
Design, Green Synthesis and Tailoring of Vitamin E TPGS Augmented Niosomal Nano-Carrier of Pyrazolopyrimidines as Potential Anti-Liver and Breast Cancer Agents with Accentuated Oral Bioavailability

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
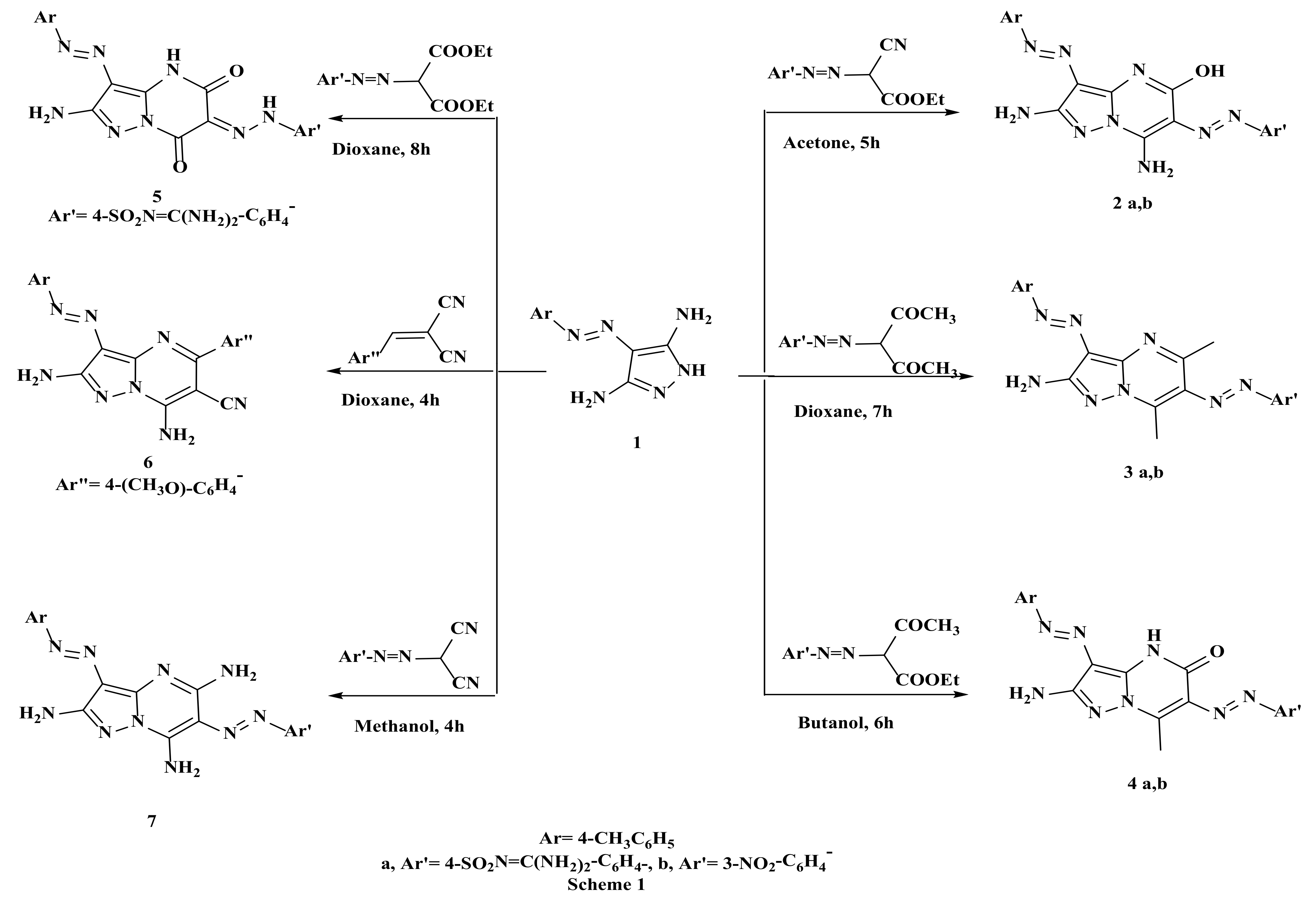
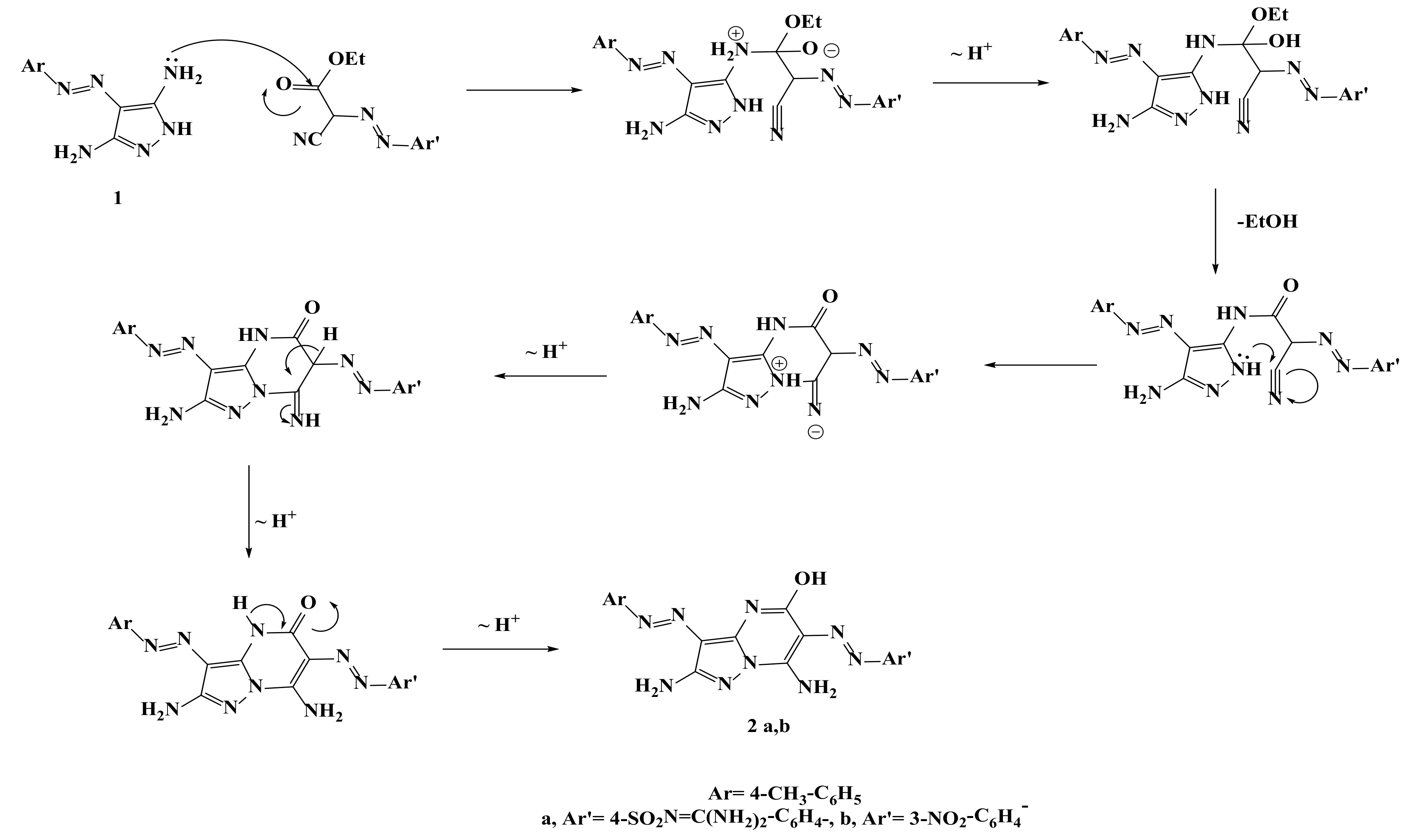
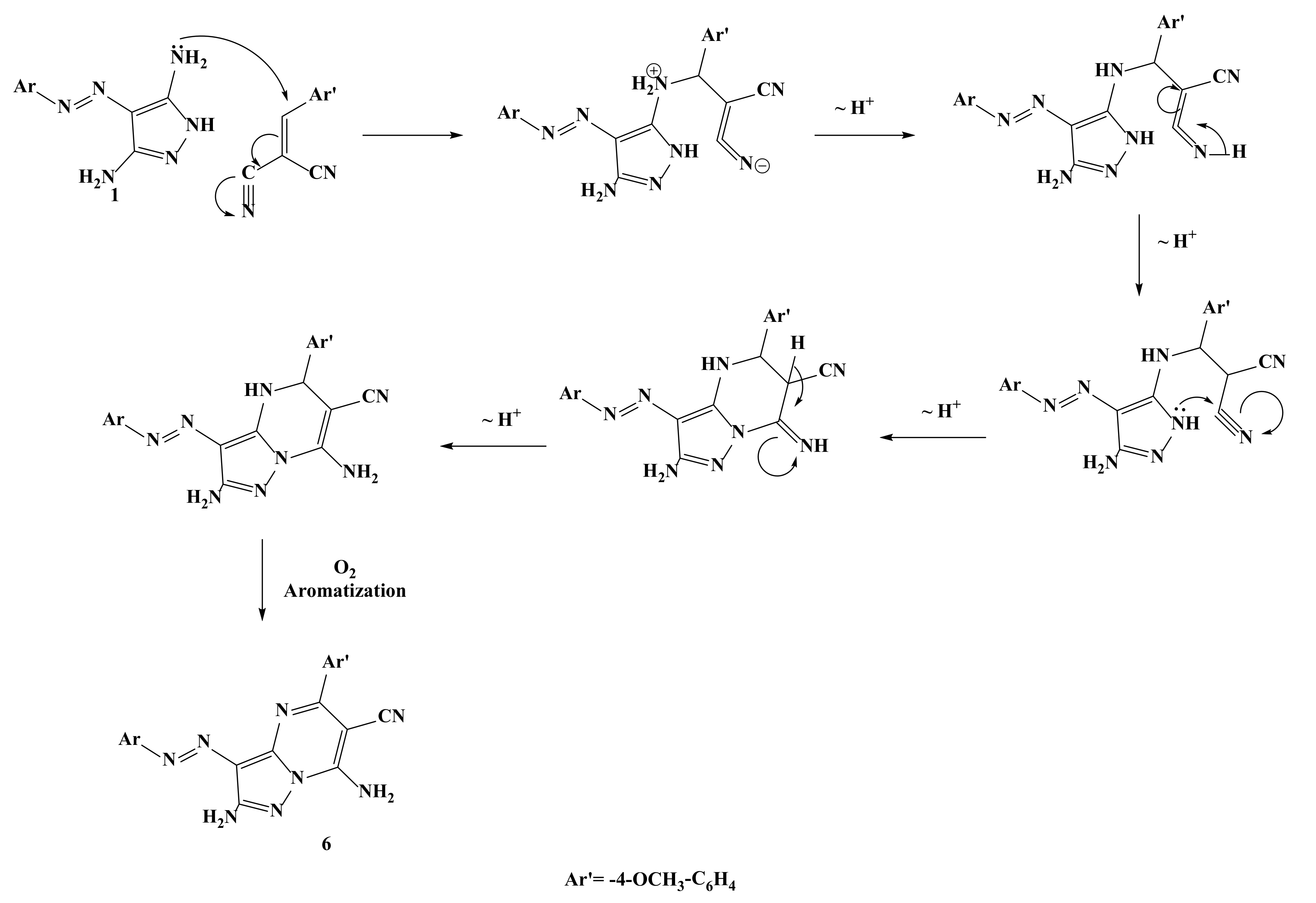
2.1. Synthesis of the Target Compounds
Comparison between Microwave and Conventional Methods
2.2. Biological Screening
2.2.1. In Vitro Cytotoxic Activities
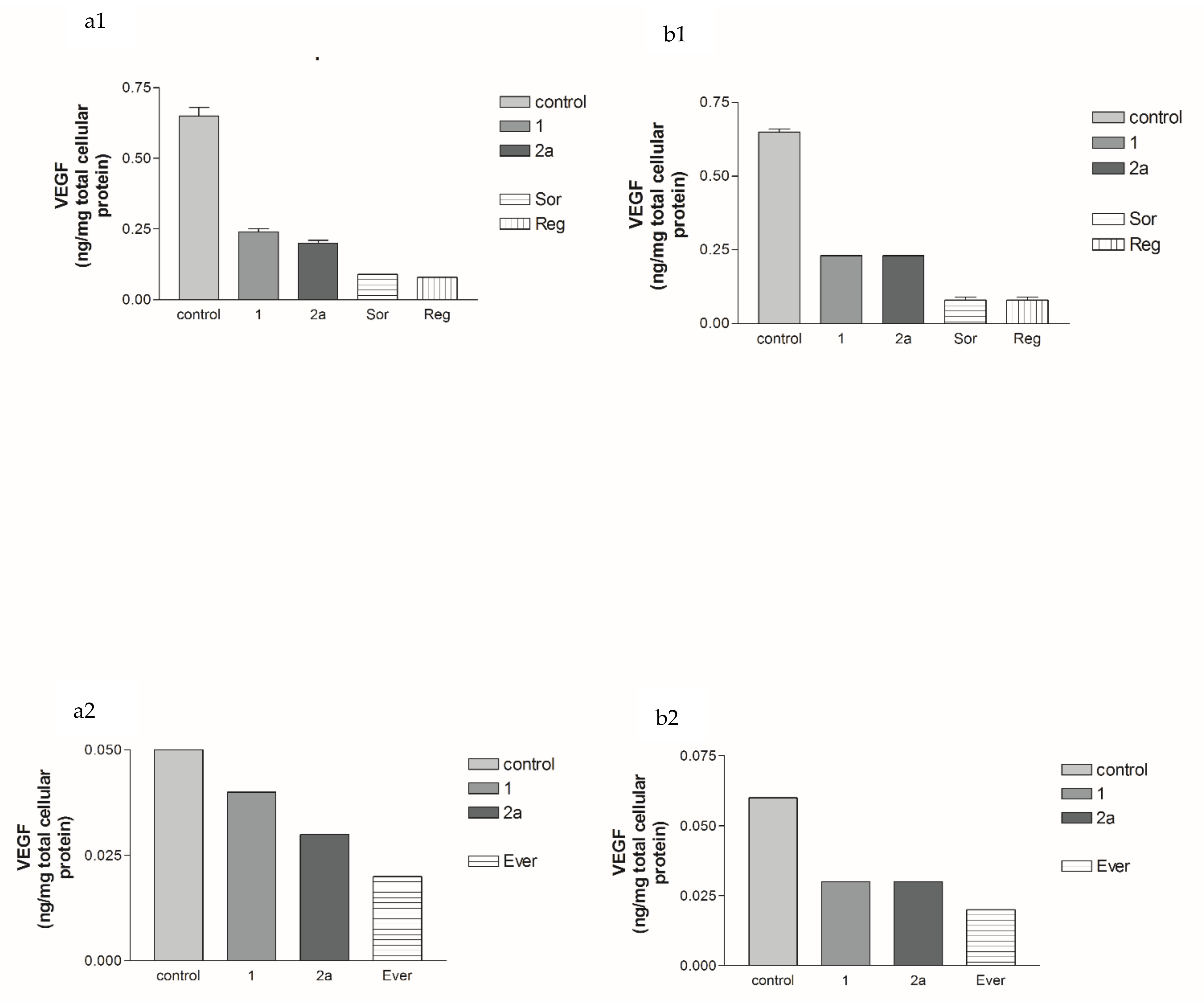
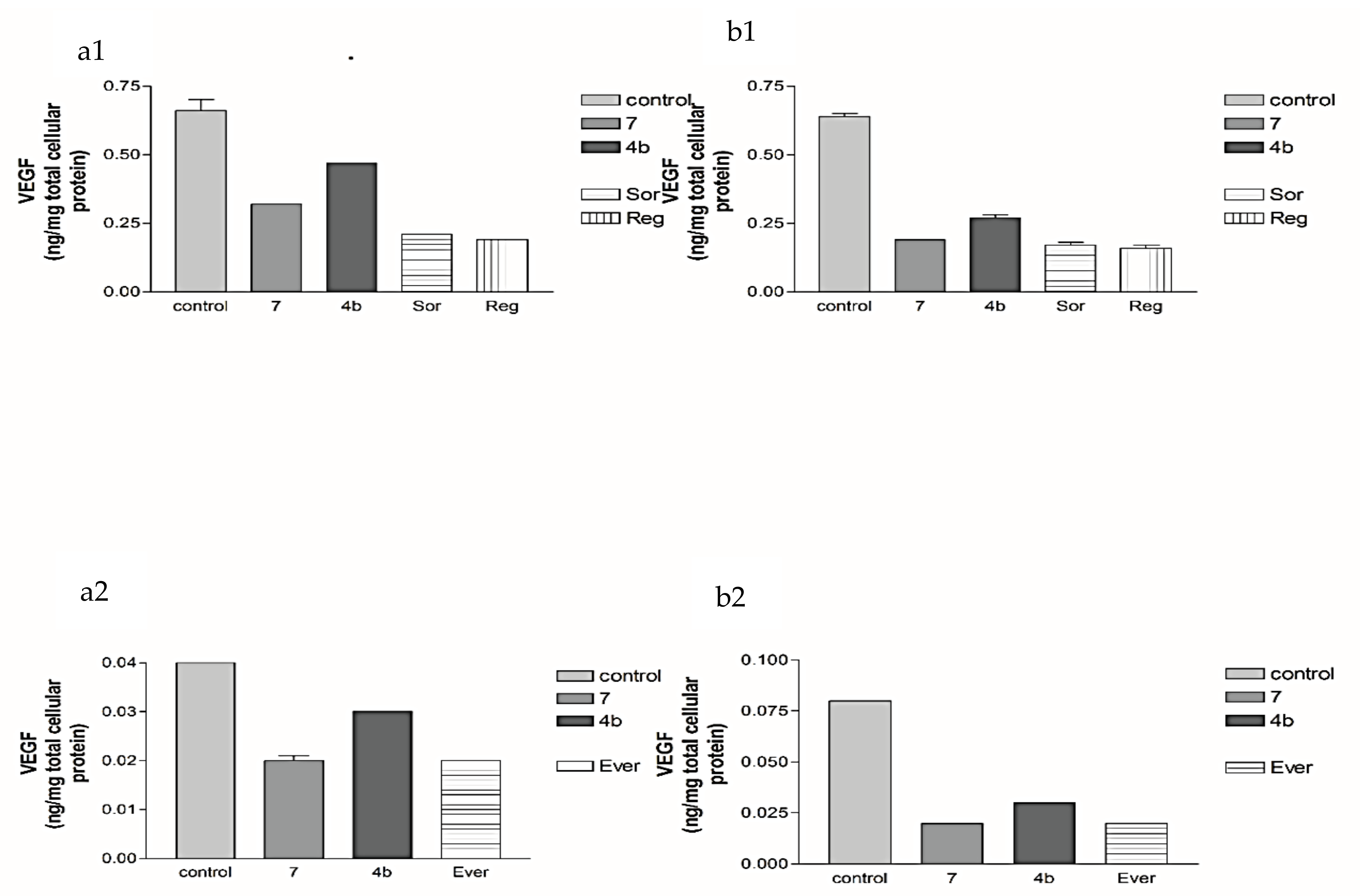
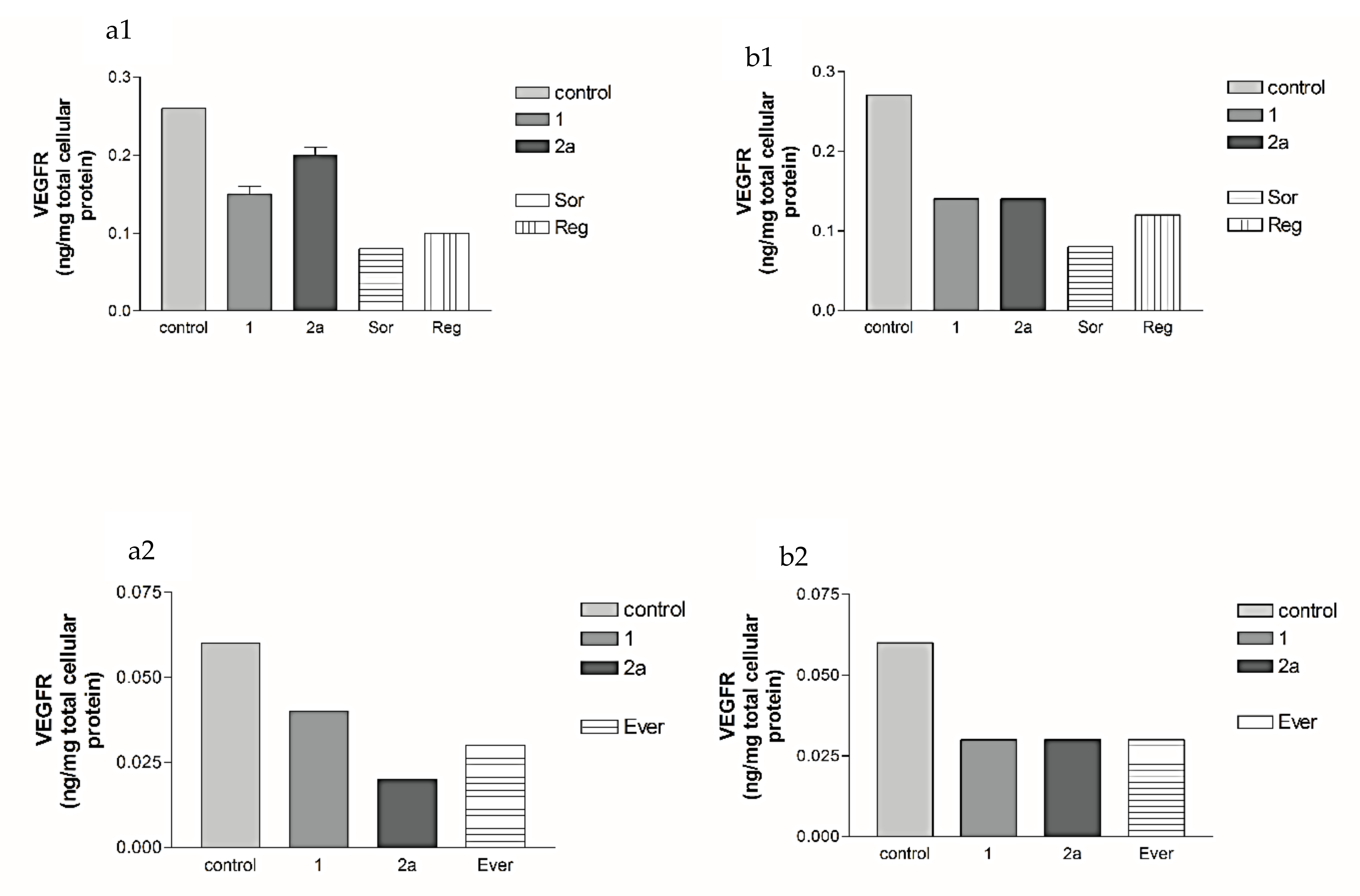
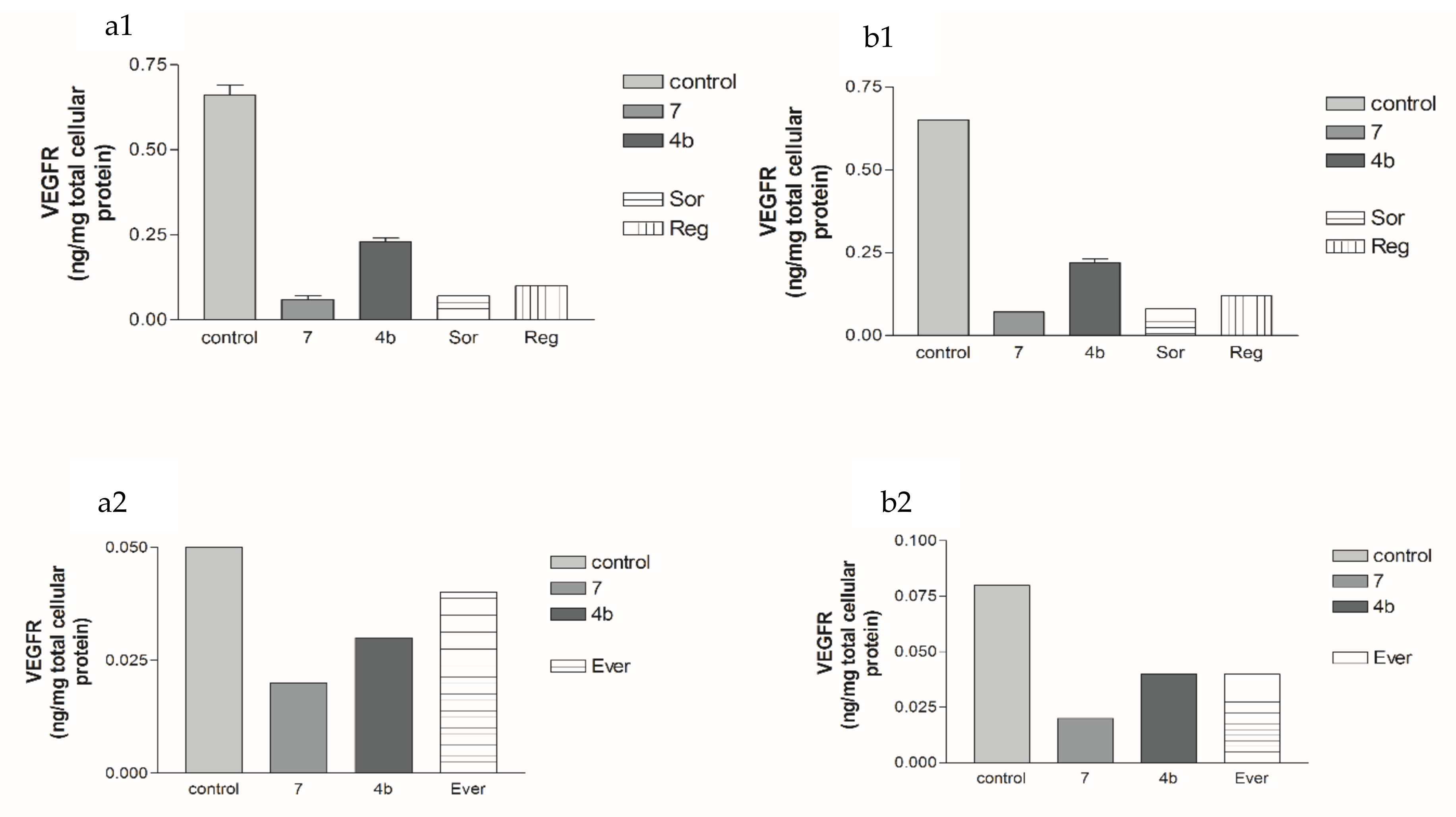
2.2.2. Effects of 1, 2a, 4b, 7, Sorafenib, Regorafenib and Everolimus on VEGF Protein Expression Levels in Both HepG2 and MCF-7 Cell Lines
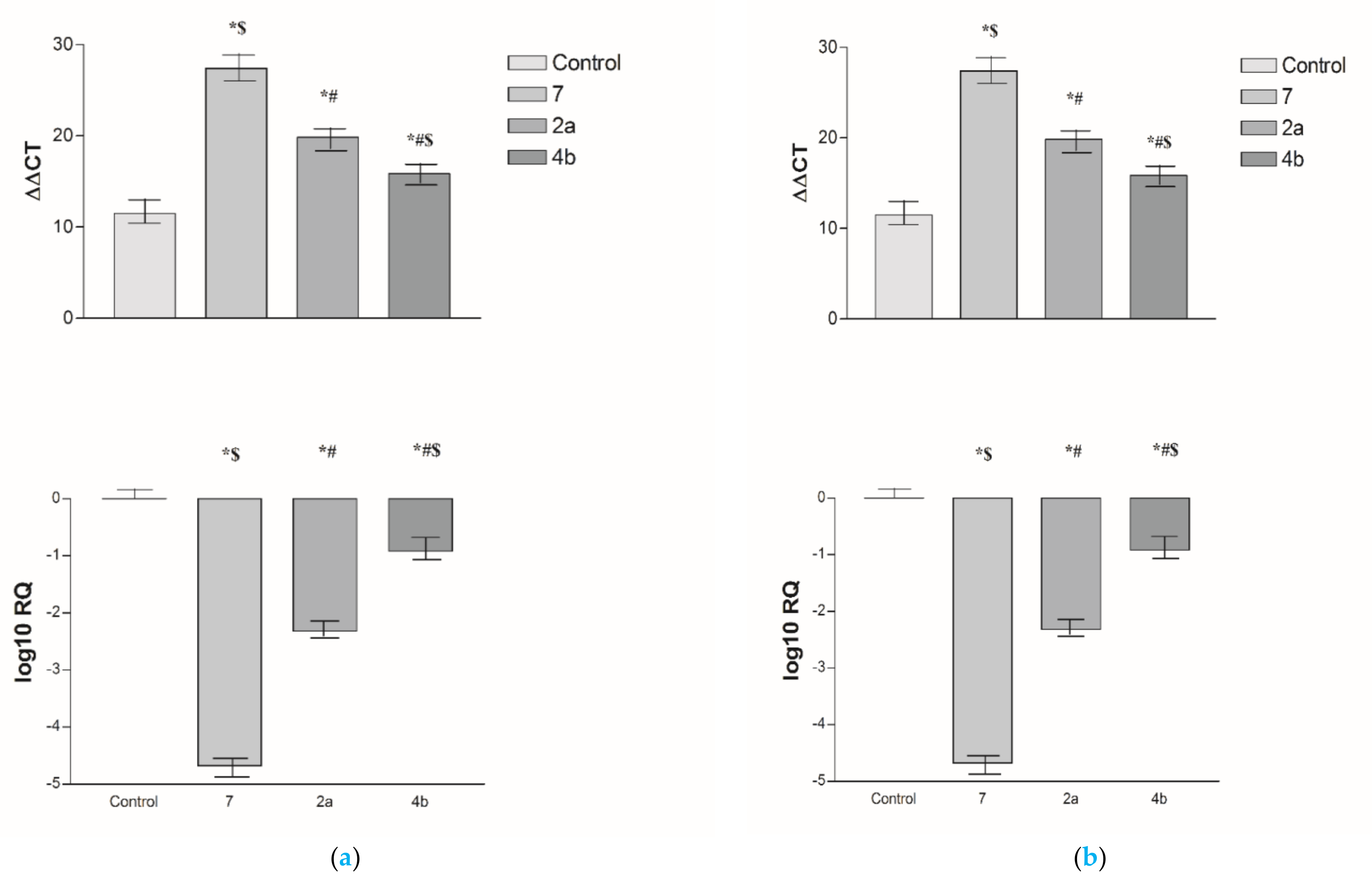
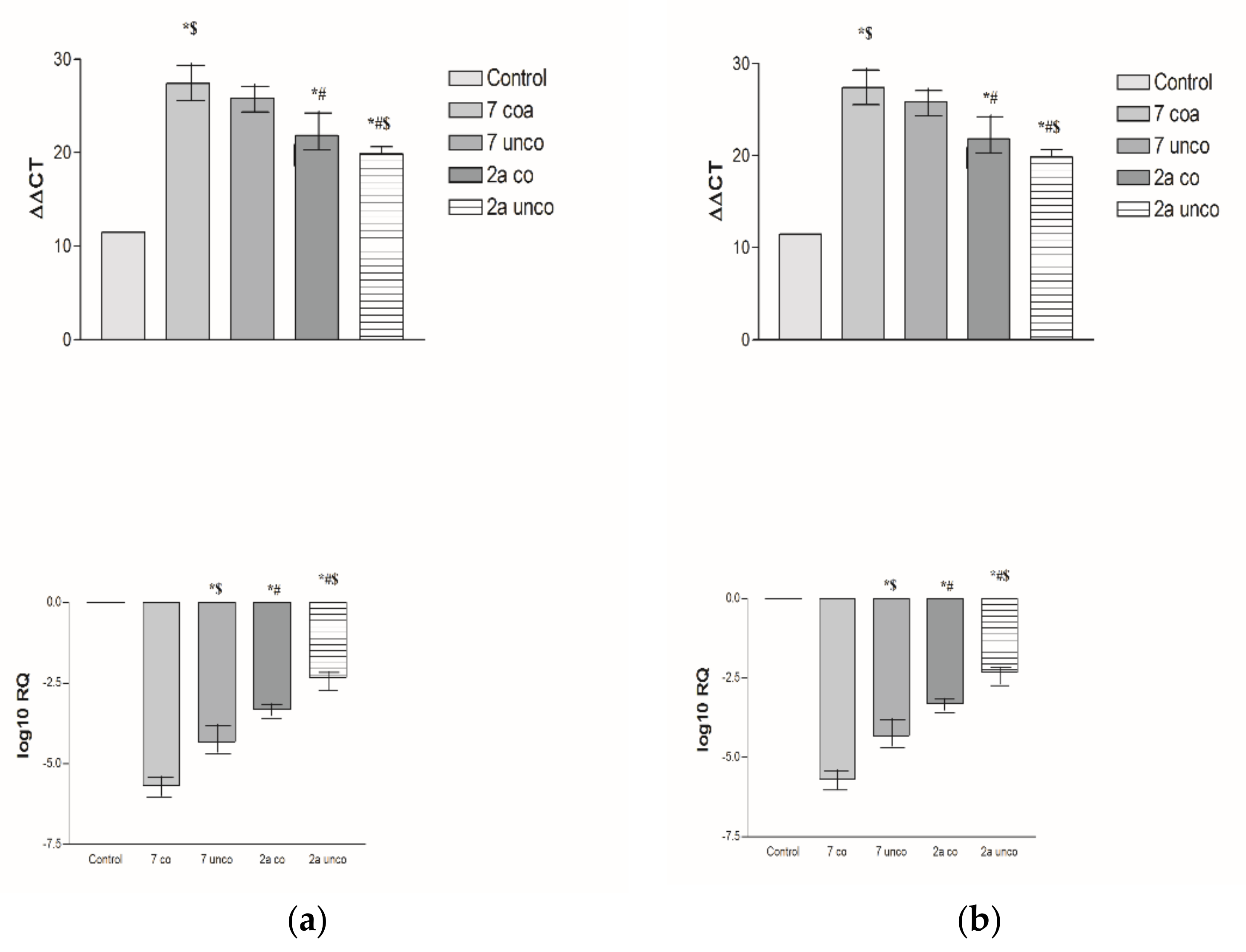
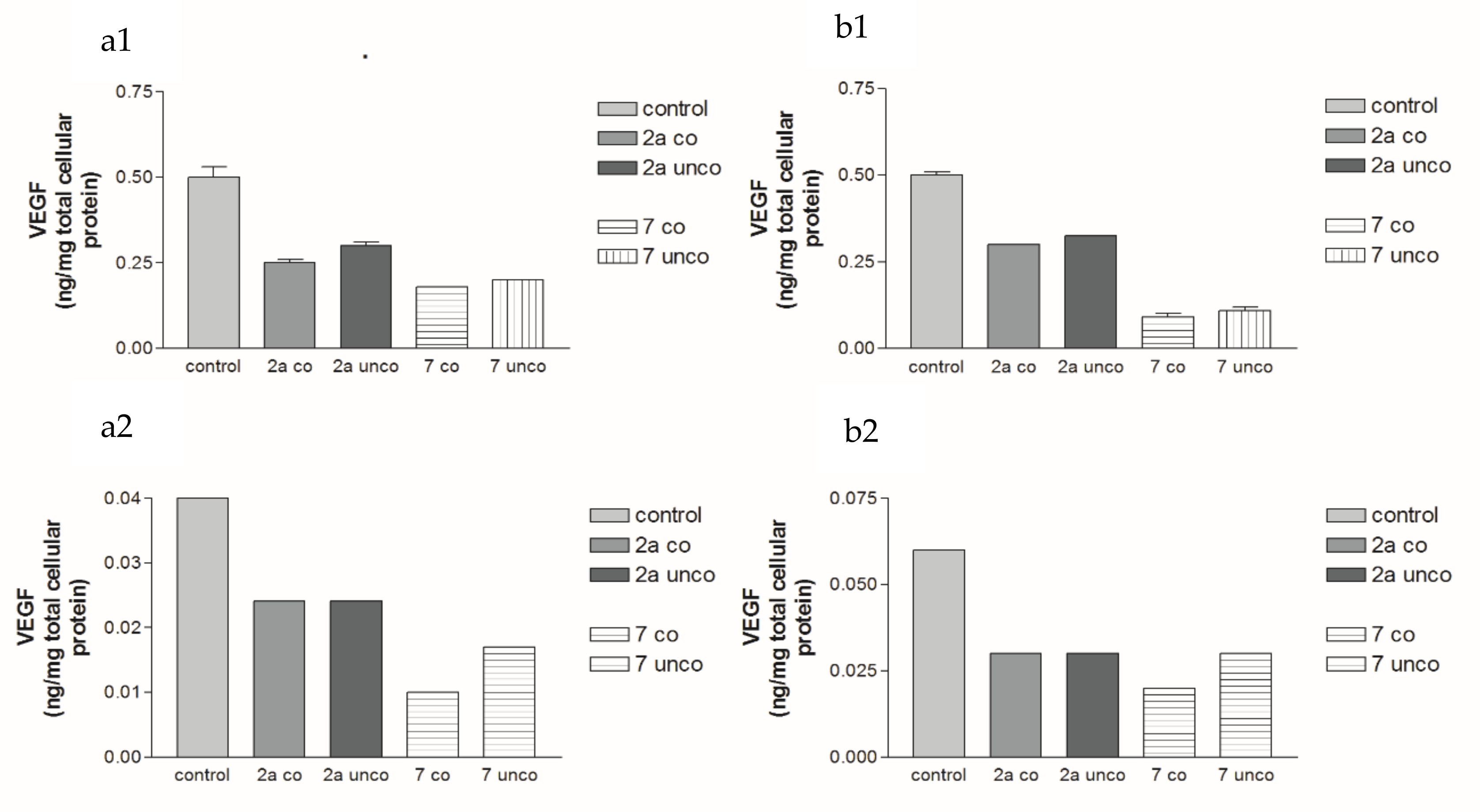
2.2.3. Effects of 2a, 4b and 7 compounds on the VEGF Gene Expression Level in Both HepG2 and MCF-7 Cell Lines
2.2.4. Structure–Activity Relationship of Synthesized Compounds
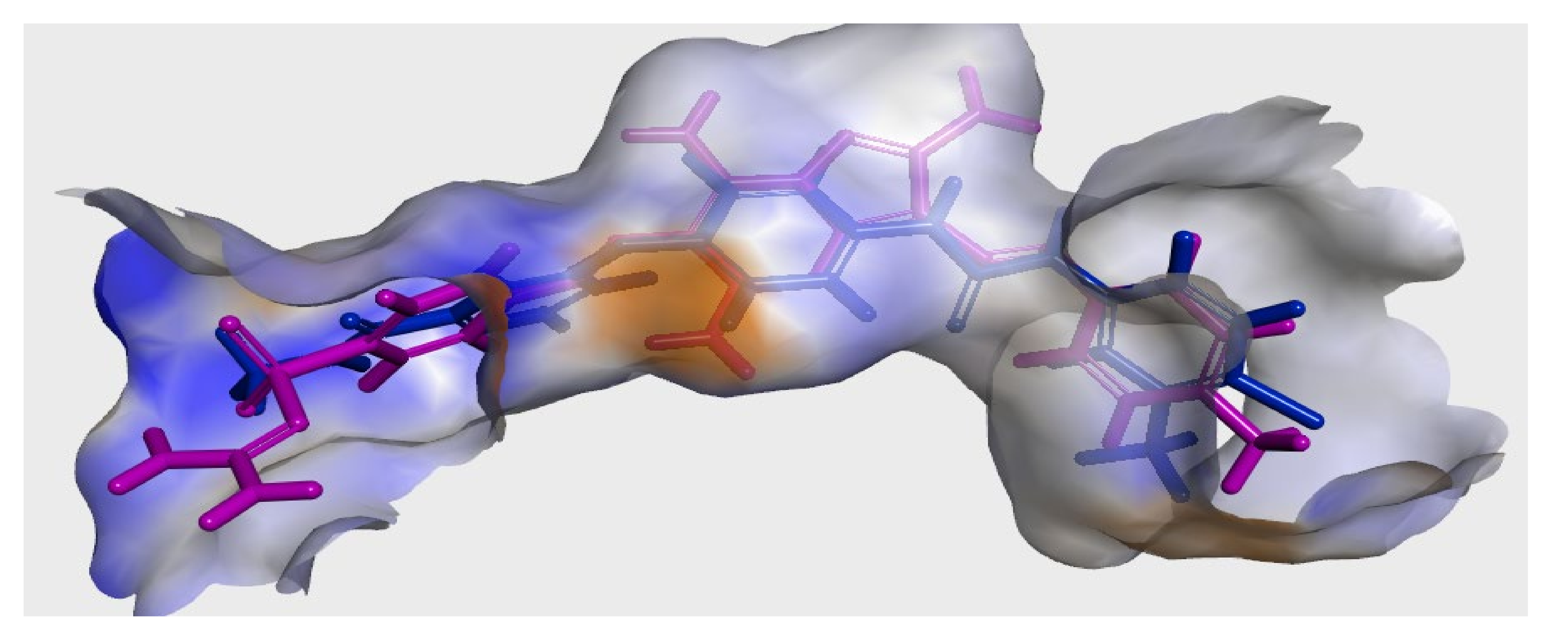
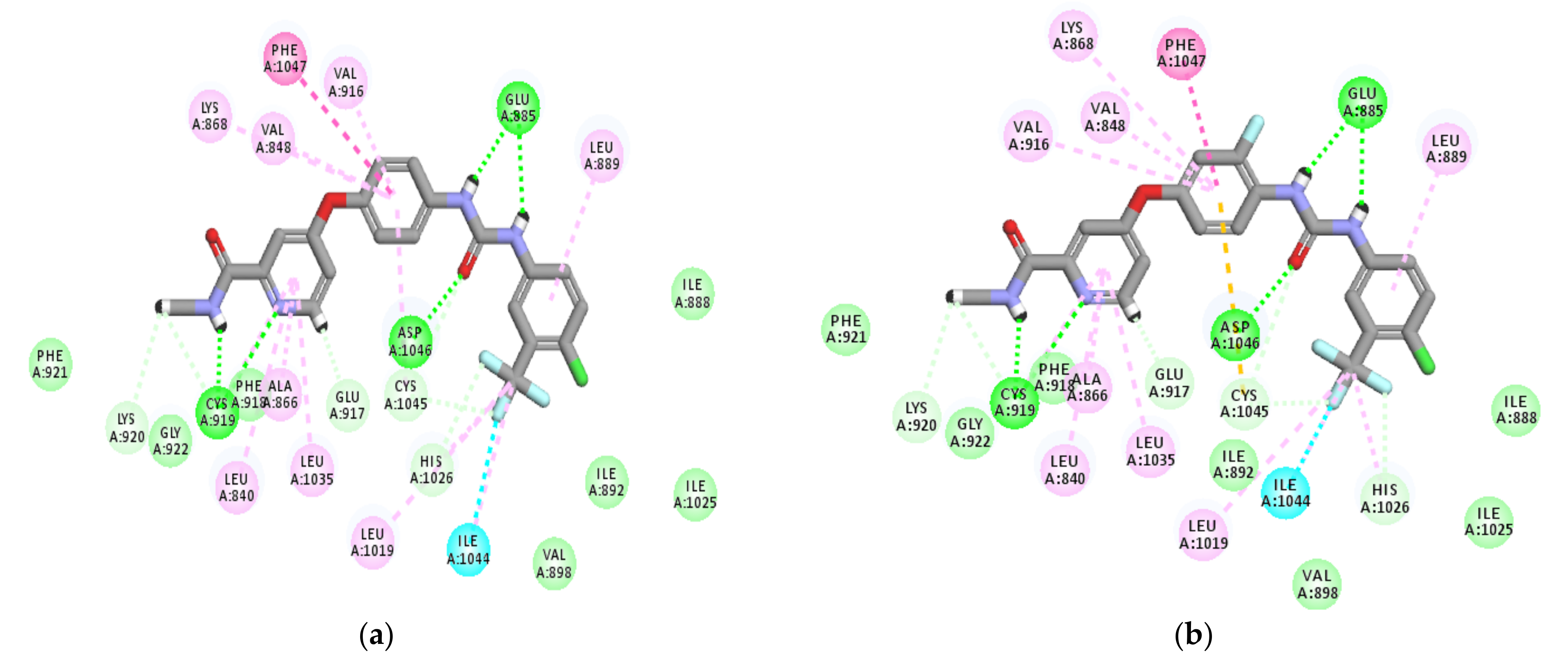
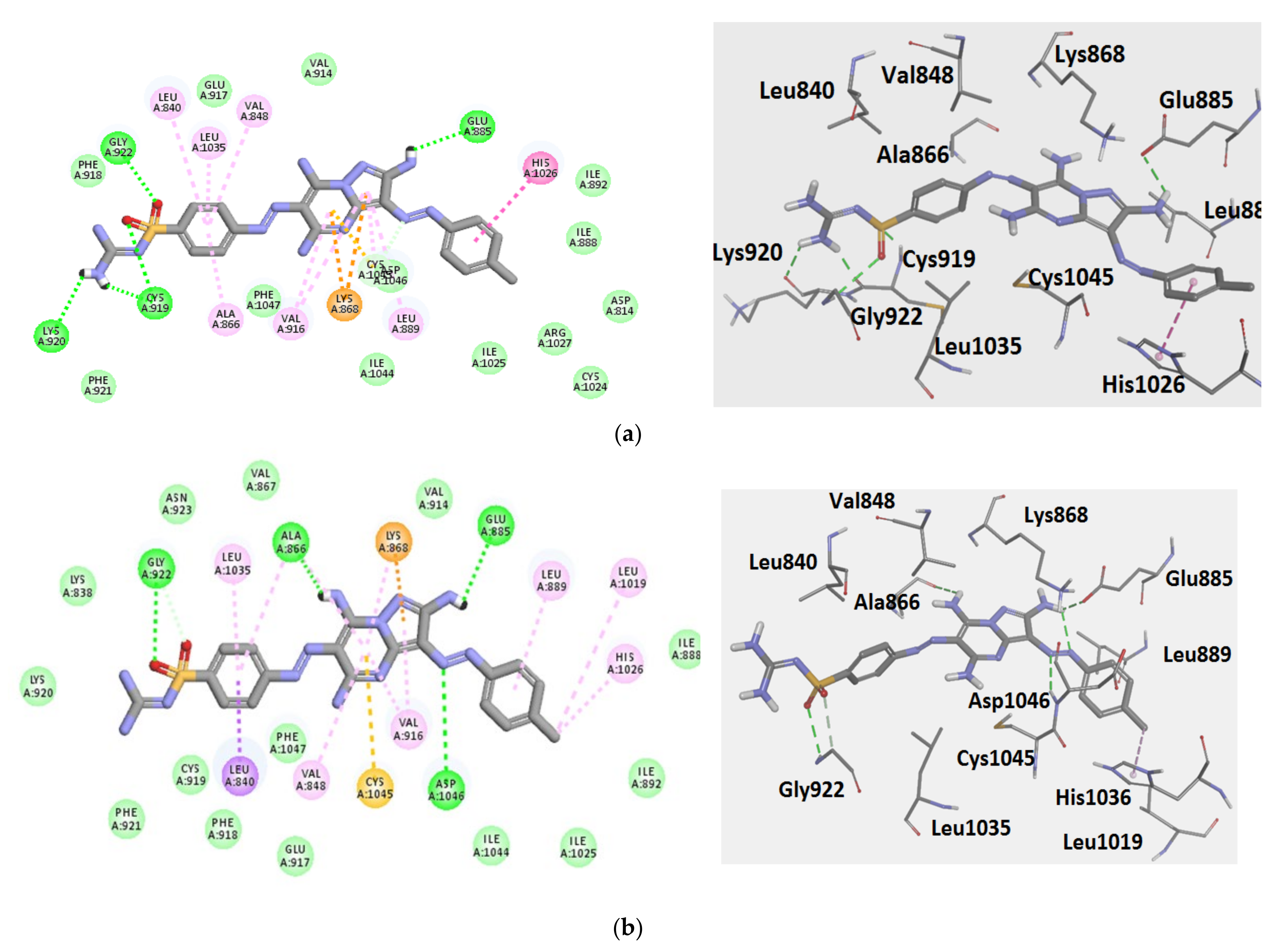
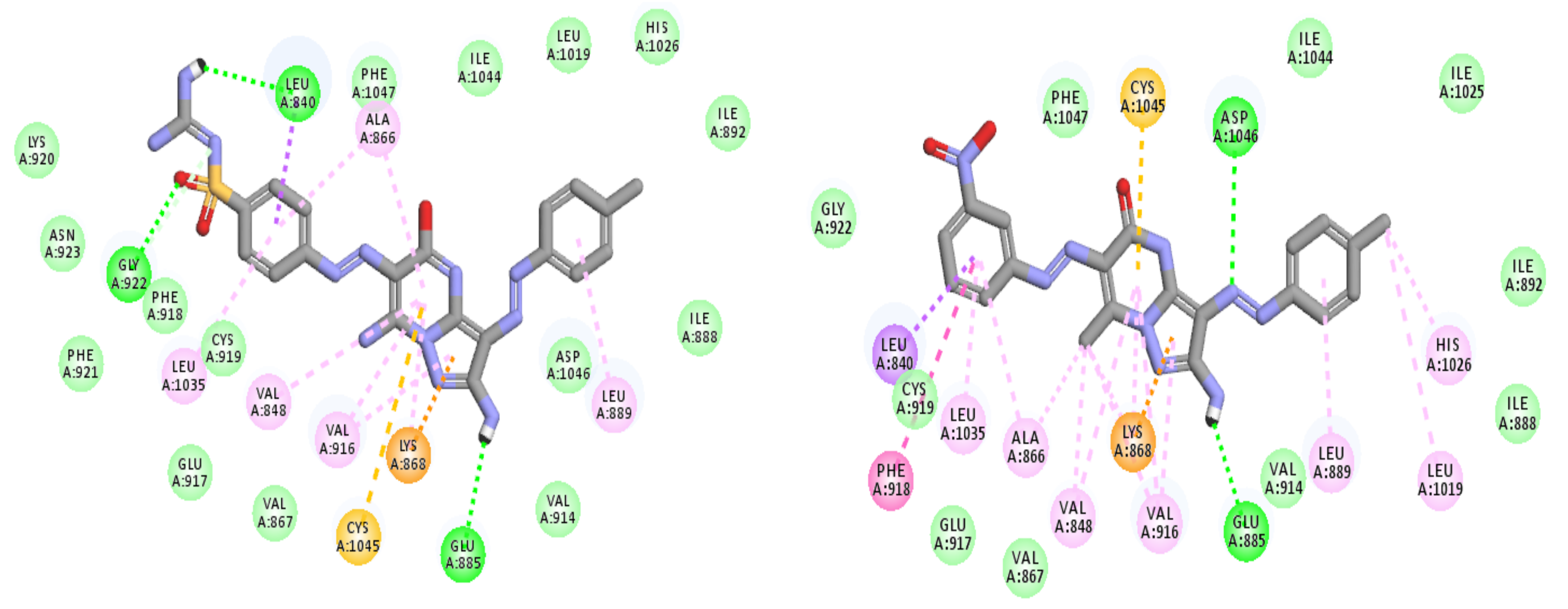
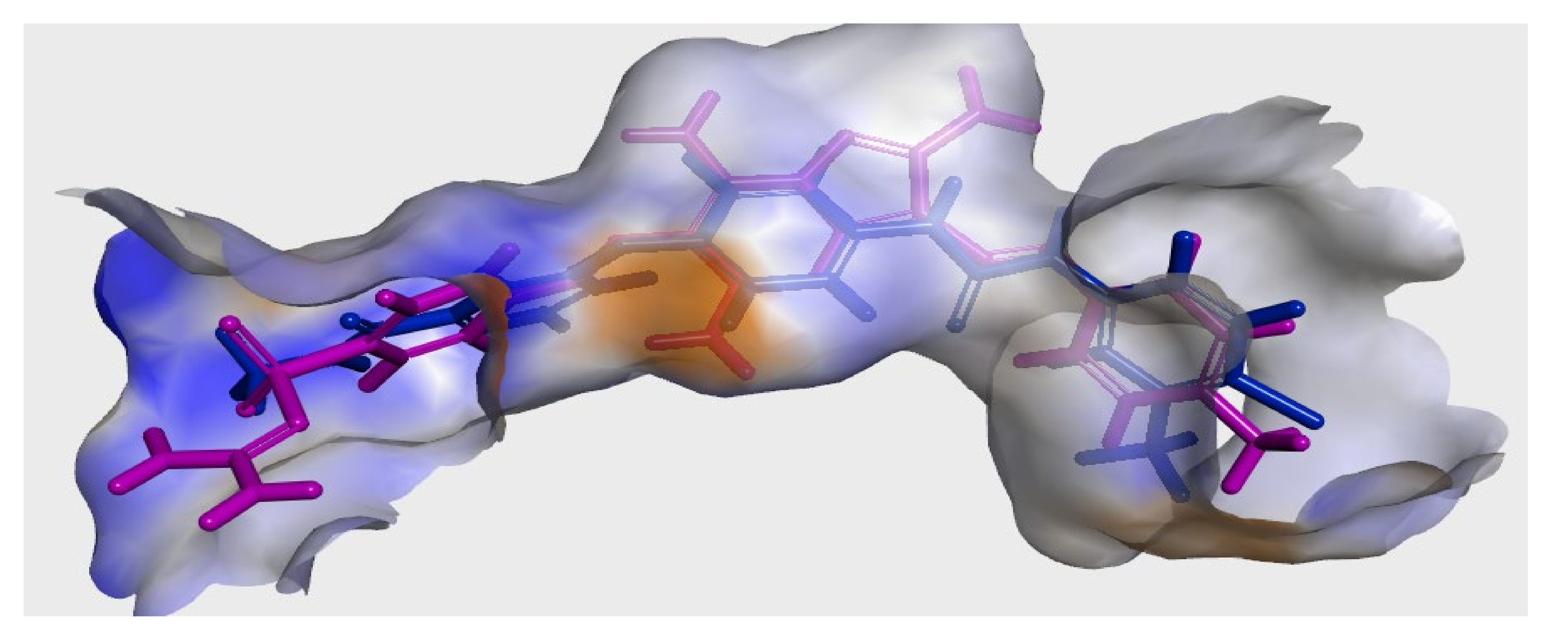
2.3. Modeling Job
2.4. Evolvement of TPGS Coated Niosomes Nano-Vesicles
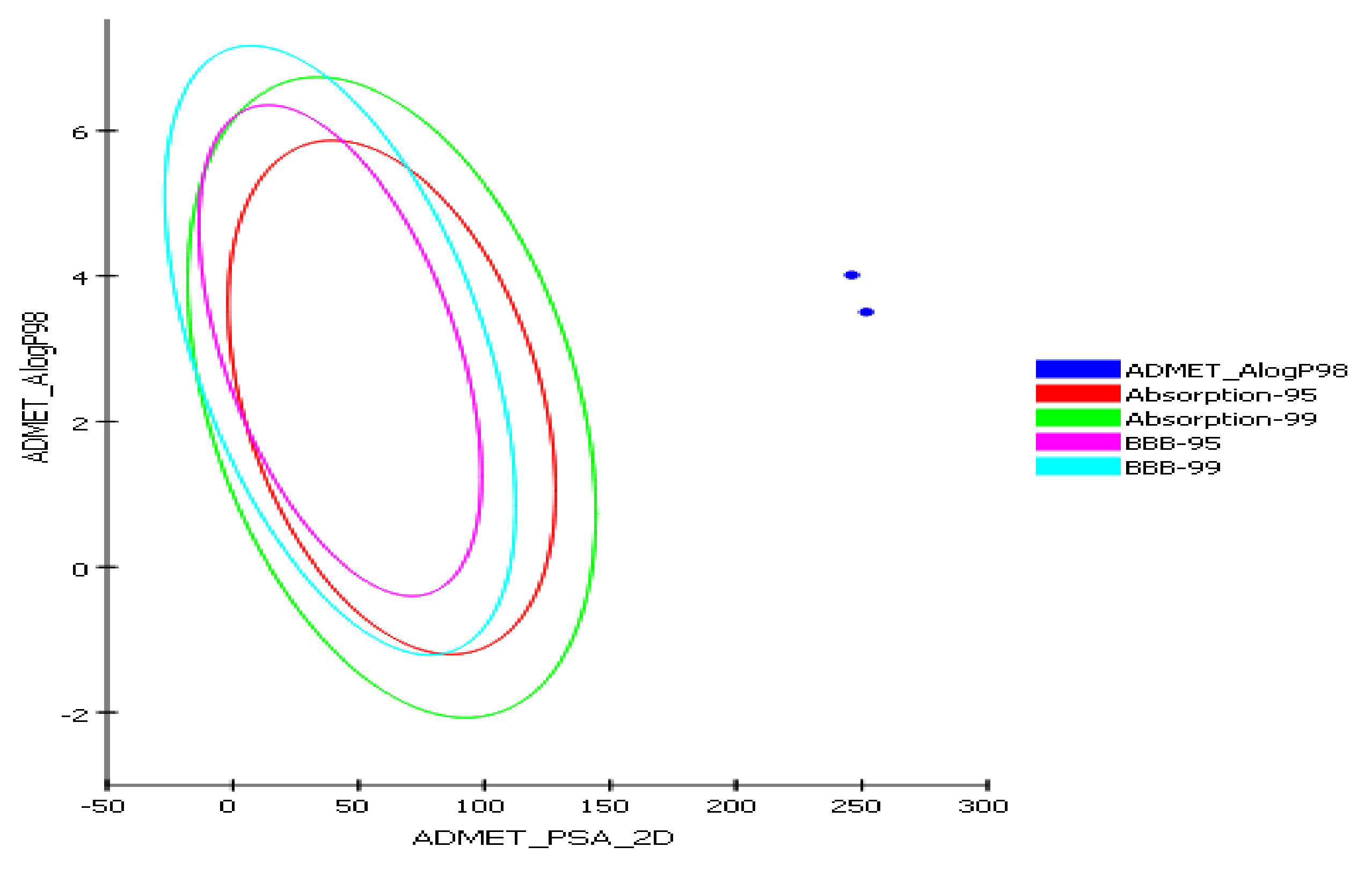
2.4.1. In Silico Predictive ADME Screening for 2a and 7
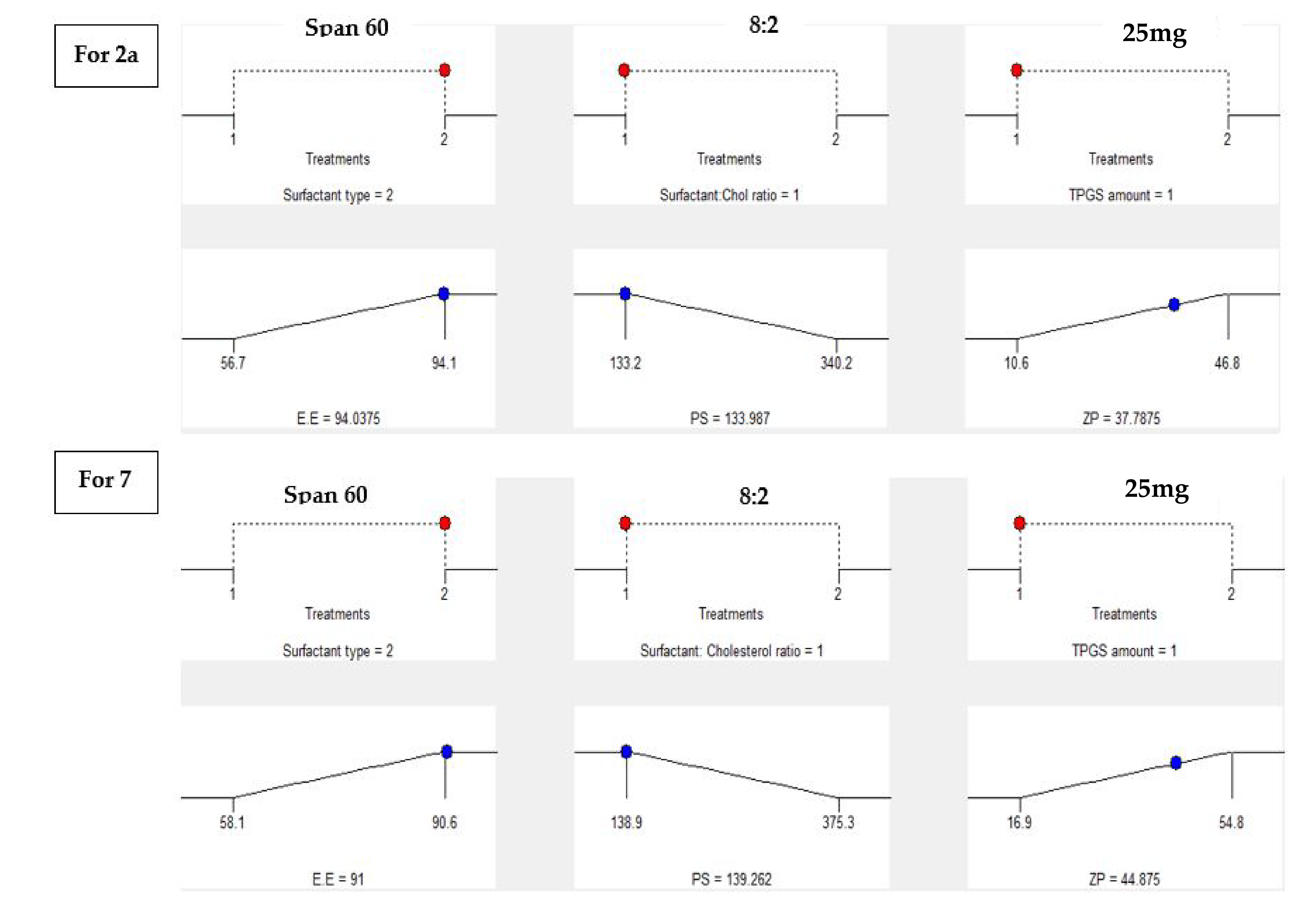
2.4.2. Construction of Experimental Design, Formulation and Statistical Assessment of Both 2a and 7 TPGS Modified Niosomes
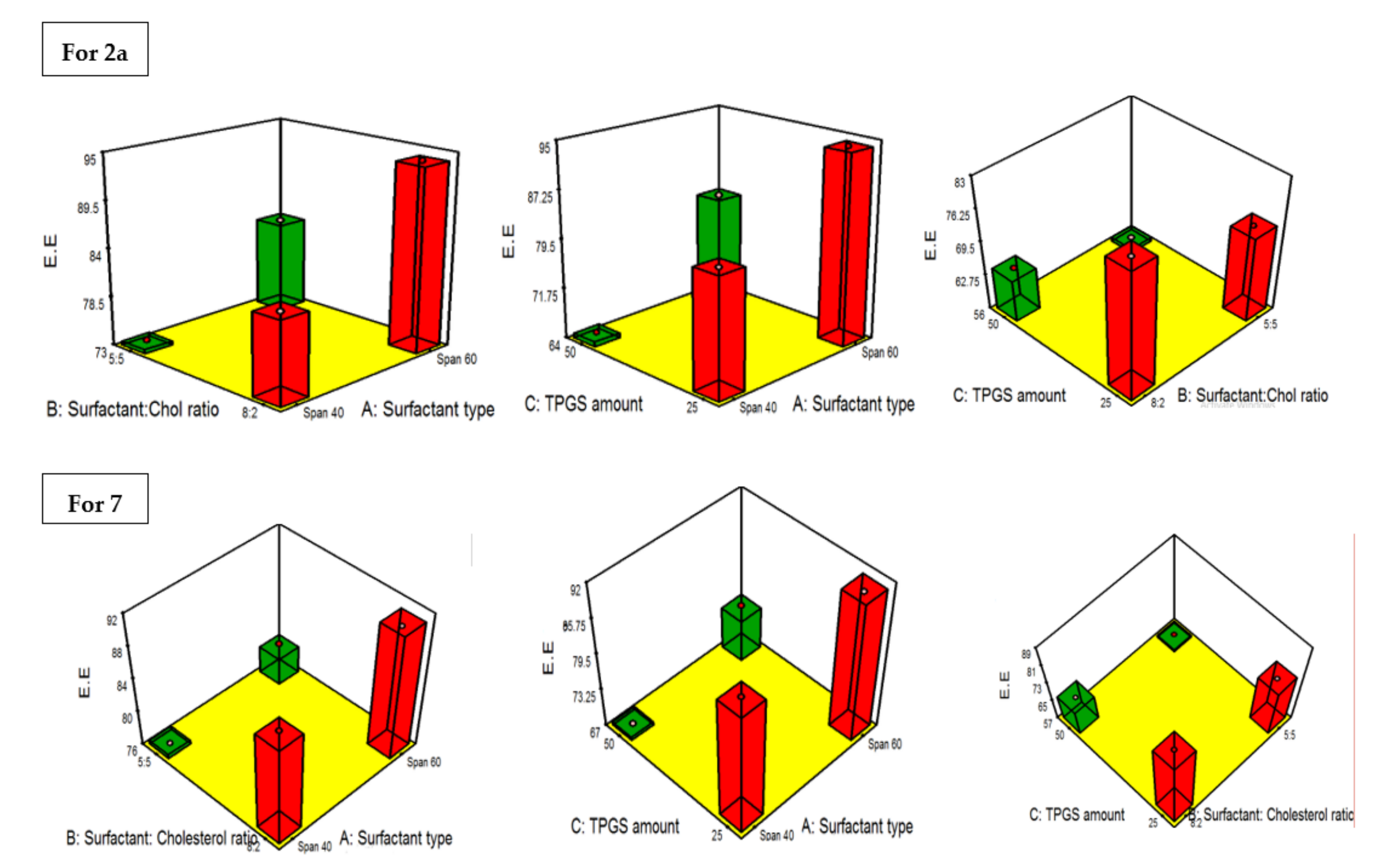
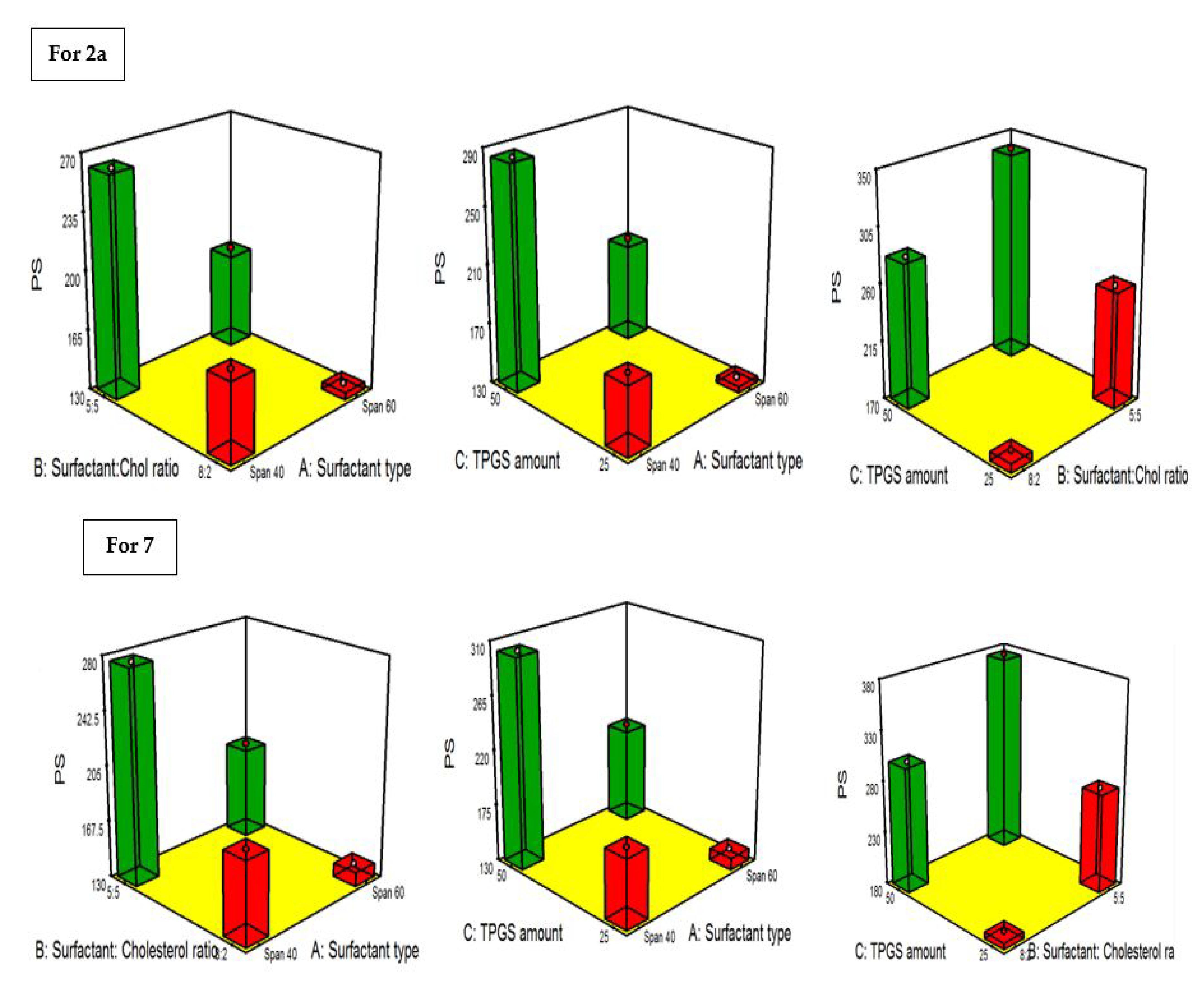
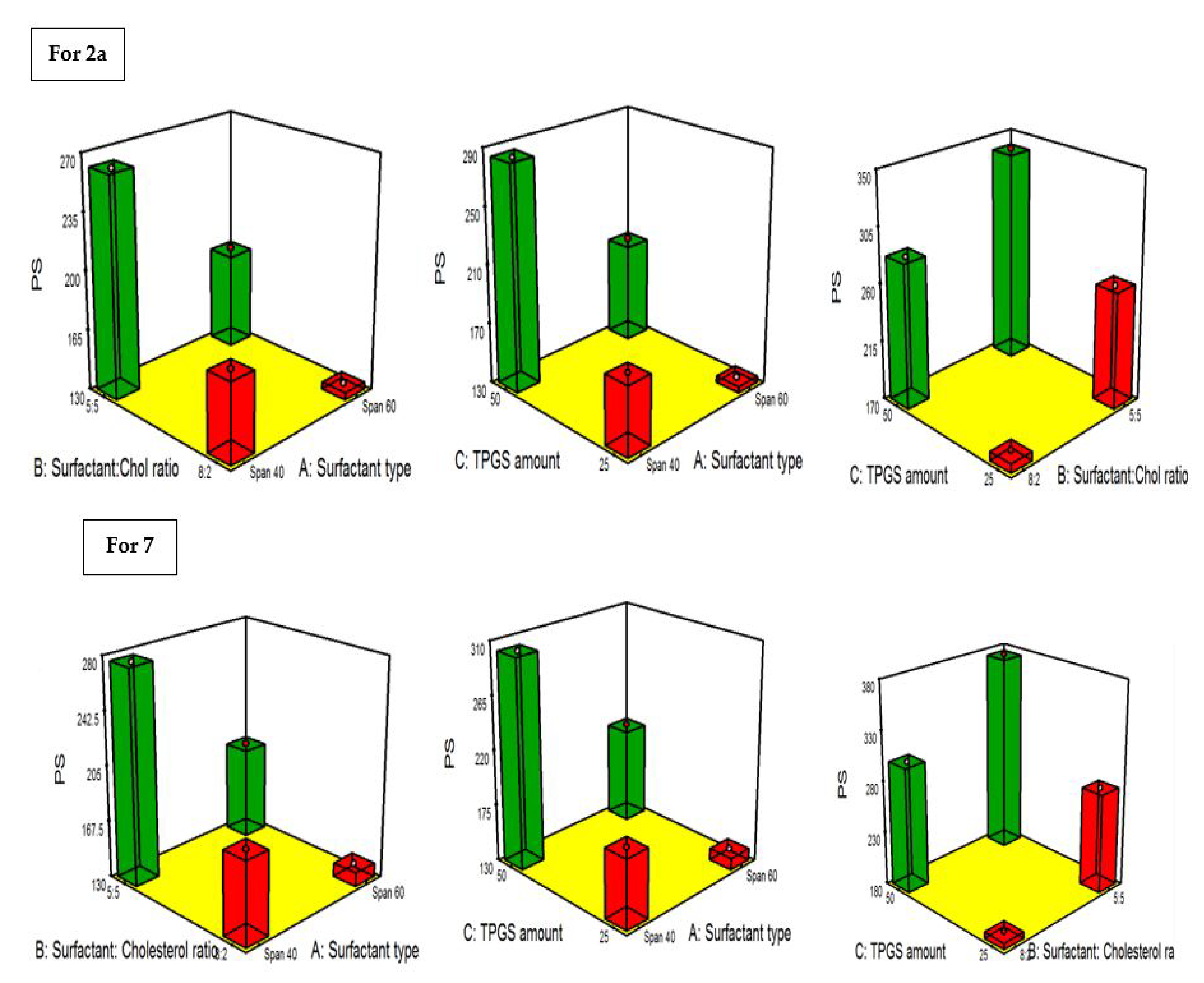
2.4.3. PDI and the Influence of the Fabrication Variables on PS
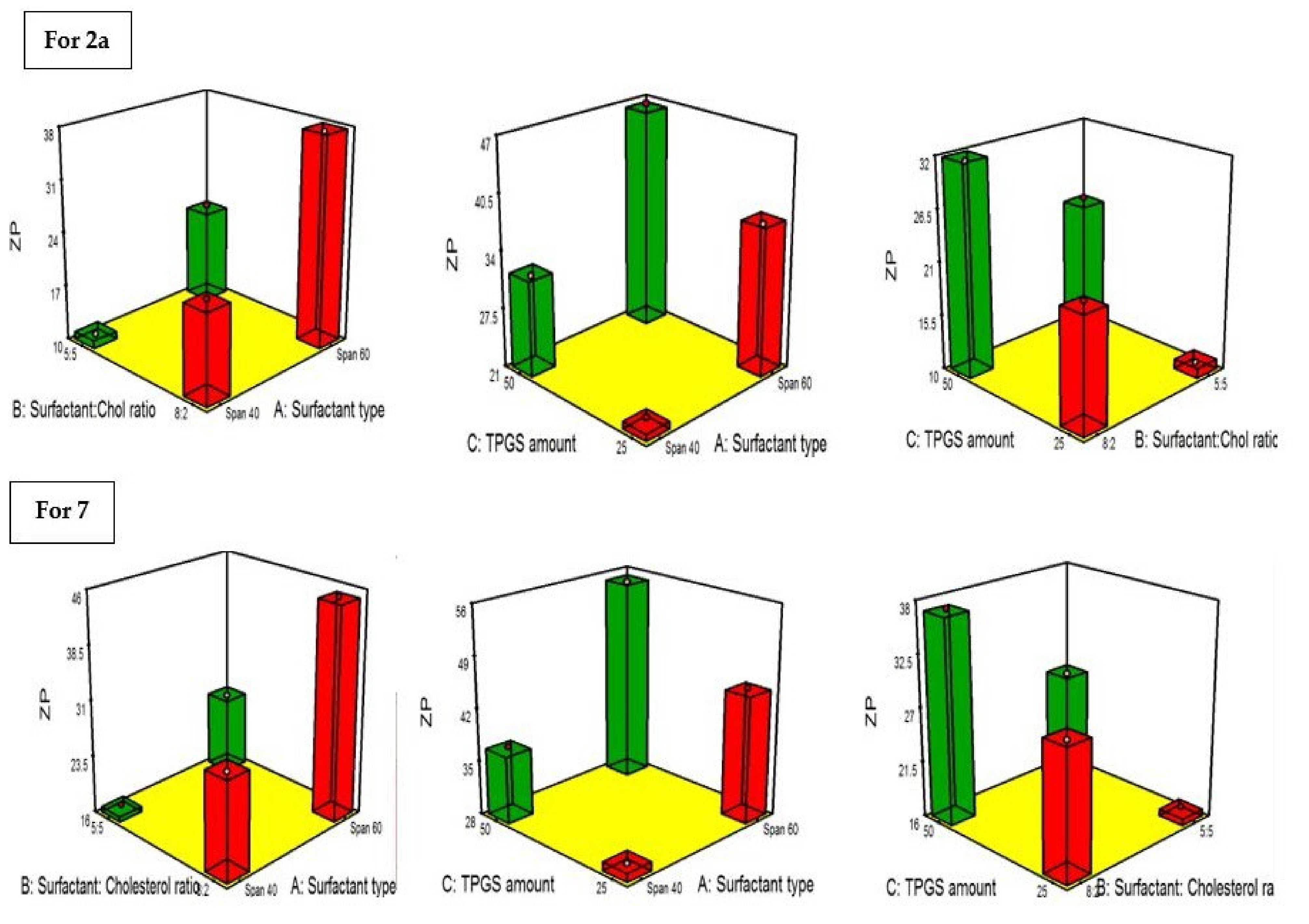
2.4.4. Influence of the Compounding Variables on ZP
2.4.5. Statistical Optimization and Validation of the Optimal of Both 2a and 7-Loaded TPGS Modified Niosomes
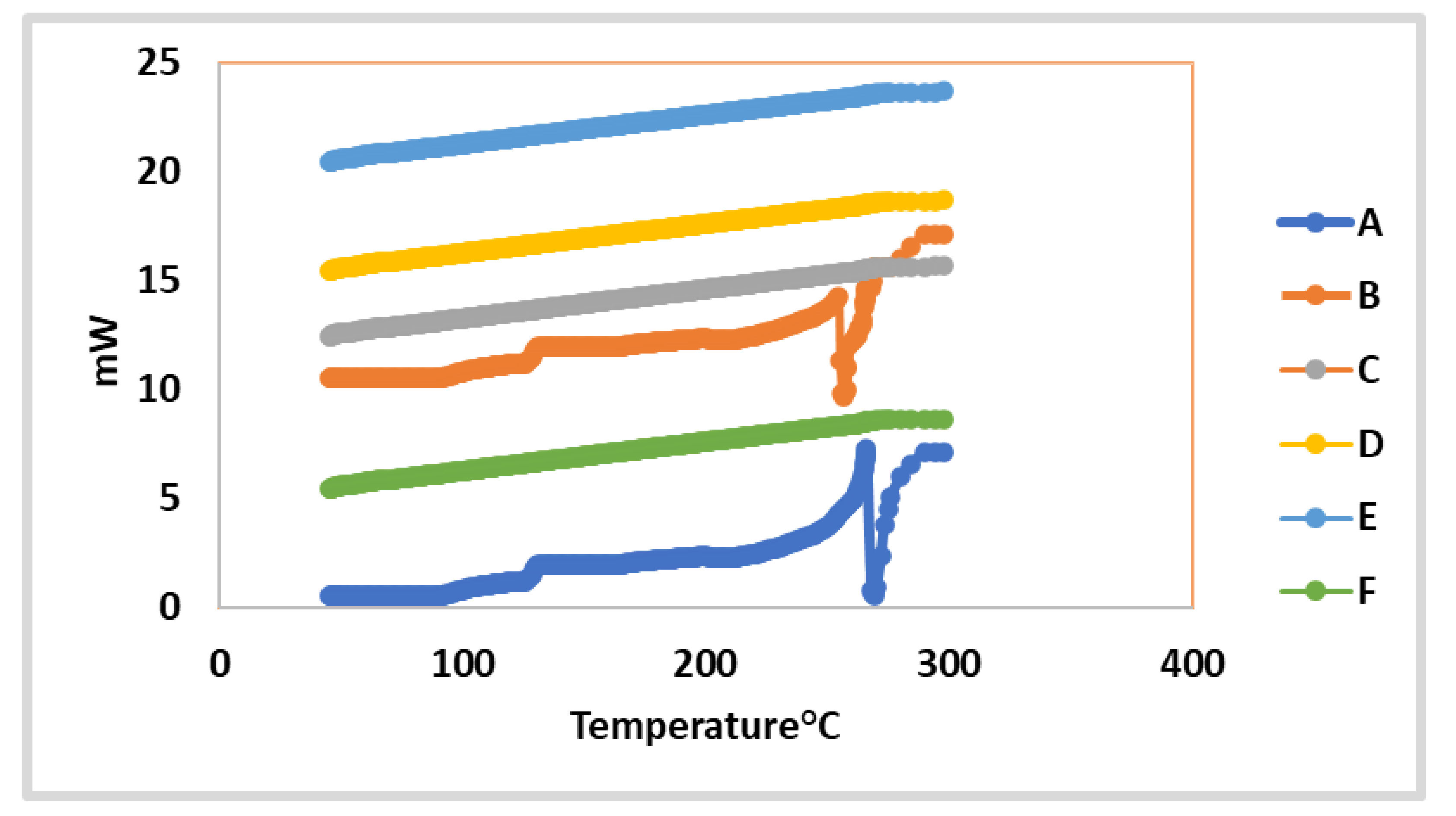
2.4.6. In vitro Investigation of the Optimized 2a and 7-Loaded TPGS-modified Niosome Differential Scanning Calorimetry (DSC)
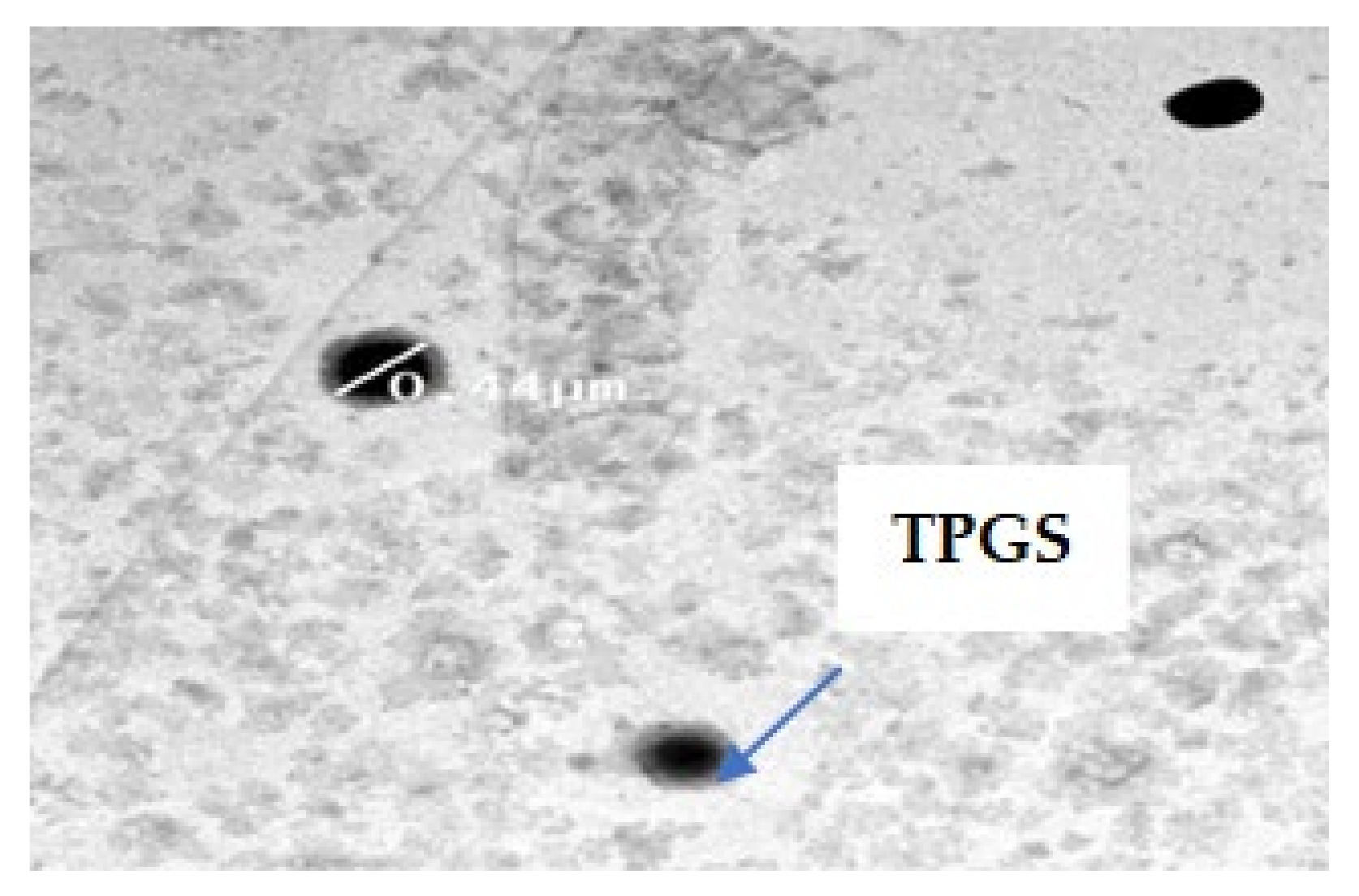
2.4.7. Transmission Electron Microscope TEM
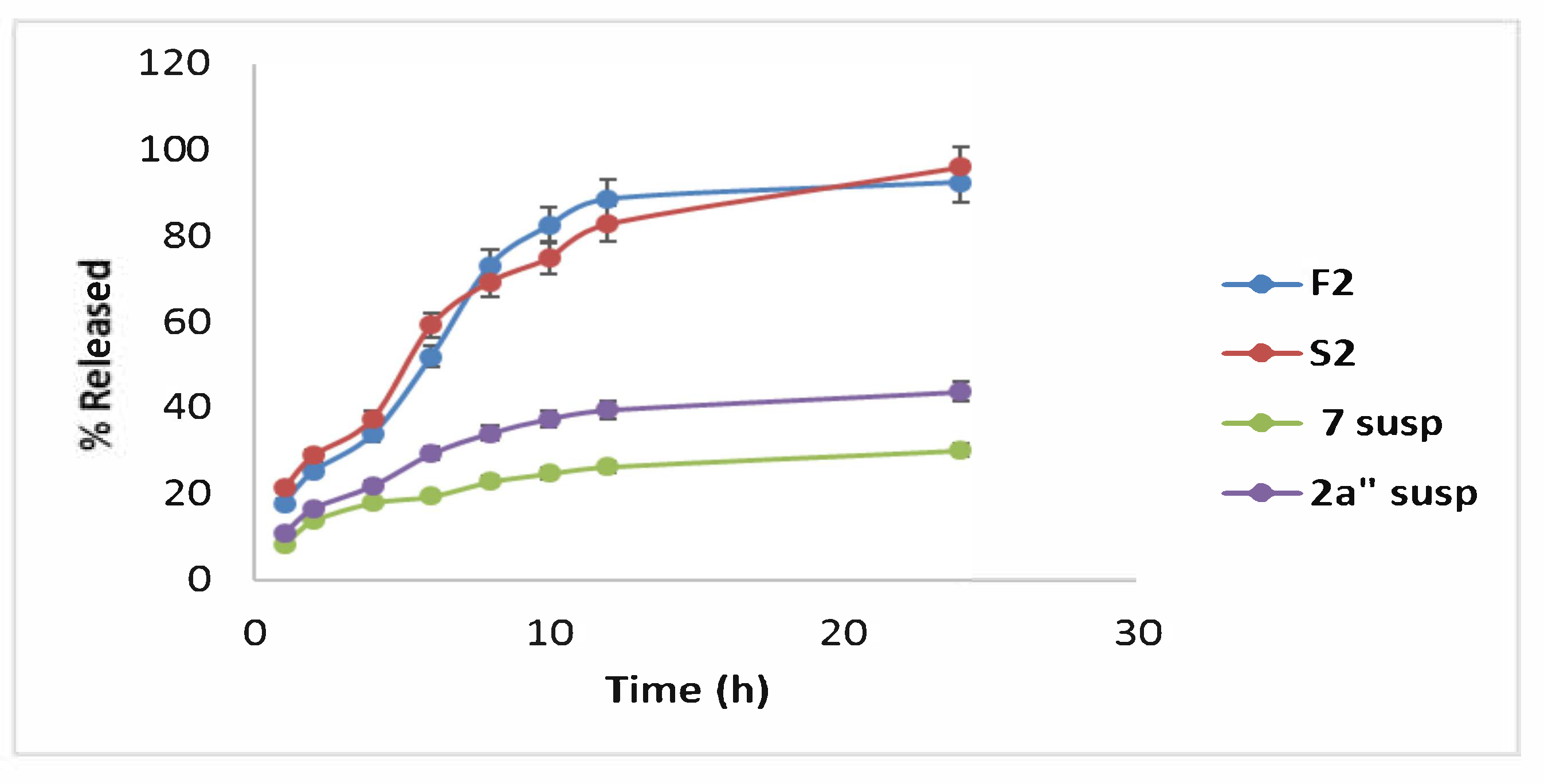
2.4.8. In Vitro Drug Release of Optimal Formula (F2 and S2) Correlated to Compound 2a and 7 Suspensions
2.5. A Comparative Study of the Optimized Formula (F2 and S2) versus the Uncoated 2a and 7-Loaded Conventional Niosomes
2.5.1. Cytotoxic Activity
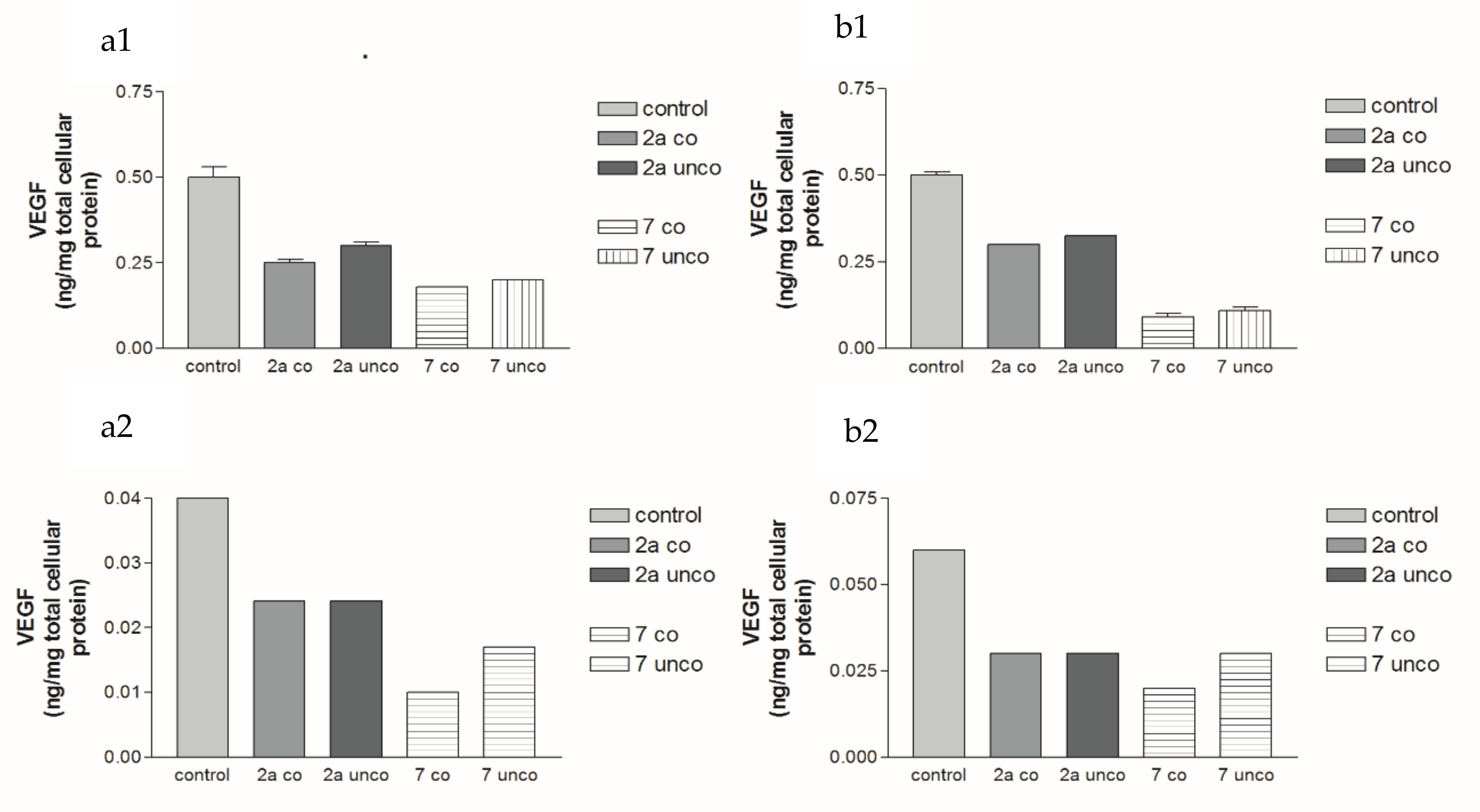
2.5.2. Effects of 2a- and 7-Loaded Niosomes (F2 and S2) on VEGF Protein Expression Levels in Both HepG2 and MCF-7 Cell Lines
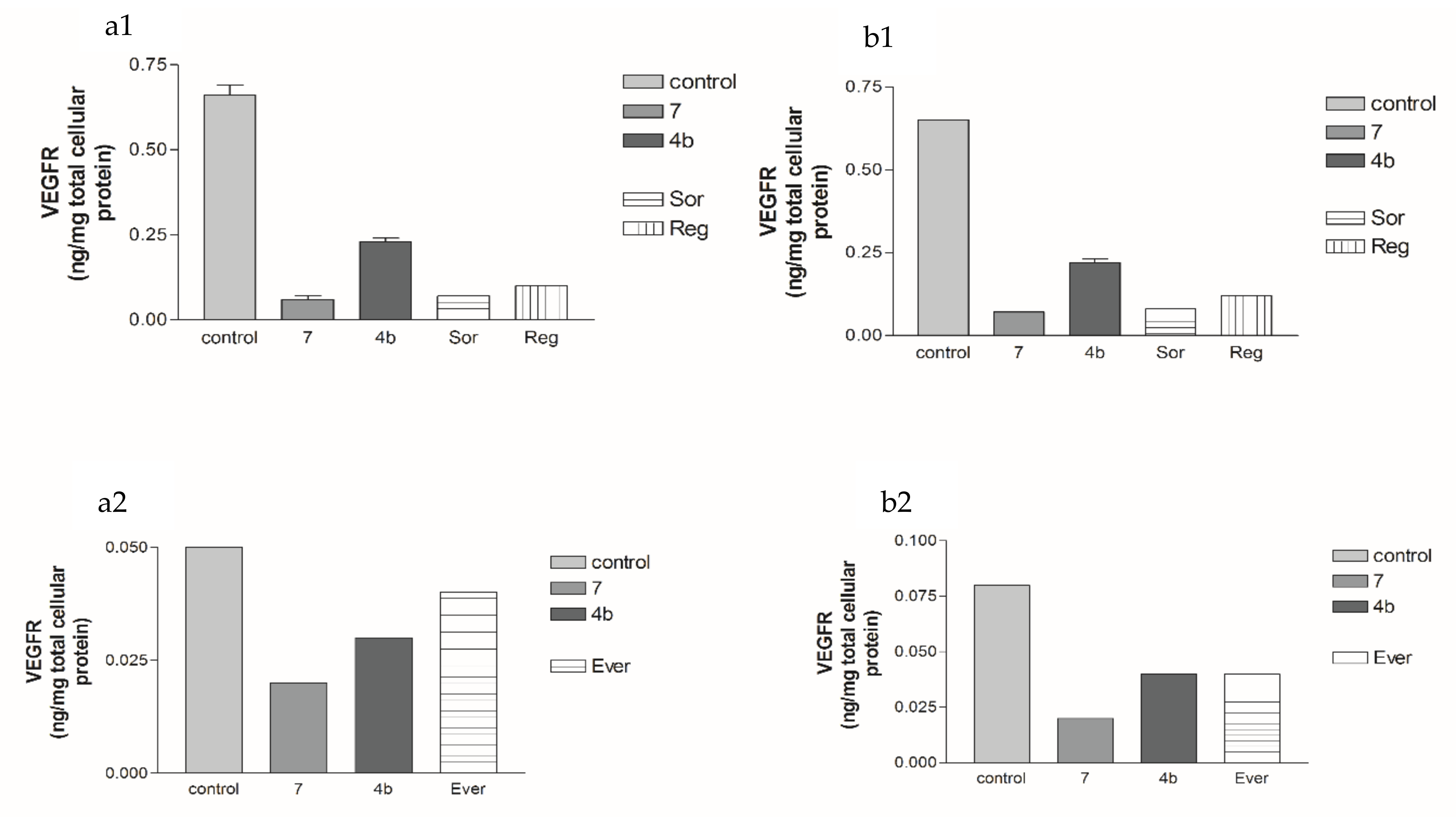
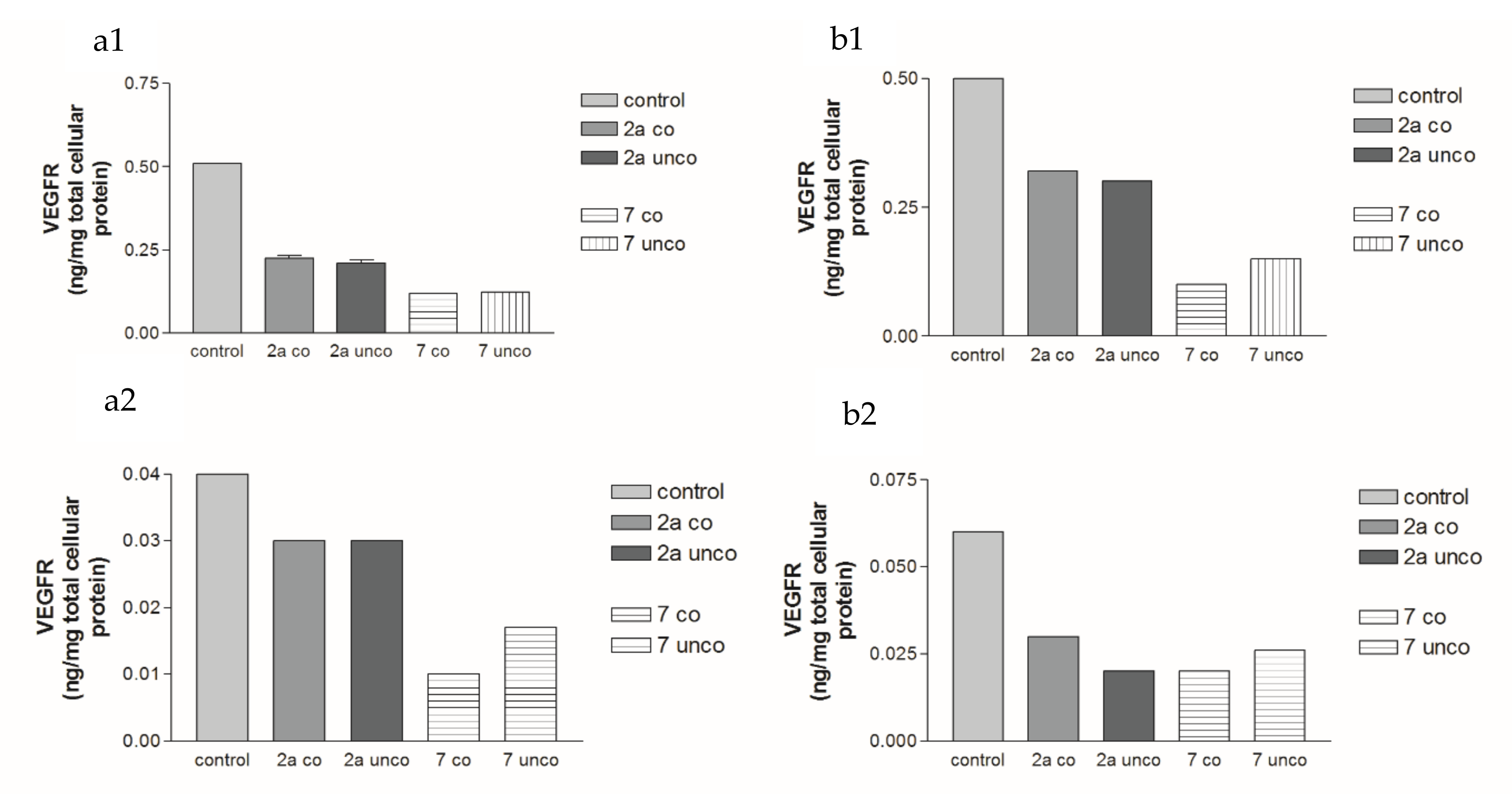
2.5.3. Effects of 2a- and 7-Loaded Niosomes (F2 and S2) on VEGFR Protein Expression Levels in Both HepG2 and MCF-7 Cell Lines
2.5.4. Effects of 2a- and 7-Loaded Niosomes (F2 and S2) on VEGF Gene Expression Level in Both HepG2 and MCF-7 Cell Lines
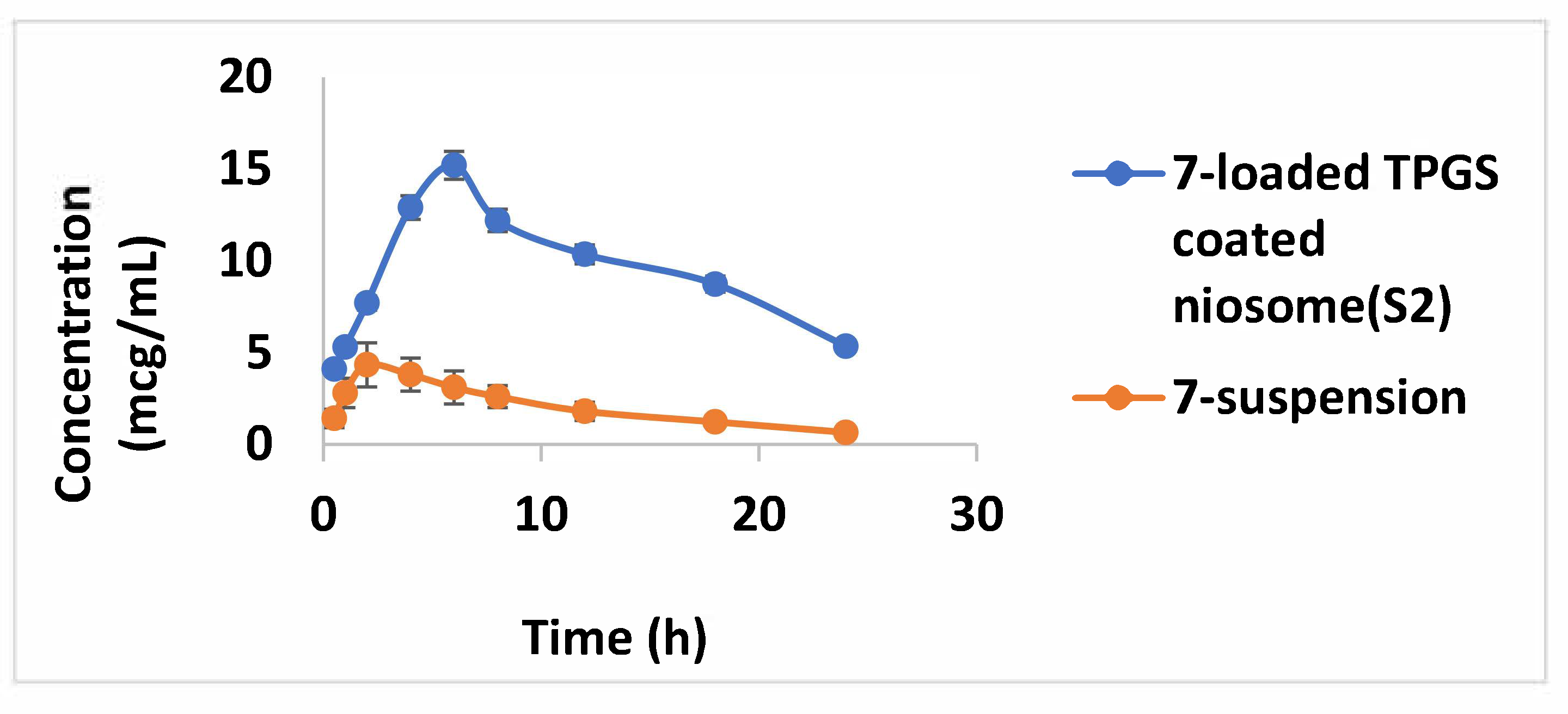
2.6. Pharmacokinetic Study
3. Materials and Methods
3.1. Chemistry
3.1.1. Materials and Methods
3.1.2. Synthesis of Compounds (2a, b), General Procedure
4-((2,7-Diamino-5-hydroxy-3-(4-tolyldiazenyl)pyrazolo[1,5-a]pyrimidin-6-yl)diazenyl)-N-(diaminomethylene)benzenesulfonamide (2a)
7-Diamino-6-((3-nitrophenyl)diazenyl)-3-(4-tolyldiazenyl)pyrazolo[1,5-a]pyrimidin-5-ol (2b)
3.1.3. Synthesis of Compounds (3a, b), General Procedure
4-((2-Amino-5,7-dimethyl-3-(4-tolyldiazenyl)pyrazolo[1,5-a]pyrimidin-6-yl)diazenyl)-N-(diaminomethylene)benzenesulfonamide (3a)
5,7-Dimethyl-6-((3-nitrophenyl)diazenyl)-3-(4-tolyldiazenyl)pyrazolo[1,5-a]pyrimidin-2-amine (3b)
3.1.4. Synthesis of Compounds (4a, b), General Procedure
4-((2-Amino-7-methyl-5-oxo-3-(4-tolyldiazenyl)-4,5-dihydropyrazolo[1,5-a]pyrimidin-6-yl)diazenyl)-N-(diaminomethylene)benzenesulfonamide (4a)
2-Amino-7-methyl-6-((3-nitrophenyl)diazenyl)-3-(4-tolyldiazenyl)pyrazolo[1,5-a]pyrimidin-5(4H)-one (4b)
3.1.5. 4-(2-(2-Amino-5,7-Dioxo-3-(4-Tolyldiazenyl)-4,5-Dihydropyrazolo[1,5-a]Pyrimidin-6(7H)-Ylidene)Hydrazinyl)-N-(Diaminomethylene)Benzenesulfonamide (5)
3.1.6. 2,7-Diamino-5-(4-Methoxyphenyl)-3-(4-Tolyldiazenyl)Pyrazolo[1,5-a]Pyrimidine-6-Carbonitrile (6)
3.1.7. N-(Diaminomethylene)-4-((2,5,7-Triamino-3-(4-Tolyldiazenyl)Pyrazolo[1,5-a]Pyrimidin-6-yl)Diazenyl)Benzenesulfonamide (7)
3.2. Biological Activity
3.2.1. Cell Lines
- HepG2 [HEPG2] (ATCC® HB-8065™, Manassas, VA, USA) epithelial hepatocellular carcinoma was derived from the liver tissue of a 15-year-old Caucasian male (a well-differentiated hepatocellular carcinoma). The human HCC cell line, HEPG-2, was obtained from the American Type Culture Collection and used to evaluate the effects of the following drugs. MCF-7(ATCC® HTB-22™, Manassas, VA, USA) breast cancer cells were derived from mammary gland or breast at a metastatic site. The human MCF-7 cell line, MCF-7, was obtained from the American Type Culture Collection and used to evaluate the effects of the following drugs.
- Caco 2 [Caco2] (ATCC® HTB 37™, Manassas, VA, USA) epithelial colorectal adenocarcinoma was derived from the colon at a metastatic site. The human Caco-2 cell line, Caco-2, was obtained from the American Type Culture Collection and used to evaluate the effects of the following drugs. A549 (ATCC® CCL-185™, Manassas, VA, USA), epithelial-like lung carcinoma, was derived from lung tissue at a metastatic site. The human A549 cell line, A549, was obtained from the American Type Culture Collection and used to evaluate the effects of the following drugs.
3.2.2. Methods
Cell Cultures
In Vitro (Growth Inhibition) Cytotoxicity Study
Determination of Total Protein by the Bradford Assay
Determination of VEGF in HEPG2 and MCF-7 Cell Culture Supernatant
Determination of Human VEGF Receptor-2 ELISA Kit (ab213476) in HEPG2 and MCF-7 Cells Culture Supernatant
Real-Time PCR for Determination of the VEGF Gene Expression Level
3.3. Molecular Docking
3.3.1. Ligand Preparation
3.3.2. Protein Preparation and Docking Process
3.4. Construction of Both 2a- and 7-Loaded TPGS-Modified Niosome
3.5. HPLC Investigation
3.6. In Vitro Investigation and Optimization of 2a- and 7-Loaded TPGS Modified Niosomes
3.6.1. Characterization of the Entrapment Efficiency Percentage (EE%)
3.6.2. Characterization of Zeta Potential, Vesicle Size and PDI
3.6.3. Construction of Experimental Design and Election of Both the Optimal 2a- and 7-Loaded TPGS Niosome
3.7. In Vitro Characterization of the Optimum 2a- or 7-Loaded TPGS Modified Niosomal Formula
3.7.1. Lyophilization of the Optimized Formula
3.7.2. Differential Scanning Calorimetry (DSC)
3.7.3. Transmission Electron Microscopy (TEM)
3.7.4. In Vitro Release Study of the Optimal Formula
3.8. Pharmacokinetic Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shibuya, M. Vascular endothelial growth factor and its receptor system: Physiological functions in angiogenesis and pathological roles in various diseases. J. Biochem. 2012, 153, 13–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaik, F.; Cuthbert, G.A.; Homer-Vanniasinkam, S.; Muench, S.P.; Ponnambalam, S.; Harrison, M.A. Structural Basis for Vascular Endothelial Growth Factor Receptor Activation and Implications for Disease Therapy. Biomolecules 2020, 10, 1673. [Google Scholar] [CrossRef]
- Reddy, V.G.; Reddy, T.S.; Jadala, C.; Reddy, M.S.; Sultana, F.; Akunuri, R.; Bhargava, S.K.; Wlodkowic, D.; Srihari, P.; Kamal, A. Pyrazolo-benzothiazole hybrids: Synthesis, anticancer properties and evaluation of antiangiogenic activity using in vitro VEGFR-2 kinase and in vivo transgenic zebrafish model. Eur. J. Med. Chem. 2019, 182, 111609. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.J.; Kulkarni, V.M. Vascular Endothelial Growth Factor Receptor (VEGFR-2)/KDR Inhibitors: Medicinal Chemistry Perspective. Med. Drug Discov. 2019, 2, 100009. [Google Scholar] [CrossRef]
- Ferrara, N.; Adamis, A.P. Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug Discov. 2016, 15, 385–403. [Google Scholar] [CrossRef] [Green Version]
- Gotink, K.J.; Verheul, H.M. Anti-angiogenic tyrosine kinase inhibitors: What is their mechanism of action? Angiogenesis 2010, 13, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, A.L.; Kang, Y.K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.-S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Zhu, Y.-J.; Zheng, B.; Wang, H.-Y.; Chen, L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol. Sin. 2017, 38, 614–622. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, S.; Carter, C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R.A.; Schwartz, B.; Simantov, R.; Kelley, S. Discovery and development of sorafenib: A multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 2006, 5, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Tak, W.Y.; Gasbarrini, A.; Santoro, A.; Colombo, M.; Lim, H.-Y.; Mazzaferro, V.M.; Wiest, R.; Reig, M.; Wagner, A.; et al. Regorafenib as second-line therapy for intermediate or advanced hepatocellular carcinoma: Multicentre, open-label, phase II safety study. Eur. J. Cancer 2013, 49, 3412–3419. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Zhang, S.; Ma, D.; Yan, D.; Zhang, G.; Cao, Y.; Wang, Z.; Wu, J.; Jiang, C. Targeting SphK2 Reverses Acquired Resistance of Regorafenib in Hepatocellular Carcinoma. Front. Oncol. 2020, 10, 694. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Jeong, K.-W.; Lee, Y.; Song, J.Y.; Kim, M.S.; Lee, G.S.; Kim, Y. Pharmacophore modeling and virtual screening studies for new VEGFR-2 kinase inhibitors. Eur. J. Med. Chem. 2010, 45, 5420–5427. [Google Scholar] [CrossRef]
- Machado, V.A.; Peixoto, D.; Costa, R.; Froufe, H.J.; Calhelha, R.C.; Abreu, R.M.; Queiroz, M.J.R. Synthesis, antiangiogenesis evaluation and molecular docking studies of 1-aryl-3-[(thieno [3, 2-b] pyridin-7-ylthio) phenyl] ureas: Discovery of a new substitution pattern for type II VEGFR-2 Tyr kinase inhibitors. Bioorganic Med. Chem. 2015, 23, 6497–6509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdallah, A.E.; Eissa, S.I.; Al Ward, M.M.S.; Mabrouk, R.R.; Mehany, A.B.; El-Zahabi, M.A.A. Design, synthesis and molecular modeling of new quinazolin-4(3H)-one based VEGFR-2 kinase inhibitors for potential anticancer evaluation. Bioorganic Chem. 2021, 109, 104695. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.R.; El Kerdawy, A.M.; George, R.F.; Georgey, H.H.; Gawad, N.M.A. Design, synthesis and in silico insights of new 7, 8-disubstituted-1, 3-dimethyl-1H-purine-2, 6 (3H, 7H)-dione derivatives with potent anticancer and multi-kinase inhibitory activities. Bioorganic Chem. 2021, 107, 104569. [Google Scholar] [CrossRef] [PubMed]
- Saleh, N.M.; El-Gaby, M.S.; El-Adl, K.; El-Sattar, N.E.A. Design, green synthesis, molecular docking and anticancer evaluations of diazepam bearing sulfonamide moieties as VEGFR-2 inhibitors. Bioorganic Chem. 2020, 104, 104350. [Google Scholar] [CrossRef]
- Elbayoumi, T.A.; Torchilin, V.P. Current trends in liposome research. Methods Mol. Biol. 2010, 605, 1–27. [Google Scholar] [PubMed]
- Muthu, M.S.; Wilson, B. Multifunctional radionanomedicine: A novel nanoplatform for cancer imaging and therapy. Nanomedicine 2010, 5, 169–171. [Google Scholar] [CrossRef] [PubMed]
- Sayed, G.H.; Azab, M.E.; Anwer, K.E.; Raouf, M.A.; Negm, N.A. Pyrazole, pyrazolone and enaminonitrile pyrazole derivatives: Synthesis, characterization and potential in corrosion inhibition and antimicrobial applications. J. Mol. Liq. 2018, 252, 329–338. [Google Scholar] [CrossRef]
- Karcı, F.; Demirçalı, A. Synthesis of disazo pyrazolo [1, 5-a] pyrimidines. Dye. Pigment. 2007, 74, 288–297. [Google Scholar] [CrossRef]
- Soliman, A.M.M.; Abd El Aleem, M.; El-Remaily, A.A.; Sultan, A.A.; Abdel-Ghany, H. Synthesis of Some Novel Imidazopyrazole and Pyrazolopyrimidine Derivatives. J. Heterocycl. Chem. 2014, 51, 1476–1481. [Google Scholar] [CrossRef]
- Abd El Aleem, M.; El-Remaily, A.A. Synthesis of pyranopyrazoles using magnetic Fe3O4 nanoparticles as efficient and reusable catalyst. Tetrahedron 2014, 70, 2971–2975. [Google Scholar]
- Sayed, A.Z.; Aboul-Fetouh, M.S.; Nassar, H.S. Synthesis, biological activity and dyeing performance of some novel azo disperse dyes incorporating pyrazolo [1, 5-a] pyrimidines for dyeing of polyester fabrics. J. Mol. Struct. 2012, 1010, 146–151. [Google Scholar] [CrossRef]
- El-Sayed, E.H.; Mohamed, K.S. Synthesis and Anti-inflammatory Evaluation of Some New Pyrazole, Pyrimidine, Pyrazolo[1,5-a]Pyrimidine, Imidazo[1,2-b]Pyrazole and Pyrazolo[5,1-b]Quinazoline Derivatives Containing Indane Moiety. Polycycl. Aromat. Compd. 2019, 41, 1077–1093. [Google Scholar] [CrossRef]
- Said, M.A.; Eldehna, W.M.; Nocentini, A.; Fahim, S.H.; Bonardi, A.; Elgazar, A.A.; Kryštof, V.; Soliman, D.H.; Abdel-Aziz, H.A.; Gratteri, P.; et al. Sulfonamide-based ring-fused analogues for CAN508 as novel carbonic anhydrase inhibitors endowed with antitumor activity: Design, synthesis, and in vitro biological evaluation. Eur. J. Med. Chem. 2020, 189, 112019. [Google Scholar] [CrossRef] [PubMed]
- Tsai, P.C.; Wang, J. Synthesis and solvatochromic properties of some disazo dyes derived from pyrazolo [1, 5-a] pyrimidine derivatives. Dye. Pigment. 2005, 64, 259–264. [Google Scholar] [CrossRef]
- Sayed, G.H.; Azab, M.E.; Anwer, K.E. Conventional and Microwave-Assisted Synthesis and Biological Activity Study of Novel Heterocycles Containing Pyran Moiety. J. Heterocycl. Chem. 2019, 56, 2121–2133. [Google Scholar] [CrossRef]
- Anwer, K.; Sayed, G.; Hassanan, H.; Azab, M. Conventional and Microwave Synthesis of Some New Pyridine Derivatives and Evaluation Their Antimicrobial and Cytotoxic Activities. Egypt. J. Chem. 2019, 62, 707–726. [Google Scholar] [CrossRef]
- Anwer, K.E.; Sayed, G.H. Conventional and microwave reactions of 1, 3-diaryl-5, 4-enaminonitrile-pyrazole derivative with expected antimicrobial and anticancer activities. J. Heterocycl. Chem. 2020, 57, 2339–2353. [Google Scholar] [CrossRef]
- McTigue, M.; Murray, B.W.; Chen, J.H.; Deng, Y.L.; Solowiej, J.; Kania, R.S. Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors. Proc. Natl. Acad. Sci. USA 2012, 109, 18281–18289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dassault Systèmes BIOVIA. Discovery Studio Modeling Environment, Release 2019; Dassault Systèmes: San Diego, CA, USA, 2019. [Google Scholar]
- Muthu, M.S.; Kulkarni, S.A.; Xiong, J.; Feng, S.-S. Vitamin E TPGS coated liposomes enhanced cellular uptake and cytotoxicity of docetaxel in brain cancer cells. Int. J. Pharm. Dec. 2011, 421, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Alemi, A.; Zavar Reza, J.; Haghiralsadat, F.; Zarei Jaliani, H.; Haghi Karamallah, M.; Hosseini, S.A.; Haghi Karamallah, S. Paclitaxel and curcumin coadministration in novel cationic PEGylated niosomal formulations exhibit enhanced synergistic antitumor efficacy. J. Nanobiotechnol. 2018, 16, 28. [Google Scholar] [CrossRef] [PubMed]
- Abdelkader, H.; Farghaly, U.; Moharram, H. Effects of surfactant type and cholesterol level on niosomes physical properties and in vivo ocular performance using timolol maleate as a model drug. J. Pharm. Investig. 2014, 44, 329–337. [Google Scholar] [CrossRef]
- Bnyan, R.; Khan, I.; Ehtezazi, T.; Saleem, I.; Gordon, S.; Neill, F.O.; Roberts, M. Surfactant Effects on Lipid-Based Vesicles Properties. J. Pharm. Sci. 2018, 107, 1237–1246. [Google Scholar] [CrossRef]
- Fouda, N.H.; Abdelrehim, R.T.; Hegazy, D.A.; Habib, B.A. Sustained ocular delivery of Dorzolamide-HCl via proniosomal gel formulation: In-vitro characterization, statistical optimization, and in-vivo pharmacodynamic evaluation in rabbits. Drug Deliv. 2018, 25, 1340–1349. [Google Scholar] [CrossRef] [Green Version]
- Zakaria, M.Y.; Fayad, E.; Althobaiti, F.; Zaki, I.; Abu Almaaty, A.H. Statistical optimization of bile salt deployed nanovesicles as a potential platform for oral delivery of piperine: Accentuated antiviral and anti-inflammatory activity in MERS-CoV challenged mice. Drug Deliv. 2021, 28, 1150–1165. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Fu, T.; Fei, W.; Han, T.; Gu, X.; Hou, Y.; Liu, Y.; Yang, J. Vitamin E D-alpha-tocopheryl polyethylene glycol 1000 succinate-conjugated liposomal docetaxel reverses multidrug resistance in breast cancer cells. J. Pharm. Pharmacol. 2019, 71, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Stetefeld, J.; McKenna, S.A.; Patel, T.R. Dynamic light scattering: A practical guide and applications in biomedical sciences. Biophys. Rev. 2016, 8, 409–427. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, T.; Sternberg, B.; Florence, A.T. Preparation and properties of vesicles (niosomes) of sorbitan monoesters (Span 20, 40, 60 and 80) and a sorbitan triester (Span 85). Int. J. Pharm. 1994, 105, 1–6. [Google Scholar] [CrossRef]
- Essa, E. Effect of formulation and processing variables on the particle size of sorbitan monopalmitate niosomes. Asian J. Pharm. 2010, 4, 227. [Google Scholar] [CrossRef]
- Cheng, B.; Pan, H.; Liu, D.; Li, D.; Li, J.; Yu, S.; Tan, G.; Pan, W. Functionalization of nanodiamond with vitamin E TPGS to facilitate oral absorption of curcumin. Int. J. Pharm. 2018, 540, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Aziz, D.E.; Abdelbary, A.A.; Elassasy, A.I. Investigating superiority of novel bilosomes over niosomes in the transdermal delivery of diacerein: In vitro characterization, ex vivo permeation and in vivo skin deposition study. J. Liposome Res. 2019, 29, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Owodeha-Ashaka, K.; Ilomuanya, M.O.; Iyire, A. Evaluation of sonication on stability-indicating properties of optimized pilocarpine hydrochloride-loaded niosomes in ocular drug delivery. Prog. Biomater. 2021, 10, 207–220. [Google Scholar] [CrossRef]
- Albash, R.; El-Nabarawi, M.A.; Refai, H.; Abdelbary, A.A. Tailoring of PEGylated bilosomes for promoting the transdermal delivery of olmesartan medoxomil: Invitro characterization, ex-vivo permeation and in-vivo assessment. Int. J. Nanomed 2019, 14, 6555–6574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pachauri, M.; Gupta, E.D.; Ghosh, P.C. Piperine loaded PEG-PLGA nanoparticles: Preparation, characterization and targeted delivery for adjuvant breast cancer chemotherapy. J. Drug Deliv. Sci. Technol. 2015, 29, 269–282. [Google Scholar] [CrossRef]
- Kalepu, S.; Nekkanti, V. Insoluble drug delivery strategies: Review of recent advances and business prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Tan, S.; Guo, Y.; Huang, J.; Chu, M.; Liu, H.; Leporatti, S.; Chen, J.; Hu, Y. pH-Sensitive Docetaxel-Loaded D -α-Tocopheryl Polyethylene Glycol Succinate–Poly(β-amino ester) Copolymer Nanoparticles for Overcoming Multidrug Resistance. Biomacromolecules 2013, 14, 2636–2646. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.M. Tetrazolium (MTT) assay for cellular viability and activity. In Polyamine Protocols; Humana Press: Totowa, NJ, USA, 1998; pp. 179–184. [Google Scholar]
- Kruger, N.J. The Bradford method for protein quantitation. In The Protein Protocols Handbook; Humana Press: Totowa, NJ, USA, 2009; pp. 17–24. [Google Scholar]
- Simpson, D.A.; Feeney, S.; Boyle, C.; Stitt, A.W. Technical brief: Retinal VEGF mRNA measured by SYBR green I fluorescence: A versatile approach to quantitative PCR. Mol. Vis. 2000, 6, 178–183. [Google Scholar] [PubMed]
- Nita, A.; Tit, D.M.; Copolovici, L.; Frunzulica, C.E.M.; Copolovici, D.M.; Bungau, S.; Iovan, C. HPLC-UV Method for Determination of Famotidine from Pharmaceutical Products. Rev. Chim. 2018, 69, 297–299. [Google Scholar] [CrossRef]
- Ammar, H.O.; Ibrahim, M.; Mahmoud, A.A.; Shamma, R.N.; El Hoffy, N.M. Non-ionic Surfactant Based In Situ Forming Vesicles as Controlled Parenteral Delivery Systems. AAPS PharmSciTech 2017, 19, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Aldawsari, H.; Ahmed, O.; Alhakamy, N.; Neamatallah, T.; Fahmy, U.; Badr-Eldin, S. Lipidic Nano-Sized Emulsomes Potentiates the Cytotoxic and Apoptotic Effects of Raloxifene Hydrochloride in MCF-7 Human Breast Cancer Cells: Factorial Analysis and In Vitro Anti-Tumor Activity Assessment. Pharmaceutics 2021, 13, 783. [Google Scholar] [CrossRef] [PubMed]
- Dubey, S.; Vyas, S.P. EMULSOMES FOR LIPOPHILIC ANTICANCER DRUG DELIVERY: DEVELOPMENT, OPTIMIZATION AND IN VITRO DRUG RELEASE KINETIC STUDY. Int. J. Appl. Pharm. 2021, 13, 114–121. [Google Scholar] [CrossRef]
- Abdelbary, G.A.; Amin, M.M.; Zakaria, M.Y.; El Awdan, S.A. Adefovir dipivoxil loaded proliposomal powders with improved hepatoprotective activity: Formulation, optimization, pharmacokinetic, and biodistribution studies. J. Liposome Res. 2017, 28, 259–274. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp No. | Time (min) | Yield (%) | YE | RME | OE | AE | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| M.W. | Con. | M.W. | Con. | M.W. | Con. | M.W. | Con. | M.W. | Con. | ||
| 2a | 2 | 300 | 93 | 66 | 46.5 | 0.22 | 74.75 | 53.05 | 81.52 | 57.85 | 91.70 |
| 2b | 3 | 300 | 94 | 78 | 31.33 | 0.26 | 72.90 | 60.50 | 80.66 | 66.94 | 90.38 |
| 3a | 4 | 420 | 91 | 64 | 22.75 | 0.1524 | 73.06 | 51.38 | 78.26 | 55.04 | 93.35 |
| 3b | 4 | 420 | 95 | 70 | 23.75 | 0.1667 | 73.70 | 54.30 | 79.88 | 58.86 | 92.26 |
| 4a | 2 | 360 | 93 | 79 | 46.5 | 0.2194 | 73.10 | 62.10 | 82.33 | 69.94 | 88.79 |
| 4b | 2 | 360 | 94 | 76 | 47 | 0.2111 | 71.20 | 57.57 | 81.77 | 66.12 | 87.07 |
| 5 | 3 | 480 | 92 | 70 | 30.67 | 0.1458 | 67.97 | 51.71 | 80.26 | 61.06 | 84.69 |
| 6 | 2 | 240 | 96 | 69 | 48 | 0.2875 | 78.30 | 56.27 | 78.69 | 56.55 | 99.5 |
| 7 | 4 | 240 | 92 | 73 | 23 | 0.3042 | 86.54 | 68.67 | 86.54 | 68.67 | 100 |
| Compound Number | HepG2 Livercarcinoma | MCF-7 Breast Cancer | CaCo-2 Colon Cancer | A-549 Lung Carcinoma |
|---|---|---|---|---|
| 1 | 20 | 50 | 18.2 | 15.1 |
| 2a | 6.2 | 20 | 12.5 | 10 |
| 4b | 12.5 | 25 | 15.4 | 14 |
| 7 | 6 | 12 | 6 | 12 |
| Sorafenib | 6 | - | - | - |
| Regorafenib | 3 | - | - | - |
| Everolimus | - | 3 | - | - |
| Comp. | VEGF 24 h | VEGF 72 h | VEGFR-2 24 h | VEGFR-2 72 h | ||||
|---|---|---|---|---|---|---|---|---|
| Conc. (µM) | % Inhibition | Conc. (µM) | % Inhibition | Conc. (µM) | % Inhibition | Conc. (µM) | % Inhibition | |
| 1 | 0.1 | 23% | 0.08 | 33.3% | 0.09 | 52.6% | 0.08 | 38.5% |
| 2a | 0.08 | 38.5% | 0.08 | 33.3% | 0.1 | 47.4% | 0.08 | 38.5% |
| 4b | 0.07 | 46% | 0.07 | 41.7% | 0.11 | 42% | 0.08 | 38.5% |
| 7 | 0.32 | 51.5% | 0.19 | 70.3% | 0.2 | 69.7% | 0.18 | 72.3% |
| Sorafenib | 0.21 | 67.7% | 0.17 | 73.8% | 0.08 | 69% | 0.08 | 70.4% |
| Regorafenib | 0.19 | 70.8% | 0.16 | 75.4% | 0.1 | 62% | 0.12 | 55.6% |
| Comp | VEGF 24 h | VEGF 72 h | VEGFR-2 24 h | VEGFR-2 72 h | ||||
|---|---|---|---|---|---|---|---|---|
| Conc. (µM) | % Inhibition | Conc. (µM) | % Inhibition | Conc. (µM) | % Inhibition | Conc. (µM) | % Inhibition | |
| 1 | 0.04 | 20% | 0.03 | 50% | 0.04 | 33.3% | 0.03 | 50% |
| 2a | 0.03 | 40% | 0.03 | 50% | 0.03 | 50% | 0.03 | 50% |
| 4b | 0.03 | 40% | 0.03 | 50% | 0.03 | 50% | 0.03 | 50% |
| 7 | 0.02 | 50% | 0.03 | 62.5% | 0.02 | 60% | 0.03 | 62.5% |
| Everolimus | 0.02 | 50% | 0.03 | 62.5% | 0.03 | 40% | 0.03 | 50% |
| Comp. No. | Binding Score (kcal/mol) | H-Bonding Interactions |
|---|---|---|
| 7 (pose A) | −43.3 | Glu885, Cys919, Lys920, Gly922 |
| 7 (pose B) | −41.3 | Glu885, Ala866, Gly922, Asp1046 |
| Sorafenib | −44.56 | Glu885, Cys919, Asp1046 |
| Regorafenib | −45.12 | Glu885, Cys919, Asp1046 |
| Parameter | Compound 2a | Compound 7 |
|---|---|---|
| BBB_Lev a | 4 | 4 |
| Absorption Lev b | 3 | 3 |
| AQ SOL Lev c | 0 | 0 |
| Alog P98 d | 4.01 | 3.5 |
| ADME_PSA_lev | 246.06 | 251.78 |
| Formula | Drug | A (Surfactant Type) | B (Surfactant: Cholesterol Ratio | C TPGS Amount) | A (EE%) | B (PS) nm | C (ZP) (mV) | PDI |
|---|---|---|---|---|---|---|---|---|
| F1 | 2a | Span 40 | 8:2 | 25 | 82.3 ± 4.1 | 177.8 ± 19.3 | −22.2 ± 4.6 | 0.34 ± 0.035 |
| F2 | 2a | Span 60 | 8:2 | 25 | 94.1 ± 4.3 | 133.2 ± 13.4 | −37.4 ± 8.2 | 0.31 ± 0.07 |
| F3 | 2a | Span 40 | 5:5 | 25 | 73.5 ± 2.9 | 261.3 ± 23.1 | −10.6 ± 1.2 | 0.29 ± 0.05 |
| F4 | 2a | Span 60 | 5:5 | 25 | 83.4 ± 3.1 | 188.4 ± 13.8 | −22.7 ± 2.9 | 0.37 ± 0.06 |
| F5 | 2a | Span 40 | 8:2 | 50 | 64.8 ± 3.9 | 283.7 ± 27.1 | −31.5 ± 4.2 | 0.28 ± 0.06 |
| F6 | 2a | Span 60 | 8:2 | 50 | 81.2 ± 3.2 | 199.5 ± 21.8 | −46.8 ± 6.4 | 0.66 ± 0.09 |
| F7 | 2a | Span 40 | 5:5 | 50 | 56.7 ± 3.1 | 340.2 ± 22.6 | −24.1 ± 3.7 | 0.32 ± 0.04 |
| F8 | 2a | Span 60 | 5:5 | 50 | 71.7 ± 3.6 | 221.4 ± 19.8 | −33.2 ± 4.3 | 0.41 ± 0.062 |
| S1 | 7 | Span 40 | 8:2 | 25 | 88.4 ± 3.7 | 186.5 ± 17.4 | −28.9 ± 2.7 | 0.32 ± 0.04 |
| S2 | 7 | Span 60 | 8:2 | 25 | 90.6 ± 5.1 | 138.9 ± 16.8 | −45.1 ± 8.9 | 0.21 ± 0.03 |
| S3 | 7 | Span 40 | 5:5 | 25 | 76.1 ± 4.2 | 276.1 ± 24.1 | −16.9 ± 2.3 | 0.41 ± 0.06 |
| S4 | 7 | Span 60 | 5:5 | 25 | 79.7 ± 2.9 | 193.4 ± 13.9 | −26.1 ± 4.4 | 0.36 ± 0.08 |
| S5 | 7 | Span 40 | 8:2 | 50 | 67.2 ± 1.9 | 301.8 ± 23.9 | −37.2 ± 7.2 | 0.53 ± 0.08 |
| S6 | 7 | Span 60 | 8:2 | 50 | 74.8 ± 3.2 | 209.3 ± 18.8 | −54.8 ± 8.1 | 0.27 ± 0.035 |
| S7 | 7 | Span 40 | 5:5 | 50 | 58.1 ± 2.8 | 375.3 ± 27.1 | −26.7 ± 6.6 | 0.44 ± 0.04 |
| S8 | 7 | Span 60 | 5:5 | 50 | 63.9 ± 2.4 | 244.8 ± 21.7 | −39.1 ± 9.2 | 0.38 ± 0.09 |
| Responses | EE (%) | PS (nm) | ZP (mV) |
|---|---|---|---|
| R2 | 0.9999 | 0.9998 | 0.9986 |
| Adjusted R2 | 0.9998 | 0.9989 | 0.9902 |
| Predicted R2 | 0.9979 | 0.9897 | 0.9104 |
| Adequate precision | 225.4 | 98.5 | 34.5 |
| Significant factors | A, B, C | A, B, C | A, B, C |
| Observed value of the optimal formula (F2) | 94.1 | 133.2 | −37.4 |
| Predicted value of the optimal formula (F2) | 94.04 | 133.98 | −37.78 |
| Absolute deviation % | 0.064 | 0.59 | 1.01 |
| R2 | 0.9986 | 0.9999 | 0.9996 |
| Adjusted R2 | 0.9902 | 0.9998 | 0.9972 |
| Predicted R2 | 0.9105 | 0.9983 | 0.9747 |
| Adequate precision | 31.46 | 245.7 | 64.4 |
| Significant factors | A, B, C | A, B, C | A, B, C |
| Observed value of the optimal formula (S2) | 90.6 | 138.9 | −45.1 |
| Predicted value of the optimal formula (S2) | 91 | 139.26 | −44.87 |
| Absolute deviation % | 0.44 | 0.26 | 0.51 |
| Compound Number | Hepg2 Liver Carcinoma | MCF-7 Breast Cancer |
|---|---|---|
| 2a-coated (F2) | 3 µM | 12 µM |
| 2a–uncoated (F2) | 3.5 µM | 15 µM |
| Pure 2a | 6.2 μM | 20 μM |
| 7-coated (S2) | 1.6 µM | 6 µM |
| 7–uncoated (S2) | 2 µM | 8 µM |
| Pure 7 | 6 μM | 12 μM |
| Compound Number | VEGF 24 h | VEGF 72 h | VEGFR 24 h | VEGFR 72 h | ||||
|---|---|---|---|---|---|---|---|---|
| Conc. (µM) | % Inhibition | Conc. (µM) | % Inhibition | Conc. (µM) | % Inhibition | Conc. (µM) | % Inhibition | |
| 2a coated (F2) | 0.08 | 46.7% | 0.08 | 42.9% | 0.065 | 56.7% | 0.075 | 42.3% |
| 2a uncoated (F2) | 0.09 | 40% | 0.09 | 35.7% | 0.067 | 55.3% | 0.078 | 40% |
| 7 coated (S2) | 0.18 | 64% | 0.09 | 80.4% | 0.12 | 76.5% | 0.1 | 80% |
| 7 uncoated (S2) | 0.2 | 60% | 0.11 | 76% | 0.122 | 76% | 0.15 | 70% |
| Compound Number | VEGF 24 h | VEGF 72 h | VEGFR 24 h | VEGFR 72 h | ||||
|---|---|---|---|---|---|---|---|---|
| Conc. (µM) | % Inhibition | Conc. (µM) | % Inhibition | Conc. (µM) | % Inhibition | Conc. (µM) | % Inhibition | |
| 2a coated (F2) | 0.03 | 40% | 0.03 | 50% | 0.03 | 35% | 0.03 | 50% |
| 2a uncoated (F2) | 0.03 | 40% | 0.03 | 50% | 0.03 | 35% | 0.02 | 66.7% |
| 7 coated (S2) | 0.01 | 75% | 0.02 | 66.7% | 0.01 | 75% | 0.02 | 66.7% |
| 7 uncoated (S2) | 0.017 | 57.5% | 0.026 | 56.7% | 0.017 | 57.5% | 0.026 | 56.7% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anwer, K.E.; El-Sattar, N.E.A.A.; Shamaa, M.M.; Zakaria, M.Y.; Beshay, B.Y. Design, Green Synthesis and Tailoring of Vitamin E TPGS Augmented Niosomal Nano-Carrier of Pyrazolopyrimidines as Potential Anti-Liver and Breast Cancer Agents with Accentuated Oral Bioavailability. Pharmaceuticals 2022, 15, 330. https://doi.org/10.3390/ph15030330
Anwer KE, El-Sattar NEAA, Shamaa MM, Zakaria MY, Beshay BY. Design, Green Synthesis and Tailoring of Vitamin E TPGS Augmented Niosomal Nano-Carrier of Pyrazolopyrimidines as Potential Anti-Liver and Breast Cancer Agents with Accentuated Oral Bioavailability. Pharmaceuticals. 2022; 15(3):330. https://doi.org/10.3390/ph15030330
Chicago/Turabian StyleAnwer, Kurls E., Nour E. A. Abd El-Sattar, Marium M. Shamaa, Mohamed Y. Zakaria, and Botros Y. Beshay. 2022. "Design, Green Synthesis and Tailoring of Vitamin E TPGS Augmented Niosomal Nano-Carrier of Pyrazolopyrimidines as Potential Anti-Liver and Breast Cancer Agents with Accentuated Oral Bioavailability" Pharmaceuticals 15, no. 3: 330. https://doi.org/10.3390/ph15030330