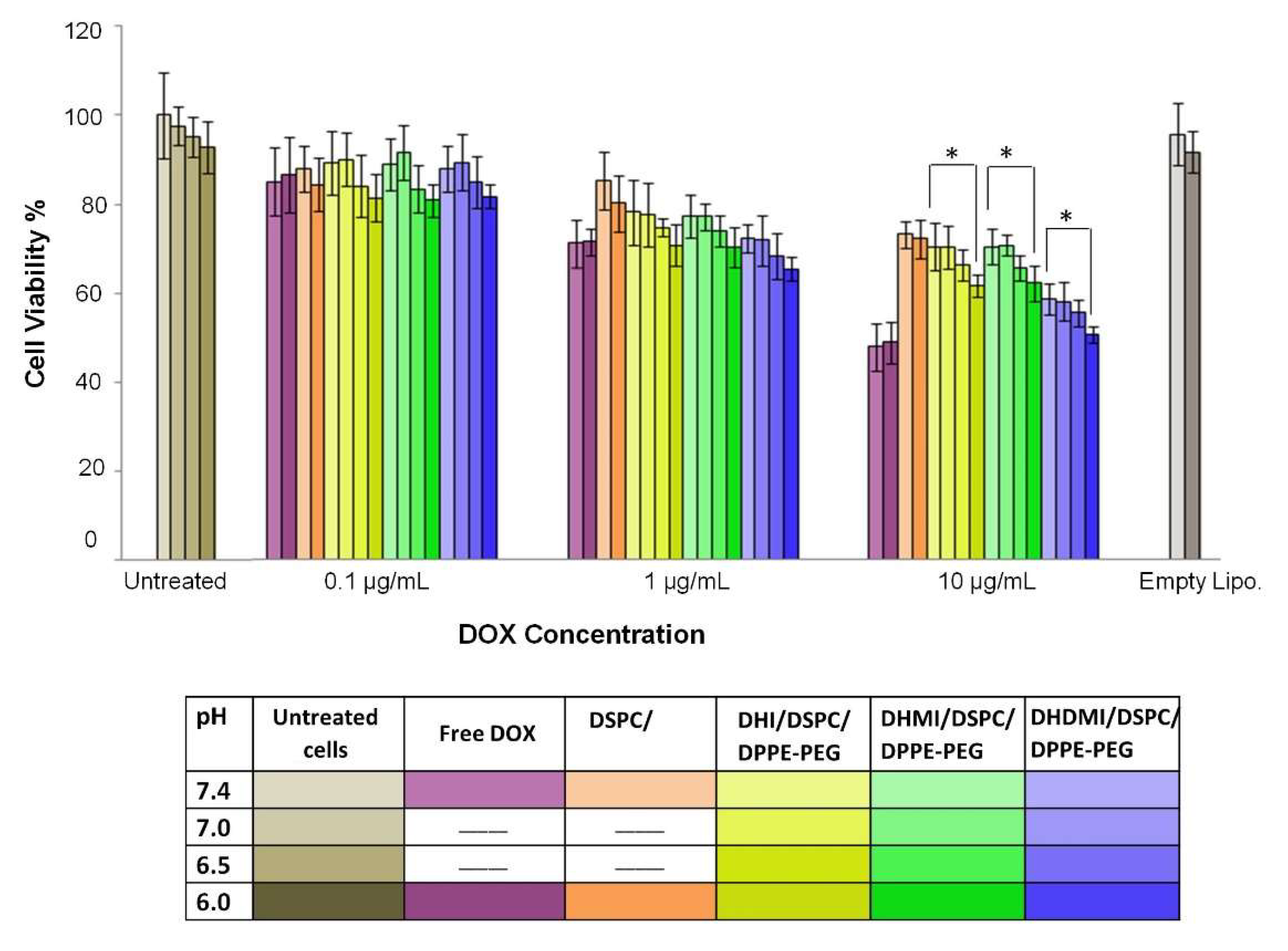
2.9. Anticancer Activity of ICL on 2D Monolayer Cells
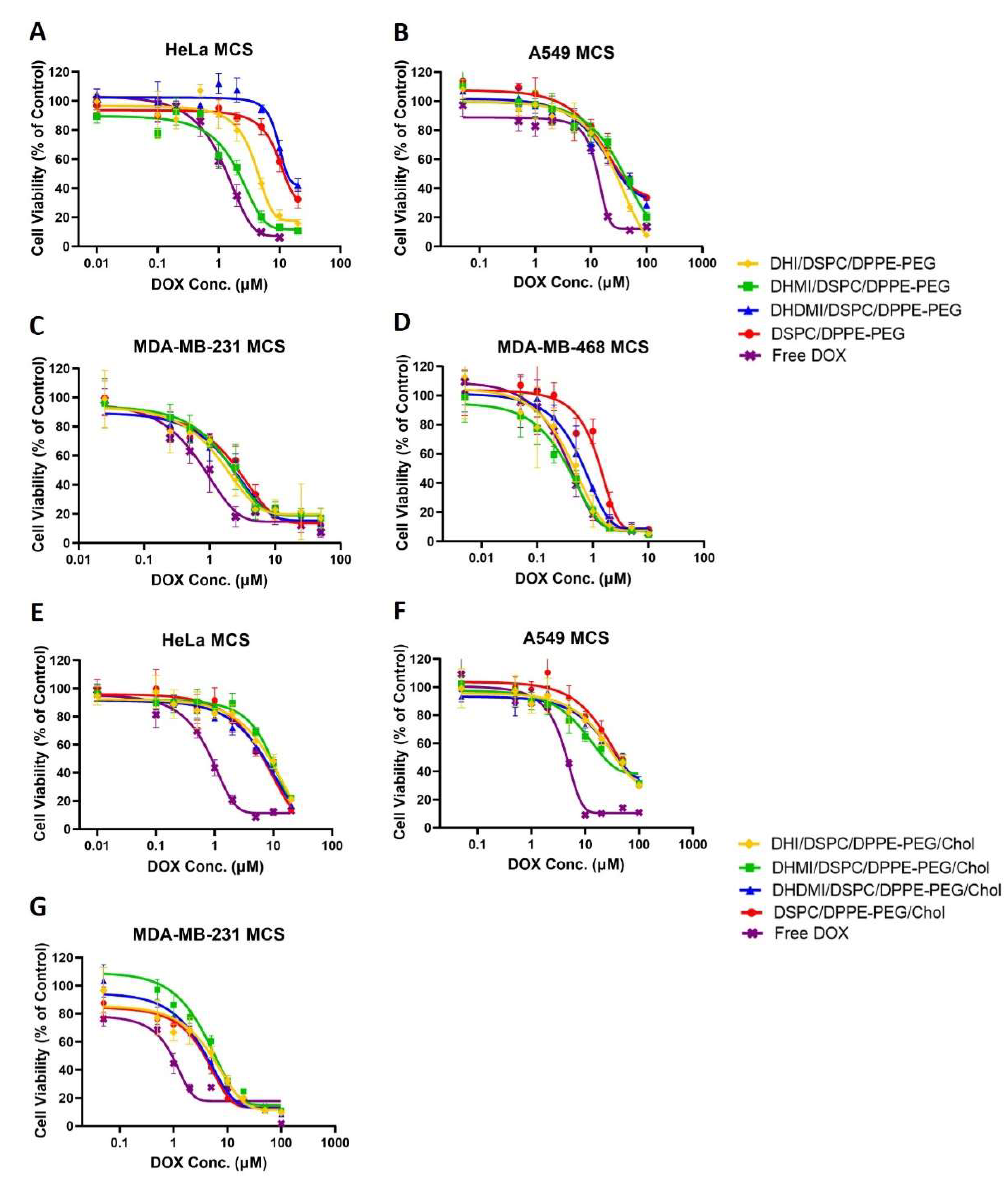
2.10. Anticancer Activity of ICL on 3D Multicellular Spheroids
3. Discussion
4. Materials and Methods
4.1. Materials
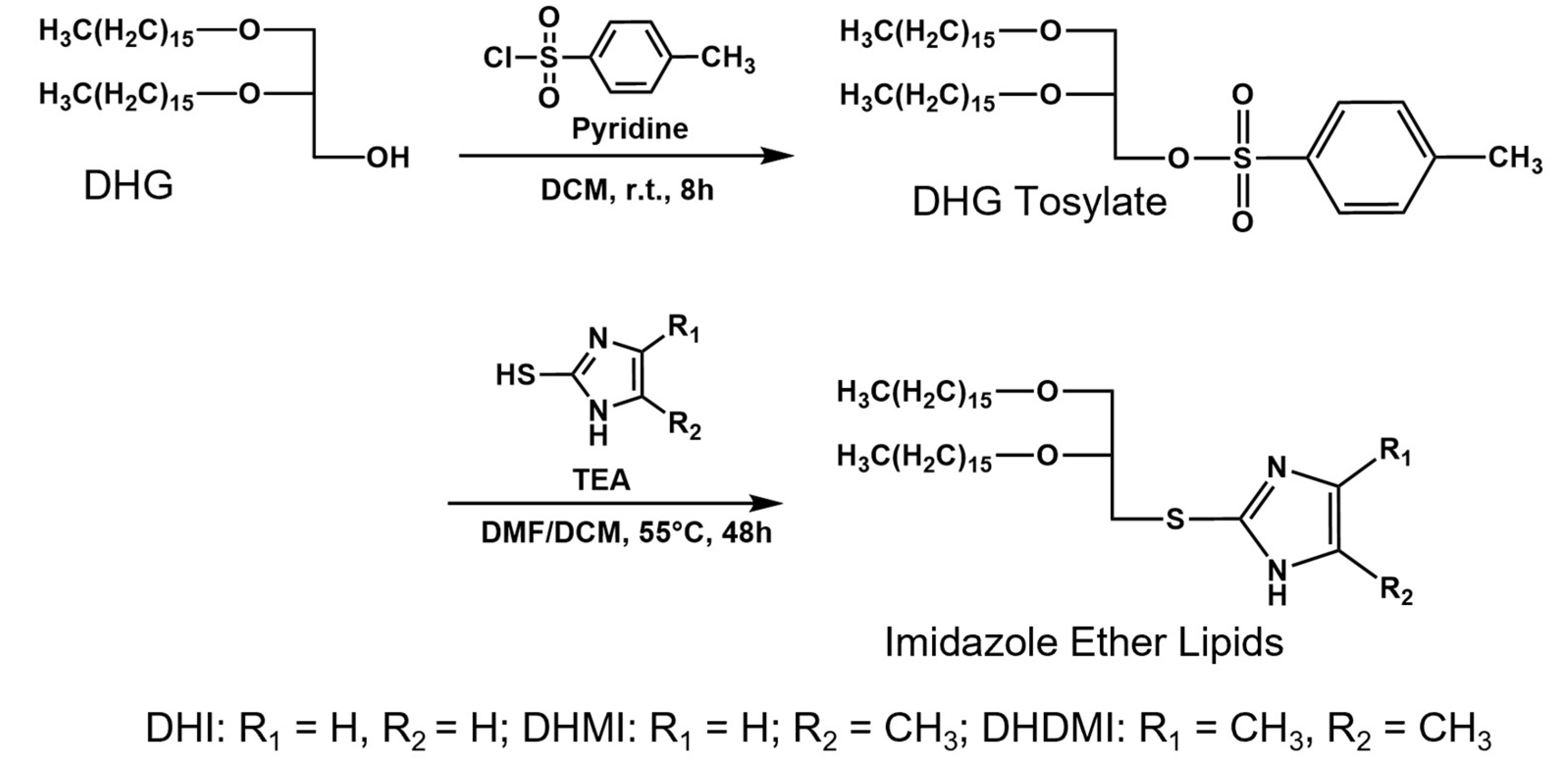
4.2. Synthesis of 2,3-di-O-Hexadecyl-1-rac-glyceryl-tosylate (DHG-Tosylate)
4.3. Synthesis of sn-2-((2,3-Dihexadecyloxypropyl)thio)-1H-imidazole (DHI)
4.4. Synthesis of sn-2-((2,3-Dihexadecyloxypropyl)thio)-5-methyl-1H-imidazole (DHMI)
4.5. Synthesis of sn-2-((2,3-Dihexadecyloxypropyl)thio)-4,5-dimethyl-1H-imidazole (DHDMI)
4.6. Preparation of ICL Formulations
4.7. Size Measurement
4.8. Quantification of Encapsulation Efficiency (EE)
4.9. Differential Scanning Calorimetry
4.10. pH-Triggered Change of ζ-Potential
4.11. pH-Dependent Interaction with Model Liposomes
4.12. pH-Dependent Drug Release
4.13. Transmission Electron Microscopy
4.14. Cell Culture
4.15. Cytotoxicity Assays on 2D Monolayer Hela Cells
4.16. Cytotoxicity Assays on 3D Multicellular Spheroids
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- U.S. Food and Drug Administration Resources on Drugs and Devices. Available online: https://clinicaltrials.gov/ct2/info/fdalinks (accessed on 8 February 2022).
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic. Bioeng. Transl. Med. 2016, 1, 10–29. [Google Scholar] [CrossRef] [PubMed]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic: An update. Bioeng. Transl. Med. 2019, 4, e10143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Beltrán-Gracia, E.; López-Camacho, A.; Higuera-Ciapara, I.; Velázquez-Fernández, J.B.; Vallejo-Cardona, A.A. Nanomedicine review: Clinical developments in liposomal applications. Cancer Nanotechnol. 2019, 10, 11. [Google Scholar] [CrossRef]
- Wang, R.; Billone, P.S.; Mullett, W.M. Nanomedicine in Action: An Overview of Cancer Nanomedicine on the Market and in Clinical Trials. J. Nanomater. 2013, 2013, 629681. [Google Scholar] [CrossRef] [Green Version]
- Gabizon, A.; Shmeeda, H.; Barenholz, Y. Pharmacokinetics of Pegylated Liposomal Doxorubicin. Clin. Pharmacokinet. 2003, 42, 419–436. [Google Scholar] [CrossRef]
- Immordino, M.L.; Dosio, F.; Cattel, L. Stealth liposomes: Review of the basic science, rationale, and clinical applications, existing and potential. Int. J. Nanomed. 2006, 1, 297–315. [Google Scholar]
- Millard, M.; Yakavets, I.; Zorin, V.; Kulmukhamedova, A.; Marchal, S.; Bezdetnaya, L. Drug delivery to solid tumors: The predictive value of the multicellular tumor spheroid model for nanomedicine screening. Int. J. Nanomed. 2017, 12, 7993–8007. [Google Scholar] [CrossRef] [Green Version]
- Vladimir, T. Tumor delivery of macromolecular drugs based on the EPR effect. Adv. Drug Deliv. Rev. 2011, 63, 131–135. [Google Scholar] [CrossRef]
- Kommineni, N.; Mahira, S.; Domb, A.J.; Khan, W. Cabazitaxel-Loaded Nanocarriers for Cancer Therapy with Reduced Side Effects. Pharmaceutics 2019, 11, 141. [Google Scholar] [CrossRef] [Green Version]
- Maeda, H. Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv. Drug Deliv. Rev. 2015, 91, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Reshetnyak, Y.K. Imaging tumor acidity: pH-low insertion peptide probe for optoacoustic tomography. Clin. Cancer Res. 2015, 21, 4502–4504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zagaynova, E.V.; Druzhkova, I.N.; Mishina, N.M.; Ignatova, N.I.; Dudenkova, V.V.; Shirmanova, M.V. Imaging of intracellular pH in tumor spheroids using genetically encoded sensor SypHer2. In Multi-Parametric Live Cell Microscopy of 3D Tissue Models; Springer: Berlin/Heidelberg, Germany, 2017; pp. 105–119. [Google Scholar]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Drummond, D.C.; Zignani, M.; Leroux, J.-C. Current status of pH-sensitive liposomes in drug delivery. Prog. Lipid Res. 2000, 39, 409–460. [Google Scholar] [CrossRef]
- Fang, Y.; Vadlamudi, M.; Huang, Y.; Guo, X. Lipid-Coated, pH-Sensitive Magnesium Phosphate Particles for Intracellular Protein Delivery. Pharm. Res. 2019, 36, 81. [Google Scholar] [CrossRef]
- Zheng, Y.; Liu, X.; Samoshina, N.M.; Samoshin, V.V.; Franz, A.H.; Guo, X. Fliposomes: Trans-2-aminocyclohexanol-based amphiphiles as pH-sensitive conformational switches of liposome membrane—A structure-activity relationship study. Chem. Phys. Lipids 2017, 210, 129–141. [Google Scholar] [CrossRef]
- He, X.; Li, J.; An, S.; Jiang, C. pH-sensitive drug-delivery systems for tumor targeting. Ther. Deliv. 2013, 4, 1499–1510. [Google Scholar] [CrossRef]
- Liu, J.; Huang, Y.; Kumar, A.; Tan, A.; Jin, S.; Mozhi, A.; Liang, X.J. pH-sensitive nano-systems for drug delivery in cancer therapy. Biotechnol. Adv. 2014, 32, 693–710. [Google Scholar] [CrossRef]
- Oya, T.A.; Ishiyama, A.A.; Nishikawa, N.A. Imidazole Compound and Liposome Containing Same. European Patent EP 3170812A1, 2017. Available online: https://data.epo.org/publication-server/document?iDocId=5335445&iFormat=0 (accessed on 8 February 2022).
- Chatin, B.; Mével, M.; Devallière, J.; Dallet, L.; Haudebourg, T.; Peuziat, P.; Colombani, T.; Berchel, M.; Lambert, O.; Edelman, A.; et al. Liposome-based Formulation for Intracellular Delivery of Functional Proteins. Mol. Ther. Nucleic Acids 2015, 4, e244. [Google Scholar] [CrossRef]
- He, J.; Xu, S.; Mixson, A.J. The Multifaceted Histidine-Based Carriers for Nucleic Acid Delivery: Advances and Challenges. Pharmaceutics 2020, 12, 774. [Google Scholar] [CrossRef]
- Ju, L.; Cailin, F.; Wenlan, W.; Pinghua, Y.; Jiayu, G.; Junbo, L. Preparation and properties evaluation of a novel pH-sensitive liposomes based on imidazole-modified cholesterol derivatives. Int. J. Pharm. 2017, 518, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Däster, S.; Amatruda, N.; Calabrese, D.; Ivanek, R.; Turrini, E.; Droeser, R.A.; Zajac, P.; Fimognari, C.; Spagnoli, G.C.; Iezzi, G.; et al. Induction of hypoxia and necrosis in multicellular tumor spheroids is associated with resistance to chemotherapy treatment. Oncotarget 2017, 8, 1725–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, C.R.; Bondurant, B.; McLean, S.D.; McGovern, K.A.; O’Brien, D.F. Liposome−Cell Interactions in Vitro: Effect of Liposome Surface Charge on the Binding and Endocytosis of Conventional and Sterically Stabilized Liposomes. Biochemistry 1998, 37, 12875–12883. [Google Scholar] [CrossRef] [PubMed]
- Brazdova, B.; Zhang, N.; Samoshin, V.V.; Guo, X. trans-2-Aminocyclohexanol as a pH-sensitive conformational switch in lipid amphiphiles. Chem. Commun. 2008, 39, 4774–4776. [Google Scholar] [CrossRef]
- Samoshina, N.M.; Liu, X.; Brazdova, B.; Franz, A.H.; Samoshin, V.V.; Guo, X. Fliposomes: pH-Sensitive Liposomes Containing a trans-2-morpholinocyclohexanol-Based Lipid That Performs a Conformational Flip and Triggers an Instant Cargo Release in Acidic Medium. Pharmaceutics 2011, 3, 379–405. [Google Scholar] [CrossRef] [Green Version]
- Nakhaei, P.; Margiana, R.; Bokov, D.O.; Abdelbasset, W.K.; Kouhbanani, M.A.J.; Varma, R.S.; Marofi, F.; Jarahian, M.; Beheshtkhoo, N. Liposomes: Structure, Biomedical Applications, and Stability Parameters with Emphasis on Cholesterol. Front. Bioeng. Biotechnol. 2021, 9, 705886. [Google Scholar] [CrossRef]
- Briuglia, M.-L.; Rotella, C.M.; McFarlane, A.; Lamprou, D.A. Influence of cholesterol on liposome stability and on in vitro drug release. Drug Deliv. Transl. Res. 2015, 5, 231–242. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Zhang, H.; McCallum, C.M.; Szoka, F.C.; Guo, X. Unsaturated Cationic Ortho Esters for Endosome Permeation in Gene Delivery. J. Med. Chem. 2007, 50, 4269–4278. [Google Scholar] [CrossRef]
- Ting-Beall, H.P. Interactions of uranyl ions with lipid bilayer membranes. J. Microsc. 1980, 118, 221–227. [Google Scholar] [CrossRef]
- Koyama, T.M.; Stevens, C.R.; Borda, E.J.; Grobe, K.J.; Cleary, D.A. Characterizing the Gel to Liquid Crystal Transition in Lipid-Bilayer Model Systems. Chem. Educ. 1999, 4, 12–15. [Google Scholar] [CrossRef]
- Anderson, M.; Moshnikova, A.; Engelman, D.M.; Reshetnyak, Y.K.; Andreev, O.A. Probe for the measurement of cell surface pH in vivo and ex vivo. Proc. Natl. Acad. Sci. USA 2016, 113, 8177–8181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cody, S.H.; Dubbin, P.N.; Beischer, A.D.; Duncan, N.D.; Hill, J.S.; Kaye, A.H.; Williams, D.A. Intracellular pH mapping with SNARF-1 and confocal microscopy. I: A quantitative technique for living tissue and isolated cells. Micron 1993, 24, 573–580. [Google Scholar] [CrossRef]
- Sun, B.; Leem, C.H.; Vaughan-Jones, R.D. Novel chloride-dependent acid loader in the guinea-pig ventricular myocyte: Part of a dual acid-loading mechanism. J. Physiol. 1996, 495, 65–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Fonseca, M.; van Winden, E.; Crommelin, D. Doxorubicin induces aggregation of small negatively charged liposomes. Eur. J. Pharm. Biopharm. 1997, 43, 9–17. [Google Scholar] [CrossRef]
- Anderson, M.; Omri, A. The Effect of Different Lipid Components on the In Vitro Stability and Release Kinetics of Liposome Formulations. Drug Deliv. 2004, 11, 33–39. [Google Scholar] [CrossRef]
- Anders, C.B.; Baker, J.D.; Stahler, A.C.; Williams, A.J.; Sisco, J.N.; Trefry, J.C.; Wooley, D.P.; Sizemore, I.E.P. Tangential Flow Ultrafiltration: A “Green” Method for the Size Selection and Concentration of Colloidal Silver Nanoparticles. J. Vis. Exp. 2012, e4167. [Google Scholar] [CrossRef] [Green Version]
- Corvera, E.; Mouritsen, O.; Singer, M.; Zuckermann, M. The permeability and the effect of acyl-chain length for phospholipid bilayers containing cholesterol: Theory and experiment. Biochim. Biophys. Acta Biomembr. 1992, 1107, 261–270. [Google Scholar] [CrossRef]
- Huang, J. Exploration of Molecular Interactions in Cholesterol Superlattices: Effect of Multibody Interactions. Biophys. J. 2002, 83, 1014–1025. [Google Scholar] [CrossRef] [Green Version]
- Magarkar, A.; Dhawan, V.; Kallinteri, P.; Viitala, T.; Elmowafy, M.; Róg, T.; Bunker, A. Cholesterol level affects surface charge of lipid membranes in saline solution. Sci. Rep. 2014, 4, 5005. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Rubinchik, S.; Wood, A.L.; Gillanders, W.E.; Dong, J.-Y.; Watson, D.K.; Cole, D.J. Bystander Effect Contributes to the Antitumor Efficacy of CaSm Antisense Gene Therapy in a Preclinical Model of Advanced Pancreatic Cancer. Mol. Ther. 2006, 13, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Cheung, B.C.; Sun, T.H.; Leenhouts, J.M.; Cullis, P.R. Loading of doxorubicin into liposomes by forming Mn2+-drug complexes. Biochim. Biophys. Acta Biomembr. 1998, 1414, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Amselem, S.; Barenholz, Y.; Gabizon, A. Optimization and Upscaling of Doxorubicin-Containing Liposomes for Clinical Use. J. Pharm. Sci. 1990, 79, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Carneiro-Da-Cunha, M.G.; Cerqueira, M.A.; Souza, B.W.; Teixeira, J.A.; Vicente, A.A. Influence of concentration, ionic strength and pH on zeta potential and mean hydrodynamic diameter of edible polysaccharide solutions envisaged for multinanolayered films production. Carbohydr. Polym. 2011, 85, 522–528. [Google Scholar] [CrossRef] [Green Version]
- Cox, M.C.; Reese, L.M.; Bickford, L.R.; Verbridge, S.S. Toward the Broad Adoption of 3D Tumor Models in the Cancer Drug Pipeline. ACS Biomater. Sci. Eng. 2015, 1, 877–894. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, S.; Guo, Q.; Kessel, S.; Rubinoff, I.; Chan, L.L.-Y.; Li, P.; Liu, Y.; Qiu, J.; Zhou, C. Optical Coherence Tomography Detects Necrotic Regions and Volumetrically Quantifies Multicellular Tumor Spheroids. Cancer Res. 2017, 77, 6011–6020. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Before DOX-Loading | After DOX-Loading | |||||
|---|---|---|---|---|---|---|
| Lipid Compositions | Molar Ratio | Size (nm) | PDI | Size (nm) | PDI | EE (%) |
| DHI/DSPC/DPPE-PEG | 25/70/5 | 114.9 ± 10.9 | 0.205 ± 0.008 | 200.8 ± 14.6 | 0.522 ± 0.047 | 56.62 ± 2.06 |
| DHMI/DSPC/DPPE-PEG | 25/70/5 | 117.9 ± 3.6 | 0.220 ± 0.033 | 189.3 ± 22.3 | 0.546 ± 0.055 | 53.18 ± 1.12 |
| DHDMI/DSPC/DPPE-PEG | 25/70/5 | 104.8 ± 3.5 | 0.176 ± 0.035 | 194.9 ± 7.0 | 0.253 ± 0.130 | 59.54 ± 0.59 |
| DSPC/DPPE-PEG | 95/5 | 101.8 ± 3.0 | 0.125 ± 0.067 | 136.9 ± 13.7 | 0.364 ± 0.085 | 57.74 ± 0.98 |
| DHI/DSPC/DPPE-PEG/Chol | 25/45/5/25 | 122.7 ± 8.19 | 0.081 ± 0.020 | 142.2 ± 9.5 | 0.113 ± 0.035 | 71.38 ± 0.61 |
| DHMI/DSPC/DPPE-PEG/Chol | 25/45/5/25 | 116.5 ± 11.9 | 0.075 ± 0.030 | 128.1 ± 8.3 | 0.078 ± 0.021 | 89.86 ± 1.27 |
| DHDMI/DSPC/DPPE-PEG/Chol | 25/45/5/25 | 114.0 ± 5.6 | 0.074 ± 0.008 | 128.6 ± 14.2 | 0.104 ± 0.020 | 92.97 ± 1.10 |
| DSPC/DPPE-PEG/Chol | 70/5/25 | 119.9 ± 5.4 | 0.066 ± 0.037 | 138.4 ± 5.6 | 0.165 ± 0.092 | 60.98 ± 1.66 |
| Liposome Membrane Composition | Lipid Molar Ratio | IC50 $ (μM) | |||
|---|---|---|---|---|---|
| Hela | A549 | MDA- MB-231 | MDA- MB-468 | ||
| DHI/DSPC/DPPE-PEG | 25/70/5 | 3.82 ± 1.13 *** | ~30 # | 1.38 ± 1.31 | 0.38 ± 0.21 ** |
| DHMI/DSPC/DPPE-PEG | 25/70/5 | 2.07 ± 1.13 *** | ~40 # | 1.77 ± 1.21 | 0.31 ± 0.15 *** |
| DHDMI/DSPC/DPPE-PEG | 25/70/5 | 9.51 ± 1.15 | ~35 # | 1.86 ± 1.24 | 0.63 ± 0.10 ** |
| DSPC/DPPE-PEG | 95/5 | 11.41 ± 1.28 | ~35 # | 2.37 ± 1.29 | 1.24 ± 0.13 |
| DHI/DSPC/DPPE-PEG/Chol | 25/45/5/25 | ~10 # | ~30 # | 5.13 ± 1.46 | - |
| DHMI/DSPC/DPPE-PEG/Chol | 25/45/5/25 | 10.38 ± 1.33 | 29.07 ± 2.73 | 3.62 ± 1.17 | - |
| DHDMI/DSPC/DPPE-PEG/Chol | 25/45/5/25 | ~10 # | 24.06 ± 1.40 | 3.26 ± 1.18 | - |
| DSPC/DPPE-PEG/Chol | 70/5/25 | ~10 # | 33.88 ± 1.62 | 3.98 ± 1.10 | - |
| Free DOX | - | 1.26 ± 0.04 | 12.59 ± 1.05 | 1.18 ± 0.29 | 0.32 ± 0.12 |
| Mol % | ||||||
|---|---|---|---|---|---|---|
| Formulations | DHI | DHMI | DHDMI | DSPC | DPPE-PEG | Chol |
| I | 25 | - | - | 70 | 5 | - |
| II | - | 25 | 70 | 5 | - | |
| III | - | - | 25 | 70 | 5 | - |
| IV | - | - | - | 95 | 5 | - |
| V | 25 | - | - | 45 | 5 | 25 |
| VI | - | 25 | - | 45 | 5 | 25 |
| VII | - | - | 25 | 45 | 5 | 25 |
| VIII | - | - | - | 70 | 5 | 25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, R.; Gyanani, V.; Zhao, S.; Lu, Y.; Guo, X. Imidazole-Based pH-Sensitive Convertible Liposomes for Anticancer Drug Delivery. Pharmaceuticals 2022, 15, 306. https://doi.org/10.3390/ph15030306
Huang R, Gyanani V, Zhao S, Lu Y, Guo X. Imidazole-Based pH-Sensitive Convertible Liposomes for Anticancer Drug Delivery. Pharmaceuticals. 2022; 15(3):306. https://doi.org/10.3390/ph15030306
Chicago/Turabian StyleHuang, Ruiqi, Vijay Gyanani, Shen Zhao, Yifan Lu, and Xin Guo. 2022. "Imidazole-Based pH-Sensitive Convertible Liposomes for Anticancer Drug Delivery" Pharmaceuticals 15, no. 3: 306. https://doi.org/10.3390/ph15030306
APA StyleHuang, R., Gyanani, V., Zhao, S., Lu, Y., & Guo, X. (2022). Imidazole-Based pH-Sensitive Convertible Liposomes for Anticancer Drug Delivery. Pharmaceuticals, 15(3), 306. https://doi.org/10.3390/ph15030306