
Dual PI3K/Akt Inhibitors Bearing Coumarin-Thiazolidine Pharmacophores as Potential Apoptosis Inducers in MCF-7 Cells

 , , and
, , and
Abstract
:1. Introduction
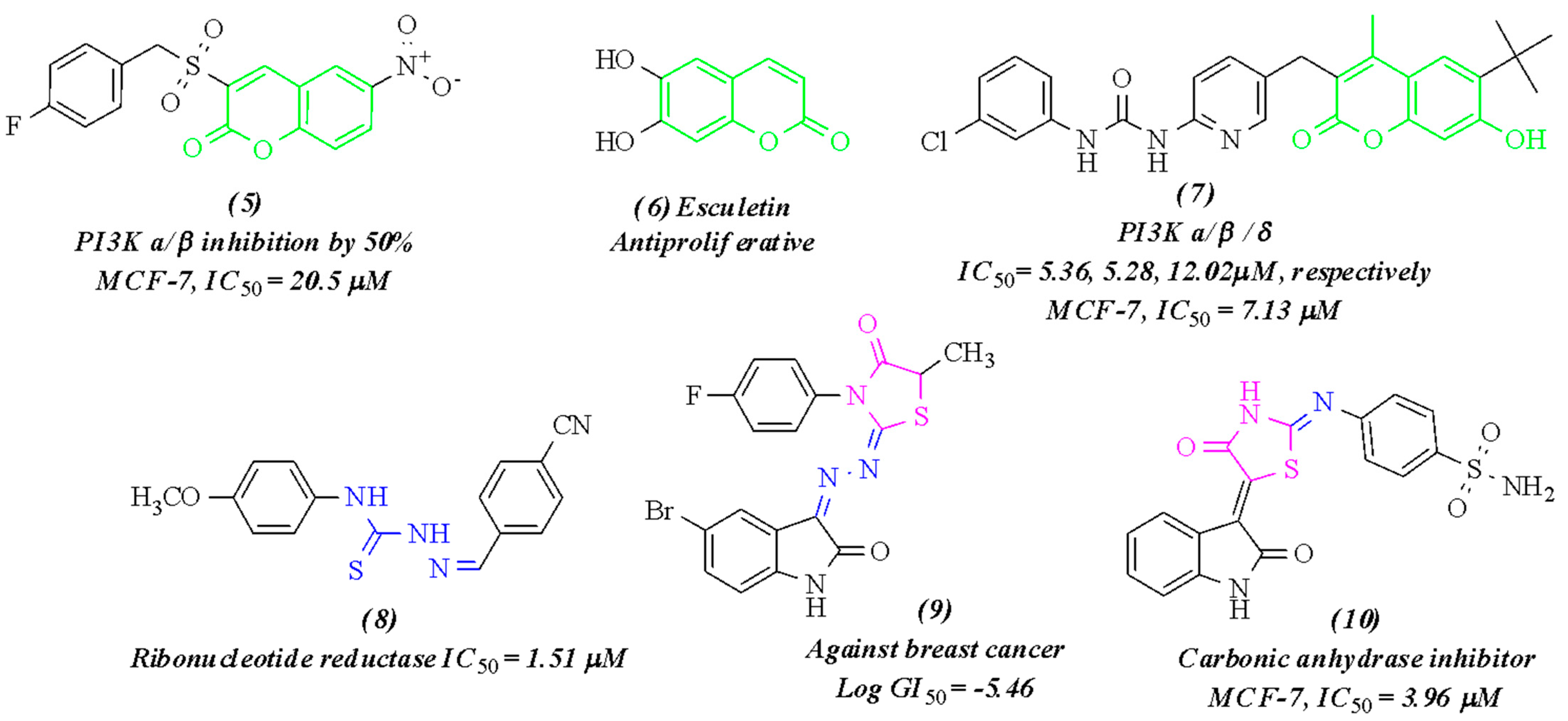
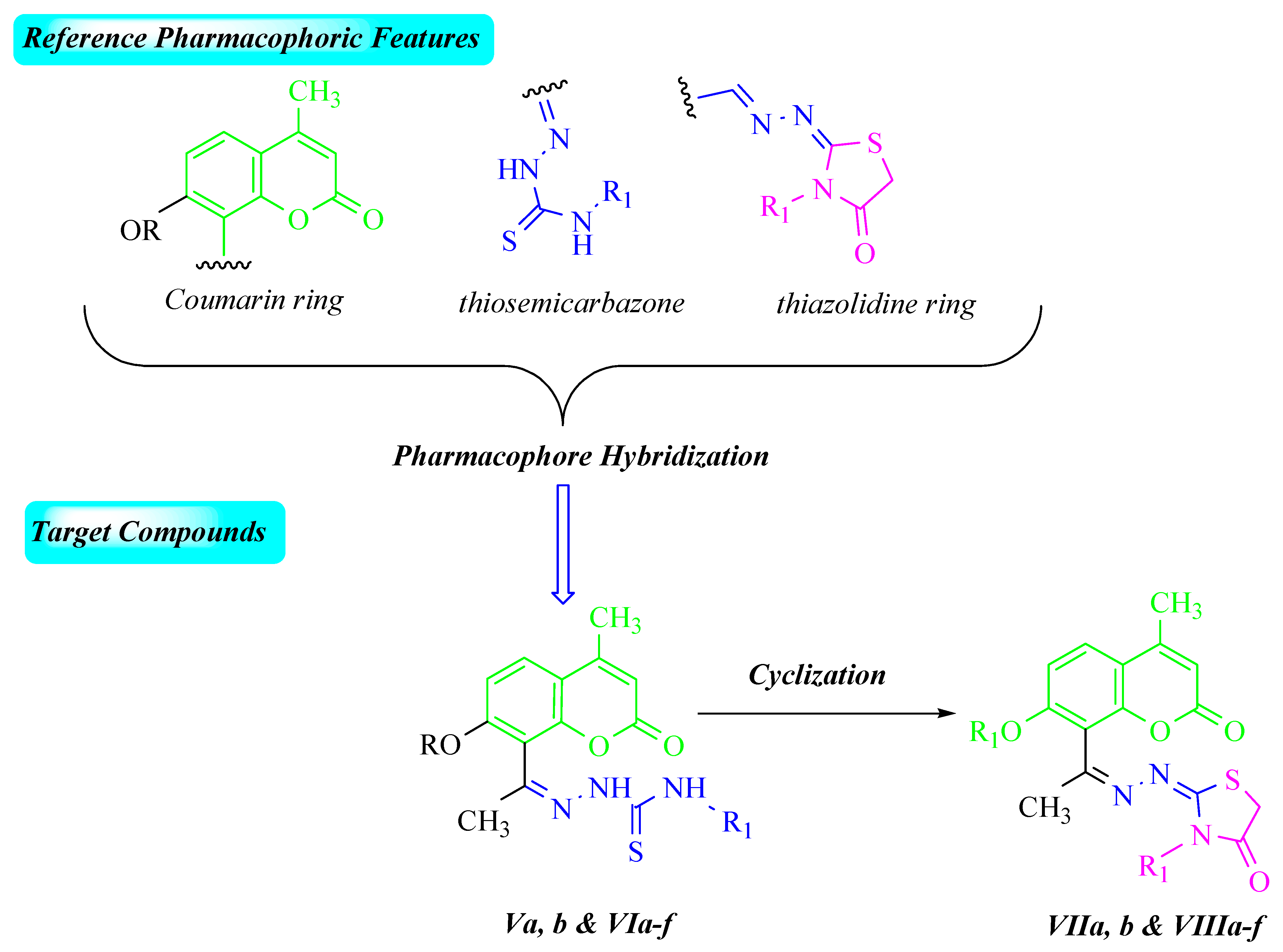
2. Results and Discussion
2.1. Chemistry
2.2. Antitumor Activity
2.2.1. Cytotoxicity Assay
2.2.2. Enzyme Inhibition Assay
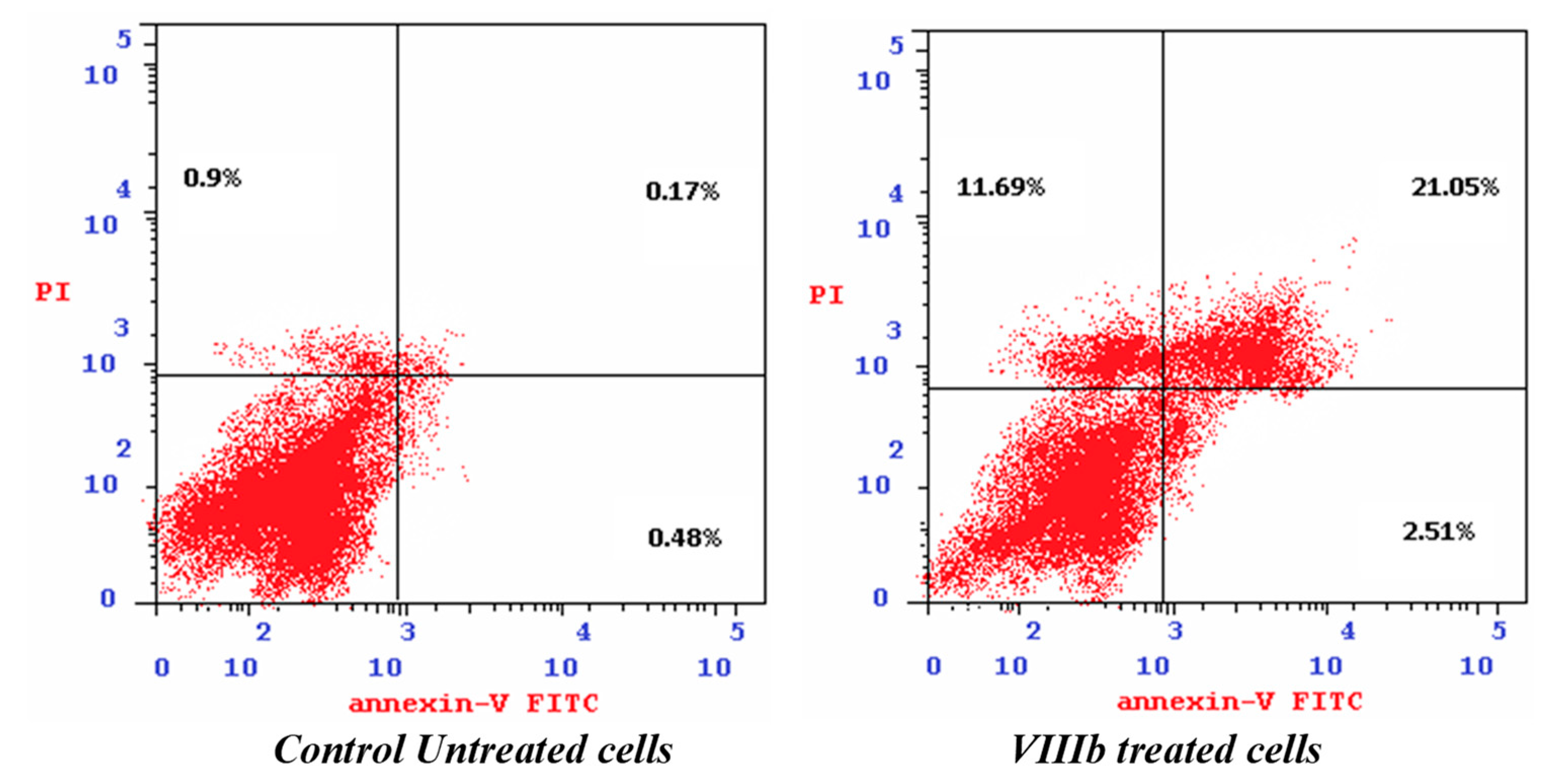
2.2.3. Cell Cycle Analysis and Apoptosis Induction
2.2.4. Caspase-9 Assay
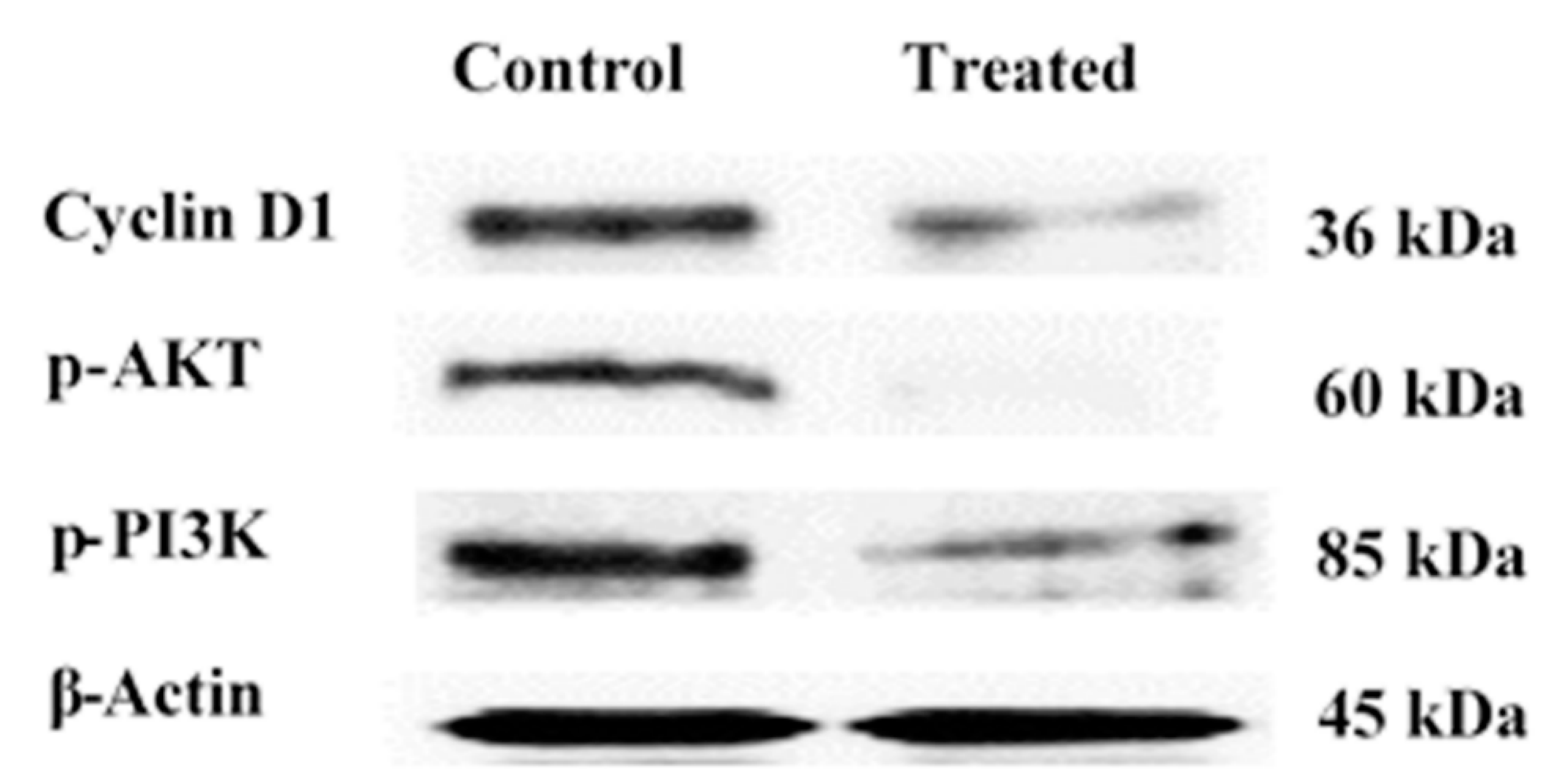
2.2.5. Western Blot
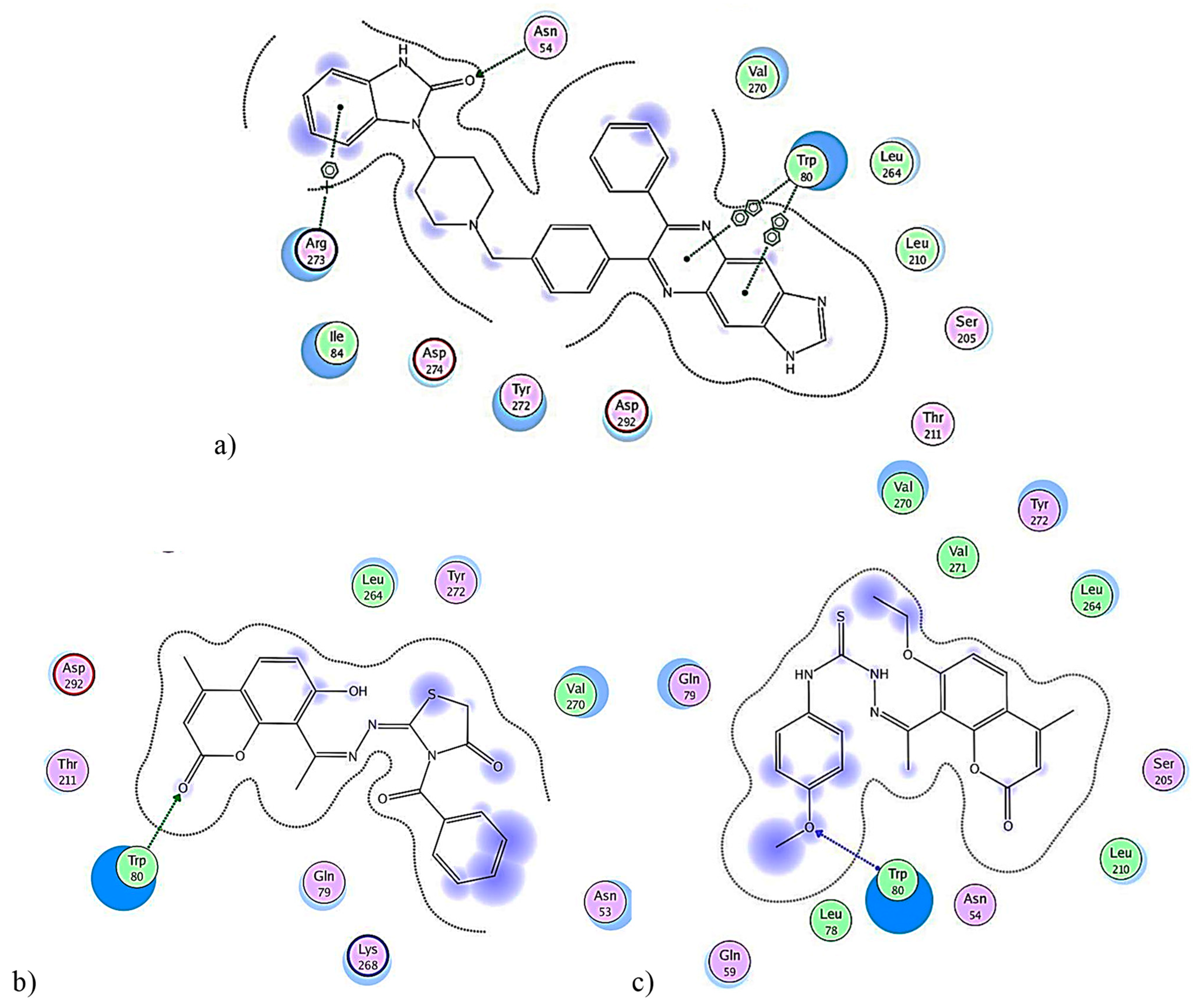
2.2.6. In Silico Molecular Simulations
3. Materials and Methods
3.1. Chemistry
General Procedures for Thiosemicarbazones V and VI Synthesis
3.2. Antitumor Activity
3.2.1. Cytotoxicity Assay
3.2.2. PI3K and Akt Enzyme Inhibition Assays
3.2.3. Cell Cycle Analysis and Apoptosis Induction
3.2.4. Determination of the Cleaved Caspase-9 Level
3.2.5. Western Blot
3.2.6. Molecular Simulation Studies
3.3. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Britt, K.L.; Cuzick, J.; Phillips, K.-A. Key steps for effective breast cancer prevention. Nat. Cancer 2020, 20, 417–436. [Google Scholar] [CrossRef] [PubMed]
- Kamińska, M.; Ciszewski, T.; Łopacka-Szatan, K.; Miotła, P.; Starosławska, E. Breast cancer risk factors. Menopausal Rev. 2015, 3, 196–202. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.A.; Jain, V.K.; Rizwanullah, M.; Ahmad, J.; Jain, K. PI3K/AKT/mTOR pathway inhibitors in triple-negative breast cancer: A review on drug discovery and future challenges. Drug Discov. Today 2019, 24, 2181–2191. [Google Scholar] [CrossRef]
- O’Donnell, J.; Massi, D.; Teng, M.W.; Mandala, M. PI3K-AKT-mTOR inhibition in cancer immunotherapy, redux. Semin. Cancer Biol. 2018, 48, 91–103. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Liu, J.; Wang, Z.; Sun, B.; Wang, J. Inhibitory effects of 5-heptadecylresorcinol on the proliferation of human MCF-7 breast cancer cells through modulating PI3K/Akt/mTOR pathway. J. Funct. Foods 2020, 69, 103946. [Google Scholar] [CrossRef]
- Franke, T.F. Akt-interacting proteins: Attractive opposites. Focus on “Carboxy-terminal modulator protein induces Akt phosphorylation and activation, thereby enhancing antiapoptotic, glycogen synthetic, and glucose uptake pathways. Am. J. Physiol. Physiol. 2007, 293, C1768–C1770. [Google Scholar] [CrossRef]
- Miricescu, D.; Totan, A.; Stanescu-Spinu, I.-I.; Badoiu, S.C.; Stefani, C.; Greabu, M. PI3K/AKT/mTOR Signaling Pathway in Breast Cancer: From Molecular Landscape to Clinical Aspects. Int. J. Mol. Sci. 2020, 22, 173. [Google Scholar] [CrossRef]
- Miller, T.W.; Rexer, B.N.; Garrett, J.T.; Arteaga, C.L. Mutations in the phosphatidylinositol 3-kinase pathway: Role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011, 13, 224. [Google Scholar] [CrossRef] [Green Version]
- Xue, G.; Zippelius, A.; Wicki, A.; Mandalà, M.; Tang, F.; Massi, D.; Hemmings, B.A. Integrated Akt/PKB Signaling in Immunomodulation and Its Potential Role in Cancer Immunotherapy. JNCI J. Natl. Cancer Inst. 2015, 107, djv171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crompton, J.G.; Sukumar, M.; Roychoudhuri, R.; Clever, D.; Gros, A.; Eil, R.L.; Tran, E.; Hanada, K.-I.; Yu, Z.; Palmer, D.C.; et al. Akt Inhibition Enhances Expansion of Potent Tumor-Specific Lymphocytes with Memory Cell Characteristics. Cancer Res. 2015, 75, 296–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, N.; Salguero, A.L.; Liu, A.; Chen, Z.; Dempsey, D.R.; Ficarro, S.B.; Alexander, W.M.; Marto, J.A.; Li, Y.; Amzel, L.M.; et al. Akt Kinase Activation Mechanisms Revealed Using Protein Semisynthesis. Cell 2018, 174, 897–907.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paplomata, E.; O’Regan, R. The PI3K/AKT/mTOR pathway in breast cancer: Targets, trials and biomarkers. Ther. Adv. Med. Oncol. 2014, 6, 154–166. [Google Scholar] [CrossRef] [Green Version]
- Dotolo, S.; Cervellera, C.; Russo, M.; Russo, G.; Facchiano, A. Virtual Screening of Natural Compounds as Potential PI3K-AKT1 Signaling Pathway Inhibitors and Experimental Validation. Molecules 2021, 26, 492. [Google Scholar] [CrossRef]
- Bahrami, A.; Khazaei, M.; Shahidsales, S.; Hassanian, S.M.; Hasanzadeh, M.; Maftouh, M.; Ferns, G.A.; Avan, A. The Therapeutic Potential of PI3K/Akt/mTOR Inhibitors in Breast Cancer: Rational and Progress. J. Cell. Biochem. 2018, 119, 213–222. [Google Scholar] [CrossRef]
- Helwa, A.A.; El-Dydamony, N.M.; Radwan, R.A.; Abdelraouf, S.M.; Abdelnaby, R.M. Novel antiproliferative agents bearing morpholinopyrimidine scaffold as PI3K inhibitors and apoptosis inducers; design, synthesis and molecular docking. Bioorganic Chem. 2020, 102, 104051. [Google Scholar] [CrossRef]
- Zhan, W.; Che, J.; Xu, L.; Wu, Y.; Hu, X.; Zhou, Y.; Cheng, G.; Hu, Y.; Dong, X.; Li, J. Discovery of pyrazole-thiophene derivatives as highly Potent, orally active Akt inhibitors. Eur. J. Med. Chem. 2019, 180, 72–85. [Google Scholar] [CrossRef]
- Yu, M.; Zeng, M.; Pan, Z.; Wu, F.; Guo, L.; He, G. Discovery of novel akt1 inhibitor induces autophagy associated death in hepatocellular carcinoma cells. Eur. J. Med. Chem. 2020, 189, 112076. [Google Scholar] [CrossRef]
- Wang, T.; Peng, T.; Wen, X.; Wang, G.; Sun, Y.; Liu, S.; Zhang, S.; Wang, L. Design, Synthesis and Preliminary Biological Evaluation of Benzylsulfone Coumarin Derivatives as Anti-Cancer Agents. Molecules 2019, 24, 4034. [Google Scholar] [CrossRef] [Green Version]
- Goel, A.; Prasad, A.K.; Parmar, V.S.; Ghosh, B.; Saini, N. 7,8-Dihydroxy-4-methylcoumarin induces apoptosis of human lung adenocarcinoma cells by ROS-independent mitochondrial pathway through partial inhibition of ERK/MAPK signaling. FEBS Lett. 2007, 581, 2447–2454. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Gu, L.; Wang, B.; Huang, W.; Zhang, Y.; Ma, Z.; Zeng, S.; Shen, Z. Discovery of novel coumarin derivatives as potent and orally bioavailable BRD4 inhibitors based on scaffold hopping. J. Enzym. Inhib. Med. Chem. 2019, 34, 808–817. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Liang, Z.; Xu, H.; Mou, Y.; Guo, C. Design, Synthesis and Cytotoxicity of Novel Dihydroartemisinin-Coumarin Hybrids via Click Chemistry. Molecules 2016, 21, 758. [Google Scholar] [CrossRef] [Green Version]
- Akkol, E.K.; Genç, Y.; Karpuz, B.; Sobarzo-Sánchez, E.; Capasso, R. Coumarins and Coumarin-Related Compounds in Pharmacotherapy of Cancer. Cancers 2020, 12, 1959. [Google Scholar] [CrossRef]
- Nepali, K.; Sharma, S.; Sharma, M.; Bedi, P.M.S.; Dhar, K.L. Rational approaches, design strategies, structure activity relationship and mechanistic insights for anticancer hybrids. Eur. J. Med. Chem. 2014, 77, 422–487. [Google Scholar] [CrossRef]
- Thakur, A.; Singla, R.; Jaitak, V. Coumarins as anticancer agents: A review on synthetic strategies, mechanism of action and SAR studies. Eur. J. Med. Chem. 2015, 101, 476–495. [Google Scholar] [CrossRef]
- Ahmed, M.F.; Almalki, A.H. Design, synthesis, antiproliferative activity, and cell cycle analysis of new thiosemicarbazone derivatives targeting ribonucleotide reductase. Arab. J. Chem. 2021, 14, 102989. [Google Scholar] [CrossRef]
- Karalı, N.; Terzioğlu, N.; Gürsoy, A. Synthesis and Primary Cytotoxicity Evaluation of New 5-Bromo-3-Substituted-Hydrazono-1H-2-Indolinones. Arch. Der Pharm. 2002, 335, 374–380. [Google Scholar] [CrossRef]
- Trotsko, N.; Przekora, A.; Zalewska, J.; Ginalska, G.; Paneth, A.; Wujec, M. Synthesis and in Vitro Antiproliferative and Antibacterial Activity of New Thiazoli-dine-2,4-Dione Derivatives. J. Enzym. Inhib. Med. Chem. 2018, 33, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Havrylyuk, D.; Roman, O.; Lesyk, R. Synthetic approaches, structure activity relationship and biological applications for pharmacologically attractive pyrazole/pyrazoline-thiazolidine-based hybrids. Eur. J. Med. Chem. 2016, 113, 145–166. [Google Scholar] [CrossRef]
- Ma, C.; Liu, Z.-P. Design and Synthesis of Coumarin Derivatives as Novel PI3K Inhibitors. Anti-Cancer Agents Med. Chem. 2017, 17, 395–403. [Google Scholar] [CrossRef]
- Eldehna, W.M.; Abo-Ashour, M.F.; Nocentini, A.; Gratteri, P.; Eissa, I.H.; Fares, M.; Ismael, O.E.; Ghabbour, H.A.; Elaasser, M.M.; Abdel-Aziz, H.A.; et al. Novel 4/3-((4-oxo-5-(2-oxoindolin-3-ylidene)thiazolidin-2-ylidene)amino) benzene-sulfonamides: Synthesis, carbonic anhydrase inhibitory activity, anticancer activity and molecular modelling studies. Eur. J. Med. Chem. 2017, 139, 250–262. [Google Scholar] [CrossRef]
- Amin, K.M.; Abou-Seri, S.M.; Abdelnaby, R.M.; Rateb, H.S.; Khalil, M.A.F.; Hussein, M.M. Synthesis and Biological Evaluation of Novel Coumarin Derivatives as Potential Antimicrobials Agents. Int. J. Pharm. Pharm. Sci. 2016, 8, 109–116. [Google Scholar]
- Fukutani, Y.; Maruoka, K.; Yamamoto, H. Stereoselective conjugate addition of organoaluminum reagents to chiral α,β-unsaturated ketals. Tetrahedron Lett. 1984, 25, 5911–5912. [Google Scholar] [CrossRef]
- Singh, S.P.; Parmar, S.S.; Raman, K.; Stenberg, V.I. Chemistry and biological activity of thiazolidinones. Chem. Rev. 1981, 81, 175–203. [Google Scholar] [CrossRef]
- Carmichael, J.; DeGraff, W.; Gazdar, A.; Minna, J.; Mitchell, J.B. Evaluation of a Tetrazolium-Based Semiautomated Color-imetric Assay: Assessment of Chemosensitivity Testing. Cancer Res. 1987, 47, 936–942. [Google Scholar]
- Takeuchi, H.; Baba, M.; Shigeta, S. An application of tetrazolium (MTT) colorimetric assay for the screening of anti-herpes simplex virus compounds. J. Virol. Methods 1991, 33, 61–71. [Google Scholar] [CrossRef]
- Badisa, R.B.; Darling-Reed, S.F.; Joseph, P.; Cooperwood, J.S.; Latinwo, L.M.; Goodman, C.B. Selective Cytotoxic Activities of Two Novel Synthetic Drugs on Human Breast Carcinoma MCF-7 Cells. Anticancer. Res. 2009, 29, 2993–2996. [Google Scholar]
- Zhang, Z.; Richmond, A. The Role of PI3K Inhibition in the Treatment of Breast Cancer, Alone or Combined with Immune Checkpoint Inhibitors. Front. Mol. Biosci. 2021, 8, 648663. [Google Scholar] [CrossRef]
- Darzynkiewicz, Z.; Bedner, E.; Smolewski, P. Flow cytometry in analysis of cell cycle and apoptosis. Semin. Hematol. 2001, 38, 179–193. [Google Scholar] [CrossRef]
- Pozarowski, P.; Darzynkiewicz, Z. Analysis of Cell Cycle by Flow Cytometry. Methods Mol. Biol. 2004, 281, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Vermes, I.; Haanen, C.; Steffens-Nakken, H.; Reutellingsperger, C. A novel assay for apoptosis Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J. Immunol. Methods 1995, 184, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Lakshmanan, I.; Batra, S. Protocol for Apoptosis Assay by Flow Cytometry Using Annexin V Staining Method. Bio-Protocol. 2013, 3, 374. [Google Scholar] [CrossRef] [PubMed]
- Brauchle, E.; Thude, S.; Brucker, S.Y.; Schenke-Layland, K. Cell death stages in single apoptotic and necrotic cells monitored by Raman microspectroscopy. Sci. Rep. 2014, 4, 4698–4707. [Google Scholar] [CrossRef] [Green Version]
- Wlodkowic, D.; Telford, W.; Skommer, J.; Darzynkiewicz, Z. Apoptosis and Beyond: Cytometry in Studies of Programmed Cell Death. Flow Cytom. 2011, 103, 55–98. [Google Scholar] [CrossRef] [Green Version]
- Olivero, O.A.; Tejera, A.M.; Fernandez, J.J.; Taylor, B.J.; Das, S.; Divi, R.L.; Poirier, M.C. Zidovudine induces S-phase arrest and cell cycle gene expression changes in human cells. Mutagenesis 2005, 20, 139–146. [Google Scholar] [CrossRef]
- Bunz, F.; Dutriaux, A.; Lengauer, C.; Waldman, T.; Zhou, S.; Brown, J.P.; Sedivy, J.M.; Kinzler, K.W.; Vogelstein, B. Requirement for p53 and p21 to Sustain G2 Arrest After DNA Damage. Science 1998, 282, 1497–1501. [Google Scholar] [CrossRef]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase Functions in Cell Death and Disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef]
- Singh, N.; Sarkar, J.; Sashidhara, K.V.; Ali, S.; Sinha, S. Anti-tumour activity of a novel coumarin–chalcone hybrid is mediated through intrinsic apoptotic pathway by inducing PUMA and altering Bax/Bcl-2 ratio. Apoptosis 2014, 19, 1017–1028. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Bae, H.; Lee, J.; Song, J.; Song, G.; Lim, W. Osthole interacts with an ER-mitochondria axis and facilitates tumor suppression in ovarian cancer. J. Cell. Physiol. 2020, 236, 1025–1042. [Google Scholar] [CrossRef] [PubMed]
- Takuwa, N.; Fukui, Y.; Takuwa, Y. Cyclin D1 Expression Mediated by Phosphatidylinositol 3-Kinase through mTOR-p70 S6K -Independent Signaling in Growth Factor-Stimulated NIH 3T3 Fibroblasts. Mol. Cell. Biol. 1999, 19, 1346–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qie, S.; Diehl, J.A. Cyclin D1, cancer progression, and opportunities in cancer treatment. Klin. Wochenschr. 2016, 94, 1313–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodges, L.C.; Cook, J.D.; Lobenhofer, E.K.; Li, L.; Bennett, L.; Bushel, P.R.; Aldaz, C.M.; Afshari, C.A.; Walker, C.L. Tamoxifen functions as a molecular agonist inducing cell cycle-associated genes in breast cancer cells. Mol. Cancer Res. 2003, 1, 300–311. [Google Scholar]
- Hui, R.; Finney, G.L.; Carroll, J.; Lee, C.S.L.; Musgrove, E.A.; Sutherland, R.L. Constitutive overexpression of cyclin D1 but not cyclin E confers acute resistance to antiestrogens in T-47D breast cancer cells. Cancer Res. 2002, 62, 6916–6923. [Google Scholar] [PubMed]
- Zhao, Y.; Zhang, X.; Chen, Y.; Lu, S.; Peng, Y.; Wang, X.; Guo, C.; Zhou, A.; Zhang, J.; Luo, Y.; et al. Crystal Structures of PI3Kα Complexed with PI103 and Its Derivatives: New Directions for Inhibitors Design. ACS Med. Chem. Lett. 2014, 5, 138–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.-I.; Voegtli, W.C.; Sturgis, H.L.; Dizon, F.P.; Vigers, G.P.A.; Brandhuber, B.J. Crystal Structure of Human AKT1 with an Allosteric Inhibitor Reveals a New Mode of Kinase Inhibition. PLoS ONE 2010, 5, e12913. [Google Scholar] [CrossRef]
- Zhao, W.; Qiu, Y.; Kong, D. Class I phosphatidylinositol 3-kinase inhibitors for cancer therapy. Acta Pharm. Sin. B 2017, 7, 27–37. [Google Scholar] [CrossRef]
- Kitchen, P.; Salman, M.M.; Halsey, A.M.; Clarke-Bland, C.; Macdonald, J.A.; Ishida, H.; Vogel, H.J.; Almutiri, S.; Logan, A.; Kreida, S.; et al. Targeting Aquaporin-4 Subcellular Localization to Treat Central Nervous System Edema. Cell 2020, 181, 784–799.e19. [Google Scholar] [CrossRef]
- Chung, K.; Huang, Y.; Chen, Y.; Juan, Y.; Hsu, C.; Nakahira, K.; Lin, M.; Wu, S.; Shih, J.; Chang, Y.; et al. Multi-kinase framework promotes proliferation and invasion of lung adenocarcinoma through activation of dynamin-related protein 1. Mol. Oncol. 2021, 15, 560–578. [Google Scholar] [CrossRef]
- Wang, H.; Zhou, W.; Zhang, J.; Li, H. Role of JNK and ERK1/2 MAPK signaling pathway in testicular injury of rats induced by di-N-butyl-phthalate (DBP). Biol. Res. 2019, 52, 41. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound No. | R | R1 | IC50 (µM) |
|---|---|---|---|
| 5-FU | - | - | 27.81 ± 1.41 |
| Va | H | Ph-CH2 | 5.13 ± 0.28 *** |
| Vb | H | Ph-C=O | 47.32 ± 2.47 |
| VIa | C2H5 | CH3 | 11.13 ± 0.58 *** |
| VIb | C2H5 | C2H5 | 38.70 ± 2.09 |
| VIc | C2H5 | CH2=CH-CH2 | 2.72 ± 0.13 *** |
| VId | C2H5 | Ph-CH2 | 2.61 ± 0.14 *** |
| VIe | C2H5 | Ph-C=O | 43.05 ± 2.25 |
| VIf | C2H5 | 4-OCH3-Ph | 1.21 ± 0.06 *** |
| VIIa | H | Ph-CH2 | 54.80 ± 2.86 |
| VIIb | H | Ph-C=O | 1.03 ± 0.05 *** |
| VIIIa | C2H5 | CH3 | 20.27 ± 1.06 ** |
| VIIIb | C2H5 | C2H5 | 57.28 ± 2.99 |
| VIIIc | C2H5 | CH2=CH-CH2 | 4.95 ± 0.26 *** |
| VIIId | C2H5 | Ph-CH2 | 79.93 ± 4.18 |
| VIIIe | C2H5 | Ph-C=O | 26.41 ± 1.38 ns |
| VIIIf | C2H5 | 4-OCH3-Ph | 11.80 ± 0.62 *** |
| Compound | IC50 (µM) MCF-10 a | IC50 (µM) MCF-7 | Selectivity Index MCF-10/MCF-7 |
|---|---|---|---|
| VIf | 20.11 ± 1.05 | 1.21 ± 0.06 | 16.61 |
| VIIb | 9.52 ± 0.60 | 1.03 ± 0.05 | 9.24 |
| 5-FU | 36.22 ± 1.89 | 27.81 ± 1.41 | 1.30 |
| Compounds | IC50 (µM) | ||
|---|---|---|---|
| PI3K-α Isoform | PI3K-γ Isoform | Akt-1 Isoform | |
| VIIb | 3.70 ± 0.19 *** | 34.70 ± 1.88 | 2.93 ± 0.15 * |
| LY294002 | 8.85 ± 0.46 | 11.5 ± 0.62 | 3.53 ± 0.18 |
| Cells | DNA Content % | |||
|---|---|---|---|---|
| G0/G1 | S | G2/M | Pre G1 | |
| VIIb-treated cells | 48.39 | 46.02 | 5.59 | 35.25 |
| Control untreated cells | 53.71 | 36.58 | 9.71 | 1.55 |
| Cells | Apoptosis % | Necrosis% | ||
|---|---|---|---|---|
| Total | Early | Late | ||
| VIIb-treated cells | 35.25 | 2.51 | 21.05 | 11.69 |
| Control untreated cells | 1.55 | 0.48 | 0.17 | 0.90 |
| Cells | Caspase-9 Level (ng/mL) | Fold |
|---|---|---|
| VIIb-treated cells | 21.61 ± 0.14 *** | 7.96 |
| Control untreated cells | 2.712 ± 0.09 | 1 |
| Compound | S-Score | Interaction Types and Residues |
|---|---|---|
| 1 X6K | −2.2807 | H-bonding, Lys802 |
| 2 VIIb | −3.7521 | H-bonding, Tyr836 |
| 3 VIf | −3.0518 | H-bonding, Lys802 |
| Compound | S-Score | Interaction Types and Residues |
|---|---|---|
| 1 IQO | −5.0984 | Hydrophobic aromatic, Trp80, Arg273; H-bonding, Asn54. |
| 2 VIIb | −2.6564 | H-bonding, Trp80 |
| 3 VIf | −4.3739 | H-bonding, Trp80 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdelnaby, R.M.; Rateb, H.S.; Ali, O.; Saad, A.S.; Nadeem, R.I.; Abou-Seri, S.M.; Amin, K.M.; Younis, N.S.; Abdelhady, R. Dual PI3K/Akt Inhibitors Bearing Coumarin-Thiazolidine Pharmacophores as Potential Apoptosis Inducers in MCF-7 Cells. Pharmaceuticals 2022, 15, 428. https://doi.org/10.3390/ph15040428
Abdelnaby RM, Rateb HS, Ali O, Saad AS, Nadeem RI, Abou-Seri SM, Amin KM, Younis NS, Abdelhady R. Dual PI3K/Akt Inhibitors Bearing Coumarin-Thiazolidine Pharmacophores as Potential Apoptosis Inducers in MCF-7 Cells. Pharmaceuticals. 2022; 15(4):428. https://doi.org/10.3390/ph15040428
Chicago/Turabian StyleAbdelnaby, Rana M., Heba S. Rateb, Omaima Ali, Ahmed S. Saad, Rania I. Nadeem, Sahar M. Abou-Seri, Kamilia M. Amin, Nancy S. Younis, and Rasha Abdelhady. 2022. "Dual PI3K/Akt Inhibitors Bearing Coumarin-Thiazolidine Pharmacophores as Potential Apoptosis Inducers in MCF-7 Cells" Pharmaceuticals 15, no. 4: 428. https://doi.org/10.3390/ph15040428
APA StyleAbdelnaby, R. M., Rateb, H. S., Ali, O., Saad, A. S., Nadeem, R. I., Abou-Seri, S. M., Amin, K. M., Younis, N. S., & Abdelhady, R. (2022). Dual PI3K/Akt Inhibitors Bearing Coumarin-Thiazolidine Pharmacophores as Potential Apoptosis Inducers in MCF-7 Cells. Pharmaceuticals, 15(4), 428. https://doi.org/10.3390/ph15040428