
Design, Synthesis, and Molecular Docking Studies of Curcumin Hybrid Conjugates as Potential Therapeutics for Breast Cancer

 ,
, 
 ,
, 
Abstract
:
1. Introduction
2. Results and Discussion
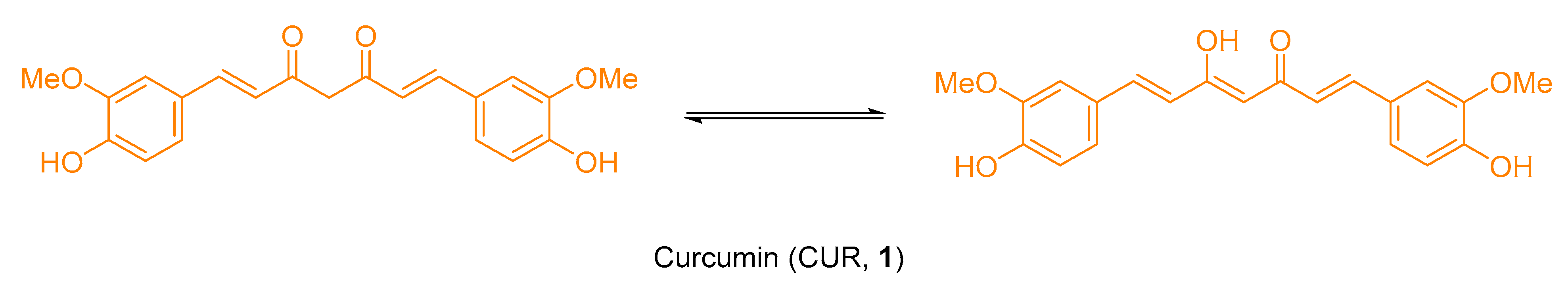
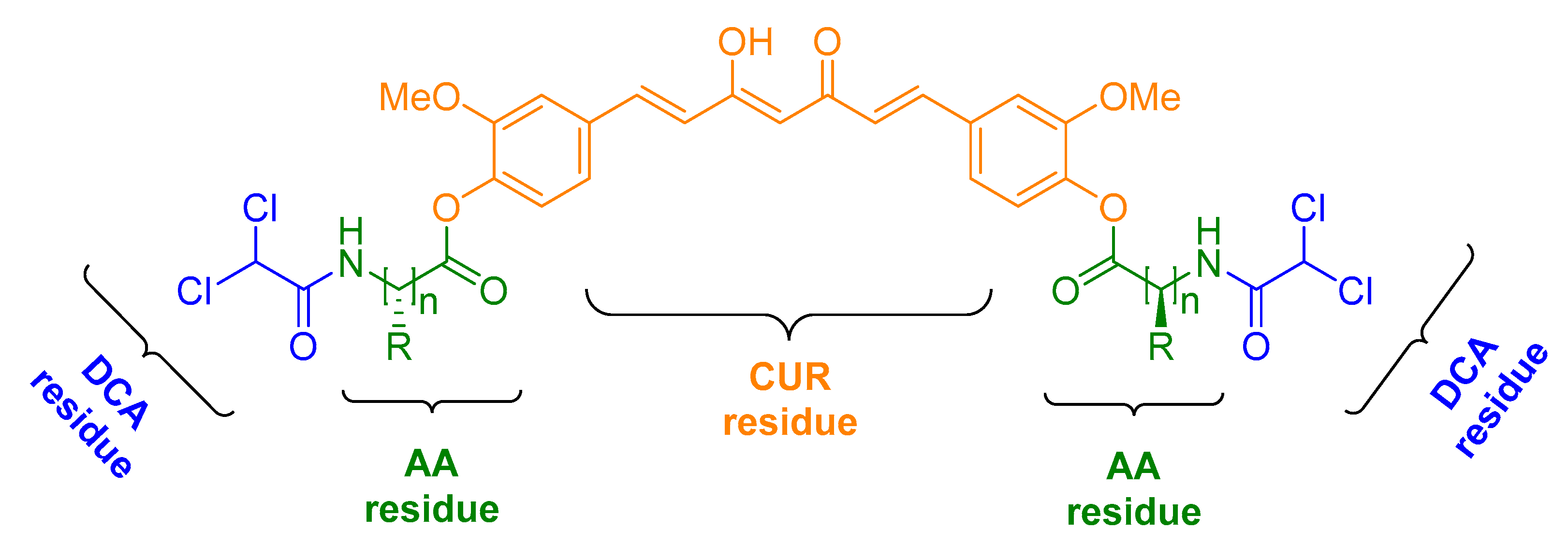
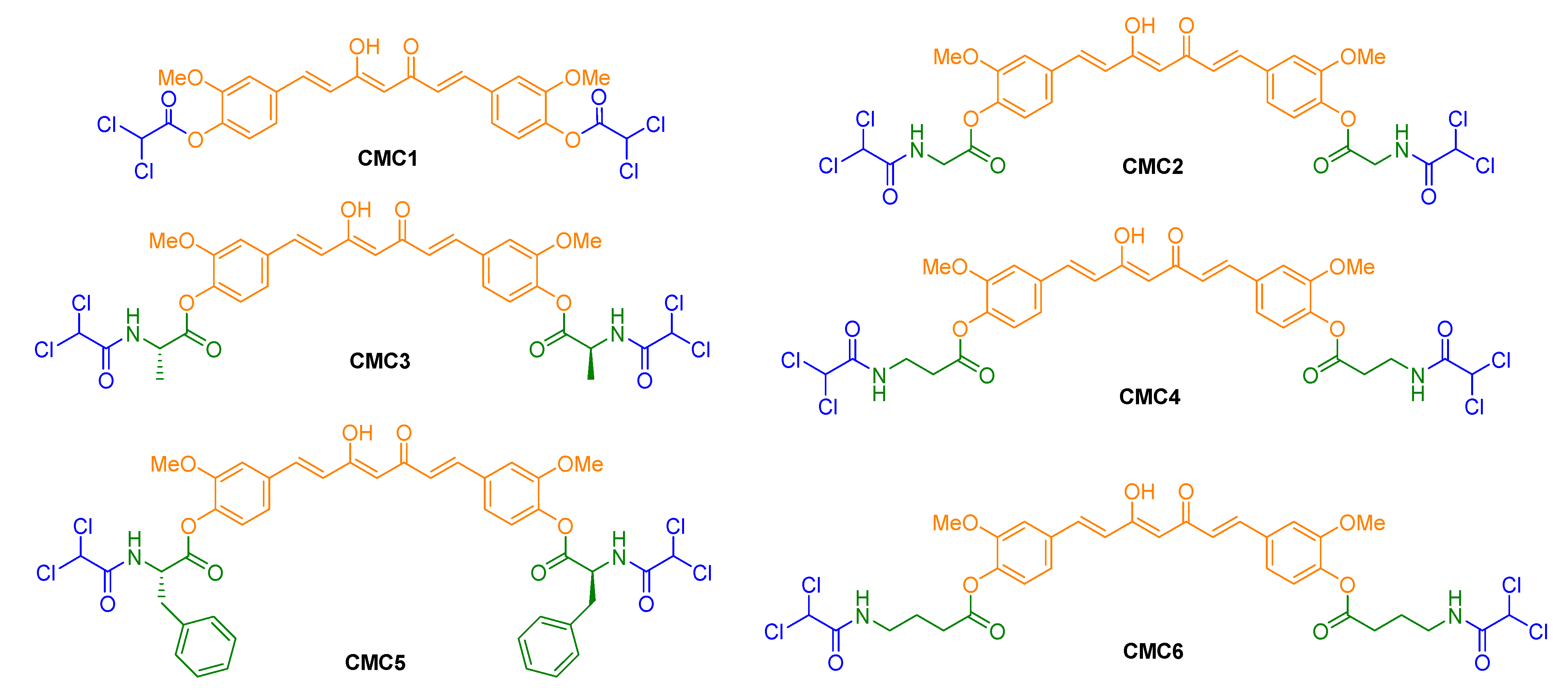
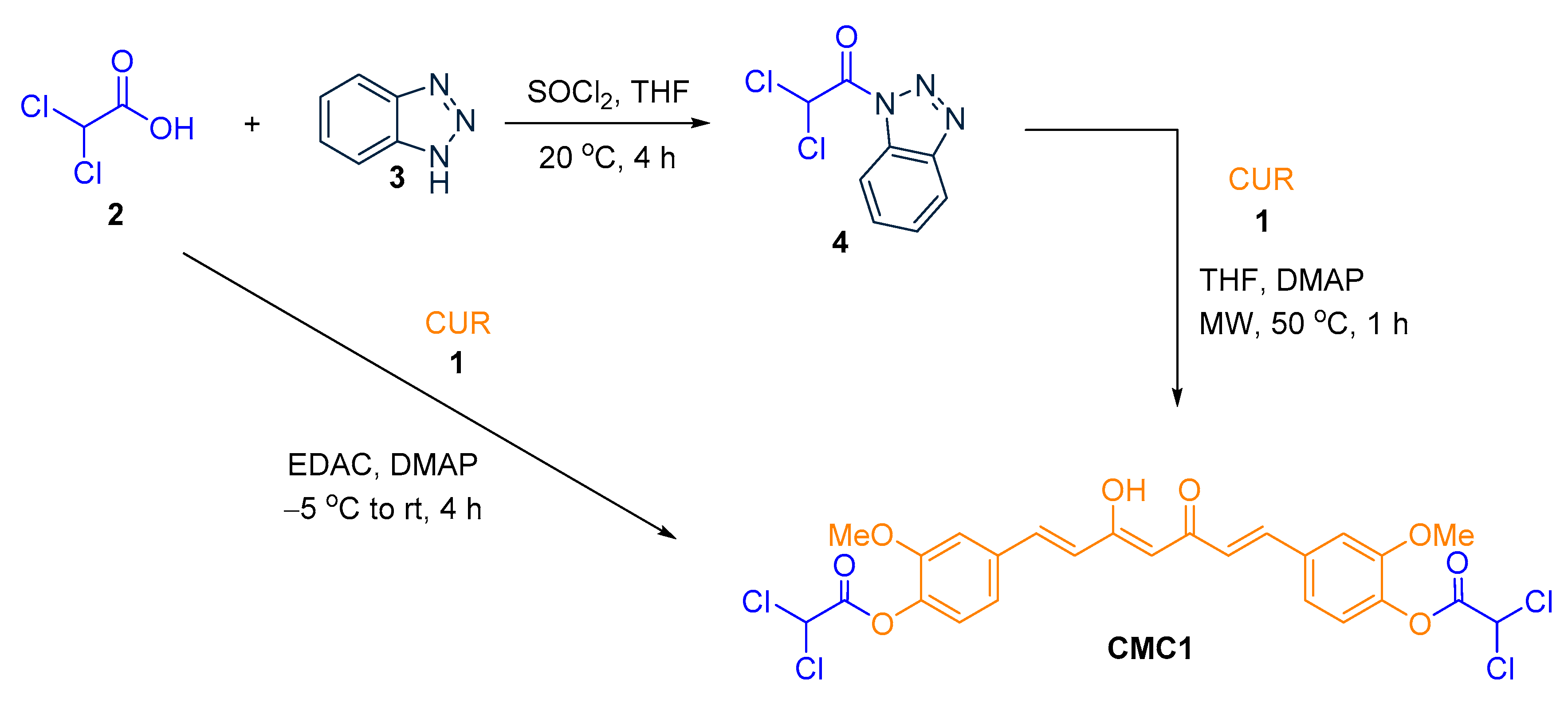
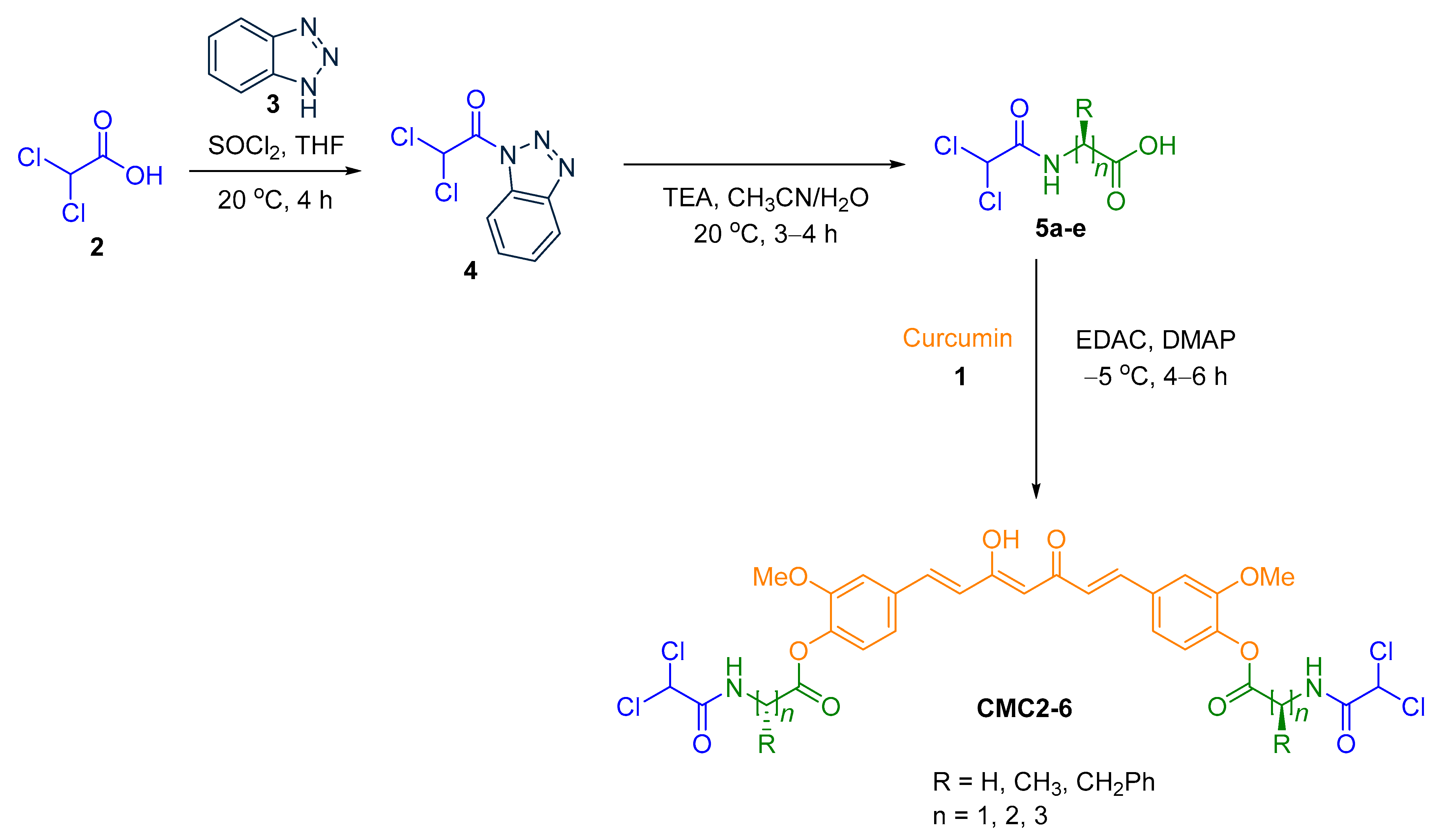
2.1. Chemistry
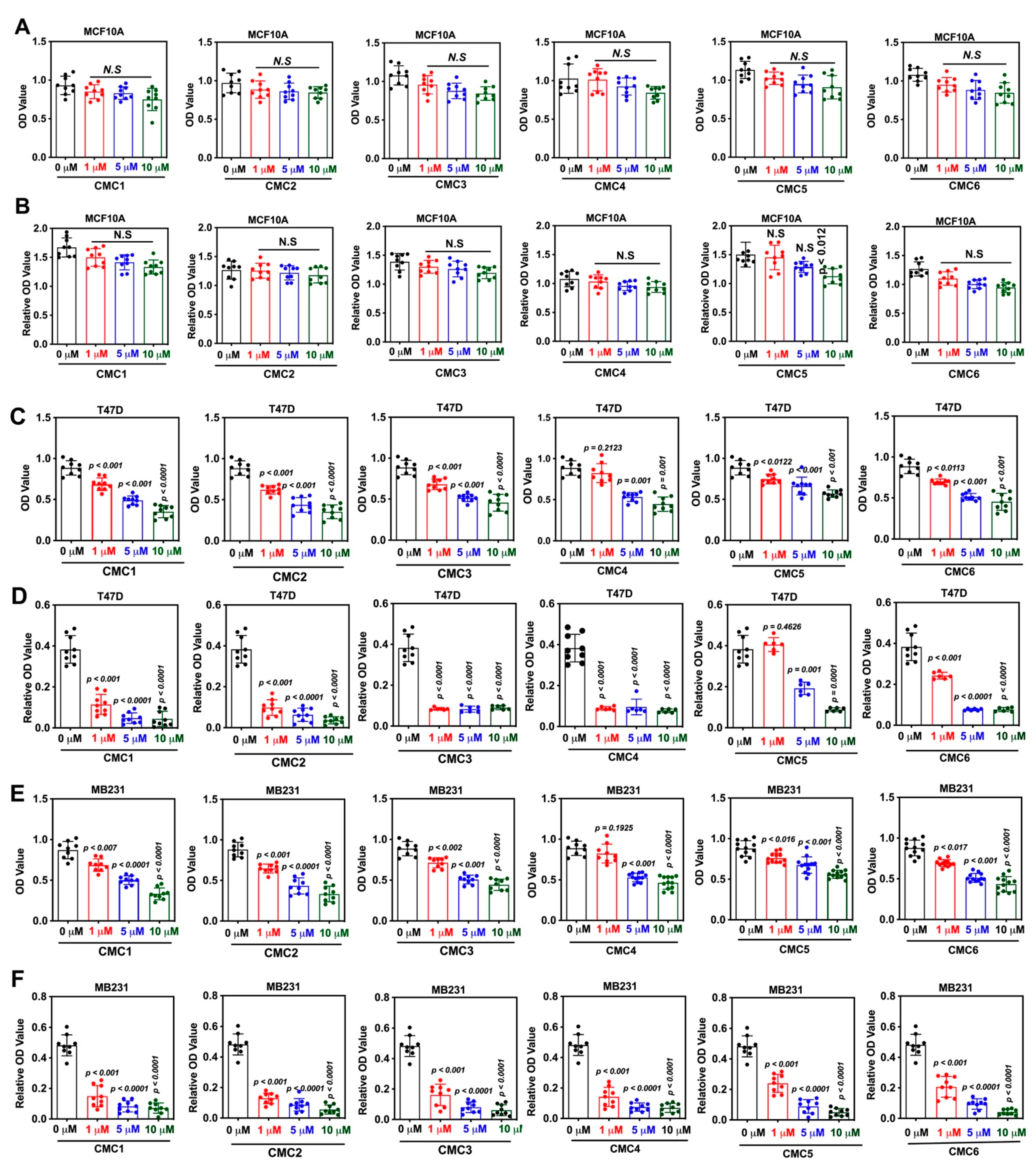
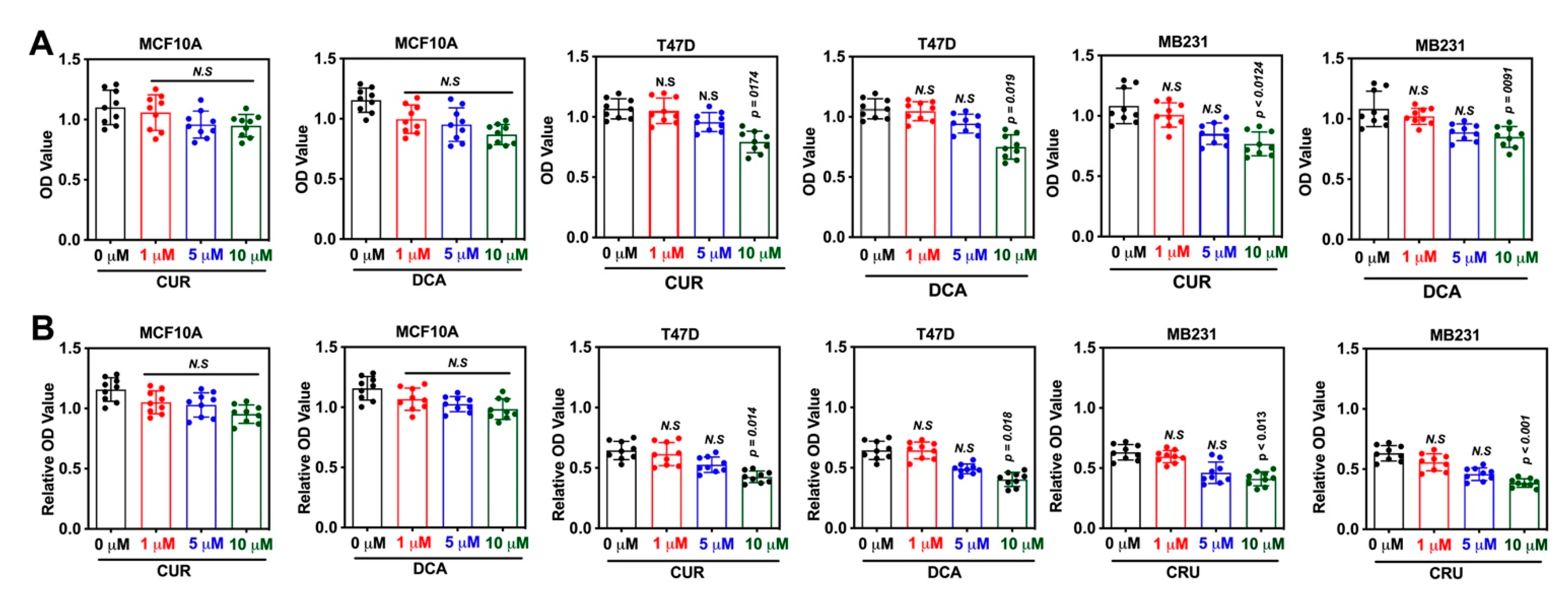
2.2. Biology
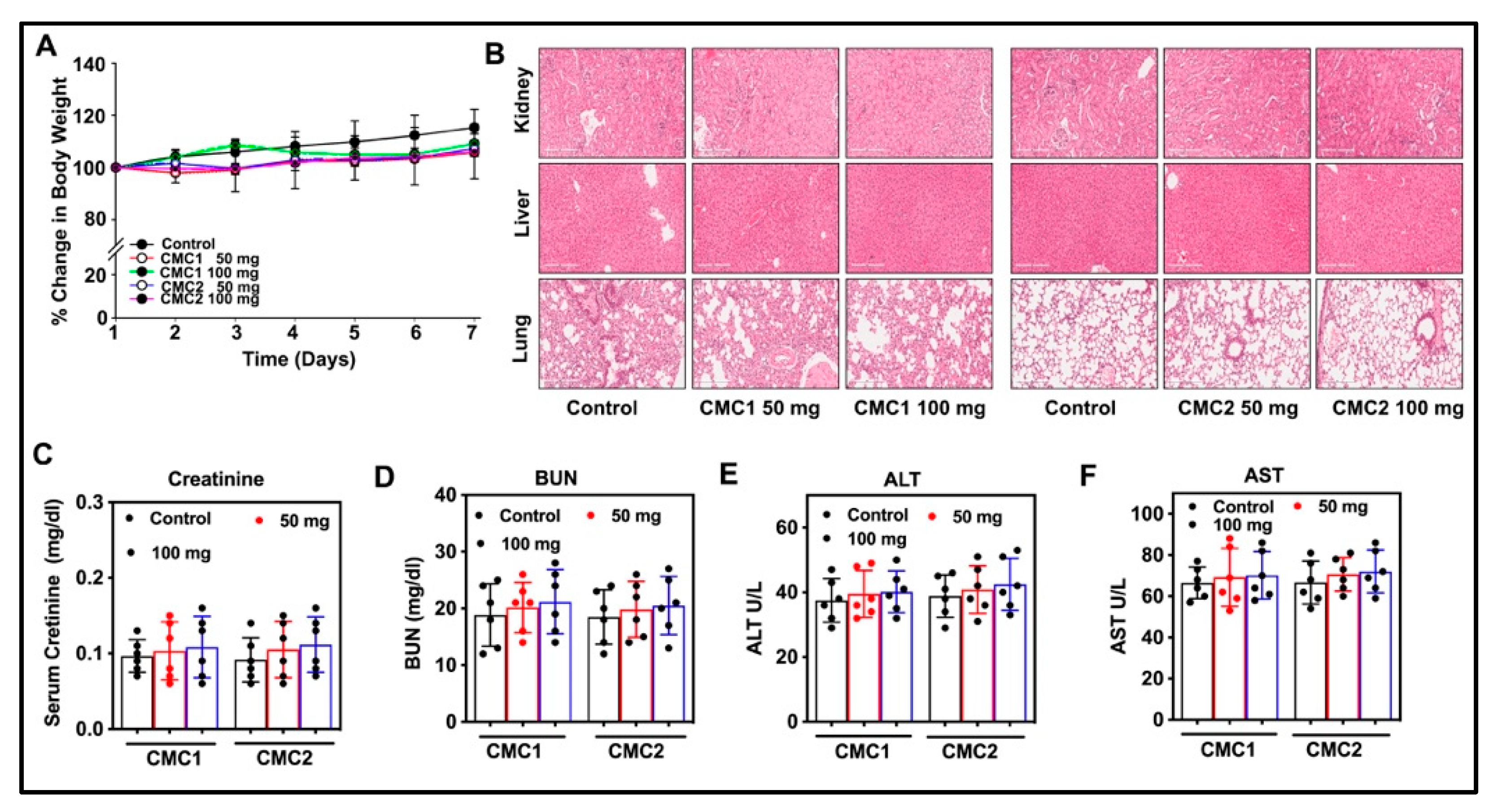
2.3. Toxicity Studies
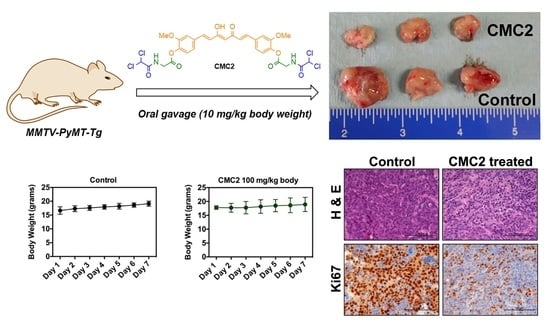
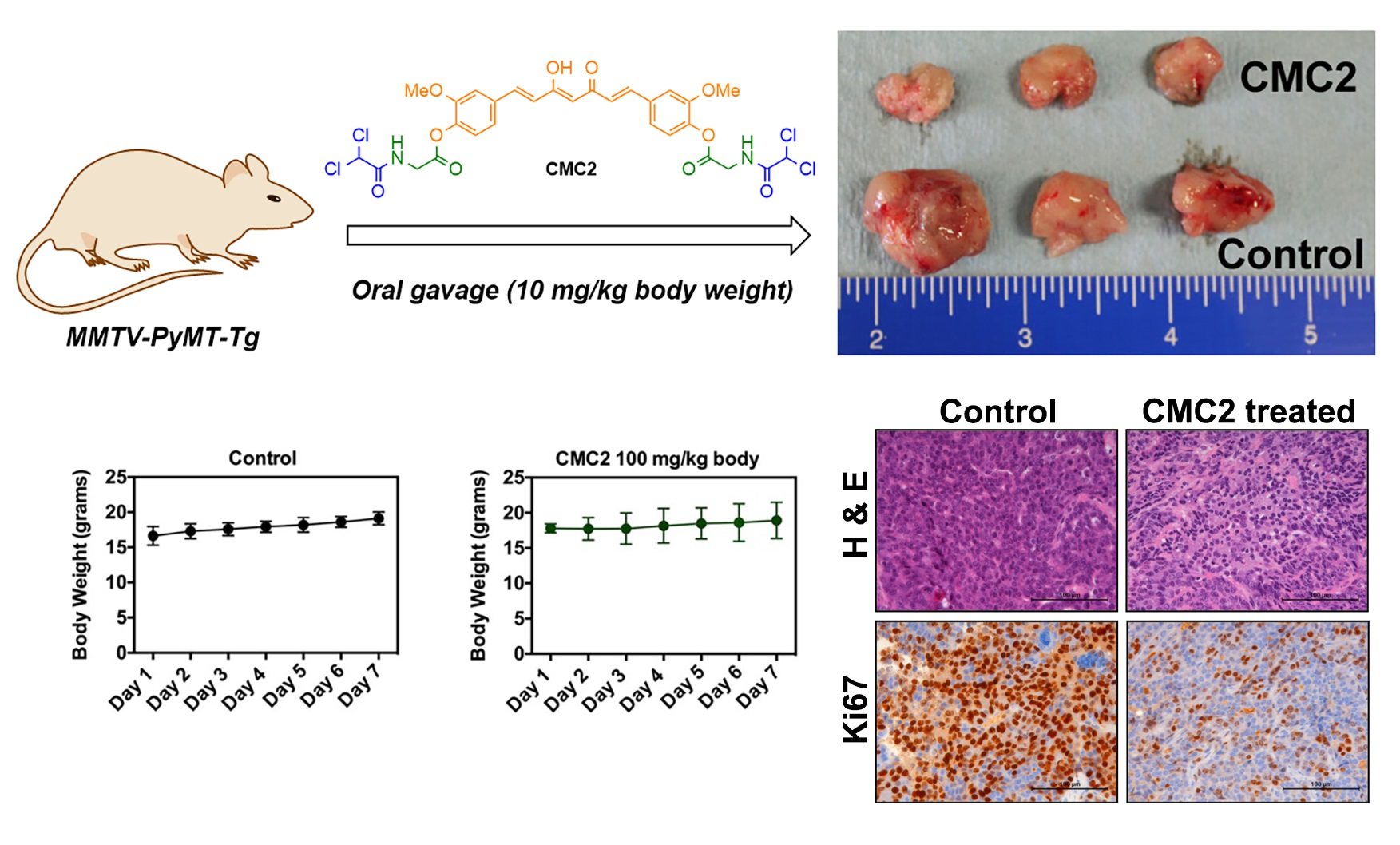
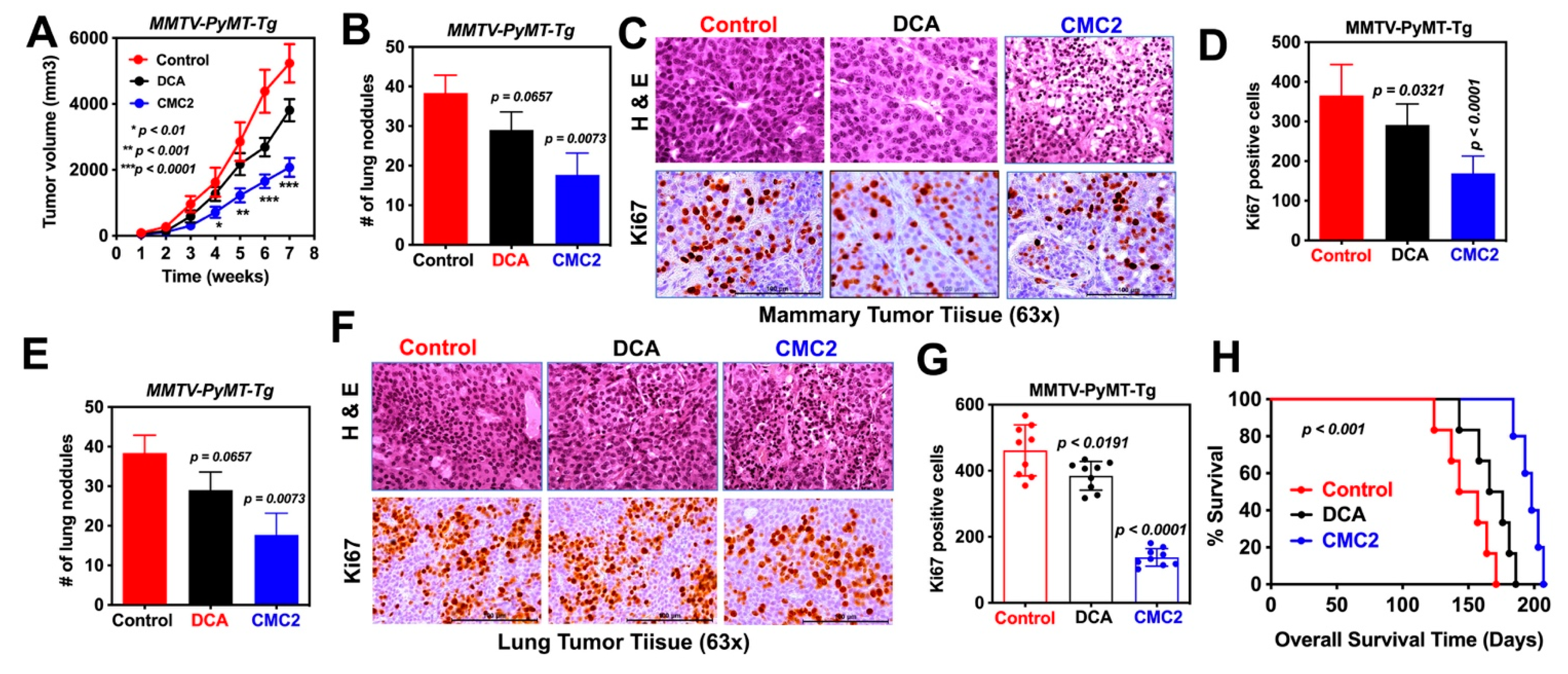
2.4. Animal Studies
2.5. Computational Studies
3. Materials and Methods
3.1. Synthesis of CUR–DCA Hybrid Conjugate (CMC1)
3.2. General Method for Preparation of CUR–DCA Hybrid Conjugates with an AA Linker (CMC2–6)
3.3. MTT Assay
3.4. Colony Formation Assay
3.5. Institutional Compliance
3.6. Cell Lines
3.7. Animals
3.8. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cai, F.-F.; Kohler, C.; Zhang, B.; Wang, M.-H.; Chen, W.-J.; Zhong, X.-Y. Epigenetic therapy for breast cancer. Int. J. Mol. Sci. 2011, 12, 4465–4476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanardi, E.; Bregni, G.; de Braud, F.; Di Cosimo, S. Better together: Targeted combination therapies in breast cancer. Semin. Oncol. 2015, 42, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Steinbrueck, A.; Sedgwick, A.C.; Brewster, J.T.; Yan, K.C.; Shang, Y.; Knoll, D.M.; Vargas-Zuniga, G.I.; He, X.P.; Tian, H.; Sessler, J.L. Transition metal chelators, pro-chelators, and ionophores as small molecule cancer chemotherapeutic agents. Chem. Soc. Rev. 2020, 49, 3726–3747. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Chen, Z. The effect of curcumin on breast cancer cells. J. Breast Cancer 2013, 16, 133–137. [Google Scholar] [CrossRef] [Green Version]
- Banik, U.; Othman, N.H.; Parasuraman, S.; Adhikary, A.K. Curcumin: The spicy modulator of breast carcinogenesis. J. Exp. Clin. Cancer Res. 2017, 36, 98. [Google Scholar] [CrossRef] [Green Version]
- Nelson, K.M.; Walters, M.A.; Dahlin, J.L.; Bisson, J.; Graham, J.; Pauli, G.F. The essential medicinal chemistry of curcumin. J. Med. Chem. 2017, 60, 1620–1637. [Google Scholar] [CrossRef]
- Jamwal, R. Bioavailable curcumin formulations: A review of pharmacokinetic studies in healthy volunteers. J. Integr. Med. 2018, 16, 367–374. [Google Scholar] [CrossRef]
- Bhuket, P.R.N.; El-Magboub, A.; Haworth, I.S.; Rojsitthisak, P. Enhancement of curcumin bioavailability via the prodrug approach: Challenges and prospects. Eur. J. Drug Metab. Pharmacokinet. 2017, 42, 341–353. [Google Scholar] [CrossRef]
- Vyas, A.; Dandawate, P.; Padhye, S.; Ahmad, A.; Sarkar, F. Perspectives on new synthetic curcumin analogs and their potential anticancer properties. Curr. Pharm. Des. 2013, 19, 2047–2069. [Google Scholar]
- Shen, L.; Liu, C.-C.; An, C.-Y.; Ji, H.-F. How does curcumin work with poor bioavailability? Clues from experimental and theoretical studies. Sci. Rep. 2016, 6, 20872. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.; Andrews, D.; Blackburn, A.C. Long-Term stabilization of stage 4 colon cancer using sodium dichloroacetate therapy. World J. Clin. Cases 2016, 4, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Florio, R.; De Lellis, L.; Veschi, S.; Verginelli, F.; di Giacomo, V.; Gallorini, M.; Natale, A.; Amoroso, R.; Cataldi, A.; Cama, A. Effects of dichloroacetate as single agent or in combination with GW6471 and metformin in paraganglioma cells. Sci. Rep. 2018, 8, 13610. [Google Scholar] [CrossRef] [PubMed]
- Parczyk, J.; Ruhnau, J.; Pelz, C.; Schilling, M.; Wu, H.; Piaskowski, N.N.; Eickholt, B.; Kühn, H.; Danker, K.; Klein, A. Dichloroacetate and PX-478 exhibit strong synergistic effects in a various number of cancer cell lines. BMC Cancer 2021, 21, 481. [Google Scholar] [CrossRef] [PubMed]
- Tataranni, T.; Piccoli, C. Dichloroacetate (DCA) and Cancer: An Overview towards Clinical Applications. Oxid. Med. Cell. Longev. 2019, 2019, 8201079. [Google Scholar] [CrossRef]
- Mey, S.D.; Dufait, I.; Jiang, H.; Corbet, C.; Wang, H.; Gucht, M.V.D.; Kerkhove, L.; Law, K.L.; Vandenplas, H.; Gevaert, T.; et al. Dichloroacetate radiosensitizes hypoxic breast cancer cells. Int. J. Mol. Sci. 2020, 21, 9367. [Google Scholar] [CrossRef]
- Stacpoole, P.W.; Martyniuk, C.J.; James, M.O.; Calcutt, N.A. Dichloroacetate-induced peripheral neuropathy. Int. Rev. Neurobiol. 2019, 145, 211–238. [Google Scholar]
- Tomeh, M.A.; Hadianamrei, R.; Zhao, X. A Review of Curcumin and Its Derivatives as Anticancer Agents. Int. J. Mol. Sci. 2019, 20, 1033. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, D.K.; Mishra, P.K. Curcumin and Its Analogues: Potential Anticancer Agents. Med. Res. Rev. 2010, 30, 818–860. [Google Scholar] [CrossRef]
- Lin, L.; Shi, Q.; Nyarko, A.K.; Bastow, K.F.; Wu, C.-C.; Su, C.-Y.; Shih, C.C.-Y.; Lee, K.-H. Antitumor Agents. 250. Design and Synthesis of New Curcumin Analogues as Potential Anti-Prostate Cancer Agents. J. Med. Chem. 2006, 49, 3963–3972. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.-Y.; Hou, Y.-C.; Yang, J.S.; Lin, H.-Y.; Chang, T.-Y.; Lee, K.-H.; Kuo, S.-C.; Hsieh, M.-T. Synthesis, Anticancer Activity, and Preliminary Pharmacokinetic Evaluation of 4,4-Disubstituted Curcuminoid 2,2-bis(Hydroxymethyl)Propionate Derivatives. Molecules 2020, 25, 479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozawa-Umeta, H.; Kishimoto, A.; Imaizumi, A.; Hashimoto, T.; Asakura, T.; Kakeya, H.; Kanai, M. Curcumin β-D-glucuronide exhibits anti–tumor effects on oxaliplatin-resistant colon cancer with less toxicity in vivo. Cancer Sci. 2020, 111, 1785–1793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackler, L., Jr.; Ozsvari, B.; Gyuris, M.; Sipos, P.; Fabian, G.; Molnar, E.; Marton, A.; Farago, N.; Mihaly, J.; Nagy, L.I.; et al. The Curcumin Analog C-150, Influencing NF-κB, UPR and Akt/Notch Pathways Has Potent Anticancer Activity In Vitro and In Vivo. PLoS ONE 2016, 11, e0149832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meunier, B. Hybrid molecules with a dual mode of action: Dream or reality? Acc. Chem. Res. 2008, 41, 69–77. [Google Scholar] [CrossRef]
- Tangutur, A.D.; Kumar, D.; Krishna, V.; Kantevari, S. Microtubule targeting agents as cancer chemotherapeutics: An overview of molecular hybrids as stabilizing and destabilizing agents. Curr. Top. Med. Chem. 2017, 17, 2523–2537. [Google Scholar] [CrossRef]
- Gontijo, V.S.; Viegas, F.P.D.; Ortiz, C.J.C.; Silva, M.F.; Damasio, C.M.; Rosa, M.C.; Campos, T.G.; Couto, D.S.; Dias, K.S.T.; Viegas, C. Molecular hybridization as a tool in the design of multi-target directed drug candidates for neurodegenerative diseases. Curr. Neuropharmacol. 2020, 18, 348–407. [Google Scholar] [CrossRef]
- Ibrahim, M.A.; Panda, S.S.; Birs, A.S.; Serrano, J.C.; Gonzalez, C.F.; Alamry, K.A.; Katritzky, A.R. Synthesis and antibacterial evaluation of amino acid-antibiotic conjugates. Bioorg. Med. Chem. Lett. 2014, 24, 1856–1861. [Google Scholar] [CrossRef]
- Panda, S.S.; Hall, C.D.; Scriven, E.; Katritzky, A.R. Aminoacyl Benzotriazolides: Versatile Reagents for the Preparation of Peptides, their Mimetics, and Conjugates. Aldrichim. Acta 2013, 46, 43–55. [Google Scholar]
- Panda, S.S.; Girgis, A.S.; Thomas, S.J.; Capito, J.E.; George, R.F.; Salman, A.; El-Manawaty, M.A.; Samir, A. Synthesis, pharmacological profile, and 2D-QSAR studies of curcumin-amino acid conjugates as potential drug candidates. Eur. J. Med. Chem. 2020, 196, 112293. [Google Scholar] [CrossRef]
- Seliem, I.A.; Panda, S.S.; Girgis, A.S.; Nagy, Y.I.; George, R.F.; Fayad, W.; Fawzy, N.G.; Ibrahim, T.S.; Al-Mahmoudy, A.M.M.; Sakhuja, R.; et al. Design, synthesis, antimicrobial, and DNA gyrase inhibitory properties of fluoroquinolone–dichloroacetic acid hybrids. Chem. Biol. Drug Des. 2020, 95, 248–259. [Google Scholar] [CrossRef]
- Thangaraju, M.; Gopal, E.; Martin, P.M.; Ananth, S.; Smith, S.B.; Prasad, P.D.; Sterneck, E.; Ganapathy, V. SLC5A8 triggers tumor cell apoptosis through pyruvate-dependent inhibition of histone deacetylases. Cancer Res. 2006, 66, 11560–11564. [Google Scholar] [CrossRef] [Green Version]
- Bridges, A.E.; Ramachandran, S.; Pathania, R.; Parwal, U.; Lester, A.; Rajpurohit, P.; Morera, D.S.; Patel, N.; Singh, N.; Korkaya, H.; et al. RAD51AP1 Deficiency reduces tumor growth by targeting stem cell self-renewal. Cancer Res. 2020, 80, 3855–3866. [Google Scholar] [CrossRef] [PubMed]
- Maglione, J.E.; Moghanaki, D.; Young, L.J.; Manner, C.K.; Ellies, L.G.; Joseph, S.O.; Nicholson, B.; Cardiff, R.D.; MacLeod, C.L. Transgenic Polyoma middle-T mice model premalignant mammary disease. Cancer Res. 2001, 61, 8298–8305. [Google Scholar] [PubMed]
- Lin, E.Y.; Jones, J.G.; Li, P.; Zhu, L.; Whitney, K.D.; Muller, W.J.; Pollard, J.W. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am. J. Pathol. 2003, 163, 2123–2126. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Ji, C.; Mayfield, J.E.; Goel, A.; Xiao, J.; Dixon, J.E.; Guo, X. Ancient drug curcumin impedes 26S proteasome activity by direct inhibition of dual-specificity tyrosine-regulated kinase 2. Proc. Natl. Acad. Sci. USA 2018, 115, 8155–8160. [Google Scholar] [CrossRef] [Green Version]
- Tandon, V.; de la Vega, L.; Banerjee, S. Emerging roles of DYRK2 in cancer. J. Biol. Chem. 2021, 296, 100233. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H.; Carter, G.T. Drug-Like property concepts in pharmaceutical design. Curr. Pharm. Des. 2009, 15, 2184–2194. [Google Scholar] [CrossRef]
- Leeson, P.D.; Bento, P.; Gaulton, A.; Hersey, A.; Manners, E.J.; Radoux, C.J.; Leach, A.R. Target-Based evaluation of “Drug-Like” properties and ligand efficiencies. J. Med. Chem. 2021, 64, 7210–7230. [Google Scholar] [CrossRef]
- Zhang, Z.; Tang, W. Drug metabolism in drug discovery and development. Acta Pharm. Sin. B 2008, 8, 721–732. [Google Scholar] [CrossRef]
- Priest, B.; Bell, I.M.; Garcia, M. Channels Role of hERG potassium channel assays in drug development. Channels 2008, 2, 87–93. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of the Compound | EC50 for MCF10A | EC50 for T47D Cells | EC50 for MB231 Cells |
|---|---|---|---|
| CMC1 | 8.982 μM | 1.648 μM | 0.4240 μM |
| CMC2 | 9.675 μM | 1.421 μM | 0.7780 μM |
| CMC3 | 9.714 μM | 1.595 μM | 0.5179 μM |
| CMC4 | 8.859 μM | 1.255 μM | 1.1320 μM |
| CMC5 | 9.604 μM | 1.245 μM | 0.8375 μM |
| CMC6 | 9.474 μM | 1.372 μM | 0.9418 μM |
| Name | CMC Docking with DYRK2 | dG kJ/mol | Score | L.E | |
|---|---|---|---|---|---|
| CUR |  |  | −24 | −29.24 | 0.22 |
| CMC1 |  |  | −27 | −22.86 | 0.18 |
| CMC2 |  |  | −67 | −33.02 | 0.35 |
| CMC3 |  |  | −21 | −16.71 | 0.11 |
| CMC4 |  |  | −49 | −25.09 | 0.25 |
| CMC6 |  |  | −58 | −21.10 | 0.28 |
| Name | Log P | Aq. Sol (log mol/L) | HERG II Inhibitor | Dev Tox | CYP2D6 Substrate | P-gp Substrate | HIA % |
|---|---|---|---|---|---|---|---|
| CUR | 3.852 | −3.878 | + | + | Med | - | 84.38 |
| CMC1 | 5.859 | −4.644 | - | - | Low | - | 81.65 |
| CMC2 | 4.092 | −4.031 | - | - | Low | + | 66.25 |
| CMC3 | 4.869 | −4.010 | - | - | Low | + | 68.18 |
| CMC4 | 4.872 | −3.700 | - | + | Low | + | 61.82 |
| CMC5 | 7.314 | −2.981 | + | + | Med | - | 81.50 |
| CMC6 | 5.65 | −3.336 | - | + | Med | + | 67.89 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panda, S.S.; Tran, Q.L.; Rajpurohit, P.; Pillai, G.G.; Thomas, S.J.; Bridges, A.E.; Capito, J.E.; Thangaraju, M.; Lokeshwar, B.L. Design, Synthesis, and Molecular Docking Studies of Curcumin Hybrid Conjugates as Potential Therapeutics for Breast Cancer. Pharmaceuticals 2022, 15, 451. https://doi.org/10.3390/ph15040451
Panda SS, Tran QL, Rajpurohit P, Pillai GG, Thomas SJ, Bridges AE, Capito JE, Thangaraju M, Lokeshwar BL. Design, Synthesis, and Molecular Docking Studies of Curcumin Hybrid Conjugates as Potential Therapeutics for Breast Cancer. Pharmaceuticals. 2022; 15(4):451. https://doi.org/10.3390/ph15040451
Chicago/Turabian StylePanda, Siva S., Queen L. Tran, Pragya Rajpurohit, Girinath G. Pillai, Sean J. Thomas, Allison E. Bridges, Jason E. Capito, Muthusamy Thangaraju, and Bal L. Lokeshwar. 2022. "Design, Synthesis, and Molecular Docking Studies of Curcumin Hybrid Conjugates as Potential Therapeutics for Breast Cancer" Pharmaceuticals 15, no. 4: 451. https://doi.org/10.3390/ph15040451
APA StylePanda, S. S., Tran, Q. L., Rajpurohit, P., Pillai, G. G., Thomas, S. J., Bridges, A. E., Capito, J. E., Thangaraju, M., & Lokeshwar, B. L. (2022). Design, Synthesis, and Molecular Docking Studies of Curcumin Hybrid Conjugates as Potential Therapeutics for Breast Cancer. Pharmaceuticals, 15(4), 451. https://doi.org/10.3390/ph15040451