
Review on the Synthesis and Therapeutic Potential of Pyrido[2,3-d], [3,2-d], [3,4-d] and [4,3-d]pyrimidine Derivatives




Abstract
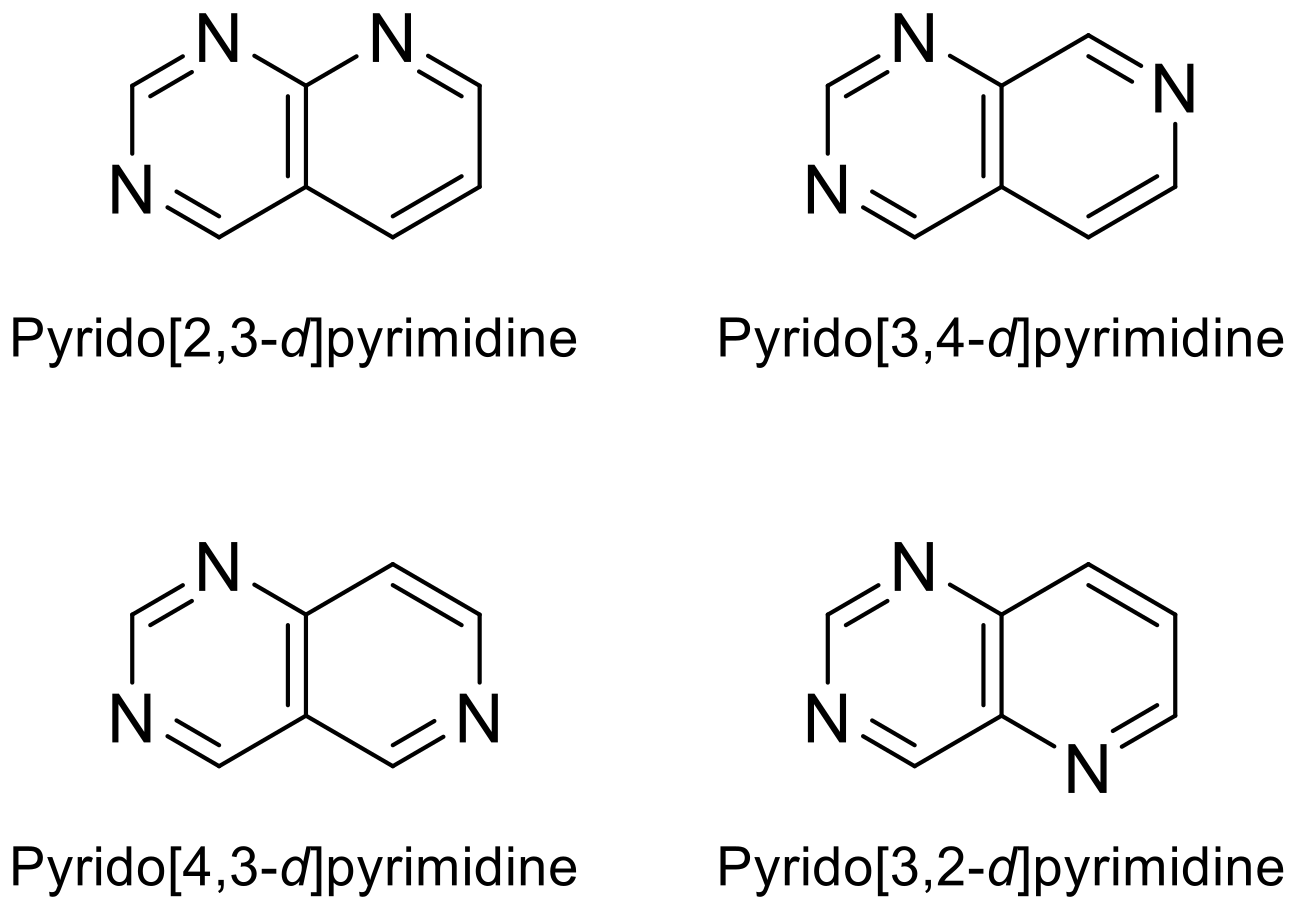
:1. Introduction: Pyridopyrimidines and Their Scaffold
2. Pyridopyrimidines: Therapeutic Potential and Synthesis
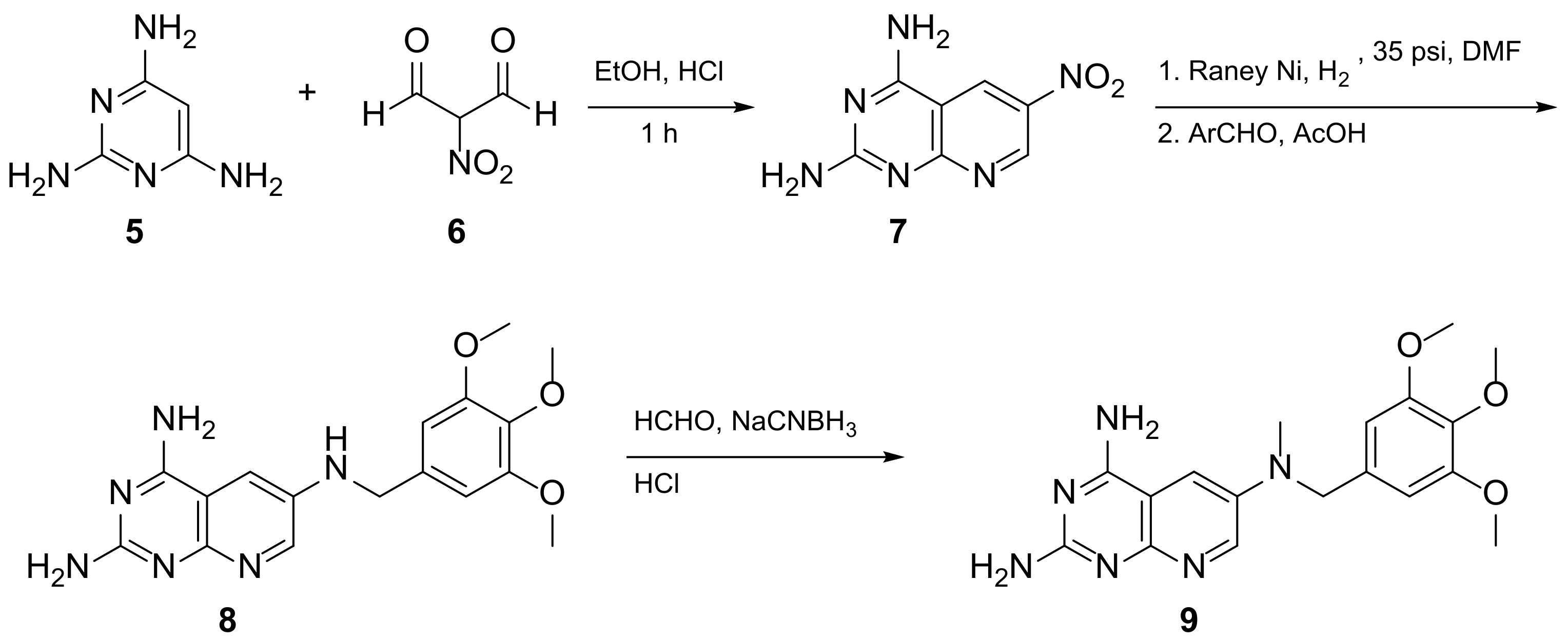
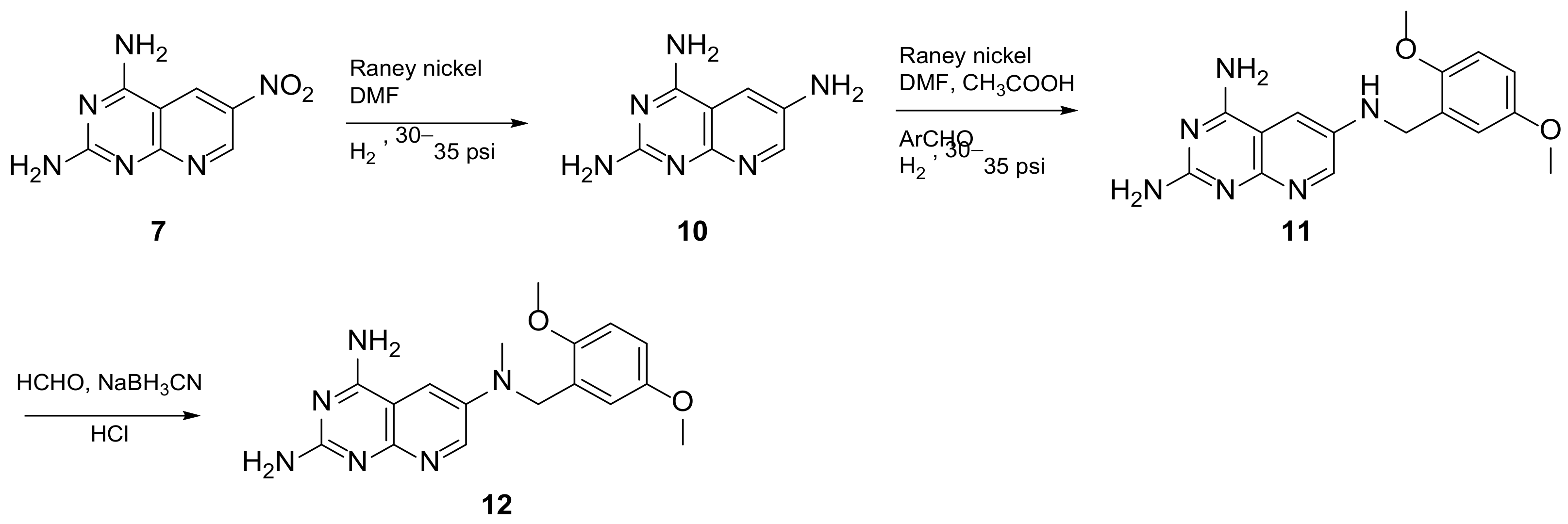
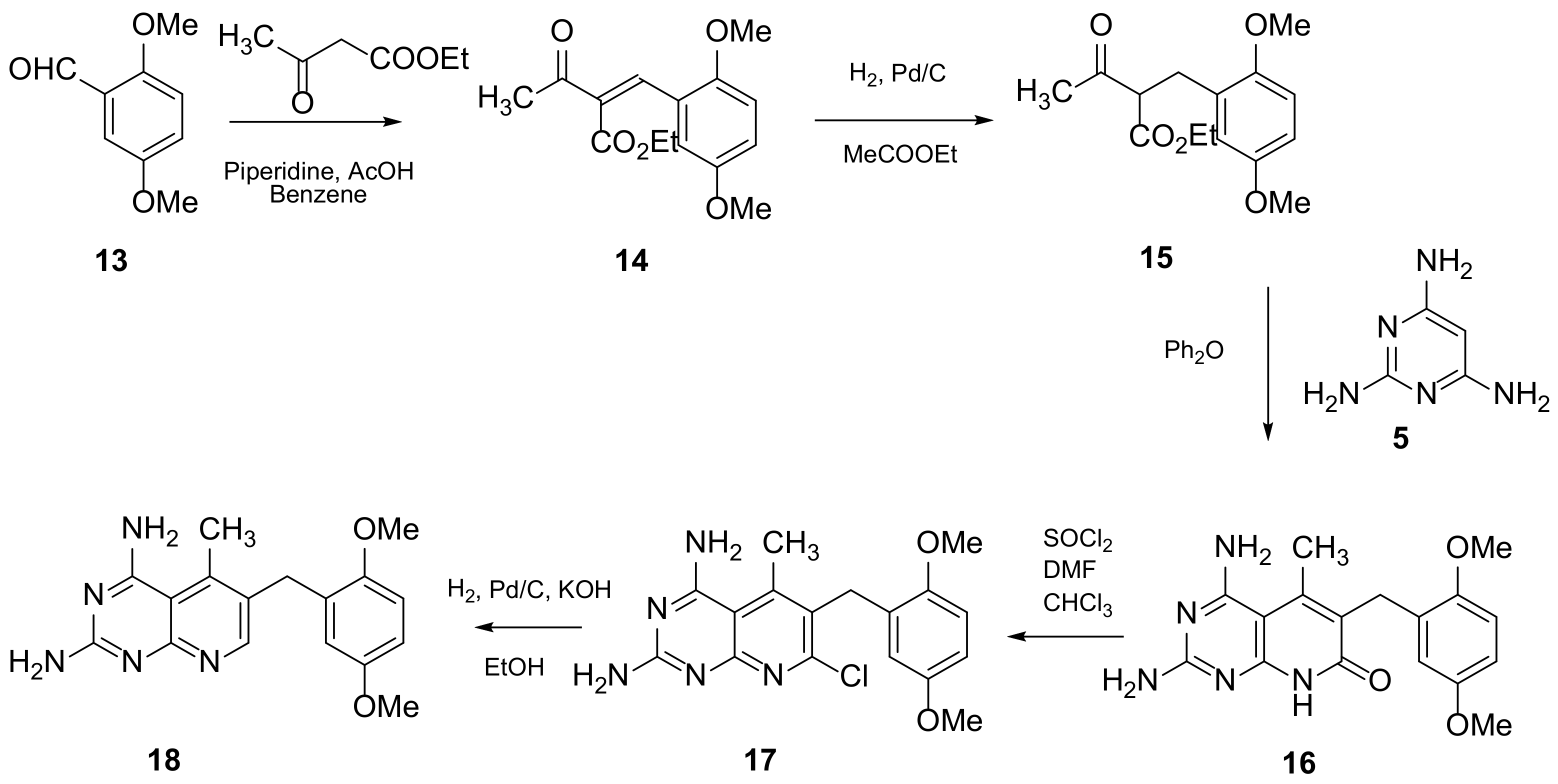
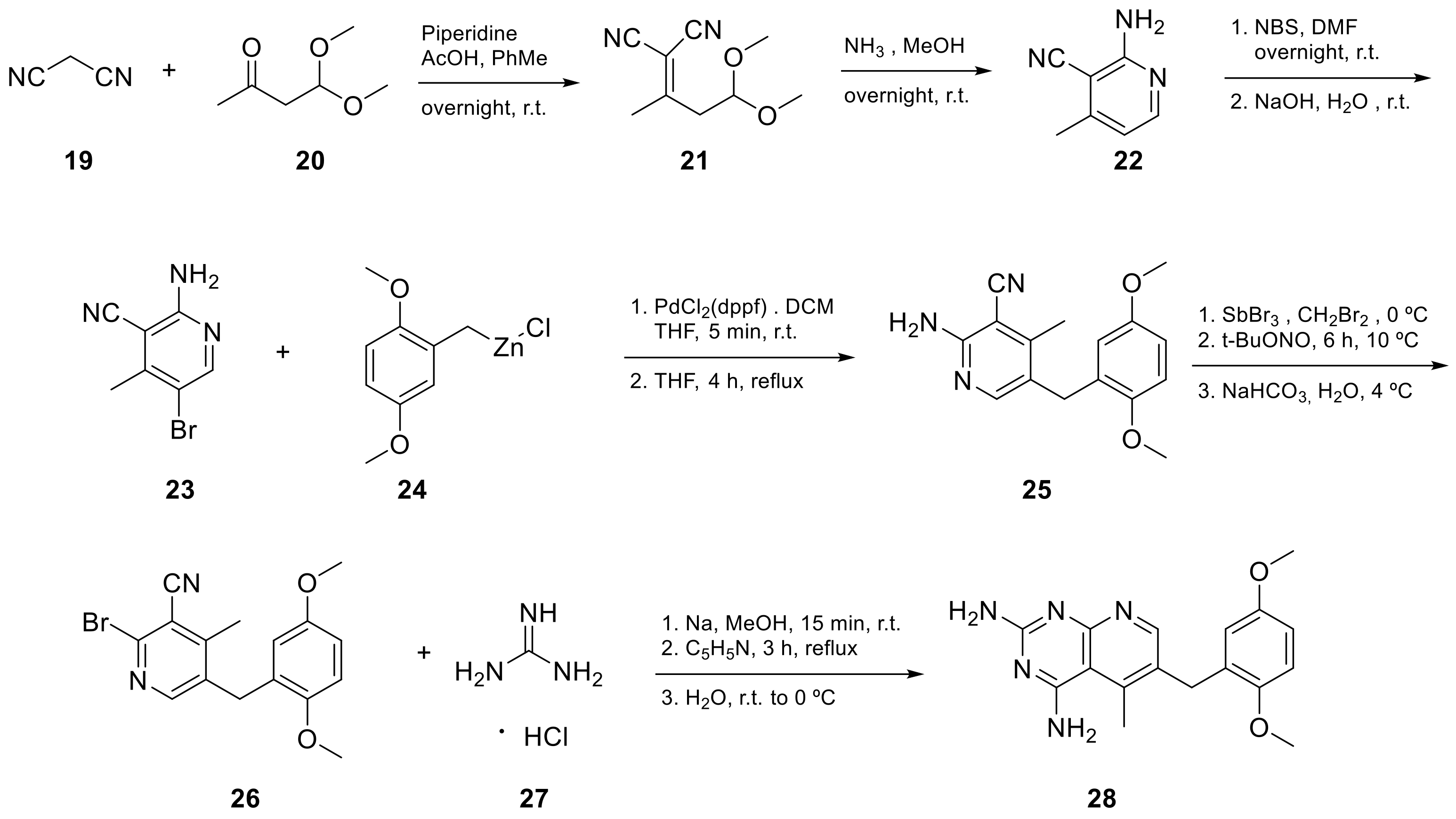
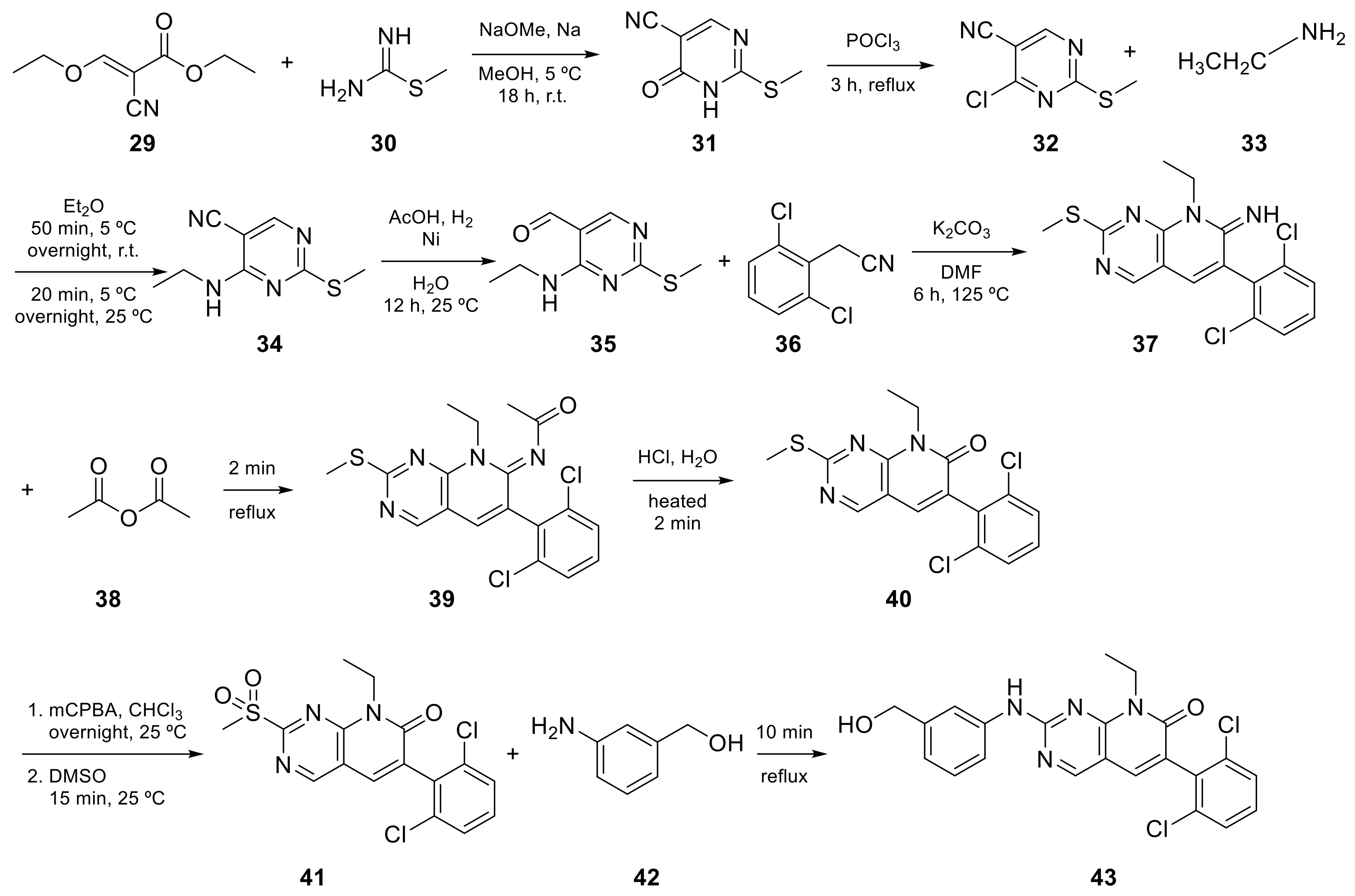
2.1. Pyrido[2,3-d]pyrimidine
2.2. Pyrido[3,4-d]pyrimidine
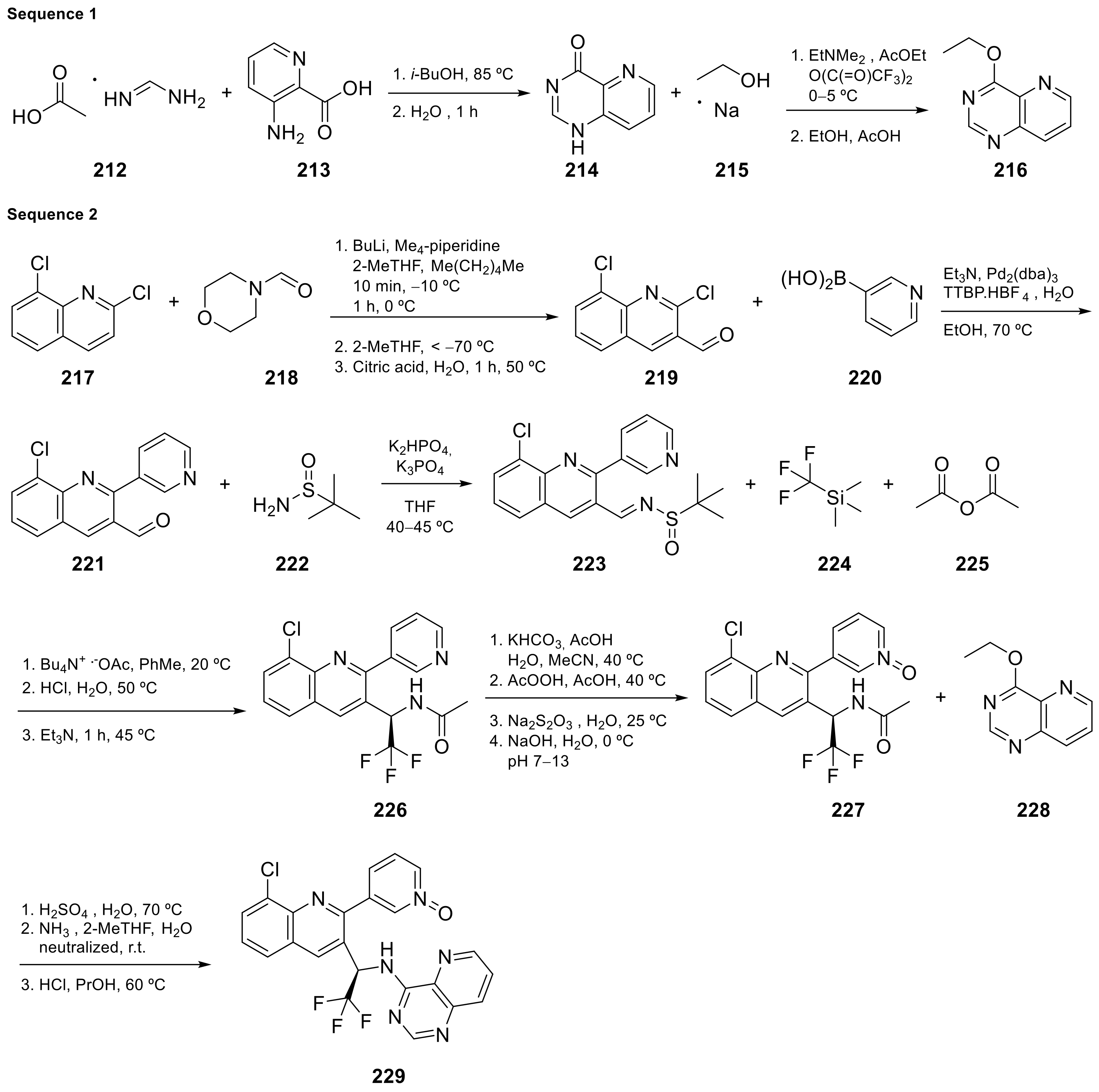
2.3. Pyrido[4,3-d]pyrimidine
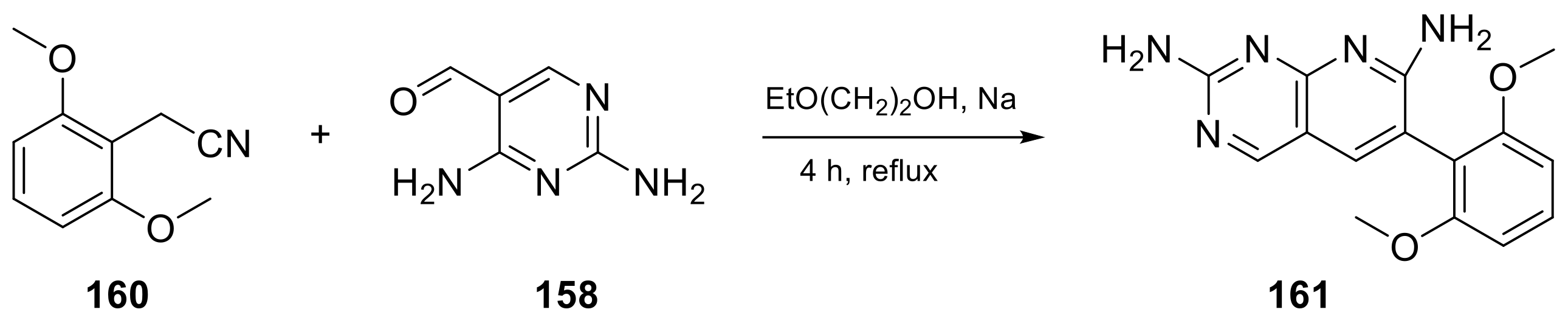
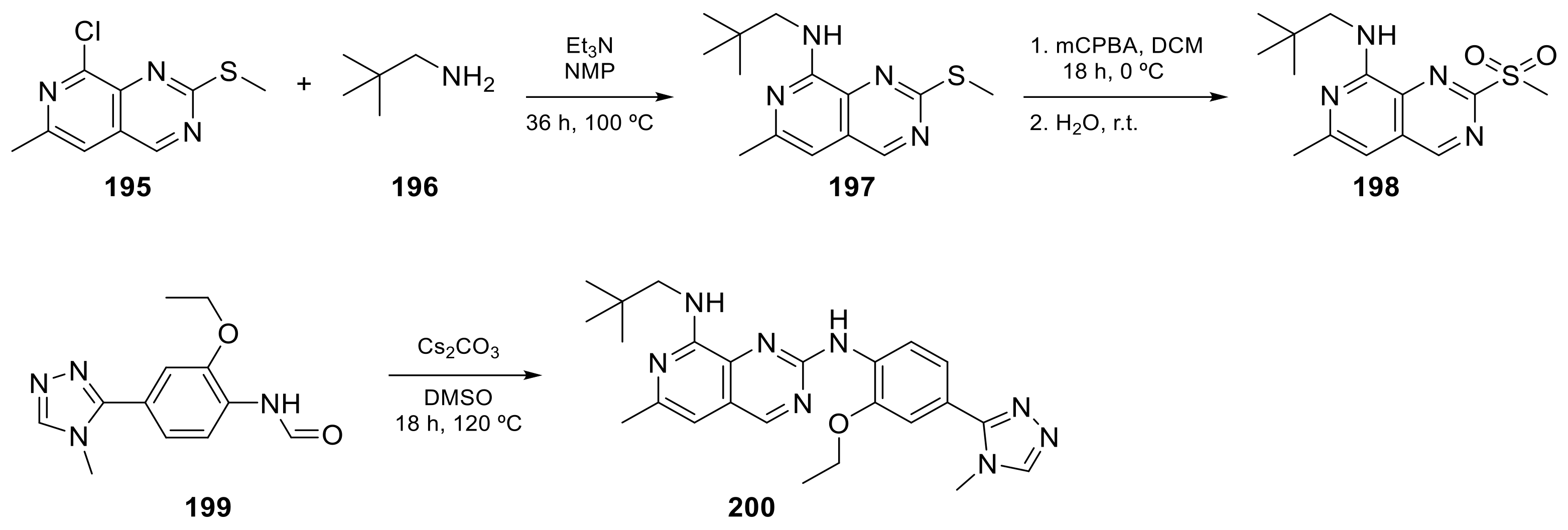
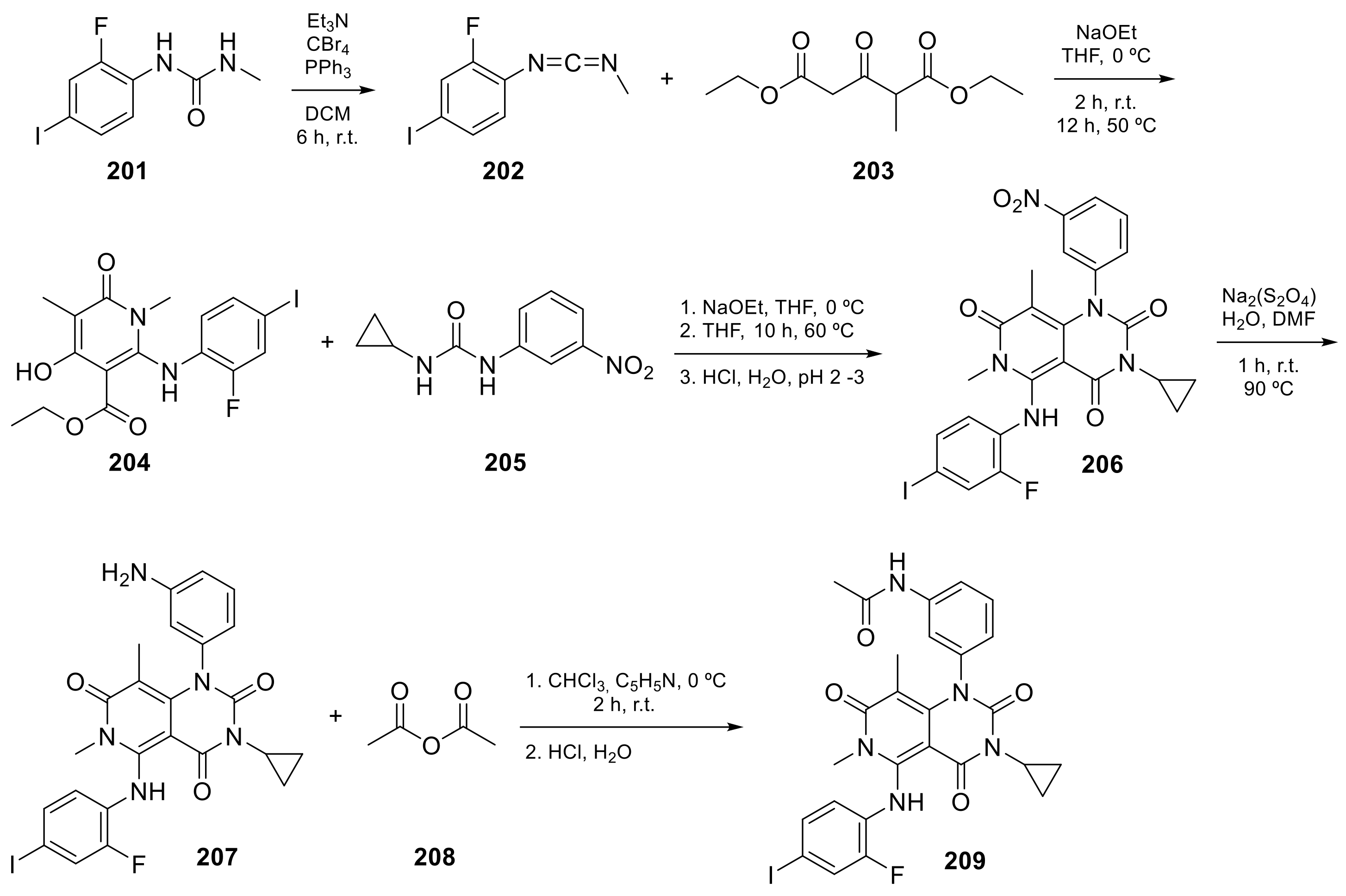
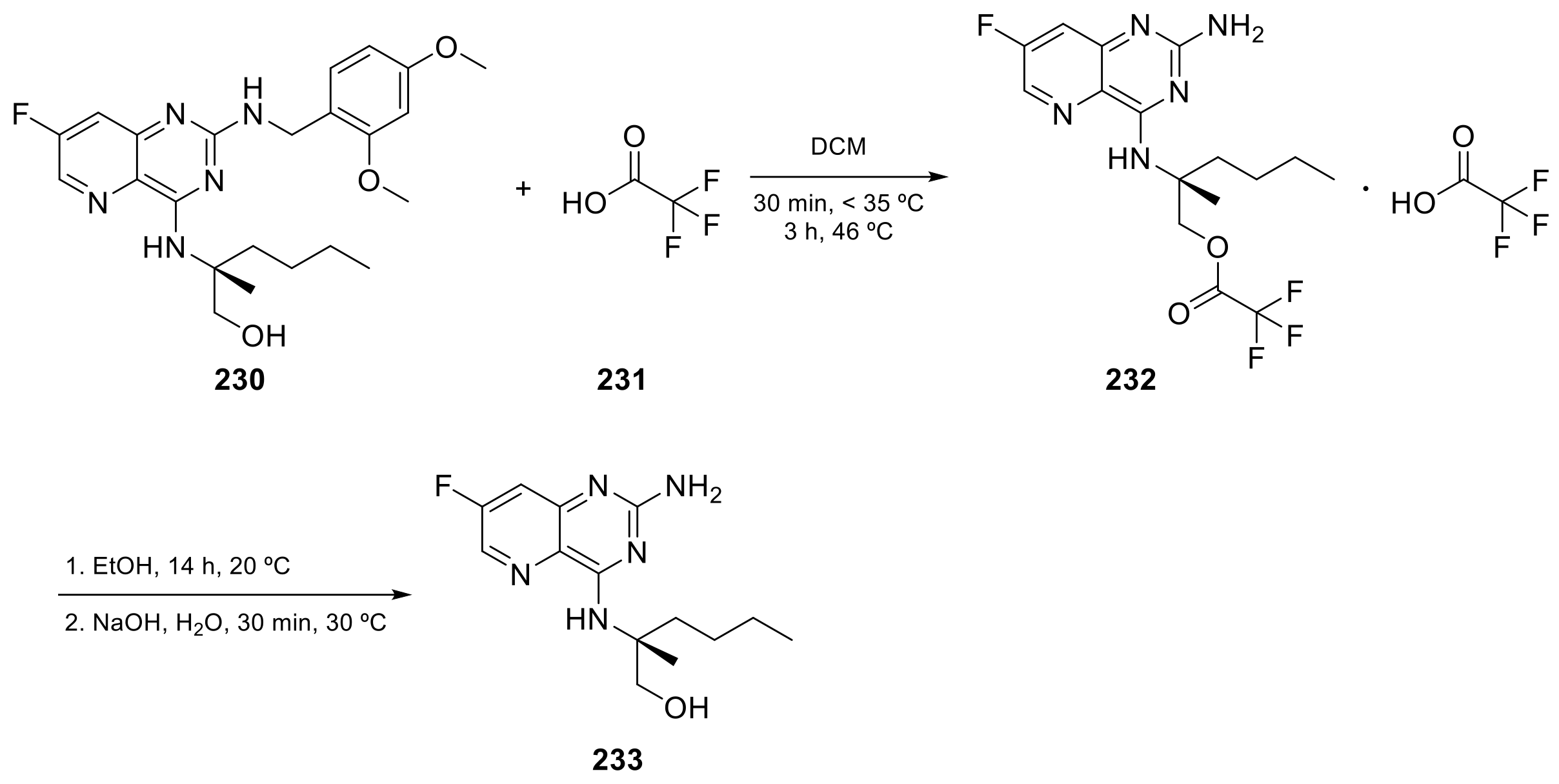
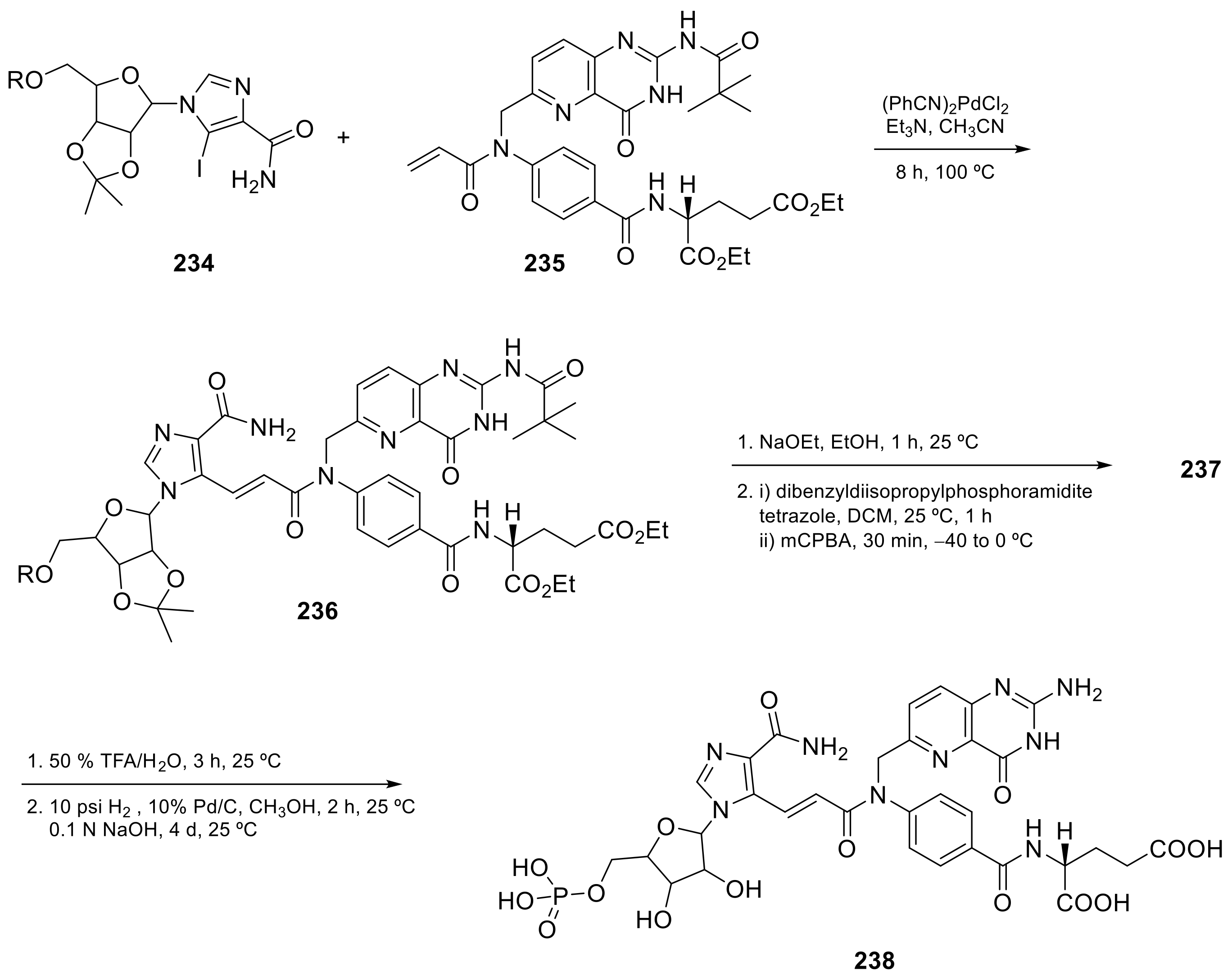
2.4. Pyrido[3,2-d]pyrimidine

| Entry | Structure | Name | Target | Ref. |
|---|---|---|---|---|
| Pyrido[2,3-d]pyrimidine | ||||
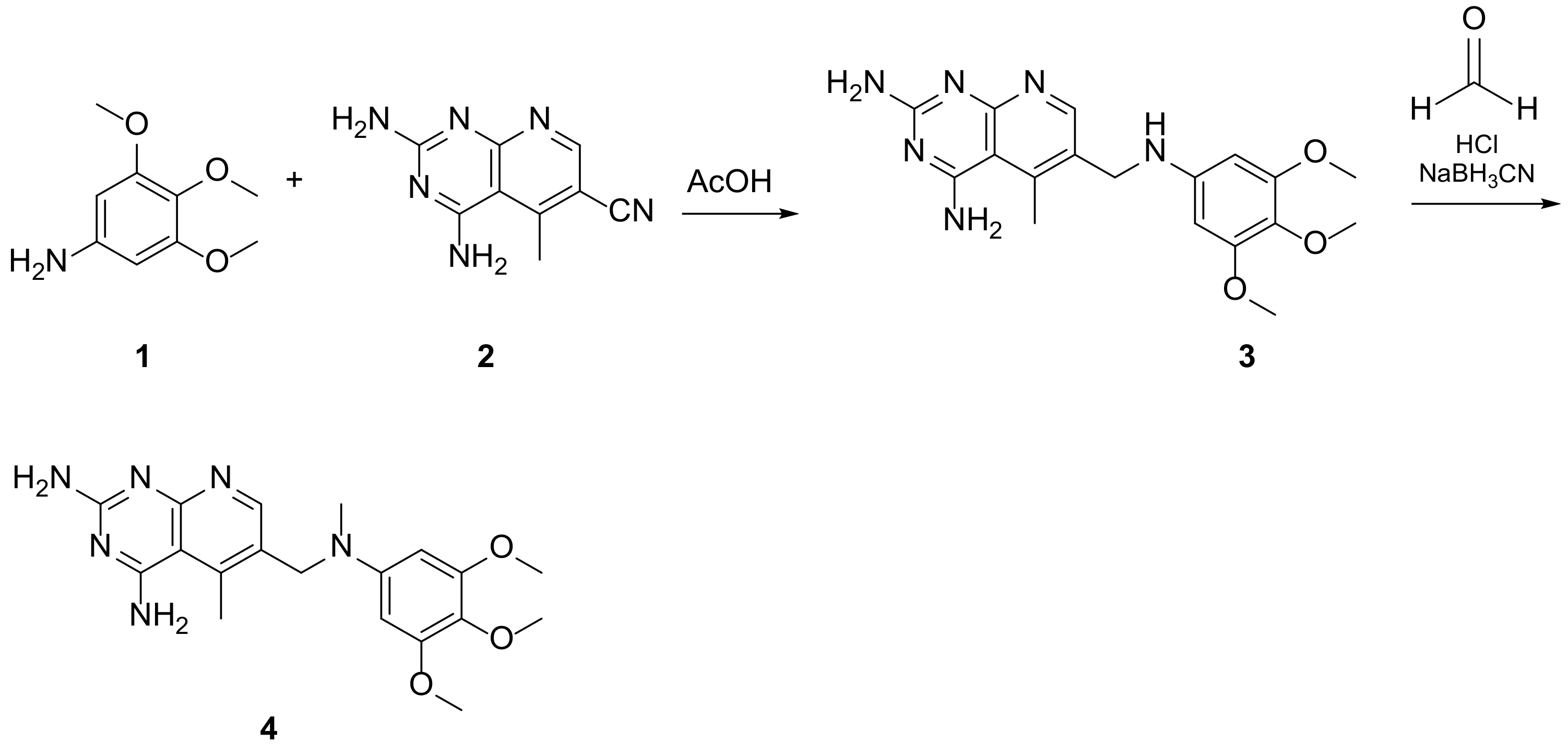
| 1 |  | 5-methyl-6-([methyl(3,4,5-trimethoxyphenyl)amino]methyl)pyrido[2,3-d]pyrimidine-2,4-diamine | DHFR Dihydrofolate reductase | [4,5,6] |
| 2 |  | N6-methyl-N6-[(3,4,5-trimethoxyphenyl)methyl]pyrido[2,3-d]pyrimidine-2,4,6-triamine | DHFR Dihydrofolate reductase | [19] |
| 3 |  | N6-[(3,5-dimethoxyphenyl)methyl]-N6-methylpyrido[2,3-d]pyrimidine-2,4,6-triamine, | DHFR Dihydrofolate reductase | [19] |
| 4 |  | N6-[(2,5-dimethoxyphenyl)methyl]-N6-methylpyrido[2,3-d]pyrimidine-2,4,6-triamine | DHFR Dihydrofolate reductase | [18] |
| 5 |  PIRITREXIM | 6-[(2,5-dimethoxyphenyl)methyl]-5-methylpyrido[2,3-d]pyrimidine-2,4-diamine | DHFR Dihydrofolate reductase | [20,21,24] |
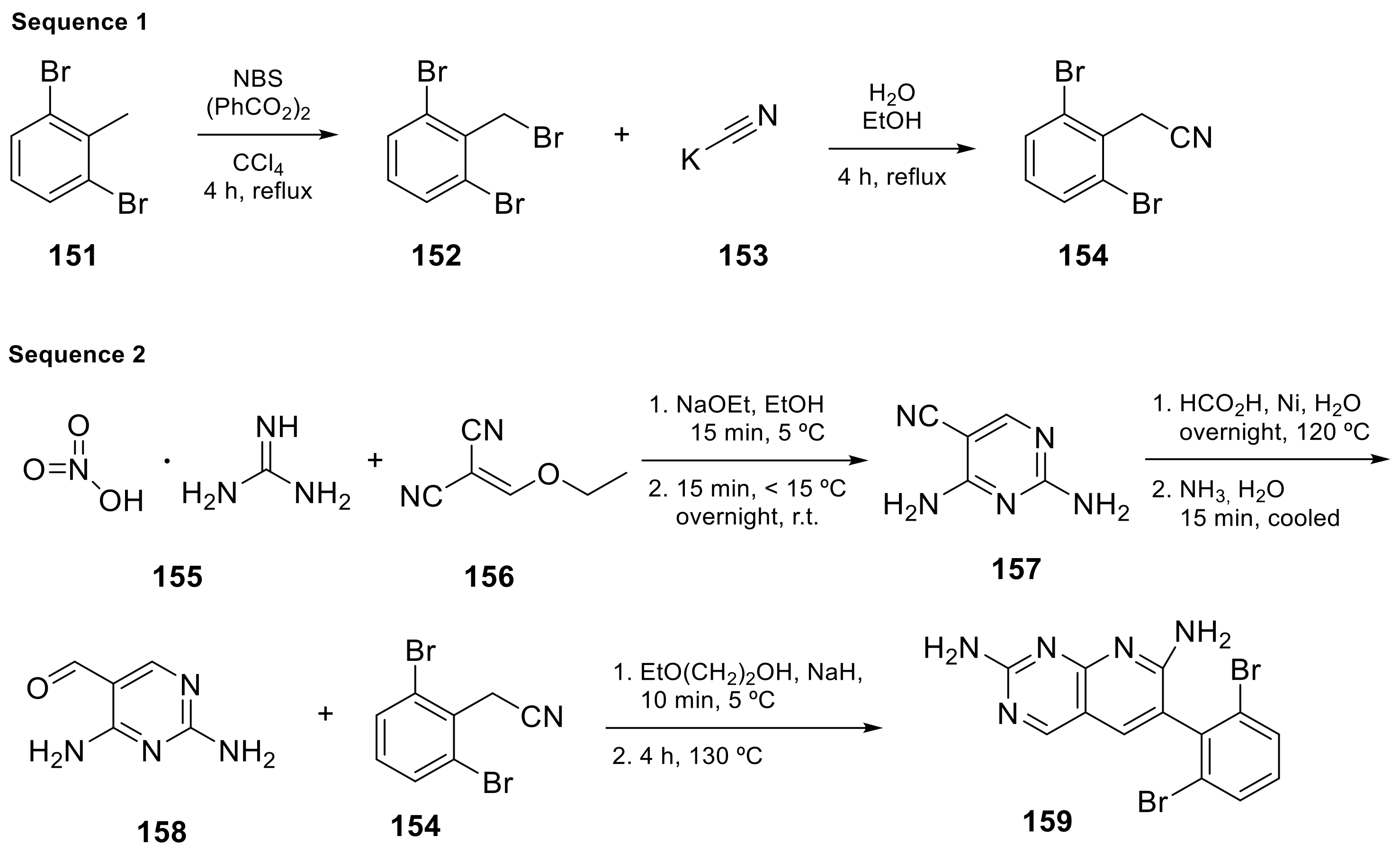
| 6 |  | 6-(2,6-dichlorophenyl)-2-([3-(hydroxymethyl)phenyl]amino)-8-ethyl-7H,8H-pyrido[2,3-d]pyrimidin-7-one | Tyrosine kinase activity | [25,26] |
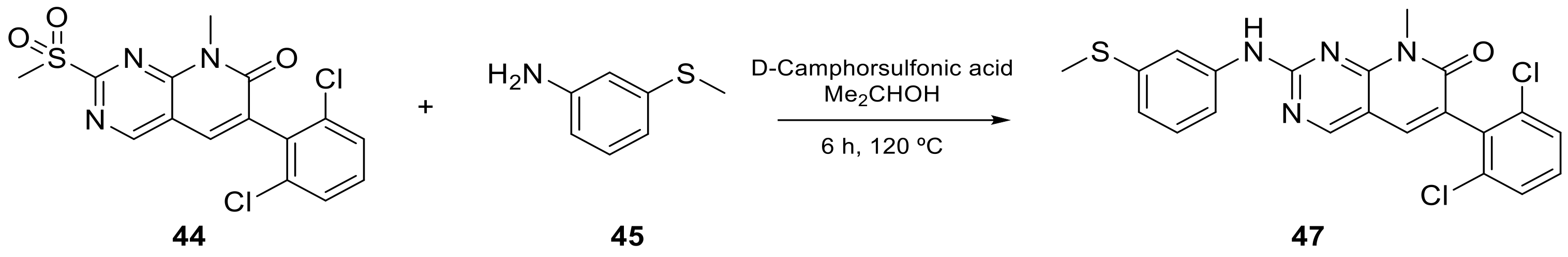
| 7 |  PD-173955 | 6-(2,6-dichlorophenyl)-8-methyl-2-([3-(methylsulfanyl)phenyl]amino)-7H,8H-pyrido[2,3-d]pyrimidin-7-one | Kinase activity: Tyrosine-protein kinase transforming protein Abl | [17] |
| 8 |  | 6-(2,4-difluorophenoxy)-8-methyl-2-[(oxan-4-yl)amino]-7H,8H-pyrido[2,3-d]pyrimidin-7-one | Kinase activity: Mitogen-activated protein kinase 14 | [27,28,29] |
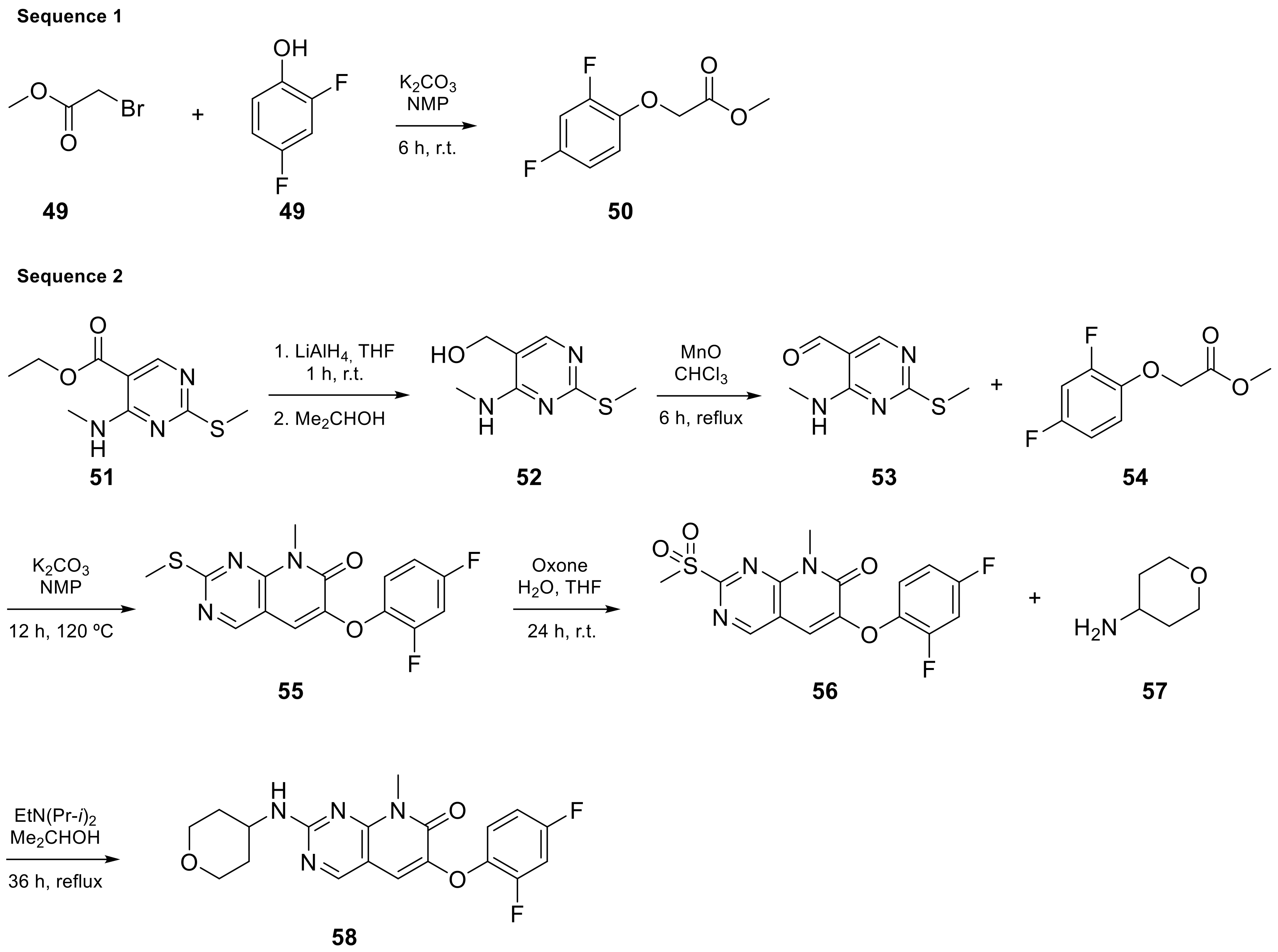
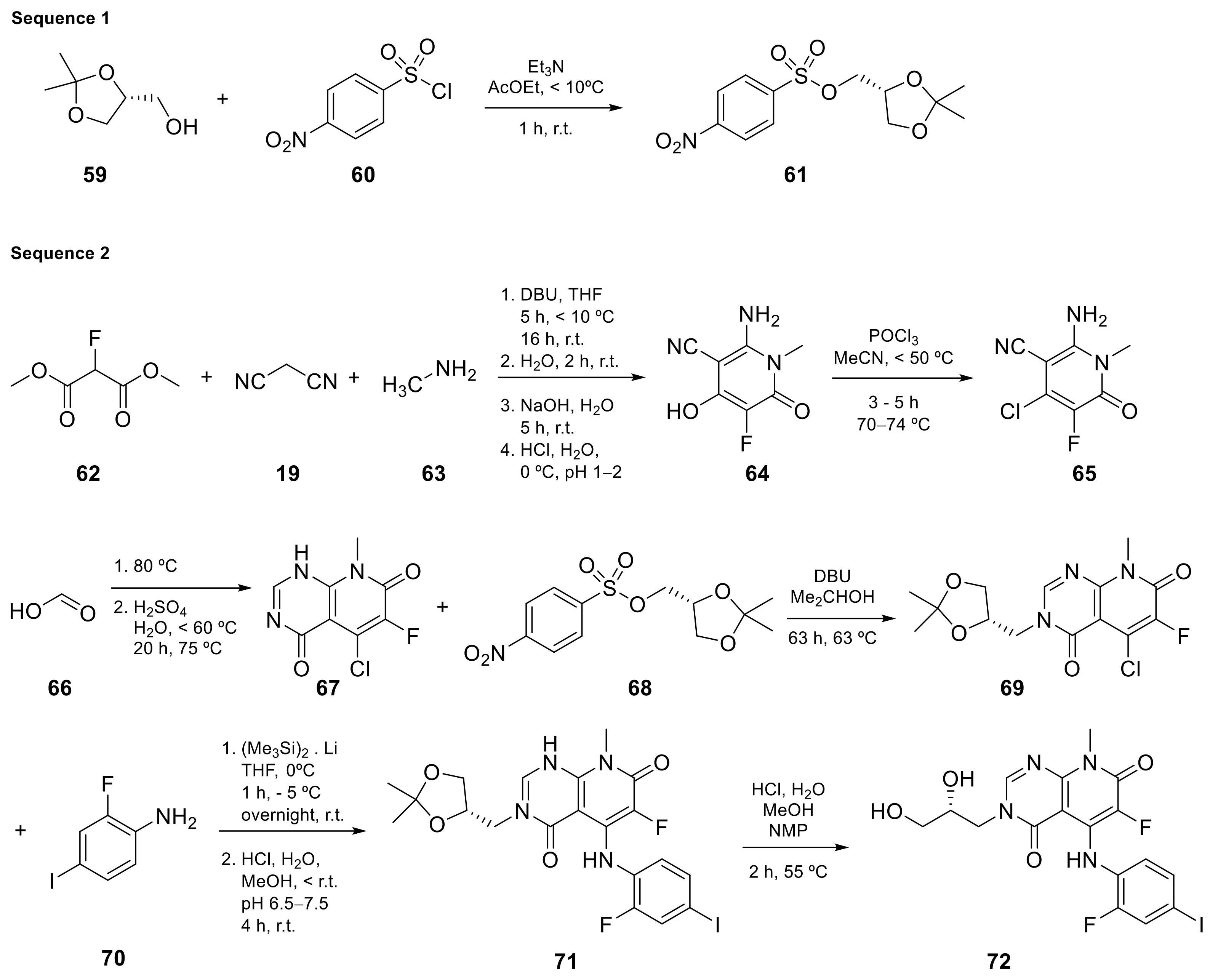
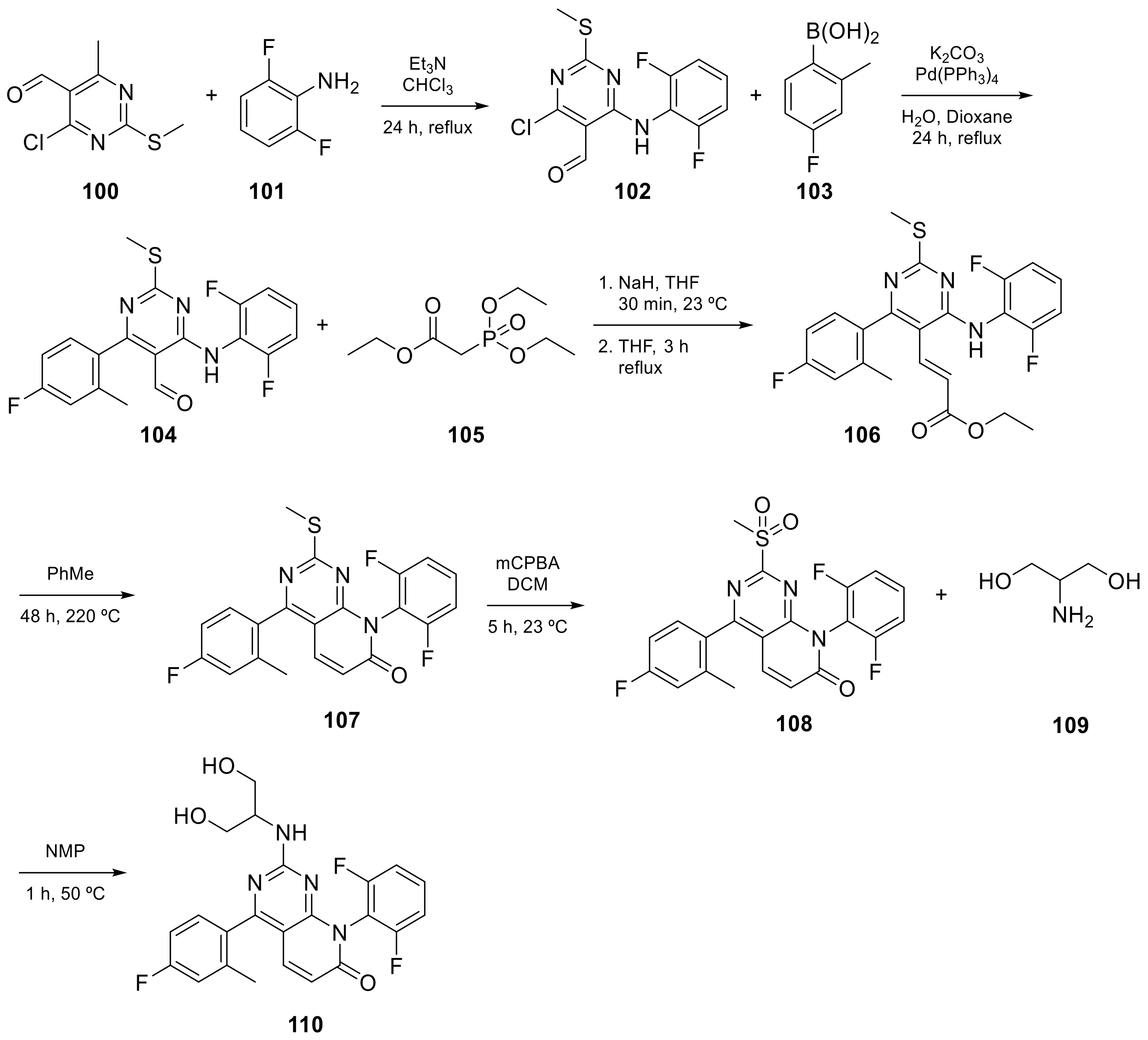
| 9 |  TAK-733 | 3-[(2R)-2,3-dihydroxypropyl]-6-fluoro-5-[(2-fluoro-4-iodophenyl)amino]-8-methyl-3H,4H,7H,8H-pyrido[2,3-d]pyrimidine-4,7-dione | Kinase activity: Against MEK and ERK | [30,31,32,33] |
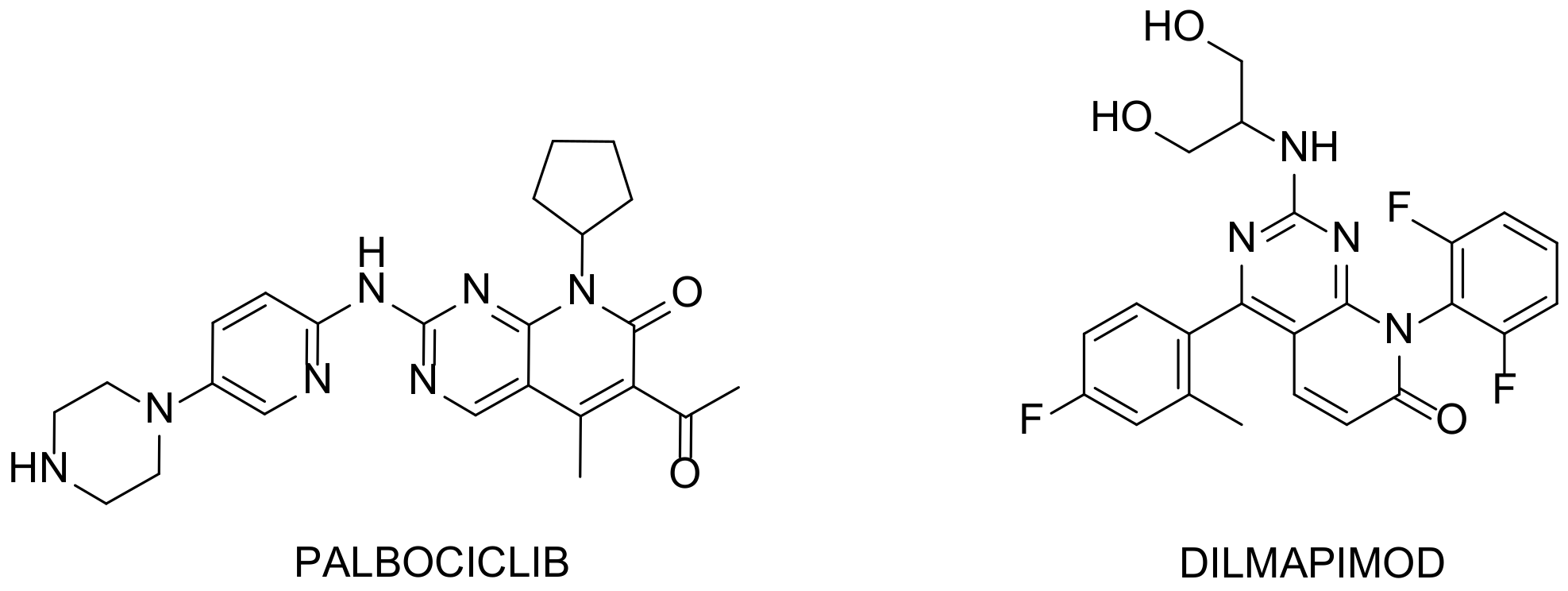
| 10 |  Palbociclib | 6-acetyl-8-cyclopentyl-5-methyl-2-([5-(piperazin-1-yl)pyridin-2-yl]amino)-7H,8H-pyrido[2,3-d]pyrimidin-7-one | Kinase activity: Cyclin-dependent kinase 4/Cyclin-dependent kinase 6 Breast cancer drug | [34,35,36,37,38] |
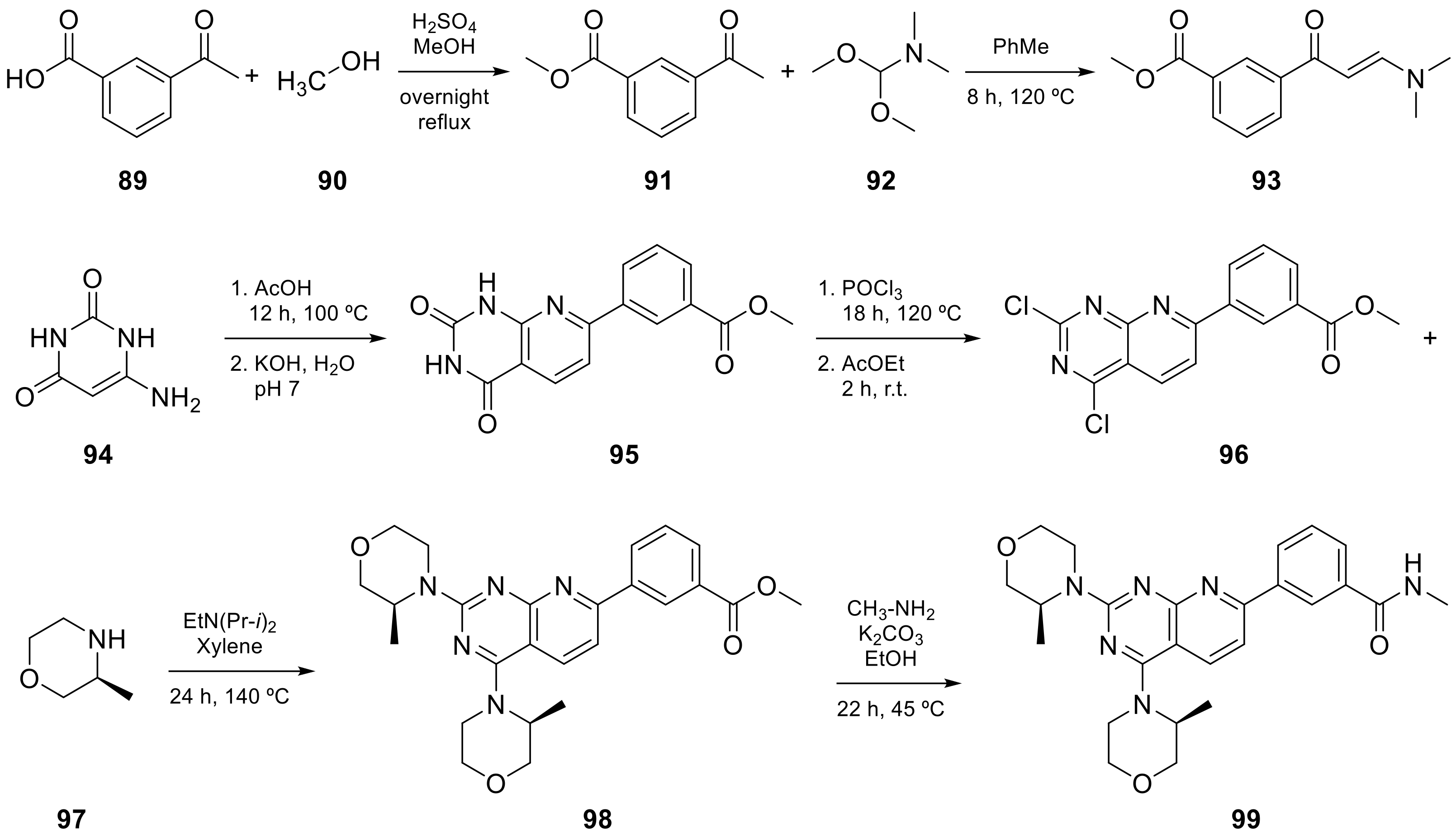
| 11 |  Vistusertib | 3-(2,4-bis[(3S)-3-methylmorpholin-4-yl]pyrido[2,3-d]pyrimidin-7-yl)-N-methylbenzamide | Kinase activity: Vistusertib (AZD2014) is a novel mTOR inhibitor | [39,40] |
| 12 |  Dilmapimod (SB-681323) | 8-(2,6-difluorophenyl)-2-[(1,3-dihydroxypropan-2-yl)amino]-4-(4-fluoro-2-methylphenyl)-7H,8H-pyrido[2,3-d]pyrimidin-7-one | Kinase activity: P38 MAPK inhibitor, Tumor necrosis factor/Interleukin-1 beta/Interleukin-6. Potential activity against rheumatoid arthritis | [41,42,43,44,45,46,47] |
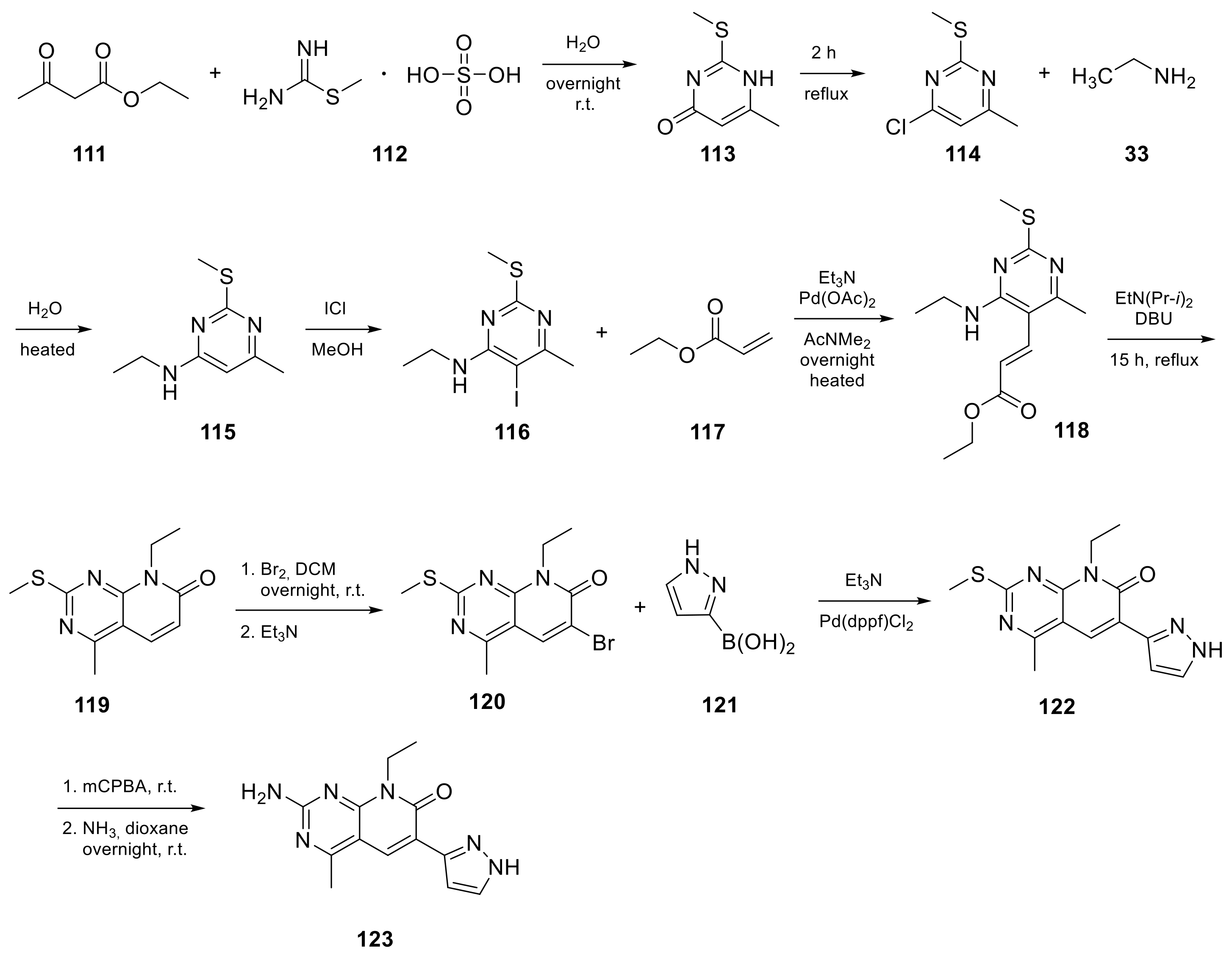
| 13 |  Voxtalisib | 2-amino-8-ethyl-4-methyl-6-(1H-pyrazol-5-yl)-7H,8H-pyrido[2,3-d]pyrimidin-7-one | Kinase activity: PI3K/mTOR Inhibitor | [48,49] |
| 14 |  AZD8055 | (5-(2,4-bis[(3S)-3-methylmorpholin-4-yl]pyrido[2,3-d]pyrimidin-7-yl)-2-methoxyphenyl)methanol | Kinase activity: Selective ATP-competitive mTOR kinase inhibitor. Induction of MEK/ERK | [50,51] |
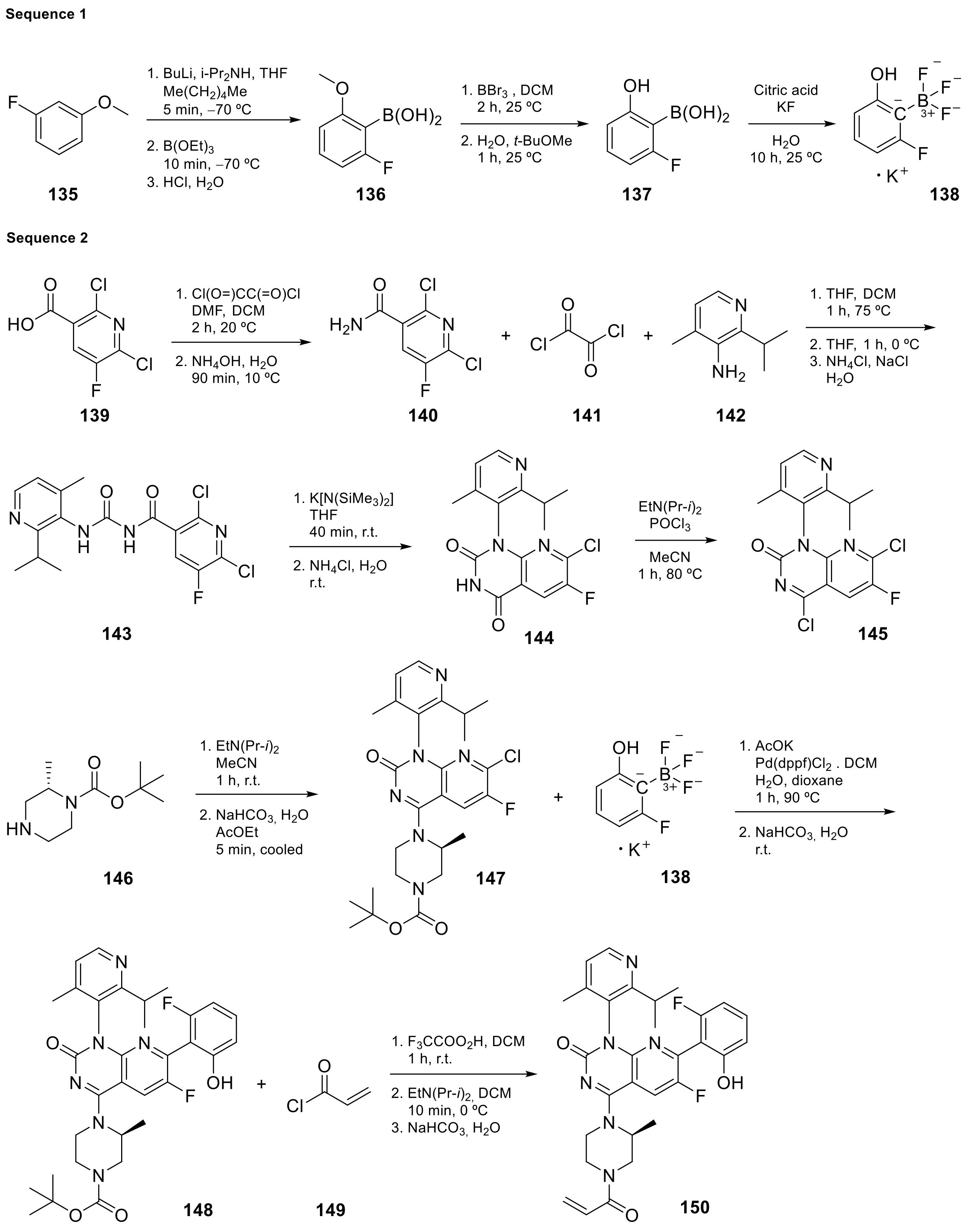
| 15 |  AMG-510 | 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1-yl]-1H,2H-pyrido[2,3-d]pyrimidin-2-one | Kinase Activity: KRAS inhibitor implicated in the RAS/MAPK pathway GTPase KRas | [52,53,54,55,56] |
| 16 |  | 6-(2,6-dibromophenyl)pyrido[2,3-d]pyrimidine-2,7-diamine | Biotin carboxylase | [57] |
| 17 |  | 6-(2,6-dimethoxyphenyl)pyrido[2,3-d]pyrimidine-2,7-diamine | Biotin carboxylase | [25,58] |
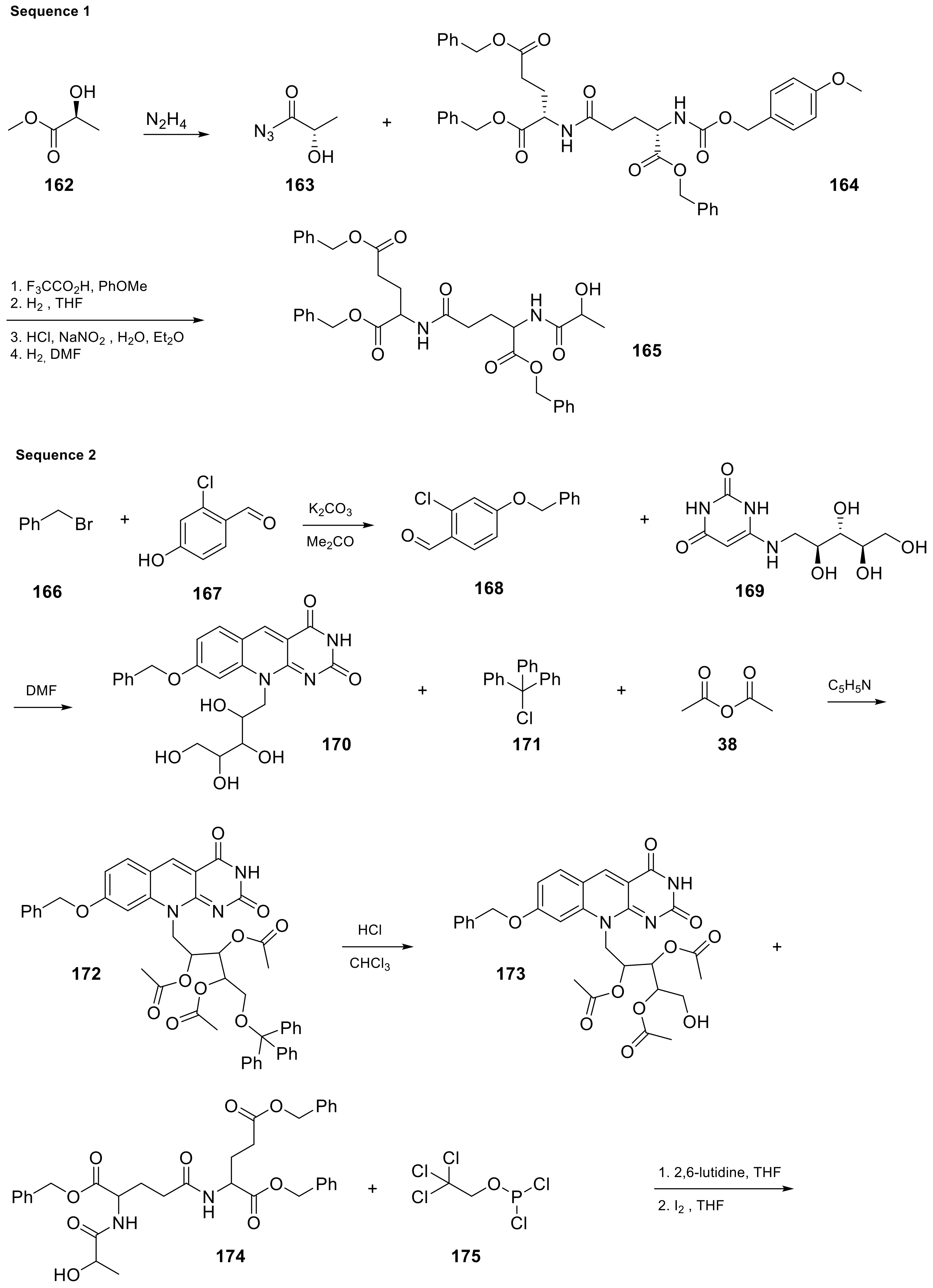
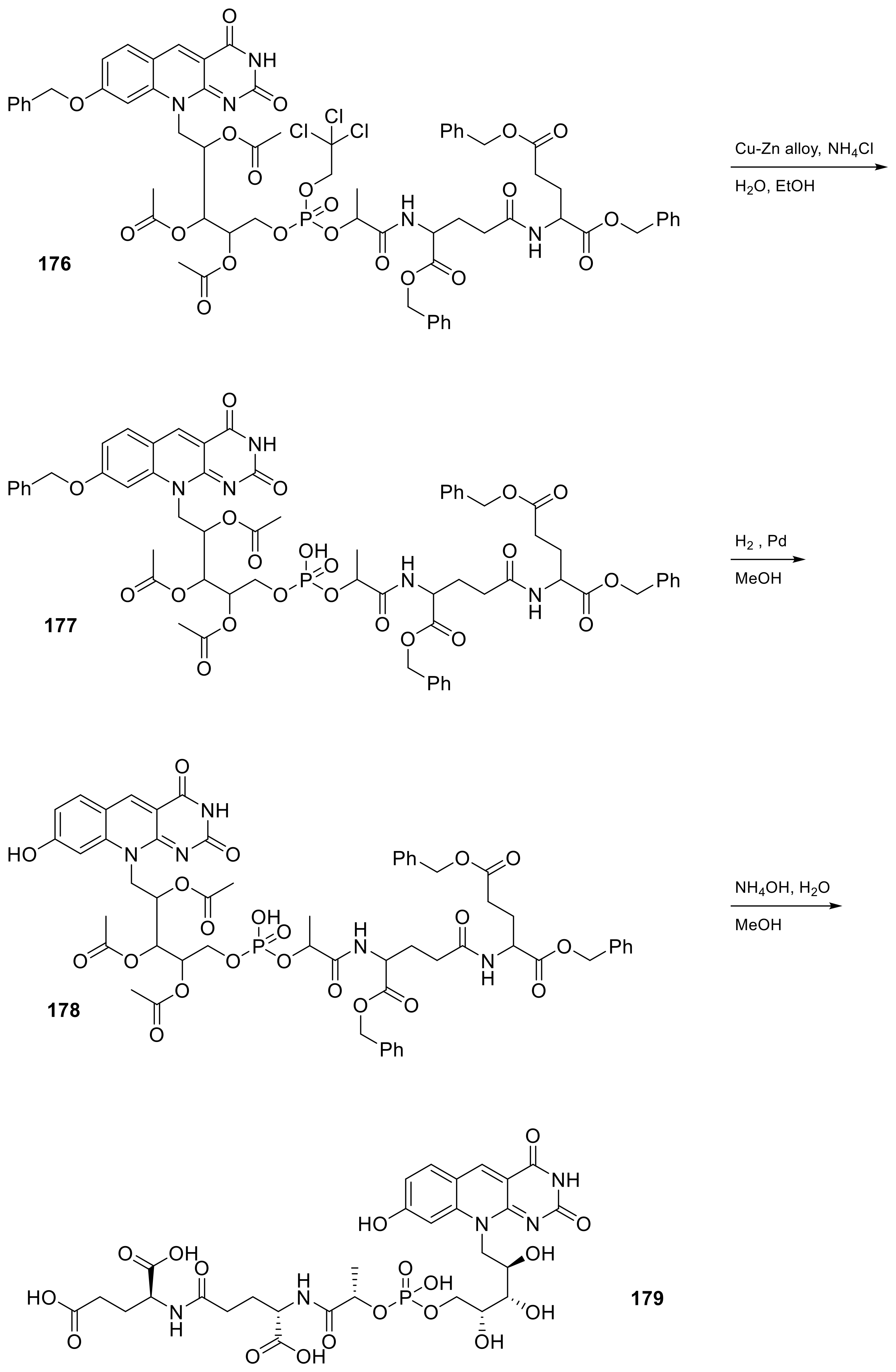
| 18 |  | (2S)-2-[(4S)-4-carboxy-4-[(2S)-2-([hydroxy(([(2R,3S,4S)-2,3,4-trihydroxy-5-(8-hydroxy-2,4-dioxo-2H,3H,4H,10H-pyrimido[4,5-b]quinolin-10-yl)pentyl]oxy))phosphoryl]oxy)propanamido]butanamido]pentanedioic acid | Methanobacterium redox coenzyme Factor 420 (F420) | [59] |
| Pyrido[3,4-d]pyrimidine | ||||
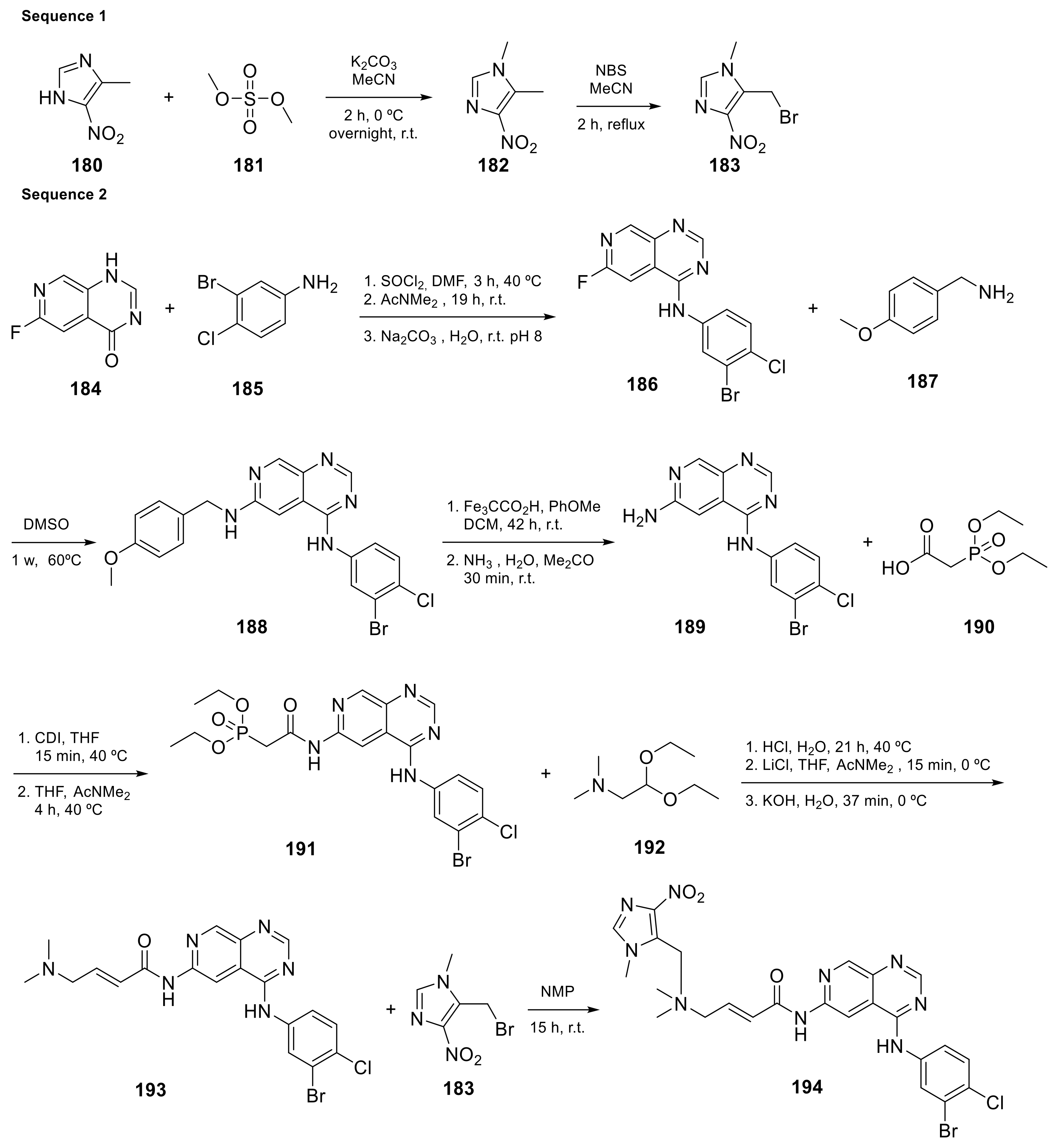
| 19 |  Tarloxotinib | [(2E)-3-((4-[(3-bromo-4-chlorophenyl)amino]pyrido[3,4-d]pyrimidin-6-yl)carbamoyl)prop-2-en-1-yl]dimethyl[(1-methyl-4-nitro-1H-imidazol-5-yl)methyl]azanium | Kinase Activity: Pan-HER kinase inibitor | [61] |
| 20 |  BOS172722 | N8-(2,2-dimethylpropyl)-N2-[2-ethoxy-4-(4-methyl-4H-1,2,4-triazol-3-yl)phenyl]-6-methylpyrido[3,4-d]pyrimidine-2,8-diamine | Kinase Activity: Dual specificity protein kinase TTK | [62,63,64,65] |
| Pyrido[4,3-d]pyrimidine | ||||
| 21 |  Trametinib | N-(3-(3-cyclopropyl-5-[(2-fluoro-4-iodophenyl)amino]-6,8-dimethyl-2,4,7-trioxo-1H,2H,3H,4H,6H,7H-pyrido[4,3-d]pyrimidin-1-yl)phenyl)acetamide | Dual specificity mitogen-activated protein kinase kinase 1/Dual specificity mitogen-activated protein kinase kinase 2 | [66,67,68,69] |
| Pyrido[3,2-d]pyrimidine | ||||
| 22 |  Seletalisib | 3-(8-chloro-3-[(1R)-2,2,2-trifluoro-1-((pyrido[3,2-d]pyrimidin-4-yl)amino)ethyl]quinolin-2-yl)pyridin-1-ium-1-olate | selective PI3Kδ inhibitor | [70,71] |
| 23 |  | (2S)-2-((2-amino-7-fluoropyrido[3,2-d]pyrimidin-4-yl)amino)-2-methylhexan-1-ol | Chronic hepatitis B TLR8 receptor | [72,73] |
| 24 |  β-DADF | (2S)-2-((4-[(2E)-N-((2-amino-4-oxo-1H,4H-pyrido[3,2-d]pyrimidin-6-yl)methyl)-3-(4-carbamoyl-1-[(2R,3R,4S,5R)-3,4-dihydroxy-5-[(phosphonooxy)methyl]oxolan-2-yl]-1H-imidazol-5-yl)prop-2-enamido]phenyl)formamido)pentanedioic acid | Bifunctional purine biosynthesis protein PURH | [74,75] |
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Yadav, P.; Shah, K. An overview on synthetic and pharmaceutical prospective of pyrido[2,3-d]pyrimidines scaffold. Chem. Biol. Drug Des. 2021, 3, 633–648. [Google Scholar] [CrossRef] [PubMed]
- Yousif, M.N.M.; El-Gazzar, A.-R.B.A.; El-Enany, M.M. Synthesis and Biological Evaluation of Pyrido[2,3-d]pyrimidines. Mini Rev. Org. Chem. 2021, 1, 43–54. [Google Scholar] [CrossRef]
- Buron, F.; Mérour, J.Y.; Akssira, M.; Guillaumet, G.; Routier, S. Recent advances in the chemistry and biology of pyridopyrimidines. Eur. J. Med. Chem. 2015, 95, 76–95. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, B.I.; Dicker, A.P.; Bertino, J.R. Dihydrofolate reductase as a therapeutic target. FASEB J. 1990, 8, 2441–2452. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, M.V.; Randazzo, O.; La Franca, M.; Barone, G.; Vignoni, E.; Rossi, D.; Collina, S. DHFR Inhibitors: Reading the Past for Discovering Novel Anticancer Agents. Molecules 2019, 6, 1140. [Google Scholar] [CrossRef] [Green Version]
- Gangjee, A.; Shi, J.; Queener, S.F.; Barrows, L.R.; Kisliuk, R.L. Synthesis of 5-methyl-5-deaza nonclassical antifolates as inhibitors of dihydrofolate reductases and as potential antipneumocystis, antitoxoplasma, and antitumor agents. J. Med. Chem. 1993, 36, 3437–3443. [Google Scholar] [CrossRef] [PubMed]
- Hawser, S.; Lociuro, S.; Islam, K. Dihydrofolate reductase inhibitors as antibacterial agents. Biochem. Pharm. 2006, 71, 941–948. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Tanis, K.Q.; Koleske, A.J.; Colicelli, J. Enhancement of ABL kinase catalytic efficiency by a direct binding regulator is independent of other regulatory mechanisms. J. Biol. Chem. 2008, 283, 31401–31407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosgrove, B.D.; Gilbert, P.M.; Porpiglia, E.; Mourkioti, F.; Lee, S.P.; Corbel, S.Y.; Llewellyn, M.E.; Delp, S.L.; Blau, H.M. Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat. Med. 2014, 20, 255–264. [Google Scholar] [CrossRef] [Green Version]
- Segalés, J.; Perdiguero, E.; Muñoz-Cánoves, P. Regulation of muscle stem cell functions: A focus on the p38 MAPK Signaling Pathway. Front. Cell Dev. Biol. 2016, 4, 91. [Google Scholar] [CrossRef] [Green Version]
- Orton, R.J.; Sturm, O.E.; Vyshemirsky, V.; Calder, M.; Gilbert, D.R.; Kolch, W. Computational modelling of the receptor-tyrosine-kinase-activated MAPK pathway. Biochem. J. 2005, 392, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Wakil, S.J.; Stoops, J.K.; Joshi, V.C. Fatty acid synthesis and its regulation. Ann. Rev. Biochem. 1983, 52, 537–579. [Google Scholar] [CrossRef] [PubMed]
- Smejkalova, H.; Erb, T.J.; Fuchs, G. Methanol assimilation in methylobacterium extorquens AM1: Demonstration of all enzymes and their regulation. PLoS ONE 2010, 5, e13001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erb, T.J.; Berg, I.A.; Brecht, V.; Muller, M.; Fuchs, G.; Alber, B.E. Synthesis of C5-dicarboxylic acids from C2-units involving crotonyl-CoA carboxylase/reductase: The ethylmalonyl-CoA pathway. Proc. Natl. Acad. Sci. USA 2007, 104, 10631–10636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khomyakova, M.; Bukmez, O.; Thomas, L.K.; Erb, T.J.; Berg, I.A. A methylaspartate cycle in haloarchaea. Science 2011, 331, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Alber, B.E. Biotechnological potential of the ethylmalonyl-CoA pathway. Appl. Microbiol. Biotechnol. 2011, 89, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Heinzlmeir, S.; Kudlinzki, D.; Sreeramulu, S.; Klaeger, S.; Gande, S.L.; Linhard, V.; Wilhelm, M.; Qiao, H.; Helm, D.; Ruprecht, B.; et al. Chemical proteomics and structural biology define EPHA2 inhibition by clinical kinase drugs. ACS Chem. Biol. 2016, 11, 3400–3411. [Google Scholar] [CrossRef] [PubMed]
- Gangjee, A.; Adair, O.O.; Pagley, M.; Queener, S.F. N9-substituted 2,4-diaminoquinazolines: Synthesis and biological evaluation of lipophilic inhibitors of pneumocystis carinii and toxoplasma gondii dihydrofolate reductase. J. Med. Chem. 2008, 51, 6195–6200. [Google Scholar] [CrossRef] [Green Version]
- Gangjee, A.; Vasudevan, A.; Queener, S.F.; Kisliuk, R.L. 2,4-Diamino-5-deaza-6-substituted pyrido[2,3-d]pyrimidine antifolates as potent and selective nonclassical inhibitors of dihydrofolate reductases. J. Med. Chem. 1996, 39, 1438–1446. [Google Scholar] [CrossRef]
- Piper, J.R.; Johnson, C.A.; Krauth, C.A.; Carter, R.L.; Hosmer, C.A.; Queener, S.F.; Borotz, S.E.; Pfefferkorn, E.R. Lipophilic antifolates as agents against opportunistic infections. 1. Agents superior to trimetrexate and piritrexim against toxoplasma gondii and pneumocystis carinii in vitro evaluations. J. Med. Chem. 1996, 39, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C.M.; Rosowsky, A. Synthesis of the lipophilic antifolate piritrexim via a palladium(0)-catalyzed cross-coupling reaction. J. Org. Chem. 2005, 70, 1364–1368. [Google Scholar] [CrossRef] [PubMed]
- Tong, L. Structure and function of biotin-dependent carboxylases. Cell. Mol. Life Sci. 2013, 5, 863–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shivaiah, K.-K.; Upton, B.; Nikolau, B.J. Kinetic, structural, and mutational analysis of Acyl-CoA carboxylase from thermobifida fusca YX. Front. Mol. Biosci. 2021, 7, 615614. [Google Scholar] [CrossRef] [PubMed]
- Grivsky, E.M.; Lee, S.; Sigel, C.W.; Duch, D.S.; Nichol, C.A. Synthesis and antitumor activity of 2,4-diamino-6-(2,5-dimethoxybenzyl)-5-methylpyrido[2,3-d]pyrimidine. J. Med. Chem. 1980, 23, 327–329. [Google Scholar] [CrossRef] [PubMed]
- Blankley, C.J.; Boschelli, D.H.; Doherty, A.M.; Hamby, J.M.; Klutchko, S.; Panek, R.L. Preparation of Pyrido[2,3-d]pyrimidines for Inhibiting Protein Tyrosine Kinase Mediated Cellular Proliferation. U.S. Patent No. 5733914 A, 31 March 1998. [Google Scholar]
- Klutchko, S.R.; Hamby, J.M.; Boschelli, D.H.; Wu, Z.; Kraker, A.J.; Amar, M.; Hartl, B.G.; Shen, C.; Klohs, W.D.; Steinkampf, R.W.; et al. 2-Substituted aminopyrido-[2,3-d]pyrimidin-7(8H)-ones. Structure-Activity Relationships against selected tyrosine kinases and in vitro and in vivo anticancer activity. J. Med. Chem. 1998, 41, 3276–3292. [Google Scholar] [CrossRef]
- Wada, T.; Penninger, J.M. Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004, 23, 2838–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, G.; Robinson, F.; Gibson, T.B.; Xu, B.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 2, 153–183. [Google Scholar]
- Keri, G.; Oerfi, L.; Greff, Z.; Banhegyi, P.; Szantai-Kis, C.; Eros, D.; Zsakai, L.; Boros, S.; Breza, N. Novel Compounds as Kinase Inhibitors and Their Use for the Regulation of Fibrotic Cell Proliferation. HU Patent Application No. 2015000620 A2, 28 July 2017. [Google Scholar]
- Adjei, A.A.; LoRusso, P.; Ribas, A.; Sosman, J.A.; Pavlick, A.; Dy, G.K.; Zhou, X.; Gangolli, E.; Kneissl, M.; Faucette, S.; et al. A phase I dose-escalation study of TAK-733, an investigational oral MEK inhibitor, in patients with advanced solid tumors. Investig. New Drugs. 2017, 1, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Dong, Q.; Dougan, D.R.; Gong, X.; Halkowycz, P.; Jin, B.; Kanouni, T.; O’Connell, S.M.; Scorah, N.; Shi, L.; Wallace, M.B.; et al. Discovery of TAK-733, a potent and selective MEK allosteric site inhibitor for the treatment of cancer. Bioorg. Med. Chem. Lett. 2011, 21, 1315–1319. [Google Scholar] [CrossRef] [PubMed]
- Micel, L.N.; Tentler, J.J.; Tan, A.C.; Selby, H.M.; Brunkow, K.L.; Robertson, K.M.; Lindsey Davis, S.; Klauck, P.J.; Pitts, T.M.; Gangolli, E.; et al. Antitumor activity of the MEK inhibitor TAK-733 against melanoma cell lines and patient-derived tumor explants. Mol. Cancer Ther. 2015, 14, 317–325. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Zhu, L.; Provencal, D.P.; Miller, T.A.; O’Bryan, C.; Langston, M.; Shen, M.; Bailey, D.; Sha, D.; Palmer, T.; et al. Process research and kilogram synthesis of an investigational, potent MEK inhibitor. Org. Process Res. Dev. 2012, 16, 1652–1659. [Google Scholar] [CrossRef]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.-A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and letrozole in advanced breast cancer. N. Engl. J. Med. 2016, 20, 1925–1936. [Google Scholar] [CrossRef]
- Turner, N.C.; Ro, J.; André, F.; Loi, S.; Verma, S.; Iwata, H.; Harbeck, N.; Loibl, S.; Bartlett, C.H.; Zhang, K.; et al. PALOMA3 study group palbociclib in hormone-receptor-positive advanced breast cancer. N. Engl. J. Med. 2015, 3, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Galbraith, M.D.; Bender, H.; Espinosa, J.M. Therapeutic targeting of transcriptional cyclin-dependent kinases. Transcription 2019, 2, 118–136. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 15, 3079–3093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, D.; Zhang, Y.; Xie, X.; Fang, L. Preparation of Palbociclib Intermediate and Method for Synthesizing Palbociclib. CN Patent No. 111675660 A, 2020. [Google Scholar]
- Lee, J.; Kim, S.T.; Kim, K.; Lee, H.; Kozarewa, I.; Mortimer, P.G.S.; Odegaard, J.I.; Harrington, E.A.; Lee, J.; Lee, T.; et al. Tumor genomic profiling guides patients with metastatic gastric cancer to targeted treatment: The VIKTORY umbrella trial. Cancer Discov. 2019, 10, 1388–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, G.; Liu, M.; Lu, J.; Meng, T. Practical synthesis of vistusertib (AZD2014), an ATP competitive mTOR inhibitor. Tetrahedron Lett. 2019, 60, 151333. [Google Scholar] [CrossRef]
- Anand, P.; Shenoy, R.; Palmer, J.E.; Baines, A.J.; Lai, R.Y.K.; Robertson, J.; Bird, N.; Ostenfeld, T.; Chizh, B.A. Clinical trial of the p38 MAP kinase inhibitor dilmapimod in neuropathic pain following nerve injury. Eur. J. Pain. 2011, 10, 1040–1048. [Google Scholar] [CrossRef]
- Christie, J.D.; Vaslef, S.; Chang, P.K.; May, A.K.; Gunn, S.R.; Yang, S.; Hardes, K.; Kahl, L.; Powley, W.M.; Lipson, D.A.; et al. A randomized dose-escalation study of the safety and anti-inflammatory activity of the p38 mitogen-activated protein kinase inhibitor dilmapimod in severe trauma subjects at risk for acute respiratory distress syndrome. Crit. Care Med. 2015, 9, 1859–1869. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Zhang, H.; Baloch, Z. Pathogenetic and therapeutic applications of tumor necrosis factor-α (TNF-α) in major depressive disorder: A systematic review. Int. J. Mol. Sci. 2016, 5, 733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanamee, E.S.; Faustman, D.L. Structural principles of tumor necrosis factor superfamily signaling. Sci. Signal. 2018, 511, eaao4910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palomo, J.; Dietrich, D.; Martin, P.; Palmer, G.; Gabay, C. The interleukin (IL)-1 cytokine family--Balance between agonists and antagonists in inflammatory diseases. Cytokine 2015, 1, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. Interleukin-6 family cytokines. Cold Spring Harb. Perspect. Biol. 2018, 2, a028415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, J.L.; Boehm, J.C.; Hall, R.; Jin, Q.; Kasparec, J.; Silva, D.J.; Taggart, J.J. Preparation of 2,4,8-Trisubstituted-8H-pyrido[2,3-d]pyrimidin-7-ones as CSBP/RK/p38 Kinase Inhibitors. WO Patent No. 2002059083 A2, 1 August 2002. [Google Scholar]
- Mehnert, J.M.; Edelman, G.; Stein, M.; Camisa, H.; Lager, J.; Dedieu, J.-F.; Ghuysen, A.-F.; Sharma, J.; Liu, L.; LoRusso, P.M. A phase I dose-escalation study of the safety and pharmacokinetics of a tablet formulation of voxtalisib, a phosphoinositide 3-kinase inhibitor, in patients with solid tumors. Investig. New Drugs 2018, 1, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Cai, T.; Zaks, T.; Romanelli, A. Compositions and Methods for Treating Cancer Using PI3K Inhibitor and Anti-CD19 Maytansinoid Immunoconjugate. WO Patent No. 2014058947 A1, 17 April 2014. [Google Scholar]
- Xu, D.-Q.; Toyoda, H.; Qi, L.; Morimoto, M.; Hanaki, R.; Iwamoto, S.; Komada, Y.; Hirayama, M. Induction of MEK/ERK activity by AZD8055 confers acquired resistance in neuroblastoma. Biochem. Biophys. Res. Commun. 2018, 3, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Pike, K.G.; Malagu, K.; Hummersone, M.G.; Menear, K.A.; Duggan, H.M.E.; Gomez, S.; Martin, N.M.B.; Ruston, L.; Pass, S.L.; Pass, M. Optimization of potent and selective dual mTORC1 and mTORC2 inhibitors: The discovery of AZD8055 and AZD2014. Bioorg. Med. Chem. Lett. 2013, 23, 1212–1216. [Google Scholar] [CrossRef] [PubMed]
- Kettle, J.G.; Cassar, D.J. Covalent inhibitors of the GTPase KRAS G12C: A review of the patent literature. Expert. Opin. Ther Pat. 2020, 2, 103–120. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xiang, S.; Kazi, A.; Sebti, S.M. The GTPase KRAS suppresses the p53 tumor suppressor by activating the NRF2-regulated antioxidant defense system in cancer cells. J. Biol. Chem. 2020, 10, 3055–3063. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-Z.; Xiao, J.-Q.; Xiao, S.-S.; Cheng, Y. KRAS: A promising therapeutic target for cancer treatment. Curr. Top. Med. Chem. 2019, 23, 2081–2097. [Google Scholar] [CrossRef] [PubMed]
- Uprety, D.; Adjei, A.A. KRAS: From undruggable to a druggable cancer target. Cancer Treat. Rev. 2020, 89, 102070. [Google Scholar] [CrossRef] [PubMed]
- Parsons, A.T.; Cochran, B.M.; Powazinik, W., IV; Caporini, M.A. Improved Synthesis of Key Intermediates of KRAS G12C Inhibitor. WO Patent No. 2020102730 A1, 22 May 2020. [Google Scholar]
- Skedelj, V.; Arsovska, E.; Tomasic, T.; Kroflic, A.; Hodnik, V.; Hrast, M.; Bester-Rogac, M.; Anderluh, G.; Gobec, S.; Bostock, J.; et al. 6-Arylpyrido [2,3-d] pyrimidines as novel ATP-competitive inhibitors of bacterial D-alanine: D-alanine ligase. PLoS ONE 2012, 7, e39922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blankley, C.J.; Doherty, A.M.; Hamby, J.M.; Panek, R.L.; Schroeder, M.C.; Showalter, H.D.H.; Connolly, C. Preparation of 6-Arylpyrido[2,3-d]pyrimidines and Naphthyridines for Inhibiting Protein Tyrosine Kinase Mediated Cellular Proliferation. U.S. Patent No. 5733913 A, 31 March 1998. [Google Scholar]
- Kimachi, T.; Kawase, M.; Matsuki, S.; Tanaka, K.; Yoneda, F. First total synthesis of coenzyme factor 420. J. Chem. Soc. Perkin Trans. 1 1990, 2, 253–256. [Google Scholar] [CrossRef]
- Estrada-Bernal, A.; Le, A.T.; Doak, A.E.; Tirunagaru, V.G.; Silva, S.; Bull, M.R.; Smaill, J.B.; Patterson, A.V.; Kim, C.; Liu, S.V.; et al. Tarloxotinib Is a Hypoxia-Activated Pan-HER kinase inhibitor active against a broad range of HER-family oncogenes. Clin. Cancer Res. 2021, 5, 1463–1475. [Google Scholar] [CrossRef] [PubMed]
- Smaill, J.B.; Patterson, A.V.; Lu, G.-L.; Lee, H.H.; Ashoorzadeh, A.; Anderson, R.F.; Wilson, W.R.; Denny, W.A.; Hsu, H.-L.A.; Maroz, A.; et al. Preparation of 4-Anilinopyrido[3,4-d]pyridine Prodrugs as Kinase Inhibitors Useful for Treatment of Cancer. U.S Patent No. 9101632 B2, 11 August 2015. [Google Scholar]
- Woodward, H.L.; Innocenti, P.; Cheung, K.-M.J.; Hayes, A.; Roberts, J.; Henley, A.T.; Faisal, A.; Mak, G.W.-Y.; Box, G.; Westwood, I.M.; et al. Introduction of a methyl group curbs metabolism of pyrido[3,4-d]pyrimidine monopolar spindle 1 (MPS1) inhibitors and enables the discovery of the phase 1 clinical candidate N2-(2-ethoxy-4-(4-methyl-4 H-1,2,4-triazol-3-yl)phenyl)-6-methyl-N8-neopentylpyrido[3,4-d]pyrimidine-2,8-diamine (BOS172722). J. Med. Chem. 2018, 18, 8226–8240. [Google Scholar] [CrossRef]
- Anderhub, S.J.; Mak, G.W.-Y.; Gurden, M.D.; Faisal, A.; Drosopoulos, K.; Walsh, K.; Woodward, H.L.; Innocenti, P.; Westwood, I.M.; Naud, S.; et al. High proliferation rate and a compromised spindle assembly checkpoint confers sensitivity to the MPS1 inhibitor BOS172722 in triple-negative breast cancers. Mol. Cancer Ther. 2019, 10, 1696–1707. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Zhang, M.; Liang, D.; Sun, W.; Zhang, C.; Jiang, M.; Liu, J.; Li, J.; Li, C.; Yang, X.; et al. Molecular design and anticancer activities of small-molecule monopolar spindle 1 inhibitors: A medicinal chemistry perspective. Eur. J. Med. Chem. 2019, 175, 247–268. [Google Scholar] [CrossRef] [PubMed]
- Kessler, A.F.; Feldheim, J.; Schmitt, D.; Feldheim, J.J.; Monoranu, C.M.; Ernestus, R.-I.; Löhr, M.; Hagemann, C. Monopolar spindle 1 kinase (MPS1/TTK) mRNA expression is associated with earlier development of clinical symptoms, tumor aggressiveness and survival of glioma patients. Biomedicines 2020, 7, 192. [Google Scholar] [CrossRef]
- Zeiser, R.; Andrlová, H.; Meiss, F. Trametinib (GSK1120212) recent results. Cancer Res. 2018, 211, 91–100. [Google Scholar] [CrossRef]
- Long, G.V.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandalà, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A.; et al. Adjuvant dabrafenib plus trametinib in stage III BRAF-mutated melanoma. N. Engl. J. Med. 2017, 19, 1813–1823. [Google Scholar] [CrossRef] [Green Version]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.T.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 8, 1263–1284. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Hai, W.; Shi, Z. Method for Synthesizing Trametinib for Treating Melanoma with Low Cost. CN Patent No. 109320513 A, 12 February 2019. [Google Scholar]
- Juarez, M.; Diaz, N.; Johnston, G.I.; Nayar, S.; Payne, A.; Helmer, E.; Cain, D.; Williams, P.; Devauchelle-Pensec, V.; Fisher, B.A.; et al. A phase 2 randomized, double-blind, placebo-controlled, proof-of-concept study of oral seletalisib in primary Sjögren’s syndrome. Rheumatology 2021, 3, 1364–1375. [Google Scholar] [CrossRef] [PubMed]
- Aerts, L.L.J.J.; Assaf, G.; Carly, N.E.; Cool, V.A.C.; Delatinne, J.-P.; Delhaye, L.J.W.; Kestemont, J.P.; Le Meur, S. Crystalline Forms of Seletalisib for Treatment of Inflammatory, Autoimmune, Cardiovascular, Neurodegenerative, Metabolic, Oncolologic, Nociceptive or Ophthalmic Conditions. WO Patent No. 2018219772 A1, 6 December 2018. [Google Scholar]
- Mackman, R.L.; Mish, M.; Chin, G.; Perry, J.K.; Appleby, T.; Aktoudianakis, V.; Metobo, S.; Pyun, P.; Niu, C.; Daffis, S.; et al. Discovery of GS-9688 (Selgantolimod) as a potent and selective oral toll-like receptor 8 agonist for the treatment of chronic hepatitis B. J. Med. Chem. 2020, 63, 10188–10203. [Google Scholar] [CrossRef] [PubMed]
- Asselin, S.M.; Badalov, P.R.; Morrison, H.G.; Regens, C.S.; Vieira, T. Preparation of Solid Forms of (R)-2-[(2-Amino-7-fluoropyrido[3,2-d]pyrimidin-4-yl)amino]-2-methylhexan-1-ol as Toll-Like Receptor Modulators. WO Patent No. 2020214663 A1, 22 October 2020. [Google Scholar]
- Wall, M.; Shim, J.H.; Benkovic, S.J. A multisubstrate adduct inhibitor of AICAR transformylase. J. Med. Chem. 1999, 42, 3421–3424. [Google Scholar] [CrossRef] [PubMed]
- Warren, M.S.; Mattia, K.M.; Marolewski, A.E.; Benkovic, S.J. The transformylase enzymes of de novo purine biosynthesis. Pure Appl. Chem. 1996, 68, 2029–2036. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campos, J.F.; Besson, T.; Berteina-Raboin, S. Review on the Synthesis and Therapeutic Potential of Pyrido[2,3-d], [3,2-d], [3,4-d] and [4,3-d]pyrimidine Derivatives. Pharmaceuticals 2022, 15, 352. https://doi.org/10.3390/ph15030352
Campos JF, Besson T, Berteina-Raboin S. Review on the Synthesis and Therapeutic Potential of Pyrido[2,3-d], [3,2-d], [3,4-d] and [4,3-d]pyrimidine Derivatives. Pharmaceuticals. 2022; 15(3):352. https://doi.org/10.3390/ph15030352
Chicago/Turabian StyleCampos, Joana F., Thierry Besson, and Sabine Berteina-Raboin. 2022. "Review on the Synthesis and Therapeutic Potential of Pyrido[2,3-d], [3,2-d], [3,4-d] and [4,3-d]pyrimidine Derivatives" Pharmaceuticals 15, no. 3: 352. https://doi.org/10.3390/ph15030352
APA StyleCampos, J. F., Besson, T., & Berteina-Raboin, S. (2022). Review on the Synthesis and Therapeutic Potential of Pyrido[2,3-d], [3,2-d], [3,4-d] and [4,3-d]pyrimidine Derivatives. Pharmaceuticals, 15(3), 352. https://doi.org/10.3390/ph15030352