
Discovery of Azaindolin-2-One as a Dual Inhibitor of GSK3β and Tau Aggregation with Potential Neuroprotective Activity

 ,
,  ,
,  ,
,  , , ,
, , , 
Abstract
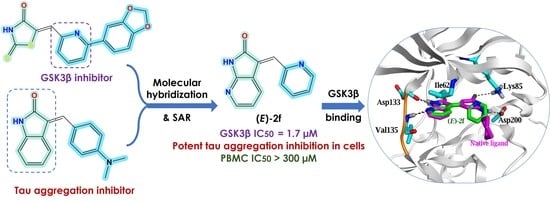
:
1. Introduction
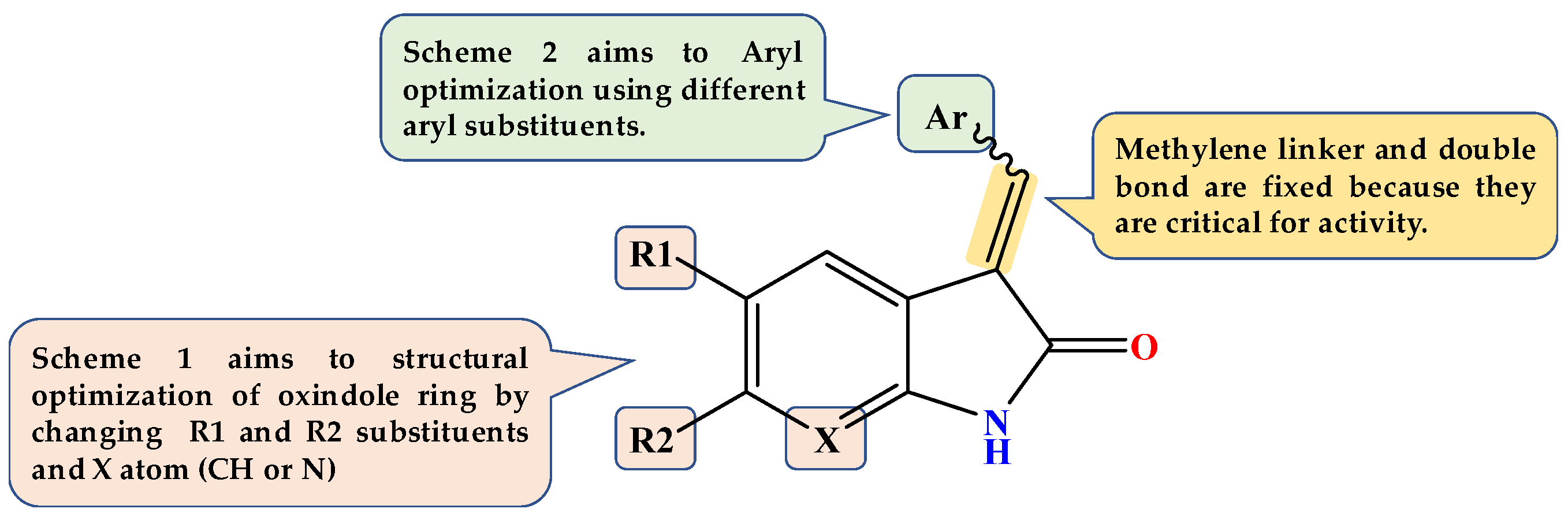
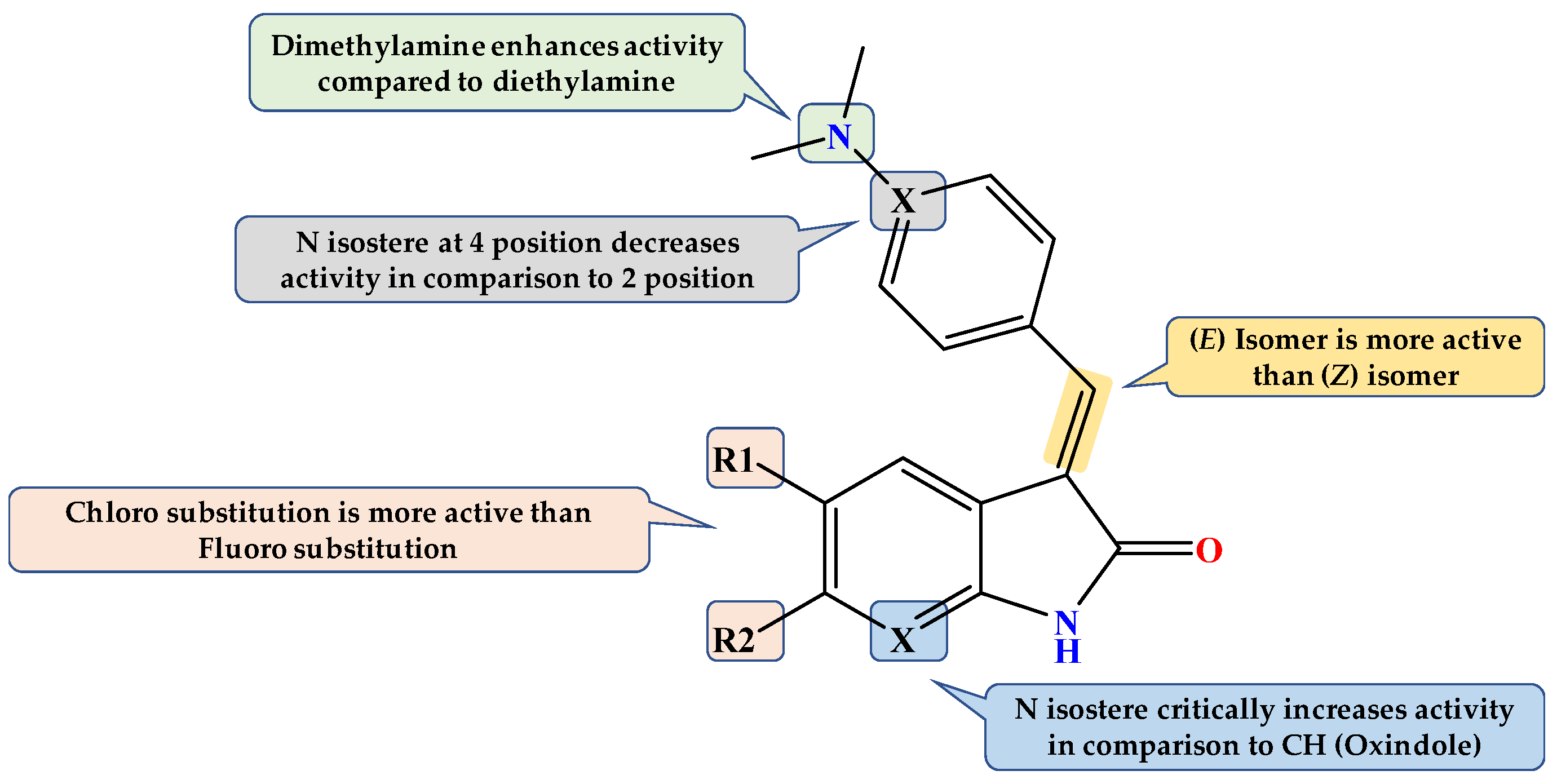
Design of Dual GSK3β/tau Aggregation Inhibitors
2. Results
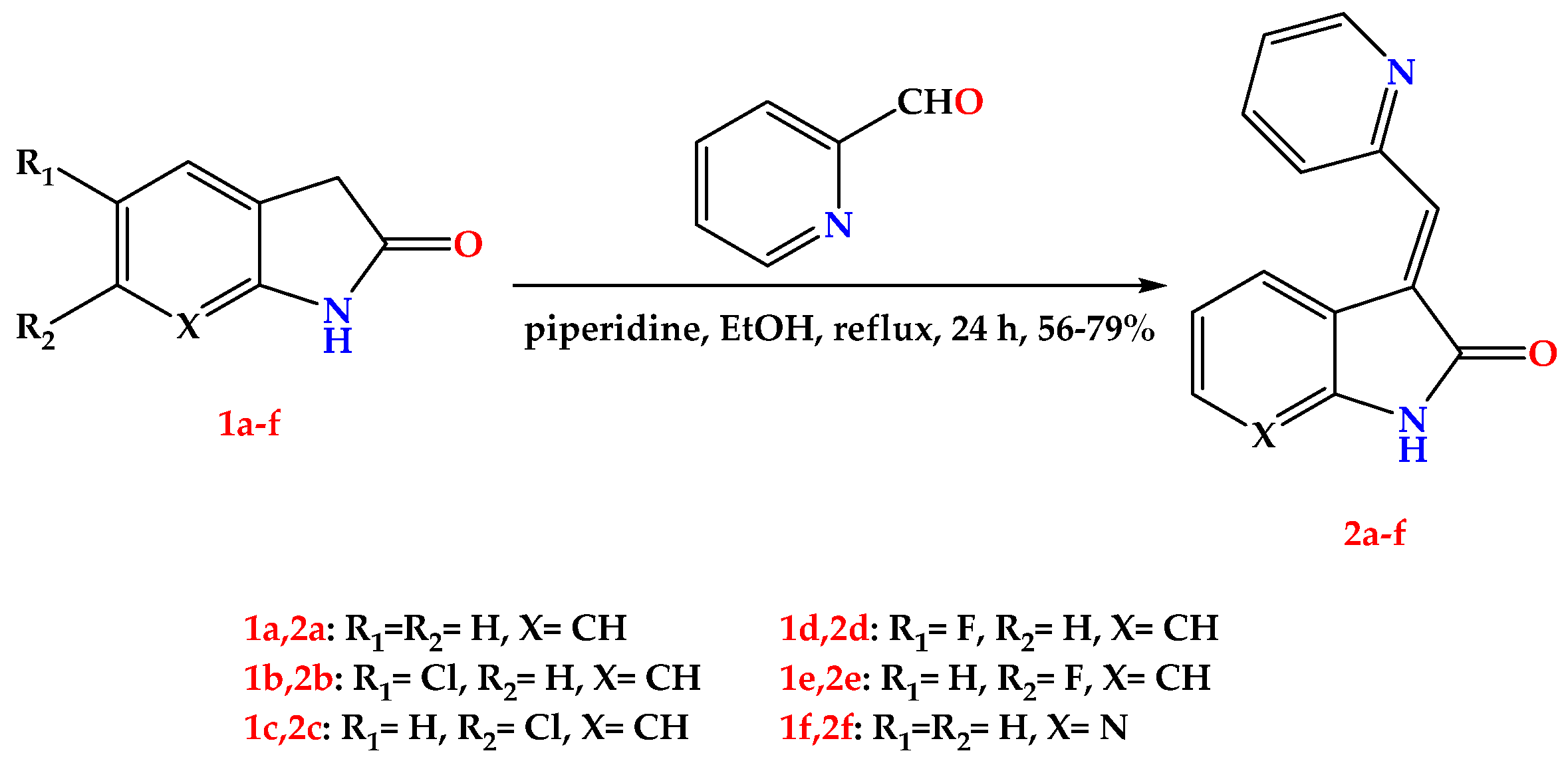
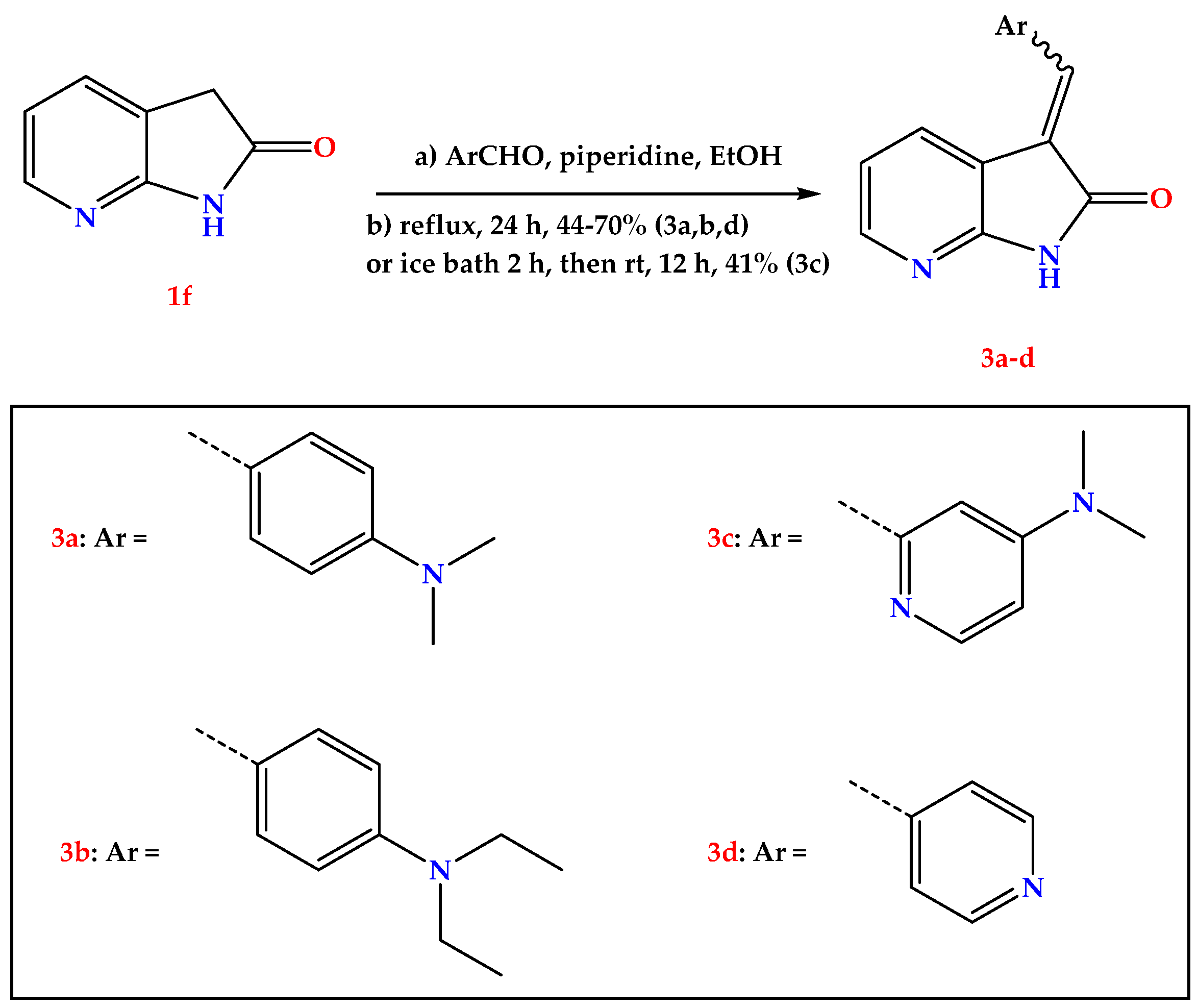
2.1. Chemistry
2.2. GSK3β Inhibition Assay
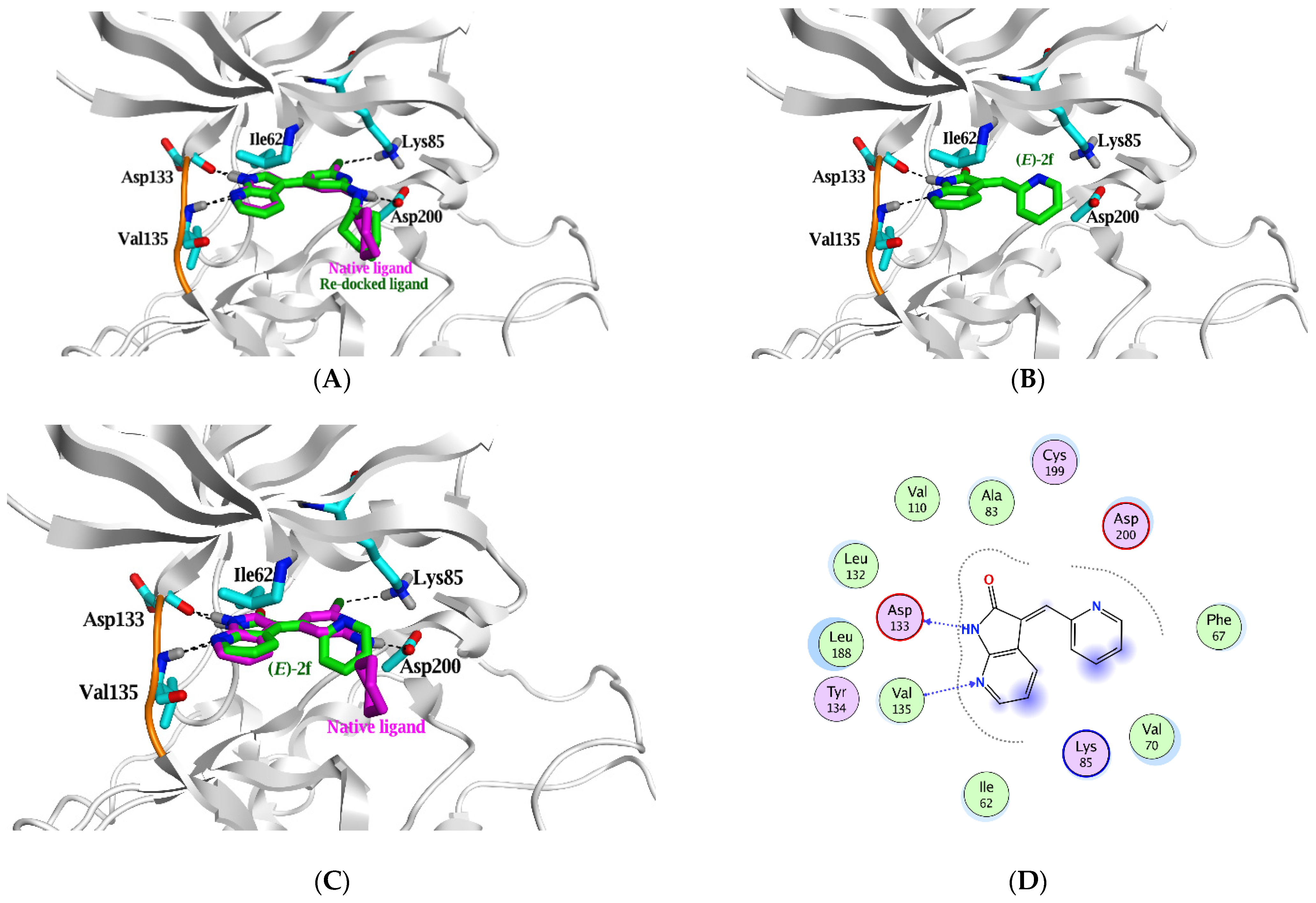
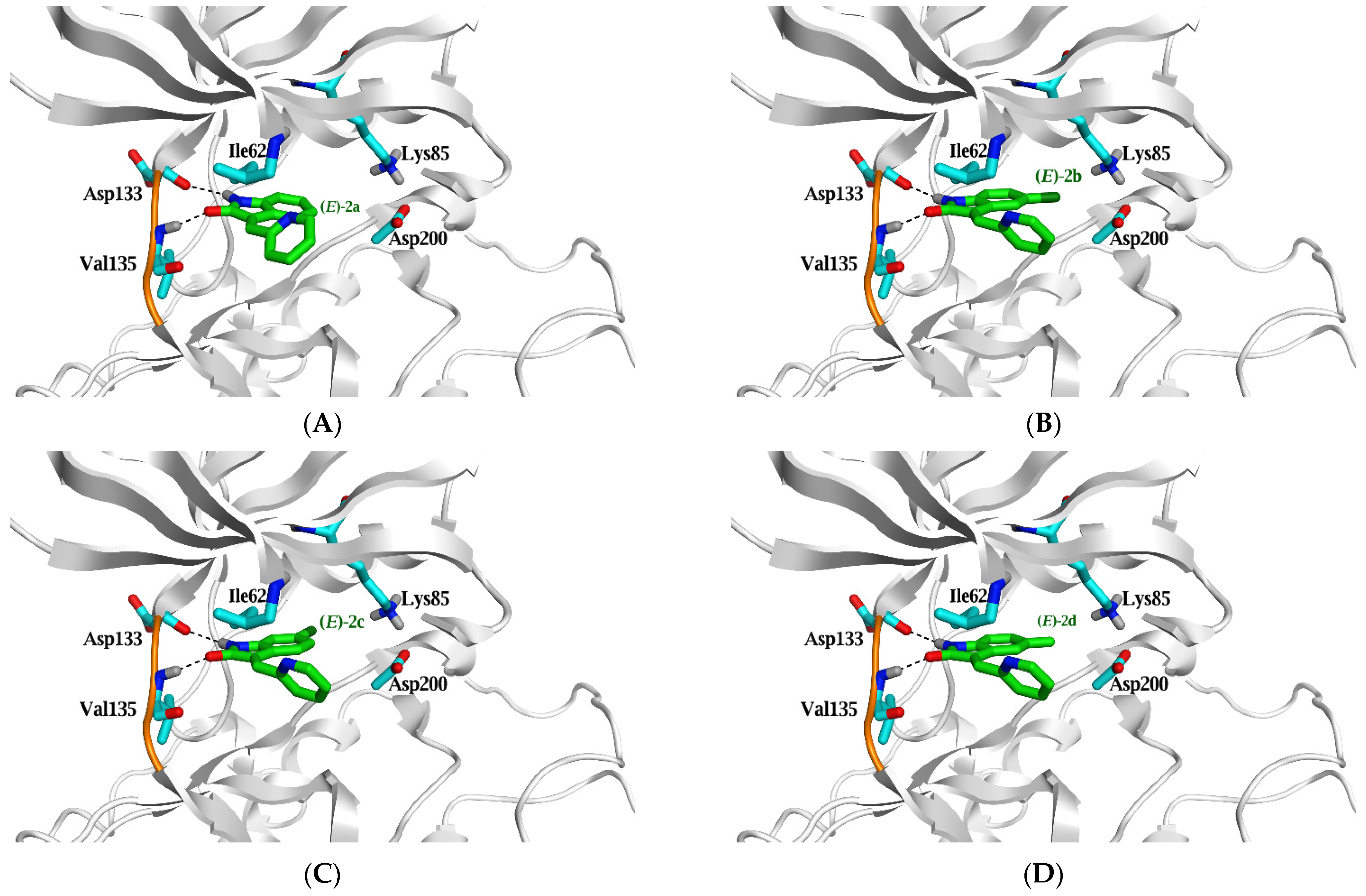
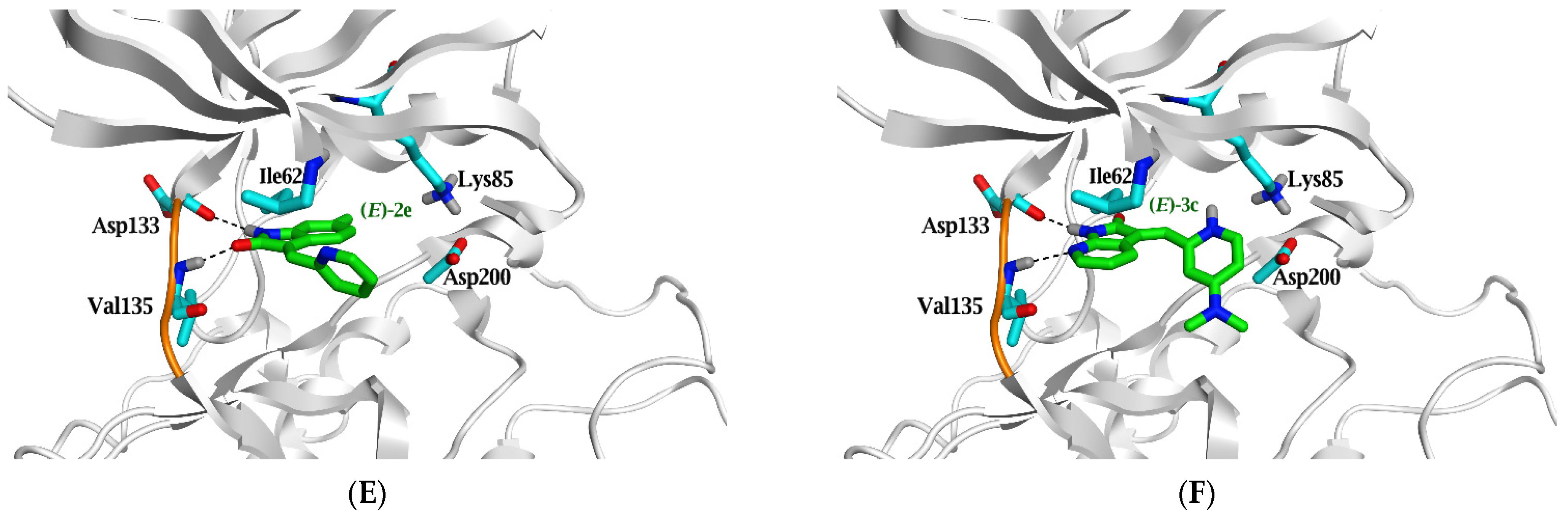
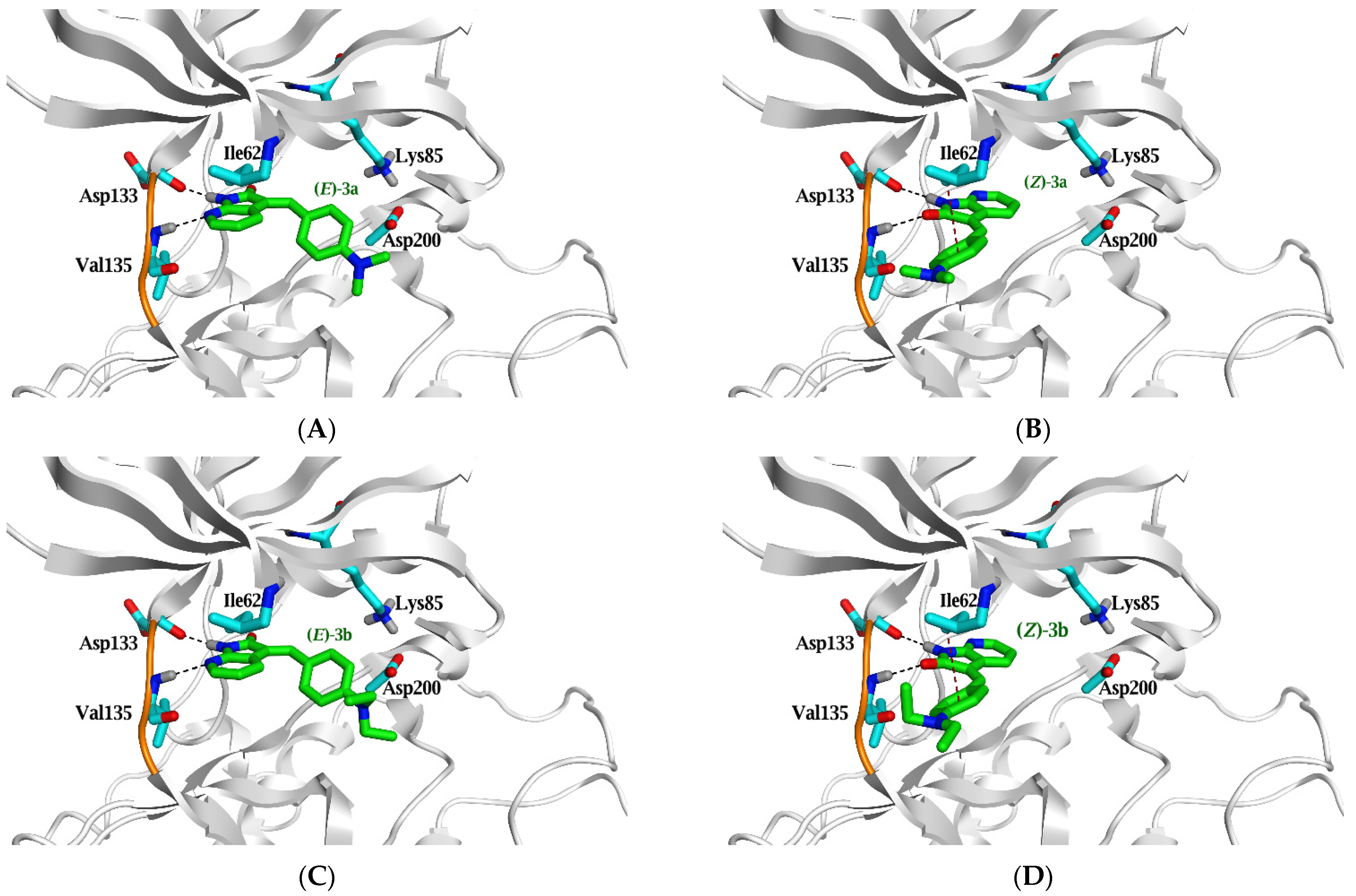
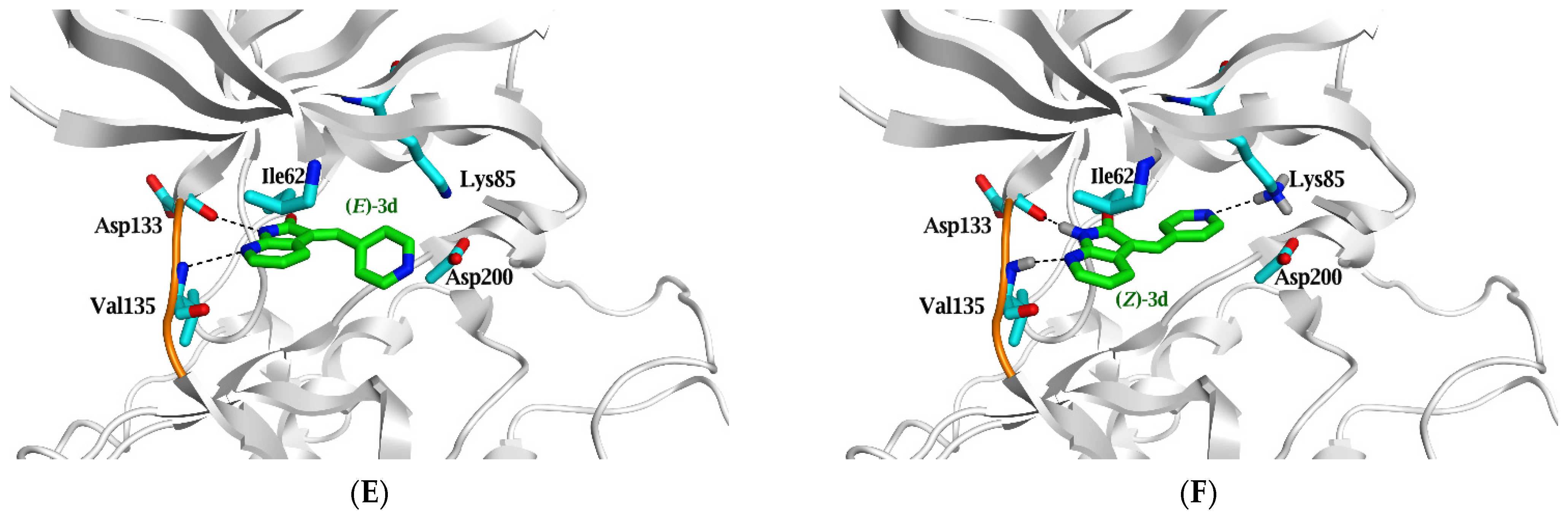
2.3. Molecular Docking Study
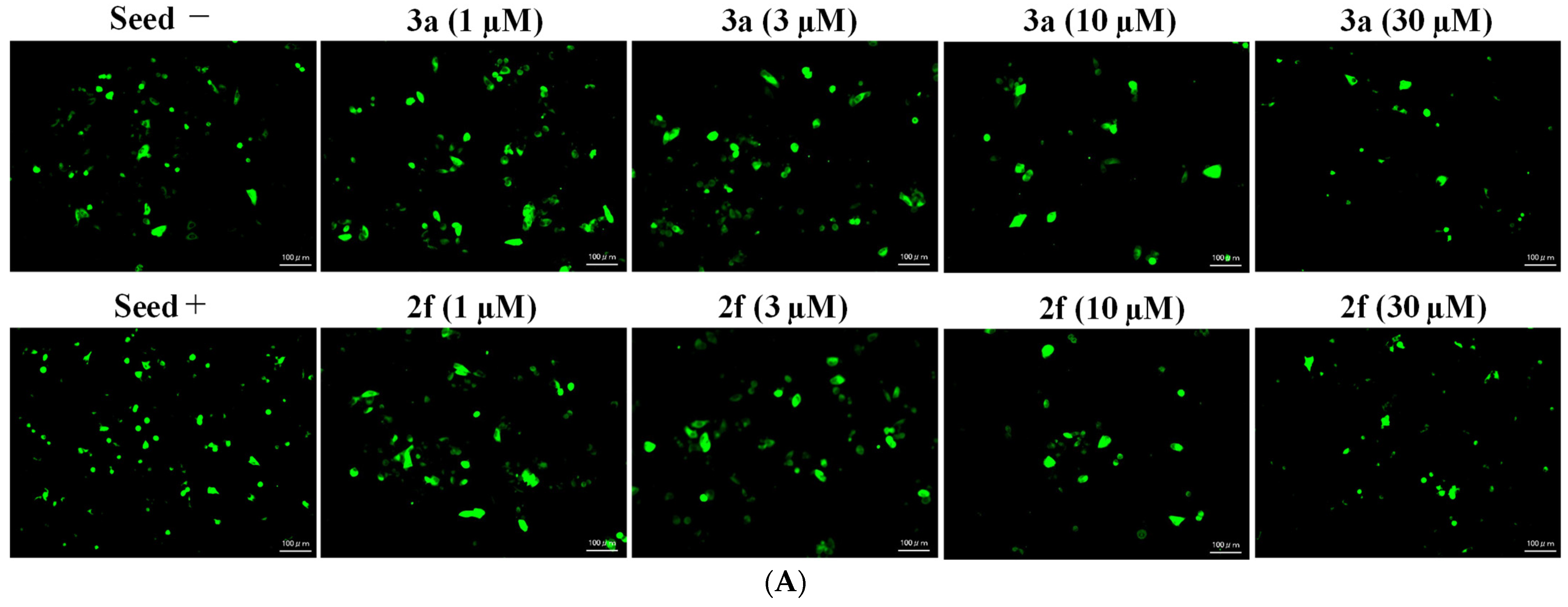
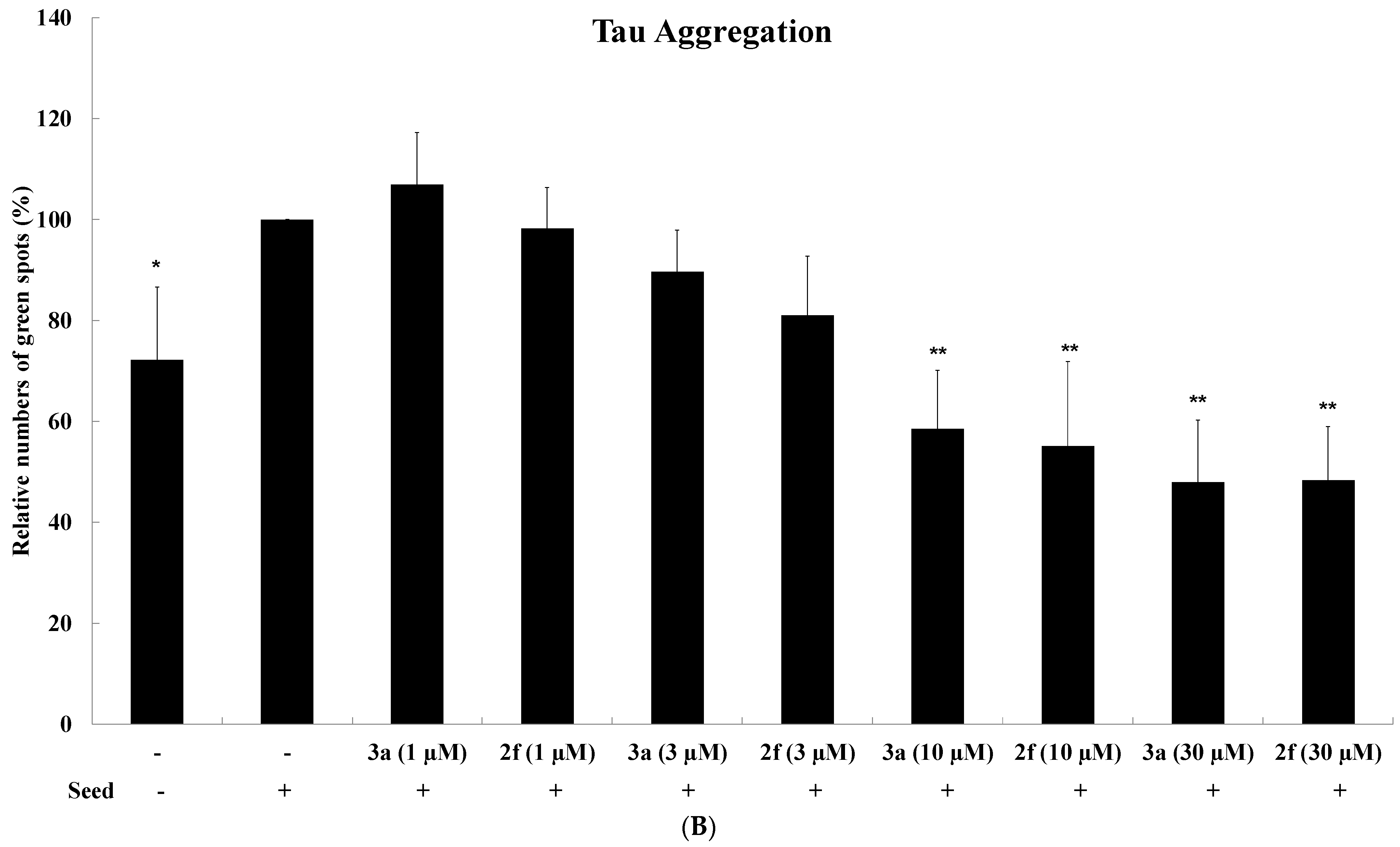
2.4. Tau Aggregation Inhibition in a Cell Model of Tauopathy and Western Blot Analysis
2.5. Selective Cytotoxicity on Cancer Cells and Normal Blood Cells
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. General Procedure for the Synthesis of Compounds 2a–f
- (E)-3-(pyridin-2-ylmethylene)indolin-2-one (2a) [27].
- (E)-5-chloro-3-(pyridin-2-ylmethylene)indolin-2-one (2b).
- (E)-6-chloro-3-(pyridin-2-ylmethylene)indolin-2-one (2c).
- (E)-5-fluoro-3-(pyridin-2-ylmethylene)indolin-2-one (2d).
- (E)-6-fluoro-3-(pyridin-2-ylmethylene)indolin-2-one (2e).
- (E)-3-(pyridin-2-ylmethylene)-1,3-dihydro-2H-pyrrolo [2,3-b]pyridin-2-one (2f).
4.1.2. General Procedure for the Synthesis of Compounds 3a–d
- (E/Z)-3-(4-(diethylamino)benzylidene)-1,3-dihydro-2H-pyrrolo [2,3-b]pyridin-2-one (3b).
- (E)-3-((5-(dimethylamino)pyridin-2-yl)methylene)-1,3-dihydro-2H-pyrrolo [2,3-b]pyridin-2-one (3c).
- (E/Z)-3-(pyridin-4-ylmethylene)-1,3-dihydro-2H-pyrrolo [2,3-b]pyridin-2-one (3d).
4.2. GSK-3β Inhibition Assay
4.3. Molecular Docking Study
4.4. Tau Aggregation Inhibition in a Cell Model of Tauopathy and Western Blot Analysis
4.5. Selective Cytotoxicity on Cancer Cells and Normal Blood Cells
4.5.1. Cells
4.5.2. MTT Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jiang, X.; Wang, Y.; Liu, C.; Xing, C.; Wang, Y.; Lyu, W.; Wang, S.; Li, Q.; Chen, T.; Chen, Y.; et al. Discovery of potent glycogen synthase kinase 3/cholinesterase inhibitors with neuroprotection as potential therapeutic agent for Alzheimer’s disease. Bioorg. Med. Chem. 2021, 30, 115940. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Deng, Y.; Qing, H. Potential Therapeutic Strategies for Alzheimer’s Disease Targeting or Beyond β-Amyloid: Insights from Clinical Trials. BioMed Res. Int. 2014, 2014, 837157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalili-Baleh, L.; Forootanfar, H.; Küçükkılınç, T.T.; Nadri, H.; Abdolahi, Z.; Ameri, A.; Jafari, M.; Ayazgok, B.; Baeeri, M.; Rahimifard, M.; et al. Design, synthesis and evaluation of novel multi-target-directed ligands for treatment of Alzheimer’s disease based on coumarin and lipoic acid scaffolds. Eur. J. Med. Chem. 2018, 152, 600–614. [Google Scholar] [CrossRef] [PubMed]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Dzamba, D.; Harantova, L.; Butenko, O.; Anderova, M. Glial Cells—The Key Elements of Alzheimer´s Disease. Curr. Alzheimer Res. 2016, 13, 894–911. [Google Scholar] [CrossRef] [PubMed]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2017, 107, 41–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbagallo, M. Type 2 diabetes mellitus and Alzheimer’s disease. World J. Diabetes 2014, 5, 889–893. [Google Scholar] [CrossRef] [Green Version]
- Haghighijoo, Z.; Zamani, L.; Moosavi, F.; Emami, S. Therapeutic potential of quinazoline derivatives for Alzheimer’s disease: A comprehensive review. Eur. J. Med. Chem. 2021, 227, 113949. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.; Zhou, D.; Gaba, V.; Liu, J.; Li, S.; Peng, X.; Xu, J.; Dhavale, D.; Bagchi, D.P.; D’Avignon, A.; et al. Design, Synthesis, and Characterization of 3-(Benzylidene)indolin-2-one Derivatives as Ligands for α-Synuclein Fibrils. J. Med. Chem. 2015, 58, 6002–6017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimes, C.A.; Jope, R.S. The multifaceted roles of glycogen synthase kinase 3β in cellular signaling. Prog. Neurobiol. 2001, 65, 391–426. [Google Scholar] [CrossRef]
- Barth, A.I.M.; Caro-Gonzalez, H.Y.; Nelson, W.J. Role of adenomatous polyposis coli (APC) and microtubules in directional cell migration and neuronal polarization. Semin. Cell Dev. Biol. 2008, 19, 245–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J. Glycogen synthase kinase 3β (GSK3β) in tumorigenesis and cancer chemotherapy. Cancer Lett. 2009, 273, 194–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J. The role of GSK3beta in the development of the central nervous system. Front. Biol. 2012, 7, 212–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.M.; Zhang, T.; Li, Q.; Huang, J.K.; Chen, R.F.; Sun, X.J. Inhibition of glycogen synthase kinase-3β by lithium chloride suppresses 6-hydroxydopamine-induced inflammatory response in primary cultured astrocytes. Neurochem. Int. 2013, 63, 345–353. [Google Scholar] [CrossRef]
- Beurel, E.; Michalek, S.M.; Jope, R.S. Innate and adaptive immune responses regulated by glycogen synthase kinase-3 (GSK3). Trends Immunol. 2010, 31, 24–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beurel, E.; Jope, R.S. Lipopolysaccharide-induced interleukin-6 production is controlled by glycogen synthase kinase-3 and STAT3 in the brain. J. Neuroinflamm. 2009, 6, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- L’Episcopo, F.; Serapide, M.F.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C.; Pluchino, S.; Marchetti, B. A Wnt1 regulated Frizzled-1/β-Cateninsignaling pathway as a candidate regulatory circuit controlling mesencephalic dopaminergic neuron-astrocyte crosstalk: Therapeutical relevance for neuron survival and neuroprotection. Mol. Neurodegener. 2011, 6, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, G.G. Tauopathies. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 145, pp. 355–368. [Google Scholar]
- Ramsay, R.R.; Popovic-Nikolicb, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Simone, A.; Tumiatti, V.; Andrisano, V.; Milelli, A. Glycogen Synthase Kinase 3β: A New Gold Rush in Anti-Alzheimer’s Disease Multitarget Drug Discovery? J. Med. Chem. 2021, 64, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Avila, J.; Wandosell, F.; Hernández, F. Role of glycogen synthase kinase-3 in Alzheimer’s disease pathogenesis and glycogen synthase kinase-3 inhibitors. Expert Rev. Neurother. 2010, 10, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Palomo, V.; Perez, D.I.; Roca, C.; Anderson, C.; Rodríguez-Muela, N.; Perez, C.; Morales-Garcia, J.A.; Reyes, J.A.; Campillo, N.E.; Perez-Castillo, A.M.; et al. Subtly Modulating Glycogen Synthase Kinase 3 β: Allosteric Inhibitor Development and Their Potential for the Treatment of Chronic Diseases. J. Med. Chem. 2017, 60, 4983–5001. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, S.; Garnier, M.; Hoessel, R.; Marko, D.; Bibb, J.A.; Snyder, G.L.; Greengard, P.; Biernat, J.; Wu, Y.-Z.; Mandelkow, E.-M.; et al. Indirubins Inhibit Glycogen Synthase Kinase-3β and CDK5/P25, Two Protein Kinases Involved in Abnormal Tau Phosphorylation in Alzheimer’s Disease. A property common to most CDK inhibitors? J. Biol. Chem. 2001, 276, 251–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Gaisina, I.; Gunosewoyo, H.; Malekiani, S.A.; Hanania, T.; Kozikowski, A.P. Structure-Guided Design of a Highly Selective Glycogen Synthase Kinase-3β Inhibitor: A Superior Neuroprotective Pyrazolone Showing Antimania Effects. ChemMedChem 2011, 6, 1587–1592. [Google Scholar] [CrossRef] [PubMed]
- Gandini, A.; Bartolini, M.; Tedesco, D.; Martinez-Gonzalez, L.; Roca, C.; Campillo, N.E.; Zaldivar-Diez, J.; Perez, C.; Zuccheri, G.; Miti, A.; et al. Tau-Centric Multitarget Approach for Alzheimer’s Disease: Development of First-in-Class Dual Glycogen Synthase Kinase 3β and Tau-Aggregation Inhibitors. J. Med. Chem. 2018, 61, 7640–7656. [Google Scholar] [CrossRef]
- Bourahla, K.; Guihéneuf, S.; Limanton, E.; Paquin, L.; Le Guével, R.; Charlier, T.; Rahmouni, M.; Durieu, E.; Lozach, O.; Carreaux, F.; et al. Design and Microwave Synthesis of New (5Z) 5-Arylidene-2-thioxo-1,3-thiazolinidin-4-one and (5Z) 2-Amino-5-arylidene-1,3-thiazol-4(5H)-one as New Inhibitors of Protein Kinase DYRK1A. Pharmaceuticals 2021, 14, 1086. [Google Scholar] [CrossRef] [PubMed]
- Lozinskaya, N.A.; Babkov, D.; Zaryanova, E.V.; Bezsonova, E.N.; Efremov, A.M.; Tsymlyakov, M.D.; Anikina, L.V.; Zakharyascheva, O.Y.; Borisov, A.V.; Perfilova, V.N.; et al. Synthesis and biological evaluation of 3-substituted 2-oxindole derivatives as new glycogen synthase kinase 3β inhibitors. Bioorg. Med. Chem. 2019, 27, 1804–1817. [Google Scholar] [CrossRef] [PubMed]
- Honson, N.S.; Johnson, R.L.; Huang, W.; Inglese, J.; Austin, C.P.; Kuret, J. Differentiating Alzheimer disease-associated aggregates with small molecules. Neurobiol. Dis. 2007, 28, 251–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, Y.; Stewart, K.D.; Florjancic, A.S.; Harlan, J.E.; Merta, P.J.; Przytulinska, M.; Soni, N.; Swinger, K.K.; Zhu, H.; Johnson, E.F.; et al. Azaindole-Based Inhibitors of Cdc7 Kinase: Impact of the Pre-DFG Residue, Val 195. ACS Med. Chem. Lett. 2013, 4, 211–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selenica, M.-L.; Jensen, H.S.; Larsen, A.K.; Pedersen, M.L.; Helboe, L.; Leist, M.; Lotharius, J. Efficacy of small-molecule glycogen synthase kinase-3 inhibitors in the postnatal rat model of tau hyperphosphorylation. J. Cereb. Blood Flow Metab. 2007, 152, 959–979. [Google Scholar] [CrossRef] [Green Version]
- Duran-Frigola, M.; Siragusa, L.; Ruppin, E.; Barril, X.; Cruciani, G.; Aloy, P. Detecting similar binding pockets to enable systems polypharmacology. PLoS Comput. Biol. 2017, 13, e1005522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prati, F.; Cavalli, A.; Bolognesi, M.L. Navigating the Chemical Space of Multitarget-Directed Ligands: From Hybrids to Fragments in Alzheimer’s Disease. Molecules 2016, 21, 466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanfar, M.A.; Hill, R.A.; Kaddoumi, A.; El Sayed, K.A. Discovery of Novel GSK-3β Inhibitors with Potent in Vitro and in Vivo Activities and Excellent Brain Permeability Using Combined Ligand- and Structure-Based Virtual Screening. J. Med. Chem. 2010, 53, 8534–8545. [Google Scholar] [CrossRef]
- Arfeen, M.; Bhagat, S.; Patel, R.; Prasad, S.; Roy, I.; Chakraborti, A.K.; Bharatam, P.V. Design, synthesis and biological evaluation of 5-benzylidene-2-iminothiazolidin-4-ones as selective GSK-3β inhibitors. Eur. J. Med. Chem. 2016, 121, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Cisek, K.; Cooper, G.; Huseby, C.; Kuret, J. Structure and Mechanism of Action of Tau Aggregation Inhibitors. Curr. Alzheimer Res. 2014, 11, 918–927. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.L.; Buist, A.; Soares, A.; Callaerts, K.; Calafate, S.; Stevenaert, F.; Daniels, J.P.; Zoll, B.E.; Crowe, A.; Brunden, K.R.; et al. The Dynamics and Turnover of Tau Aggregates in Cultured Cells. J. Biol. Chem. 2016, 291, 13175–13193. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, G.; Matsumoto, K.; Kimura, T.; Suhara, T.; Higuchi, M.; Sahara, N.; Mori, N. Tau Fibril Formation in Cultured Cells Compatible with a Mouse Model of Tauopathy. Int. J. Mol. Sci. 2018, 19, 1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciftci, H.I.; Radwan, M.O.; Sever, B.; Hamdy, A.K.; Emirdağ, S.; Ulusoy, N.G.; Sozer, E.; Can, M.; Yayli, N.; Araki, N.; et al. EGFR-Targeted Pentacyclic Triterpene Analogues for Glioma Therapy. Int. J. Mol. Sci. 2021, 22, 10945. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Shi, R.; Gu, J.; Tung, Y.C.; Zhou, Y.; Zhou, D.; Wu, R.; Chu, D.; Jin, N.; Deng, K.; et al. Alzheimer’s disease brain contains tau fractions with differential prion-like activities. Acta Neuropathol. Commun. 2021, 9, 28. [Google Scholar] [CrossRef]
- Cieri, D.; Vicario, M.; Vallese, F.; D’Orsi, B.; Berto, P.; Grinzato, A.; Catoni, C.; De Stefani, D.; Rizzuto, R.; Brini, M.; et al. Tau localises within mitochondrial sub-compartments and its caspase cleavage affects ER-mitochondria interactions and cellular Ca2+ handling. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2018, 1864, 3247–3256. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Koga, R.; Fujino, H.; Shimagaki, K.; Ciftci, H.I.; Kamo, M.; Tateishi, H.; Otsuka, M.; Fujita, M. Zinc-binding site of human immunodeficiency virus 2 Vpx prevents instability and dysfunction of the protein. J. Gen. Virol. 2017, 98, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Akari, H.; Sakurai, A.; Yoshida, A.; Chiba, T.; Tanaka, K.; Strebel, K.; Adachi, A. Expression of HIV-1 accessory protein Vif is controlled uniquely to be low and optimal by proteasome degradation. Microbes Infect. 2004, 6, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Sever, B.; Altıntop, M.; Özdemir, A.; Çiftçi, G.A.; Ellakwa, D.; Tateishi, H.; Radwan, M.; Ibrahim, M.; Otsuka, M.; Fujita, M.; et al. In Vitro and In Silico Evaluation of Anticancer Activity of New Indole-Based 1,3,4-Oxadiazoles as EGFR and COX-2 Inhibitors. Molecules 2020, 25, 5190. [Google Scholar] [CrossRef] [PubMed]
- Ciftci, H.I.; Radwan, M.O.; Ozturk, S.E.; Ulusoy, N.G.; Sozer, E.; Ellakwa, D.E.; Ocak, Z.; Can, M.; Ali, T.; Abd-Alla, H.I.; et al. Design, Synthesis and Biological Evaluation of Pentacyclic Triterpene Derivatives: Optimization of Anti-ABL Kinase Activity. Molecules 2019, 24, 3535. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | ΔG in Kcal/mol (RMSD) | IC50 (µM) | Ligand | Interaction Parameters | |||
|---|---|---|---|---|---|---|---|
| GSK3β | Atoms | Interaction | AA Residue | δ(Å) | E (Kcal/mol) | ||
| Native ligand | --- | 0.058 * | N21 | H-donor | Asp 133 | 2.78 | −4.5 |
| N22 | H-donor | Asp 200 | 3.02 | −2.0 | |||
| CL23 | H-donor | Glu 97 | 3.18 | −0.6 | |||
| N19 | H-acceptor | Val 135 | 3.07 | −2.2 | |||
| CL23 | H-acceptor | Lys 85 | 3.6 | −0.8 | |||
| Re-docked | −7.4 (1.4) | N21 | H-donor | Asp 133 | 2.9 | −4.4 | |
| N22 | H-donor | Asp 200 | 3.3 | −1.3 | |||
| CL23 | H-donor | Glu 97 | 3.36 | −0.5 | |||
| N19 | H-acceptor | Val 135 | 3.19 | −2.1 | |||
| CL23 | H-acceptor | Lys 85 | 3.72 | −0.7 | |||
| (E)-2a | −5.2 (1.6) | >10 | N7 | H-donor | Asp 133 | 3.58 | −0.5 |
| O10 | H-acceptor | Val 135 | 2.98 | −3.3 | |||
| (E)-2b | −6.0 (1.5) | >10 | N7 | H-donor | Asp 133 | 3.16 | −3.2 |
| O10 | H-acceptor | Val 135 | 2.98 | −3.5 | |||
| (E)-2c | −5.9 (0.8) | >10 | N7 | H-donor | Asp 133 | 3.34 | −1.9 |
| O10 | H-acceptor | Val 135 | 3.03 | −3.3 | |||
| (E)-2d | −5.7 (1.9) | >10 | N7 | H-donor | Asp 133 | 3.2 | −3.1 |
| O10 | H-acceptor | Val 135 | 3 | −3.4 | |||
| (E)-2e | −5.6 (1.1) | >10 | N7 | H-donor | Asp 133 | 3.27 | −2.5 |
| O10 | H-acceptor | Val 135 | 3.01 | −3.3 | |||
| (E)-2f | −6.0 (1.7) | 1.7 | N7 | H-donor | Asp 133 | 2.97 | −4.8 |
| N3 | H-acceptor | Val 135 | 3.29 | −2.7 | |||
| (E)-3a | −6.3 (0.8) | 1.9 | N7 | H-donor | Asp 133 | 2.93 | −4.8 |
| N3 | H-acceptor | Val 135 | 3.16 | −3.2 | |||
| (Z)-3a | −5.8 (1.6) | N1 | H-donor | Asp 133 | 3.45 | −1.6 | |
| O10 | H-acceptor | Val 135 | 3.43 | −1.2 | |||
| 6-ring | π-H | Ile 62 | 3.84 | −0.5 | |||
| (E)-3b | −6.5 (1.4) | 3 | N7 | H-donor | Asp 133 | 2.97 | −4.7 |
| N3 | H-acceptor | Val 135 | 3.2 | −3.0 | |||
| (Z)-3b | −6.1 (1.8) | N1 | H-donor | Asp 133 | 3.45 | −1.6 | |
| O10 | H-acceptor | Val 135 | 3.4 | −1.4 | |||
| 6-ring | π-H | Ile 62 | 3.88 | −0.6 | |||
| (E)-3c | −6.4 (1.8) | 3.2 | N7 | H-donor | Asp 133 | 3 | −4.8 |
| N3 | H-acceptor | Val 135 | 3.27 | −2.7 | |||
| (E)-3d | −5.9 (1.7) | 5.9 | N7 | H-donor | Asp 133 | 2.96 | −4.8 |
| N3 | H-acceptor | Val 135 | 3.26 | −2.8 | |||
| (Z)-3d | −5.8 (1.2) | N1 | H-donor | Asp 133 | 3.25 | −1.6 | |
| N8 | H-acceptor | Val 135 | 3.4 | −0.9 | |||
| N15 | H-acceptor | Lys 85 | 3.54 | −1.2 | |||
| 6-ring | π-H | Asp 200 | 3.53 | −0.5 | |||
| IC50 (µM) after 72 h Drug Treatment | |||||
|---|---|---|---|---|---|
| Compound | K562 | U251 | HCT-116 | A375 | PBMC |
| (E)-2f | 13.80 | 28.2 | 12.8 | 14.00 | >300 |
| 3a | 4.7 | 10.3 | 9.8 | >100 | 94.55 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, T.F.S.; Ciftci, H.I.; Radwan, M.O.; Roshdy, E.; Shawky, A.M.; Abourehab, M.A.S.; Tateishi, H.; Otsuka, M.; Fujita, M. Discovery of Azaindolin-2-One as a Dual Inhibitor of GSK3β and Tau Aggregation with Potential Neuroprotective Activity. Pharmaceuticals 2022, 15, 426. https://doi.org/10.3390/ph15040426
Ali TFS, Ciftci HI, Radwan MO, Roshdy E, Shawky AM, Abourehab MAS, Tateishi H, Otsuka M, Fujita M. Discovery of Azaindolin-2-One as a Dual Inhibitor of GSK3β and Tau Aggregation with Potential Neuroprotective Activity. Pharmaceuticals. 2022; 15(4):426. https://doi.org/10.3390/ph15040426
Chicago/Turabian StyleAli, Taha F. S., Halil I. Ciftci, Mohamed O. Radwan, Eslam Roshdy, Ahmed M. Shawky, Mohammed A. S. Abourehab, Hiroshi Tateishi, Masami Otsuka, and Mikako Fujita. 2022. "Discovery of Azaindolin-2-One as a Dual Inhibitor of GSK3β and Tau Aggregation with Potential Neuroprotective Activity" Pharmaceuticals 15, no. 4: 426. https://doi.org/10.3390/ph15040426
APA StyleAli, T. F. S., Ciftci, H. I., Radwan, M. O., Roshdy, E., Shawky, A. M., Abourehab, M. A. S., Tateishi, H., Otsuka, M., & Fujita, M. (2022). Discovery of Azaindolin-2-One as a Dual Inhibitor of GSK3β and Tau Aggregation with Potential Neuroprotective Activity. Pharmaceuticals, 15(4), 426. https://doi.org/10.3390/ph15040426