
Natural Phaeosphaeride A Derivatives Overcome Drug Resistance of Tumor Cells and Modulate Signaling Pathways

 , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
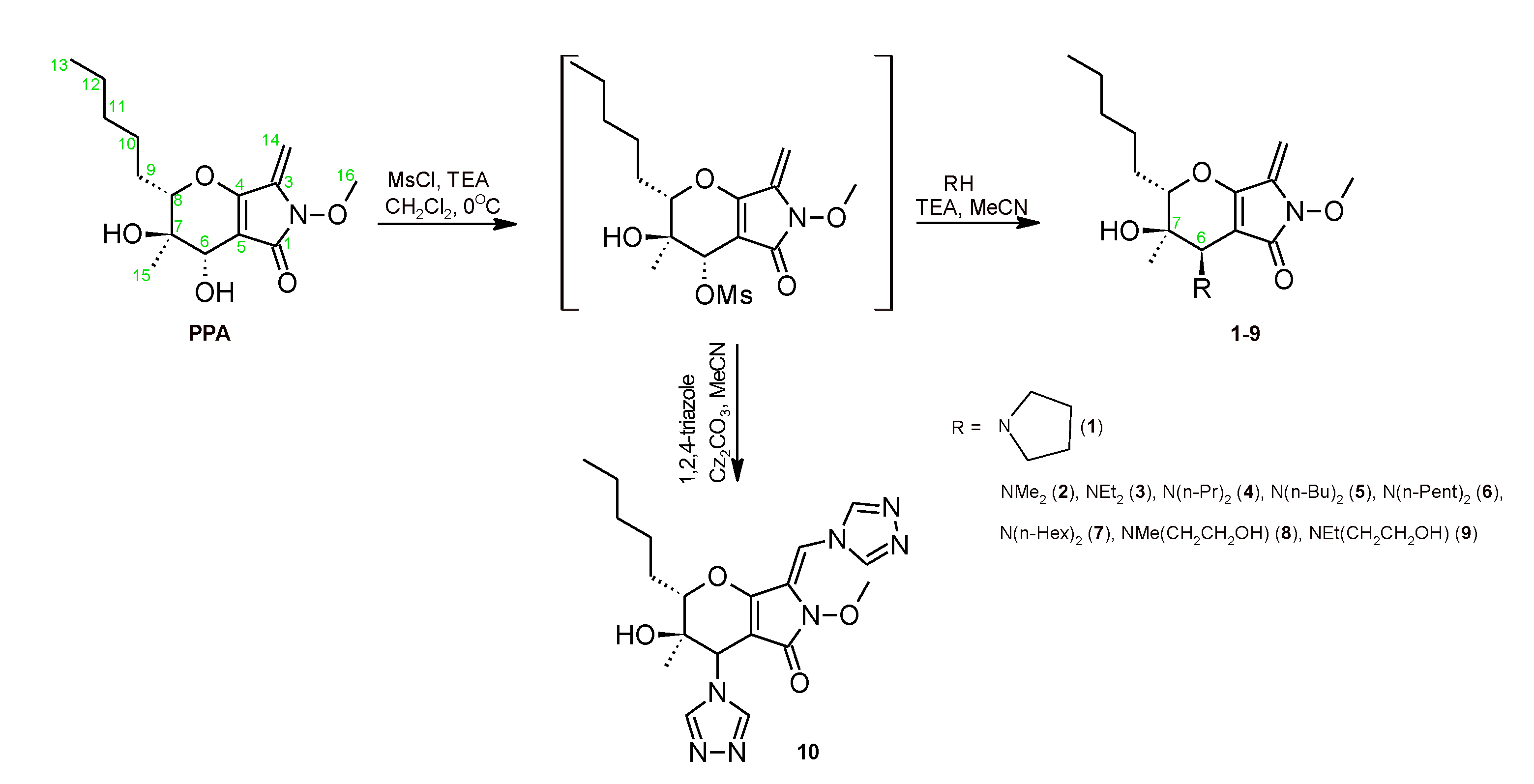
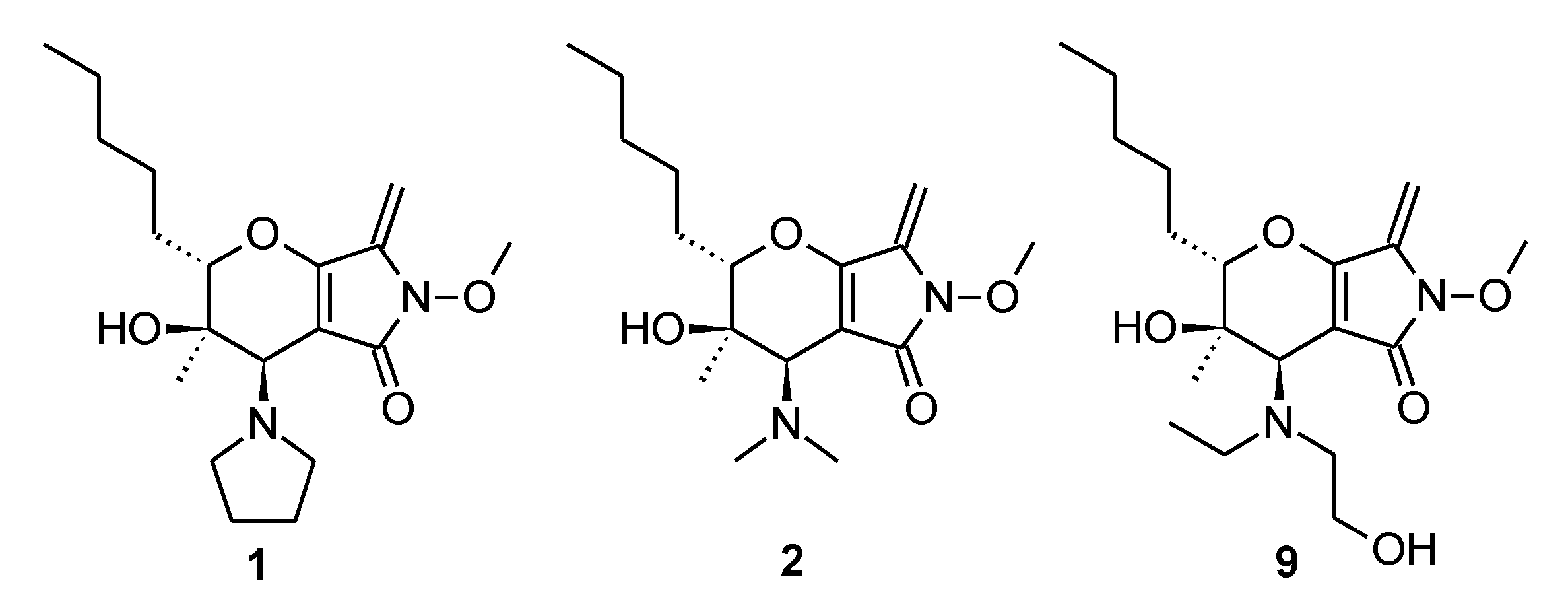
2.1. Chemistry
2.2. Biological Activities
2.2.1. Assessment of the Cytotoxicity Level of Compounds on a Panel of Tumor Cell Cultures
Assessment of the Cytotoxicity Level on Cultures of Tumor Cells
Assessment of the Cytotoxicity Level in Primary Cultures of Soft Tissue Sarcomas
Overcoming Drug Resistance of Tumor Cells
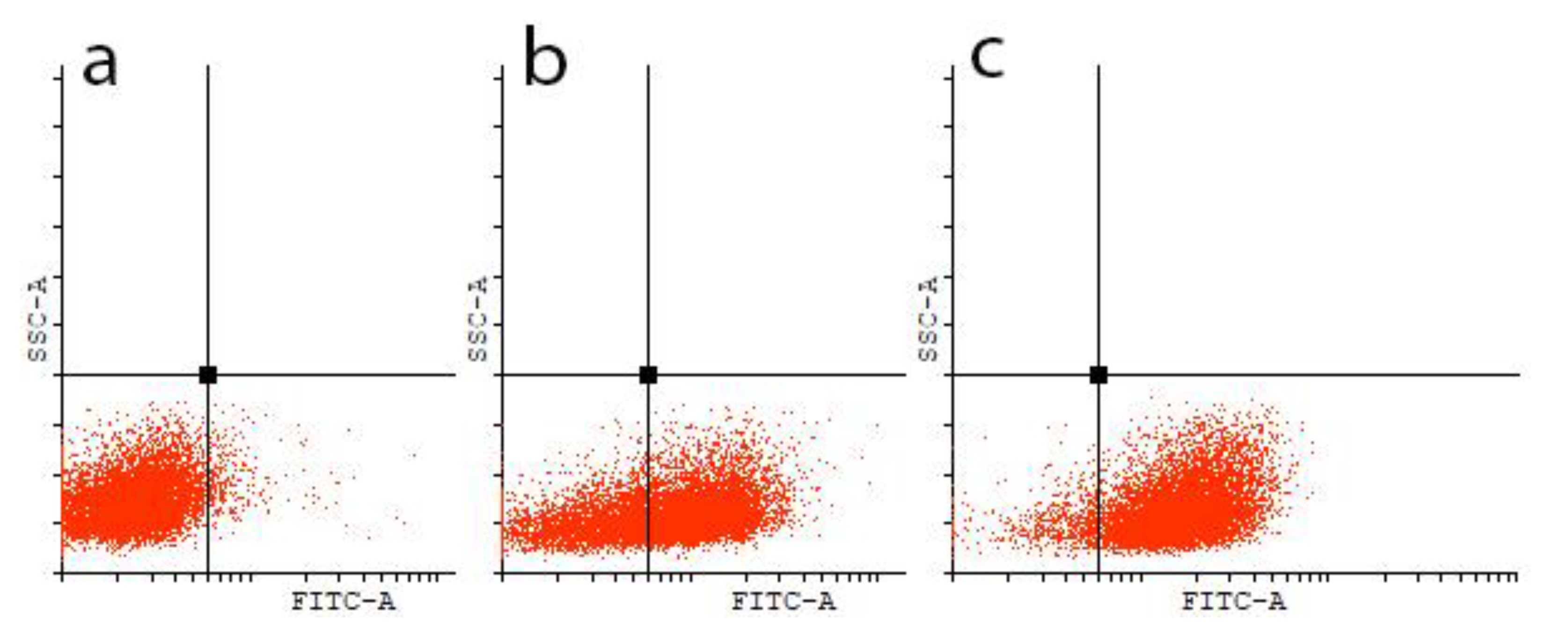
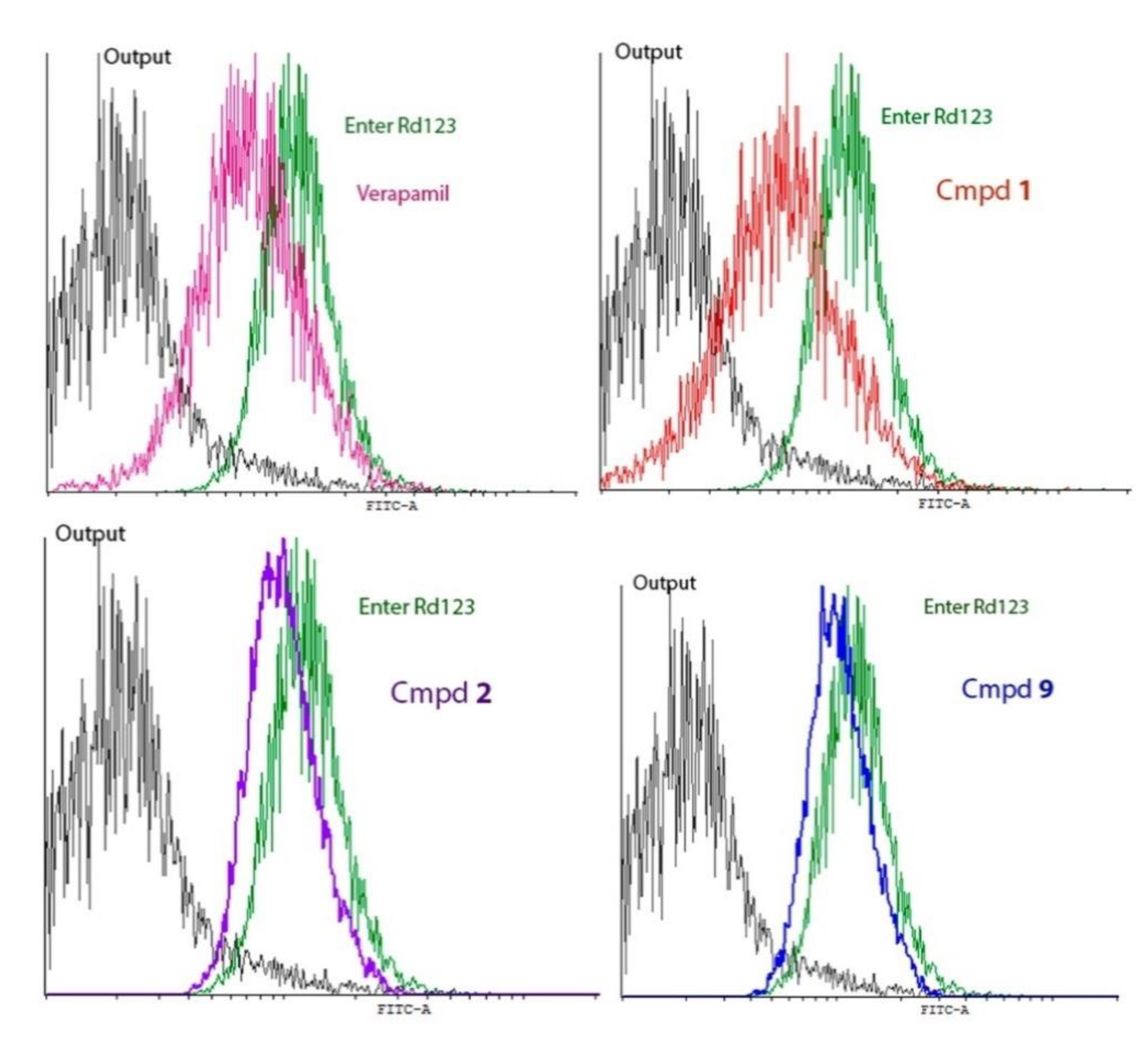
Interaction of the Obtained Compounds with the P-Glycoprotein
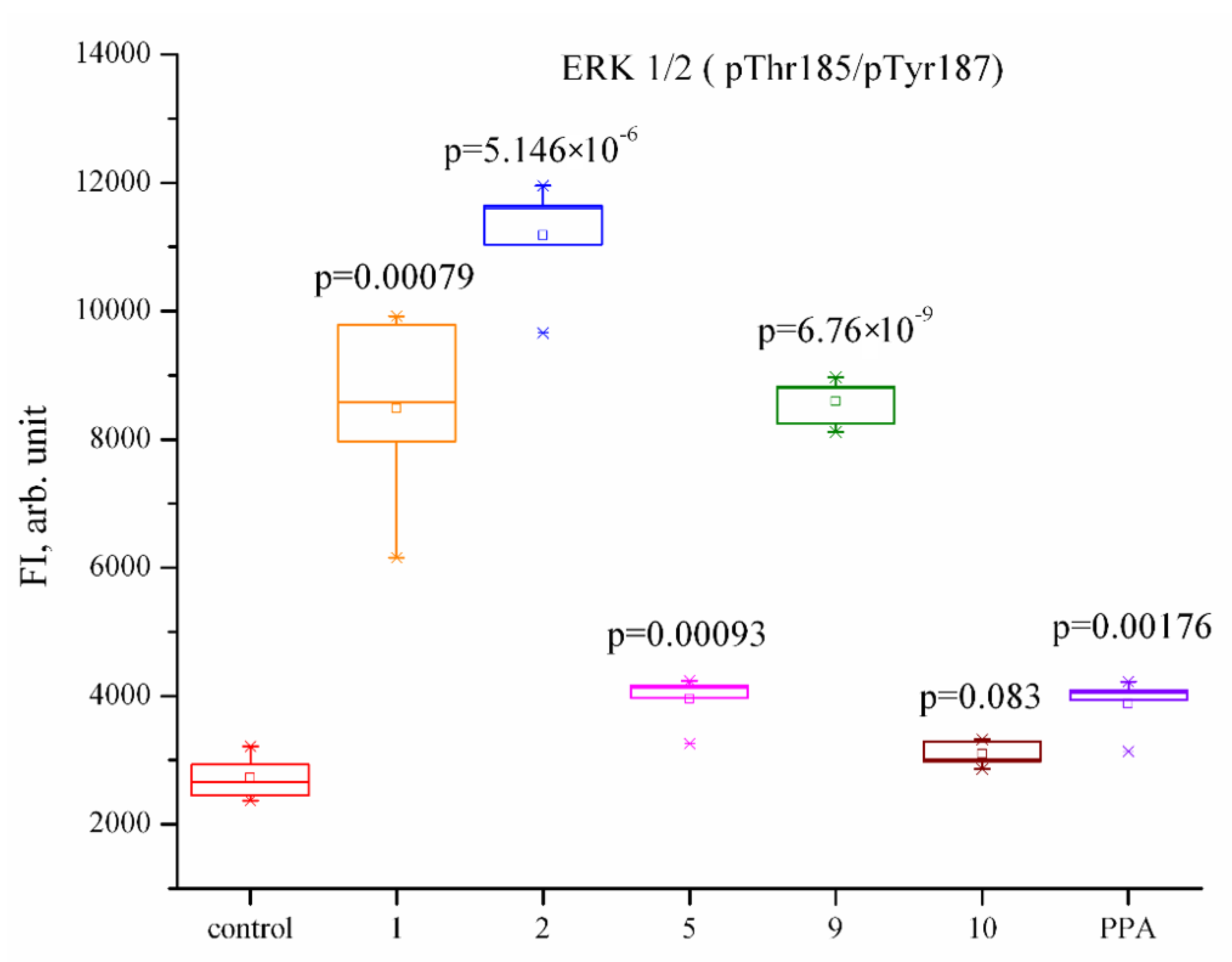
2.2.2. Influence of Phaeosphaeride A Derivatives on the Key Signaling Pathways
3. Materials and Methods
3.1. Chemistry
- General Procedure for the Synthesis of Compounds 2–9
- A mixture of the crude (2S,3S,4S)-3-hydroxy-6-methoxy-3-methyl-7-methylene-5-oxo-2-pentyl-2,3,4,5,6,7-hexahydropyrano[2,3-c]pyrrol-4-yl methane sulfonate (1 mmol), the corresponding secondary amine (1.5 mmol), and triethylamine (3 mmol) was stirred in dry acetonitrile (2 mL) at room temperature until the consumption of the starting material was complete as judged by TLC analysis (24 h). The reaction mixture was quenched with water and extracted with EtOAc (2 × 20 mL). The organic extract was washed with brine, dried over magnesium sulfate, and concentrated in vacuo. The crude product was purified by flash chromatography (DCM/methanol).
- (2S,3R,4R)-4-(dimethylamino)-3-hydroxy-6-methoxy-3-methyl-7-methylene-2-pentyl-3,4,6,7-tetrahydropyrano[2,3-c]pyrrol-5(2H)-one (2). Yield 45%, yellow oil. −158.11 (c 1.11, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 5.26 (br s, 1H), 5.03 (d, J = 1.5 Hz, 1H), 4.99 (d, J = 1.5 Hz, 1H), 3.93 (s, 3H), 3.65–3.63 (m, 1H), 3.00 (s, 1H), 2.44 (br s, 6H), 2.00–1.91 (m, 1H), 1.69–1.53 (m, 2H), 1.44–1.31 (m, 5H), 1.04 (s, 3H), 0.91 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 167.18 (s), 158.35 (s), 137.11 (s), 101.74 (s), 91.58 (s), 83.24 (s), 68.47 (s), 64.58 (s), 62.74 (s), 31.86 (s), 28.42 (s), 26.59 (s), 22.69 (s), 20.18 (s), 14.19 (s). IR (KBr) 3430, 2955, 2930, 2859, 1723, 1635, 1436, 1370, 1265, 1149, 986, 775 cm−1. HRMS [M + H]+calcd for C17H29N2O4 325.21218, found 325.21206.
- (2S,3R,4R)-4-(diethylamino)-3-hydroxy-6-methoxy-3-methyl-7-methylene-2-pentyl-3,4,6,7-tetrahydropyrano[2,3-c]pyrrol-5(2H)-one (3). Yield 50%, yellow oil. −182.36 (c 1.15, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 5.23 (s, 1H), 5.03 (d, J = 1.5 Hz, 1H), 4.98 (d, J = 1.5 Hz, 1H), 3.93 (s, 3H), 3.56–3.54 (m, 1H), 3.15 (s, 1H), 3.06–2.27 (m, 4H), 2.02–1.92 (m, 1H), 1.72–1.54 (m, 2H), 1.49–1.30 (m, 5H), 1.09 (br s, 6H), 1.05 (s, 3H), 0.92 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 167.05 (s), 158.53 (s), 137.16 (s), 102.33 (s), 91.43 (s), 83.26 (s), 67.93 (s), 64.52 (s), 60.14 (s), 48.97 (s), 45.76 (s), 31.85 (s), 28.49 (s), 26.62 (s), 22.69 (s), 19.98 (s), 14.20 (s). IR (KBr) 3207, 2961, 2932, 2860, 1723, 1635, 1468, 1379, 1268, 1149, 978, 764 cm−1. HRMS [M + H]+calcd for C19H33N2O4 353.24348, found 353.24324.
- (2S,3R,4R)-4-(dipropylamino)-3-hydroxy-6-methoxy-3-methyl-7-methylene-2-pentyl-3,4,6,7-tetrahydropyrano[2,3-c]pyrrol-5(2H)-one (4). Yield 40%, yellow oil. −205.07 (c 2.01, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 5.27 (s, 1H), 5.03 (d, J = 1.4 Hz, 1H), 4.99 (d, J = 1.4 Hz, 1H), 3.93 (s, 3H), 3.58 (m, 1H), 3.14 (s, 1H), 2.98 (br s, 1H), 2.46 (m, 3H), 1.98–1.90 (m, 1H), 1.64–1.34 (m, 11H), 1.04 (s, 3H), 0.92–0.89 (m, 9H). 13C NMR (101 MHz, CDCl3) δ 167.08 (s), 158.51 (s), 137.19 (s), 102.36 (s), 91.48 (s), 83.27 (s), 83.17 (s), 68.20 (s), 64.53 (s), 61.00 (s), 58.02 (s), 54.07 (s), 31.76 (s), 28.40 (s), 26.45 (s), 22.67 (s), 22.28 (s), 21.88 (s), 20.16 (s), 14.19 (s), 11.90 (s). IR (KBr) 3464, 2960, 2932, 2873, 1724, 1635, 1436, 1378, 1149, 1072, 914 cm−1. HRMS [M + H]+calcd for C21H37N2O4 381.27478, found 381.27435.
- (2S,3R,4R)-4-(dibutylamino)-3-hydroxy-6-methoxy-3-methyl-7-methylene-2-pentyl-3,4,6,7-tetrahydropyrano[2,3-c]pyrrol-5(2H)-one (5). Yield 39%, yellow oil. −178.71 (c 1.8, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 5.28 (s, 1H), 5.03 (d, J = 1.5 Hz, 1H), 4.99 (d, J = 1.5 Hz, 1H), 3.93 (s, 3H), 3.59–3.57 (m, 1H), 3.14 (s, 1H), 2.99 (br s, 1H), 2.70–2.26 (m, 3H), 1.99–1.91 (m, 1H), 1.67–1.31 (m, 15H), 1.05 (s, 3H), 1.05–0.90 (m, 9H). 13C NMR (101 MHz, CDCl3) δ 167.12 (s), 158.53 (s), 137.20 (s), 102.37 (s), 91.59 (s), 91.47 (s), 83.30 (s), 83.20 (s), 68.15 (s), 64.61 (s), 64.54 (s), 60.99 (s), 60.94 (s), 31.76 (s), 28.41 (s), 26.57 (s), 22.68 (s), 20.63 (s), 20.18 (s), 14.24 (s). IR (KBr) 3437, 2958, 2931, 2860, 1724, 1634, 1435, 1378, 1150, 1076, 1033, 914 cm−1. HRMS [M + H]+calcd for C23H41N2O4 409.30608, found 409.30580.
- (2S,3R,4R)-4-(dipentylamino)-3-hydroxy-6-methoxy-3-methyl-7-methylene-2-pentyl-3,4,6,7-tetrahydropyrano[2,3-c]pyrrol-5(2H)-one (6). Yield 40%, yellow oil. −179.86 (c 1.45, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 5.29 (s, 1H), 5.03 (d, J = 1.4 Hz, 1H), 4.99 (d, J = 1.4 Hz, 1H), 3.93 (s, 3H), 3.59–3.57 (m, 1H), 3.14 (s, 1H), 2.99 (br s, 1H), 2.70–2.22 (m, 3H), 2.00–1.91 (m, 1H), 1.62–1.25 (m, 19H), 1.05 (s, 3H), 0.93–0.89 (m, 9H). 13C NMR (101 MHz, CDCl3) δ 167.10 (s), 158.51 (s), 137.19 (s), 102.37 (s), 91.46 (s), 83.27 (s), 83.16 (s), 68.15 (s), 64.53 (s), 60.98 (s), 60.92 (s), 55.98 (s), 52.11 (s), 31.73 (s), 29.62 (s), 29.00 (s), 28.41 (s), 26.54 (s), 22.76 (s), 22.68 (s), 20.18 (s), 14.19 (s). IR (KBr) 3485, 2957, 2930, 2860, 2072, 1724, 1634, 1436, 1378, 1150, 1079, 915 cm−1. HRMS [M + H]+calcd for C25H45N2O4 437.33738, found 437.33701.
- (2S,3R,4R)-4-(dihexylamino)-3-hydroxy-6-methoxy-3-methyl-7-methylene-2-pentyl-3,4,6,7-tetrahydropyrano[2,3-c]pyrrol-5(2H)-one (7). Yield 41%, yellow oil. −185.98 (c 0.97, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 5.28 (s, 1H), 5.03 (d, J = 1.4 Hz, 1H), 4.98 (d, J = 1.4 Hz, 1H), 3.93 (s, 3H), 3.60–3.57 (m, 1H), 3.13 (s, 1H), 2.98 (br s, 1H), 2.67–2.29 (m, 3H), 1.99–1.91 (m, 1H), 1.65–1.29 (m, 23H), 1.04 (s, 3H), 0.93–0.86 (m, 9H). 13C NMR (101 MHz, CDCl3) δ 167.11 (s), 158.51 (s), 137.20 (s), 102.39 (s), 91.44 (s), 83.30 (s), 83.20(s), 68.15 (s), 64.61 (s), 64.53 (s), 60.99 (s), 60.94 (s), 56.15 (s), 52.34 (s), 31.91 (s), 31.75 (s), 29.28 (s), 28.80 (s), 28.42 (s), 27.12 (s), 26.57 (s), 22.77 (s), 22.69 (s), 20.18 (s), 14.18 (s). IR (KBr) 3474, 2957, 2929, 2858, 2075, 1724, 1634, 1435, 1378, 1151, 1081, 914 cm−1. HRMS [M + H]+calcd for C27H49N2O4 465.36868, found 465.36853.
- (2S,3R,4R)-3-hydroxy-4-[(2-hydroxyethyl)(methyl)amino]-6-methoxy-3-methyl-7-methylene-2-pentyl-3,4,6,7-tetrahydropyrano[2,3-c]pyrrol-5(2H)-one (8). Yield 63%, yellow oil. −151.76 (c 2.1, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 5.10 (d, J = 1.7 Hz, 1H), 5.05 (d, J = 1.6 Hz, 1H), 4.82 (br s, 1H), 4.01–3.95 (m, 4H), 3.65–3.62 (m, 2H), 3.50 (s, 1H), 3.09–2.90 (m, 2H), 2.42 (s, 3H), 2.02–1.93 (m, 1H), 1.69–1.57 (m, 2H), 1.44–1.33 (m, 5H), 1.06 (s, 3H), 0.92 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 167.72 (s), 158.61 (s), 136.70 (s), 101.63 (s), 92.74 (s), 83.18 (s), 69.57 (s), 64.76 (s), 61.20 (s), 59.22 (s), 39.75 (s), 32.06 (s), 31.83 (s), 29.84 (s), 28.23 (s), 26.54 (s), 22.68 (s), 19.75 (s), 14.17 (s). IR (KBr) 3420, 2956, 2929, 2859, 1719, 1635, 1438, 1378, 1193, 1081, 1021, 951, 913 cm−1. HRMS [M + H]+calcd for C19H30N2O5 355.22275, found 355.22267.
- (2S,3R,4R)-4-[ethyl(2-hydroxyethyl)amino]-3-hydroxy-6-methoxy-3-methyl-7-methylene-2-pentyl-3,4,6,7-tetrahydropyrano[2,3-c]pyrrol-5(2H)-one (9). Yield 45%, yellow oil. −197.38 (c 1.91, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 5.10 (s, 1H), 5.05 (s, 1H), 4.71 (br s, 1H), 3.94 (br s, 4H), 3.62–3.59 (m, 1H), 3.19–2.52 (m, 4H), 2.02–1.90 (m, 1H), 1.69–1.55 (m, 4H), 1.45–1.32 (m, 5H), 1.07 (br s, 6H), 0.91 (t, J = 6.8 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 167.70 (s), 158.60 (s), 136.76 (s), 102.19 (s), 92.59 (s), 83.07 (s), 69.10 (s), 64.70 (s), 60.78 (s), 59.36 (s), 55.50 (s), 50.96 (s), 31.81 (s), 28.25 (s), 26.56 (s), 22.67 (s), 19.72 (s), 14.39 (s), 14.19 (s). IR (KBr) 3430, 2958, 2931, 2860, 1720, 1634, 1438, 1438, 1379, 1336, 1151, 1054, 990, 913, 743 cm−1. HRMS [M + H]+calcd for C19H33N2O5 369.23840, found 369.23828.
3.2. Biological Assay Methods
3.2.1. Assessment of the Level of Compounds Cytotoxicity in a Panel of Tumor Cell Cultures
Tumor Cell Cultures
MTT Test
Flow Cytometry
Evaluation of the Release of Rhodamine 123 (Rd123) from Cells with an Overexpression of P-Glycoprotein
Statistical Analysis
3.2.2. Evaluation of the Effect of Phaeosphaeride A Derivatives on Key Signaling Pathways
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stavrovskaya, A.A.; Stromskaya, T.P. Transport proteins of the ABC family and multidrug resistance of tumor cells. Biochemistry 2008, 73, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Dewanjee, S.; Dua, T.K.; Bhattacharjee, N.; Das, A.; Gangopadhyay, M.; Khanra, R.; Joardar, S.; Riaz, M.; Feo, V.; Zia-Ul-Haq, M. Natural products as alternative choices for P-glycoprotein (P-gp) inhibition. Molecules 2017, 22, 871. [Google Scholar] [CrossRef]
- Zhou, S.F. Structure, function and regulation of P-glycoprotein and its clinical relevance in drug disposition. Xenobiotica 2008, 38, 802–832. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Chevillard, S.; Vielh, P.; Vallidire, P.; Robert, J.; Marie, J.P. A study of the expression of MDR1 gene in solid tumors. Initial results of a multicenter evaluation. Bull. Cancer 1996, 83, 626–633. [Google Scholar] [PubMed]
- Adamska, A.; Falasca, M. ATP-binding cassette transporters in progression and clinical outcome of pancreatic cancer: What is the way forward? World J. Gastroenterol. 2018, 24, 3222–3238. [Google Scholar] [CrossRef]
- Binkhathlan, Z.; Lavasanifar, A. P-glycoprotein inhibition as a therapeutic approach for overcoming multidrug resistance in cancer: Current status and future perspectives. Curr. Cancer Drug Targets 2013, 13, 326–346. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Pan, G. Drug Transporters in Drug Disposition, Effects and Toxicity. In Advances in Experimental Medicine and Biology; Springer: Singapore, 2019; Volume 1141. [Google Scholar]
- Gonçalves, B.M.F.; Cardoso, D.S.P.; Ferreira, U.M.-J. Overcoming Multidrug Resistance: Flavonoid and Terpenoid Nitrogen-Containing Derivatives as ABC Transporter Modulators. Molecules 2020, 25, 3364. [Google Scholar] [CrossRef]
- Kobayashi, K.; Kobayashi, Y.; Nakamura, M.; Tamura, O.; Kogen, H. Establishment of Relative and Absolute Configurations of Phaeosphaeride A: Total Synthesis of ent-Phaeosphaeride A. J Org. Chem. 2015, 2015, 1243–1248. [Google Scholar] [CrossRef]
- Abzianidze, V.; Poluektova, E.; Bolshakova, K.; Panikorovskii, T.; Bogachenkov, A.; Berestetskiy, A. Crystal structure of natural phaeosphaeride A. Acta Crystallogr. 2015, E71, o625–o626. [Google Scholar] [CrossRef] [PubMed]
- Wake, M.S.; Watson, C.J. STAT3 the oncogene—Still eluding therapy? FEBS J. 2015, 282, 2600–2611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, P.A.; Grandis, J.R. STAT3 signaling: Anticancer strategies and challenges. Mol. Interv. 2011, 11, 18–26. [Google Scholar] [CrossRef] [Green Version]
- Maloney, K.N.; Hao, W.; Xu, J.; Gibbons, J.; Hucul, J.; Roll, D.; Brady, S.F.; Schroeder, F.C.; Clardy, J. Phaeosphaeride A, an inhibitor of STAT3-dependent signaling isolated from an endophytic fungus. Org. Lett. 2006, 8, 4067–4070. [Google Scholar] [CrossRef]
- Shao, H.; Cheng, H.Y.; Cook, R.G.; Tweardy, D.J. Identification and characterization of signal transducer and activator of transcription 3 recruitment sites within the epidermal growth factor receptor. Cancer Res. 2003, 63, 3923–3930. [Google Scholar] [PubMed]
- Chatzimpaloglou, A.; Yavropoulou, M.P.; Rooij, K.E.; Biedermann, R.; Mueller, U.; Kaskel, S.; Sarli, V. Total Synthesis and Biological Activity of the Proposed Structure of Phaeosphaeride A. J. Org. Chem. 2012, 77, 9659–9667. [Google Scholar] [CrossRef] [PubMed]
- Chatzimpaloglou, A.; Kolosov, M.; Eckols, T.K.; Tweardy, D.J.; Sarli, V. Synthetic and Biological Studies of Phaeosphaerides. J. Org. Chem. 2014, 79, 4043–4054. [Google Scholar] [CrossRef] [PubMed]
- Abzianidze, V.; Prokofieva, D.; Chisty, L.; Bolshakova, K.; Berestetskiy, A.; Panikorovskii, T.; Bogachenkov, A.; Holder, A. Synthesis of natural phaeosphaeride A derivatives and an in vitro evaluation of their anti-cancer potential. Bioorg. Med. Chem. Lett. 2015, 25, 5566–5569. [Google Scholar] [CrossRef]
- Abzianidze, V.; Beltyukov, P.; Zakharenkova, S.; Moiseeva, N.; Mejia, J.; Holder, A.; Trishin, Y.; Berestetskiy, A.; Kuznetsov, V. Synthesis and Biological Evaluation of Phaeosphaeride A Derivatives as Antitumor Agents. Molecules 2018, 23, 3043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abzianidze, V.; Zakharenkova, S.; Moiseeva, N.; Beltyukov, P.; Polukeev, V.; Dubrovskii, Y.; Kuznetsov, V.; Trishin, Y.; Mejia, J.; Holder, A. Towards lead compounds as anti-cancer agents via new phaeosphaeride A derivatives. Bioorg. Med. Chem. Lett. 2019, 29, 59–61. [Google Scholar] [CrossRef]
- Zakharenkova, S.; Abzianidze, V.; Moiseeva, N.; Lukina, D.; Chistyi, L.; Krivorotov, D.; Trishin, Y. Antitumor activity of phaeosphaeride A modified with nitrogen heterocyclic groups. Mendeleev Commun. 2021, 31, 662–663. [Google Scholar] [CrossRef]
- Barbosa, R.; Acevedo, L.A.; Marmorstein, R. The MEK/ERK Network as a Therapeutic Target in Human Cancer. Mol. Cancer Res. 2021, 19, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Yip, H.Y.K.; Papa, A. Signaling Pathways in Cancer: Therapeutic Targets, Combinatorial Treatments, and New Developments. Cells 2021, 10, 659. [Google Scholar] [CrossRef] [PubMed]
- Panda, M.; Biswal, B.K. Cell signaling and cancer: A mechanistic insight into drug resistance. Mol. Biol. Rep. 2019, 46, 5645–5659. [Google Scholar] [CrossRef] [PubMed]
- Senga, S.S.; Grose, R.P. Hallmarks of cancer—The new testament. Open Biol. 2021, 11, 200358. [Google Scholar] [CrossRef] [PubMed]
- D’yakonov, V.A.; Makarov, A.A.; Dzhemileva, L.U.; Ramazanov, I.R.; Makarova, E.K.; Dzhemilev, U.M. Natural trienoic acids as anticancer agents: First stereoselective synthesis, cell cycle analysis, induction of apoptosis, cell signaling and mitochondrial targeting studies. Cancers 2021, 13, 1808. [Google Scholar] [CrossRef]
- Filimonova, K.S.; Rogovskaya, N.Y.; Beltyukov, P.P.; Babakov, V.N. Assessing hepatoprotective effects of antioxidants on amiodarone-induced cytotoxicity in human hepatoma HepaRG cell line. Med. Extrem. Situat. 2021, 3, 43–51. [Google Scholar] [CrossRef]
- Gamboa, A.C.; Gronchi, A.; Cardona, K. Soft-tissue sarcoma in adults: An update on the current state of histiotype-specific management in an era of personalized medicine. CA Cancer J. Clin. 2020, 70, 200–229. [Google Scholar] [CrossRef] [Green Version]
- Galland, F.; Seady, M.; Taday, J.; Smaili, S.S.; Gonçalves, C.A.; Leite, M.C. Astrocyte culture models: Molecular and function characterization of primary culture, immortalized astrocytes and C6 glioma cells. Neurochem. Int. 2019, 131, 104538. [Google Scholar] [CrossRef] [PubMed]
- Turkson, J.; Bowman, T.; Adnane, J.; Zhang, Y.; Djeu, J.Y.; Sekharam, M.; Frank, D.; Holzman, L.; Wu, J.; Sebti, S.; et al. Requirement for Ras/Rac1-mediated p38 and c-Jun N-terminal kinase signaling in Stat3 transcriptional activity induced by the Src oncoprotein. Mol. Cell. Biol. 1999, 19, 7519–7528. [Google Scholar] [CrossRef] [Green Version]
- Ki, Y.; Parka, J.; Lee, J.; Shin, I.; Koh, H. JNK and p38 MAPK regulate oxidative stress and the inflammatory response in chlorpyrifos-induced apoptosis. Toxicol. Lett. 2013, 218, 235–245. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, Q.; Evers, B.M.; Chung, D.H. Signal transduction pathways involved in oxidative stress-induced intestinal epithelial cell apoptosis. Pediatr. Res. 2005, 58, 1192–1197. [Google Scholar] [CrossRef] [Green Version]
- Corre, I.; Paris, F.; Huot, J. The p38 pathway, a major pleiotropic cascade that transduces stress and metastatic signals in endothelial cells. Oncotarget 2017, 8, 55684. [Google Scholar] [CrossRef] [PubMed]
- Saint-Ruf, C.; Nardeux, P.; Estrade, S.; Brouty-Boye, D.; Lavialle, C.; Rhim, J.S.; Cassingena, R. Accelerated malignant conversion of human HBL-100 cells by the v-Ki-ras oncogene. Exp. Cell Res. 1988, 176, 60–67. [Google Scholar] [CrossRef]
- Moiseeva, N.I.; Laletina, L.A.; Fetisov, T.I.; Makhmudova, L.F.; Manikaylo, A.E.; Fomina, L.Y.; Burov, D.A.; Lesovaya, E.A.; Bokhyan, B.Y.; Zinovieva, V.Y.; et al. Analysis of Multiple Drug Resistance Mechanism in Different Types of Soft Tissue Sarcomas: Assessment of the Expression of ABC-Transporters, MVP, YB-1, and Analysis of Their Correlation with Chemosensitivity of Cancer Cells. Int. J. Mol. Sci. 2022, 23, 3183. [Google Scholar] [CrossRef]
- Mechetner, E.B.; Roninson, I.B. Efficient inhibition of P-glycoprotein-mediated multidrug resistance with a monoclonal antibody. Proc. Natl. Acad. Sci. USA 1992, 89, 5824–5828. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HCT-116 | MCF-7 | PC3 | A549 | HEK293 | |
|---|---|---|---|---|---|
| Etoposide | 21.0 ± 9.9 | 8.9 ± 2.3 | 27.1 ± 3.2 | 65.3 ± 5.2 | 2.0 ± 0.5 |
| 1 | 7.6 ± 0.9 | 3.9 ± 0.8 | 7.6 ± 0.9 | 14.0 ± 0.1 | 5.1 ± 0.7 |
| 2 | 4.2 ± 0.2 | 3.0 ± 0.8 | 5.9 ± 3.0 | 9.6 ± 2.1 | 4.2 ± 0.3 |
| 5 | 12.0 ± 0.9 | 7.4 ± 0.5 | 9.4 ± 1.0 | 23.4 ± 2.3 | 8.2 ± 0.9 |
| 8 | 7.5 ± 1.0 | 1.8 ± 0.4 | 3.3 ± 0.6 | 16.3 ± 0.9 | 3.8 ± 0.2 |
| 9 | 3.9 ± 0.1 | 2.9 ± 1.0 | 7.8 ± 0.1 | 8.9 ± 0.2 | 3.8 ± 0.4 |
| 10 | not active | 49.0 ± 5.3 | not active | not active | 41.0 ± 2.1 |
| NCI-H929 | THP-1 | K562 | RPMI8226 | Jurkat | |
|---|---|---|---|---|---|
| Etoposide | 1.4 ± 0.5 | 1.8 ± 0.2 | 5.5 ± 3.0 | 7.2 ± 1.9 | 0.9 ± 0.3 |
| 1 | 0.7 ± 0.2 | 1.5 ± 0.4 | 3.8 ± 0.6 | 1.6 ± 0.3 | 1.5 ± 0.4 |
| 2 | 0.5 ± 0.1 | 0.9 ± 0.3 | 2.4 ± 0.7 | 1.6 ± 0.5 | 1.1 ± 0.1 |
| 5 | 2.9 ± 0.3 | 8.2 ± 0.6 | 6.5 ± 0.5 | 6.3 ± 0.3 | 3.7 ± 0.2 |
| 8 | 0.9 ± 0.3 | 1.8 ± 0.2 | 3.4 ± 0.3 | 1.5 ± 0.4 | 1.0 ± 0.3 |
| 9 | 0.6 ± 0.2 | 1.7 ± 0.2 | 3.7 ± 0.5 | 1.8 ± 0.2 | 1.3 ± 0.4 |
| 10 | 5.6 ± 0.7 | 42.0 ± 5.2 | 45.0 ± 5.4 | 51.0 ± 0.2 | 94.0 ± 22.6 |
| CMT № 1 | CMT № 2 | CMT № 3 | |
|---|---|---|---|
| Histological Subtype | Extraskeletal Ewing’s Sarcoma | Malignant Schwannoma | Epithelial Sarcoma (Metastasis) |
| 1 | 2.6 ± 1.1 | 2.5 ± 0.9 | 4.8 ± 0.9 |
| 2 | 1.6 ± 0.7 | 1.9 ± 0.5 | 7.8 ± 0.6 |
| 8 | 3.9 ± 0.8 | 2.3 ± 0.8 | 4.8 ± 0.5 |
| 9 | 2.9 ± 1.1 | 4.1 ± 1.1 | 13 ± 0.9 |
| Compound | K562 | K562/i-S9 | K562/i-S9_Dox |
|---|---|---|---|
| Doxorubicin, µM | 0.36 ± 0.17 | 3.9 ± 1.6 | 10.3 ± 1.2 |
| 1, µM | 3.8 ± 0.6 | 2.4 ± 0.6 | 3.1 ± 0.8 |
| 2, µM | 2.4 ± 0.7 | 1.6 ± 0.5 | 2.4 ± 0.9 |
| 9, µM | 3.7 ± 0.5 | 2.8 ± 0.6 | 3.3 ± 0.6 |
| Compound | HBL-100 | HBL-100/Dox | p Value |
|---|---|---|---|
| Doxorubicin, µM | 0.23 ± 0.03 | 128.0 ± 7.4 | p < 0.0001 |
| Etoposide, µM | 4.7 ± 0.9 | 111.3 ± 8.3 | p < 0.0001 |
| 1, µM | 6.2 ± 0.8 | 7.6 ± 0.6 | 0.26 |
| 2, µM | 4.9 ± 1.0 | 6.6 ± 1.2 | 0.31 |
| 9, µM | 6.5 ± 0.8 | 8.4 ± 0.4 | 0.06 |
| Sample | % Luminous Cells | Compounds vs. Release of Rh123 | Compounds vs. Verapamil’s Rh 123 (Release) |
|---|---|---|---|
| Rh 123 entry | 99.8 ± 0.3 | ||
| Rh123 release | 17.1 ± 6.6 | ||
| Rh 123 (release) + verapamil | 89.7 ± 8.1 | p < 0.0001 | |
| Rh123 (release) + 1 | 73.4 ± 6.7 | p < 0.0001 | 0.025 |
| Rh 123 (release) + 2 | 82.4 ± 3.0 | p < 0.0001 | 0.13 |
| Rh 123 (release) + 9 | 80.2 ± 6.2 | p < 0.0001 | 0.08 |
| Compounds | 1 | 2 | 5 | 9 | 10 | PPA |
|---|---|---|---|---|---|---|
| IC50, µM | 3.7 | 1.9 | >50 | 4.6 | >50 | >50 |
| Cmpd | Analyte (Fluorescence Intensity, Rel. Units (M ± mx)) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Akt (pSer473) | CREB (pSer133) | ERK ½ (pThr185/pTyr187) | JNK (pThr183/pTyr185) | NF-kB (pSer536) | p38 (pThr180/pTyr182) | p70S6K (pThr389/412) | STAT3 (pSer727) | STAT5 (pTyr694/699) | |
| Control | 76.4 ± 16.9 | 154.5 ± 34.2 | 839.7 ± 248.9 | 50.9 ± 10.5 | 15.0 ± 1.5 | 567.7 ± 266.6 | 39.9 ± 7.2 | 70.7 ± 18.3 | 44.2 ± 10.2 |
| 1 | 112.6 ± 12.0 | 329.9 ± 48.4 *↑ | 4650.1 ± 509.0 *↑ | 239.9 ± 23.9 *↑ | 18.6 ± 1.5 | 2349.9 ± 755.5 | 51.7 ± 4.8 | 163.0 ± 20.7 *↑ | 115.4 ± 28.3 |
| 2 | 132.4 ± 17.9 | 610.0 ± 113.0 *↑ | 5074.6 ± 770.1 *↑ | 559.2 ± 31.5 *↑ | 18.2 ± 2.1 | 2795.4 ± 572.0 *↑ | 56.9 ± 9.9 | 272.3 ± 50.5 *↑ | 179.2 ± 12.6 *↑ |
| 5 | 100.9 ± 11.7 | 202.1 ± 26.3 | 1880.4 ± 81.7 *↑ | 83.1 ± 8.3 *↑ | 17.1 ± 0.8 | 1245.9 ± 342.8 | 70.6 ± 9.2 *↑ | 123.4 ± 8.3 *↑ | 75.7 ± 2.7 *↑ |
| 9 | 88.8 ± 10.7 | 339.8 ± 53.3 *↑ | 3276.9 ± 700.0 *↑ | 117.9 ± 15.1 *↑ | 13.9 ± 1.2 | 657.1 ± 287.5 | 38.8 ± 7.4 | 151.5 ± 28.7 | 69.6 ± 15.6 |
| 10 | 142.1 ± 9.5 *↑ | 584.8 ± 105.5 *↑ | 1864.9 ± 149.5 *↑ | 157.2 ± 15.4 *↑ | 15.4 ± 0.5 | 1783.6 ± 181.9 *↑ | 53.4 ± 4.0 | 178.9 ± 13.4 *↑ | 115.2 ± 16.9 *↑ |
| PPA | 123.5 ± 4.9 *↑ | 291.0 ± 32.8 *↑ | 995.4 ± 46.5 | 132.8 ± 9.1 *↑ | 19.3 ± 3.0 | 1459.1 ± 117.6 *↑ | 46.1 ± 2.9 | 128.8 ± 2.4 *↑ | 82.5 ± 3.5 *↑ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abzianidze, V.; Moiseeva, N.; Suponina, D.; Zakharenkova, S.; Rogovskaya, N.; Laletina, L.; Holder, A.A.; Krivorotov, D.; Bogachenkov, A.; Garabadzhiu, A.; et al. Natural Phaeosphaeride A Derivatives Overcome Drug Resistance of Tumor Cells and Modulate Signaling Pathways. Pharmaceuticals 2022, 15, 395. https://doi.org/10.3390/ph15040395
Abzianidze V, Moiseeva N, Suponina D, Zakharenkova S, Rogovskaya N, Laletina L, Holder AA, Krivorotov D, Bogachenkov A, Garabadzhiu A, et al. Natural Phaeosphaeride A Derivatives Overcome Drug Resistance of Tumor Cells and Modulate Signaling Pathways. Pharmaceuticals. 2022; 15(4):395. https://doi.org/10.3390/ph15040395
Chicago/Turabian StyleAbzianidze, Victoria, Natalia Moiseeva, Diana Suponina, Sofya Zakharenkova, Nadezhda Rogovskaya, Lidia Laletina, Alvin A. Holder, Denis Krivorotov, Alexander Bogachenkov, Alexander Garabadzhiu, and et al. 2022. "Natural Phaeosphaeride A Derivatives Overcome Drug Resistance of Tumor Cells and Modulate Signaling Pathways" Pharmaceuticals 15, no. 4: 395. https://doi.org/10.3390/ph15040395
APA StyleAbzianidze, V., Moiseeva, N., Suponina, D., Zakharenkova, S., Rogovskaya, N., Laletina, L., Holder, A. A., Krivorotov, D., Bogachenkov, A., Garabadzhiu, A., Ukolov, A., & Kosorukov, V. (2022). Natural Phaeosphaeride A Derivatives Overcome Drug Resistance of Tumor Cells and Modulate Signaling Pathways. Pharmaceuticals, 15(4), 395. https://doi.org/10.3390/ph15040395