
Development and Characterization of Eudragit® EPO-Based Solid Dispersion of Rosuvastatin Calcium to Foresee the Impact on Solubility, Dissolution and Antihyperlipidemic Activity


 , ,
, ,  , , , and
, , , and
Abstract
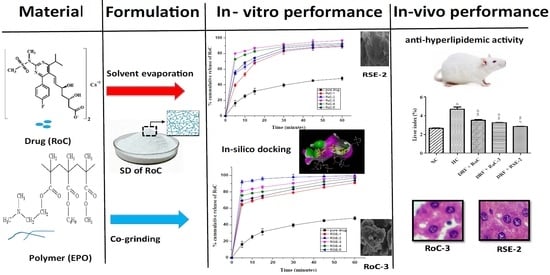
1. Introduction
2. Results
2.1. Saturation Solubility Studies
2.2. Gibb’s Free Energy Calculation
2.3. Percentage Yield and Drug Loading
2.4. FTIR Analysis
2.5. Thermal Analysis
2.6. PXRD Analysis
2.7. SEM Analysis
2.8. In Vitro Dissolution Study
2.9. Docking Studies for In Silico Prediction of Solubility
2.10. Pharmacodynamic Studies
2.10.1. Influence on the Gain of Body Weight and Liver Index
2.10.2. Biochemical Analysis of Serum Lipid Levels
2.10.3. Biochemical Analysis of Liver Functions (LFTs)
2.10.4. Macroscopic and Microscopic Examination of Liver
2.11. Cell Viability Assay
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. In Silico Docking Studies
4.3. Methods for Preparation of SDs
4.4. Saturation Solubility Studies
4.5. Gibbs-Free-Energy (ΔG°tr) Analysis
4.6. Determination of Percentage Yield and Drug Content
4.7. FTIR for Structural Analysis
4.8. DSC Analysis
4.9. PXRD Analysis
4.10. Scanning Electron Microscopy (SEM)
4.11. In Vitro Drug Release
4.12. Pharmacodynamics (PD) Study in Rats
4.12.1. Experimental Animals
4.12.2. Experimental Protocol
4.12.3. Sample Collection
4.12.4. Serum Biochemical Analysis
4.12.5. Histopathological Examination
4.12.6. Statistical Analysis
4.13. In Vitro Cell Viability Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Polymeric Amorphous Solid Dispersions: A Review of Amorphization, Crystallization, Stabilization, Solid-State Characterization, and Aqueous Solubilization of Biopharmaceutical Classification System Class II Drugs. J. Pharm. Sci. 2016, 105, 2527–2544. [Google Scholar] [CrossRef] [PubMed]
- Giri, T.K.; Kumar, K.; Alexander, A.; Ajazuddin; Badwaik, H.; Tripathi, D.K. A novel and alternative approach to controlled release drug delivery system based on solid dispersion technique. Bull. Fac. Pharmacy, Cairo Univ. 2012, 50, 147–159. [Google Scholar] [CrossRef]
- Singh, A.; Worku, Z.A.; Van Den Mooter, G. Oral formulation strategies to improve solubility of poorly water-soluble drugs. Expert Opin. Drug Deliv. 2011, 8, 1361–1378. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.G.; Li, J.J.; Williams, G.R.; Zhao, M. Electrospun amorphous solid dispersions of poorly water-soluble drugs: A review. J. Control. Release 2018, 292, 91–110. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Kaushik, A.; Almeer, R.; Habibur Rahman, M.; Abdel-Daim, M.M.; Kaushik, D. Improved pharmacodynamic potential of rosuvastatin by self-nanoemulsifying drug delivery system: An in vitro and in vivo evaluation. Int. J. Nanomed. 2021, 16, 905–924. [Google Scholar] [CrossRef]
- Van Ngo, H.; Nguyen, P.K.; Van Vo, T.; Duan, W.; Tran, V.T.; Tran, P.H.L.; Tran, T.T.D. Hydrophilic-hydrophobic polymer blend for modulation of crystalline changes and molecular interactions in solid dispersion. Int. J. Pharm. 2016, 513, 148–152. [Google Scholar] [CrossRef]
- Al-Heibshy, F.N.S.; Başaran, E.; Arslan, R.; Öztürk, N.; Vural, İ.; Demirel, M. Preparation, characterization and pharmacokinetic evaluation of rosuvastatin calcium incorporated cyclodextrin-polyanhydride nanoparticles. Drug Dev. Ind. Pharm. 2019, 45, 1635–1645. [Google Scholar] [CrossRef]
- Miyako, Y.; Khalef, N.; Matsuzaki, K.; Pinal, R. Solubility enhancement of hydrophobic compounds by cosolvents: Role of solute hydrophobicity on the solubilization effect. Int. J. Pharm. 2010, 393, 48–54. [Google Scholar] [CrossRef]
- Pradhan, R.; Kim, S.Y.; Yong, C.S.; Kim, J.O. Preparation and characterization of spray-dried valsartan-loaded Eudragit® E PO solid dispersion microparticles. Asian J. Pharm. Sci. 2016, 11, 744–750. [Google Scholar] [CrossRef]
- Sarfraz, R.M.; Ahmad, M.; Mahmood, A.; Minhas, M.U.; Yaqoob, A. Development and Evaluation of Rosuvastatin Calcium Based Microparticles for Solubility Enhancement: An In Vitro Study. Adv. Polym. Technol. 2017, 36, 433–441. [Google Scholar] [CrossRef]
- Tekade, A.R.; Yadav, J.N. A review on solid dispersion and carriers used therein for solubility enhancement of poorly water soluble drugs. Adv. Pharm. Bull. 2020, 10, 359–369. [Google Scholar] [CrossRef]
- Dudhipala, N.; Veerabrahma, K. Improved anti-hyperlipidemic activity of Rosuvastatin Calcium via lipid nanoparticles: Pharmacokinetic and pharmacodynamic evaluation. Eur. J. Pharm. Biopharm. 2017, 110, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zou, M.; Piao, H.; Liu, Y.; Tang, B.; Gao, Y.; Ma, N.; Cheng, G. Characterization and pharmacokinetic study of aprepitant solid dispersions with soluplus®. Molecules 2015, 20, 11345–11356. [Google Scholar] [CrossRef] [PubMed]
- Alshehri, S.; Imam, S.S.; Hussain, A.; Altamimi, M.A.; Alruwaili, N.K.; Alotaibi, F.; Alanazi, A.; Shakeel, F. Potential of solid dispersions to enhance solubility, bioavailability, and therapeutic efficacy of poorly water-soluble drugs: Newer formulation techniques, current marketed scenario and patents. Drug Deliv. 2020, 27, 1625–1643. [Google Scholar] [CrossRef]
- Tambe, A.; Pandita, N. Enhanced solubility and drug release profile of boswellic acid using a poloxamer-based solid dispersion technique. J. Drug Deliv. Sci. Technol. 2018, 44, 172–180. [Google Scholar] [CrossRef]
- Das, T.; Mehta, C.H.; Nayak, U.Y. Multiple approaches for achieving drug solubility: An in silico perspective. Drug Discov. Today 2020, 25, 1206–1212. [Google Scholar] [CrossRef]
- Vo, C.L.N.; Park, C.; Lee, B.J. Current trends and future perspectives of solid dispersions containing poorly water-soluble drugs. Eur. J. Pharm. Biopharm. 2013, 85, 799–813. [Google Scholar] [CrossRef]
- Zhai, X.; Li, C.; Lenon, G.B.; Xue, C.C.L.; Li, W. Preparation and characterisation of solid dispersions of tanshinone IIA, cryptotanshinone and total tanshinones. Asian J. Pharm. Sci. 2017, 12, 85–97. [Google Scholar] [CrossRef]
- Van Drooge, D.J.; Hinrichs, W.L.J.; Visser, M.R.; Frijlink, H.W. Characterization of the molecular distribution of drugs in glassy solid dispersions at the nano-meter scale, using differential scanning calorimetry and gravimetric water vapour sorption techniques. Int. J. Pharm. 2006, 310, 220–229. [Google Scholar] [CrossRef]
- Mai, N.N.S.; Nakai, R.; Kawano, Y.; Hanawa, T. Enhancing the Solubility of Curcumin Using a Solid Dispersion System with Hydroxypropyl-β-Cyclodextrin Prepared by Grinding, Freeze-Drying, and Common Solvent Evaporation Methods. Pharmacy 2020, 8, 203. [Google Scholar] [CrossRef]
- Bennett, R.C.; Brough, C.; Miller, D.A.; O’Donnell, K.P.; Keen, J.M.; Hughey, J.R.; Williams, R.O.; McGinity, J.W. Preparation of amorphous solid dispersions by rotary evaporation and KinetiSol Dispersing: Approaches to enhance solubility of a poorly water-soluble gum extract. Drug Dev. Ind. Pharm. 2015, 41, 382–397. [Google Scholar] [CrossRef] [PubMed]
- Ziaee, A.; O’Dea, S.; Howard-Hildige, A.; Padrela, L.; Potter, C.; Iqbal, J.; Albadarin, A.B.; Walker, G.; O’Reilly, E.J. Amorphous solid dispersion of ibuprofen: A comparative study on the effect of solution based techniques. Int. J. Pharm. 2019, 572, 118816. [Google Scholar] [CrossRef] [PubMed]
- Islam, N.; Irfan, M.; Abbas, N.; Syed, H.K.; Iqbal, M.S.; Khan, I.u.; Rasul, A.; Inam, S.; Hussain, A.; ul Amin Mohsin, N.; et al. Enhancement of solubility and dissolution rate of ebastine fast-disintegrating tablets by solid dispersion method. Trop. J. Pharm. Res. 2020, 19, 1797–1805. [Google Scholar] [CrossRef]
- Smeets, A.; Koekoekx, R.; Clasen, C.; Van den Mooter, G. Amorphous solid dispersions of darunavir: Comparison between spray drying and electrospraying. Eur. J. Pharm. Biopharm. 2018, 130, 96–107. [Google Scholar] [CrossRef]
- Gala, U.; Chauhan, H. Principles and applications of Raman spectroscopy in pharmaceutical drug discovery and development. Expert Opin. Drug Discov. 2015, 10, 187–206. [Google Scholar] [CrossRef]
- Kalani, M.M.; Nourmohammadi, J.; Negahdari, B.; Rahimi, A.; Sell, S.A. Electrospun core-sheath poly(vinyl alcohol)/silk fibroin nanofibers with Rosuvastatin release functionality for enhancing osteogenesis of human adipose-derived stem cells. Mater. Sci. Eng. C 2019, 99, 129–139. [Google Scholar] [CrossRef]
- Hirpara, M.R.; Manikkath, J.; Sivakumar, K.; Managuli, R.S.; Gourishetti, K.; Krishnadas, N.; Shenoy, R.R.; Jayaprakash, B.; Rao, C.M.; Mutalik, S. Long circulating PEGylated-chitosan nanoparticles of rosuvastatin calcium: Development and in vitro and in vivo evaluations. Int. J. Biol. Macromol. 2018, 107, 2190–2200. [Google Scholar] [CrossRef]
- Beg, S.; Raza, K.; Kumar, R.; Chadha, R.; Katare, O.P.; Singh, B. Improved intestinal lymphatic drug targeting via phospholipid complex-loaded nanolipospheres of rosuvastatin calcium. RSC Adv. 2016, 6, 8173–8187. [Google Scholar] [CrossRef]
- Alshora, D.H.; Ibrahim, M.A.; Elzayat, E.; Almeanazel, O.T.; Alanazi, F. Rosuvastatin calcium nanoparticles: Improving bioavailability by formulation and stabilization codesign. PLoS ONE 2018, 13, e0200218. [Google Scholar] [CrossRef]
- Weerapol, Y.; Limmatvapirat, S.; Nunthanid, J.; Konthong, S.; Suttiruengwong, S.; Sriamornsak, P. Development and characterization of nifedipine-amino methacrylate copolymer solid dispersion powders with various adsorbents. Asian J. Pharm. Sci. 2017, 12, 335–343. [Google Scholar] [CrossRef]
- Guo, S.; Wang, G.; Wu, T.; Bai, F.; Xu, J.; Zhang, X. Solid dispersion of berberine hydrochloride and Eudragit® S100: Formulation, physicochemical characterization and cytotoxicity evaluation. J. Drug Deliv. Sci. Technol. 2017, 40, 21–27. [Google Scholar] [CrossRef]
- Xie, T.; Gao, W.; Taylor, L.S. Impact of Eudragit EPO and hydroxypropyl methylcellulose on drug release rate, supersaturation, precipitation outcome and redissolution rate of indomethacin amorphous solid dispersions. Int. J. Pharm. 2017, 531, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Fule, R.; Meer, T.; Sav, A.; Amin, P. Solubility and dissolution rate enhancement of lumefantrine using hot melt extrusion technology with physicochemical characterisation. J. Pharm. Investig. 2013, 43, 305–321. [Google Scholar] [CrossRef]
- Butt, S.; Hasan, S.M.F.; Hassan, M.M.; Alkharfy, K.M.; Neau, S.H. Directly compressed rosuvastatin calcium tablets that offer hydrotropic and micellar solubilization for improved dissolution rate and extent of drug release. Saudi Pharm. J. 2019, 27, 619–628. [Google Scholar] [CrossRef]
- Kerdsakundee, N.; Mahattanadul, S.; Wiwattanapatapee, R. Development and evaluation of gastroretentive raft forming systems incorporating curcumin-Eudragit® EPO solid dispersions for gastric ulcer treatment. Eur. J. Pharm. Biopharm. 2015, 94, 513–520. [Google Scholar] [CrossRef]
- Li, J.; Lee, I.W.; Shin, G.H.; Chen, X.; Park, H.J. Curcumin-Eudragit® e PO solid dispersion: A simple and potent method to solve the problems of curcumin. Eur. J. Pharm. Biopharm. 2015, 94, 322–332. [Google Scholar] [CrossRef]
- Gangurde, A.B.; Kundaikar, H.S.; Javeer, S.D.; Jaiswar, D.R.; Degani, M.S.; Amin, P.D. Enhanced solubility and dissolution of curcumin by a hydrophilic polymer solid dispersion and its insilico molecular modeling studies. J. Drug Deliv. Sci. Technol. 2015, 29, 226–237. [Google Scholar] [CrossRef]
- Chen, Y.; Huang, W.; Chen, J.; Wang, H.; Zhang, S.; Xiong, S. The Synergetic Effects of Nonpolar and Polar Protic Solvents on the Properties of Felodipine and Soluplus in Solutions, Casting Films, and Spray-Dried Solid Dispersions. J. Pharm. Sci. 2018, 107, 1615–1623. [Google Scholar] [CrossRef]
- Zhang, Q.; Pu, Y.; Wang, B.; Wang, Y.; Dong, T.T.; Guo, T.; Zhang, T.; Cai, Z.; Chaparala, A. Characterization, molecular docking, and in vitro dissolution studies of solid dispersions of 20(S)-protopanaxadiol. Molecules 2017, 22, 274. [Google Scholar] [CrossRef]
- Al-Shdefat, R.; Anwer, M.K.; Fayed, M.H.; Alsulays, B.B.; Tawfeek, H.M.; Abdel-Rahman, R.F.; Soliman, G.A. Preparation and evaluation of spray dried rosuvastatin calcium-PVP microparticles for the improvement of serum lipid profile. J. Drug Deliv. Sci. Technol. 2020, 55, 101342. [Google Scholar] [CrossRef]
- Pawar, J.N.; Shete, R.T.; Gangurde, A.B.; Moravkar, K.K.; Javeer, S.D.; Jaiswar, D.R.; Amin, P.D. Development of amorphous dispersions of artemether with hydrophilic polymers via spray drying: Physicochemical and in silico studies. Asian J. Pharm. Sci. 2016, 11, 385–395. [Google Scholar] [CrossRef]
- Beg, S.; Alam, M.N.; Ahmad, F.J.; Singh, B. Chylomicron mimicking nanocolloidal carriers of rosuvastatin calcium for lymphatic drug targeting and management of hyperlipidemia. Colloids Surfaces B Biointerfaces 2019, 177, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Mehmood, H.Q.; Faran, S.A.; Chaudhry, M.A.; Khalid, S.H.; Khan, I.U.; Hassan, W.; Ashfaq, R.; Asghar, S. An assessment of bioavailability of acrylate based pH-sensitive complexes of lovastatin. Pak. J. Pharm. Sci. 2019, 32, 1129–1136. [Google Scholar] [PubMed]
- El-Nahas, A.E.; Allam, A.N.; Abdelmonsif, D.A.; El-Kamel, A.H. Silymarin-Loaded Eudragit Nanoparticles: Formulation, Characterization, and Hepatoprotective and Toxicity Evaluation. AAPS PharmSciTech 2017, 18, 3076–3086. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhou, J.Y.; Chang, J.H.; Liu, X.G.; Xue, H.F.; Wang, R.X.; Li, Z.S.; Li, C.S.; Wang, J.; Liu, C.Z. Soluplus-mediated diosgenin amorphous solid dispersion with high solubility and high stability: Development, characterization and oral bioavailability. Drug Des. Devel. Ther. 2020, 14, 2959–2975. [Google Scholar] [CrossRef]
- Wang, R.; Zhou, H.; Siu, S.W.I.; Gan, Y.; Wang, Y.; Ouyang, D. Comparison of three molecular simulation approaches for cyclodextrin-ibuprofen complexation. J. Nanomater. 2015, 16, 267. [Google Scholar] [CrossRef]
- Guan, J.; Liu, Q.; Zhang, X.; Zhang, Y.; Chokshi, R.; Wu, H.; Mao, S. Alginate as a potential diphase solid dispersion carrier with enhanced drug dissolution and improved storage stability. Eur. J. Pharm. Sci. 2018, 114, 346–355. [Google Scholar] [CrossRef]
- Veseli, A.; Žakelj, S.; Kristl, A. A review of methods for solubility determination in biopharmaceutical drug characterization. Drug Dev. Ind. Pharm. 2019, 45, 1717–1724. [Google Scholar] [CrossRef]
- Linn, M.; Collnot, E.M.; Djuric, D.; Hempel, K.; Fabian, E.; Kolter, K.; Lehr, C.M. Soluplus® as an effective absorption enhancer of poorly soluble drugs in vitro and in vivo. Eur. J. Pharm. Sci. 2012, 45, 336–343. [Google Scholar] [CrossRef]
- Fule, R.; Meer, T.; Amin, P.; Dhamecha, D.; Ghadlinge, S. Preparation and characterisation of lornoxicam solid dispersion systems using hot melt extrusion technique. J. Pharm. Investig. 2014, 44, 41–59. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Luo, Y.; Yao, Q.; Zhong, Y.; Tian, B.; Tang, X. Extruded Soluplus/SIM as an oral delivery system: Characterization, interactions, in vitro and in vivo evaluations. Drug Deliv. 2016, 23, 1902–1911. [Google Scholar] [CrossRef] [PubMed]
- Kamel, A.O.; Mahmoud, A.A. Enhancement of human oral bioavailability and in vitro antitumor activity of rosuvastatin via spray dried self-nanoemulsifying drug delivery system. J. Biomed. Nanotechnol. 2013, 9, 26–39. [Google Scholar] [CrossRef]
- Sarfraz, R.M.; Ahmad, M.; Mahmood, A.; Minhas, M.U.; Yaqoob, A. Fabrication and evaluation of rosuvastatin calcium fast-disintegrating tablets using β-cyclodextrin and superdisintegrants. Trop. J. Pharm. Res. 2015, 14, 1961–1968. [Google Scholar] [CrossRef]
- Shamma, R.N.; Basha, M. Soluplus®: A novel polymeric solubilizer for optimization of Carvedilol solid dispersions: Formulation design and effect of method of preparation. Powder Technol. 2013, 237, 406–414. [Google Scholar] [CrossRef]
- Patil-Gadhe, A.; Pokharkar, V. Pulmonary targeting potential of rosuvastatin loaded nanostructured lipid carrier: Optimization by factorial design. Int. J. Pharm. 2016, 501, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.J.; Ahn, H.I.; Park, J.Y.; Ho, M.J.; Lee, D.R.; Cho, H.R.; Park, J.S.; Choi, Y.S.; Kang, M.J. Improved oral absorption of tacrolimus by a solid dispersion with hypromellose and sodium lauryl sulfate. Int. J. Biol. Macromol. 2016, 83, 282–287. [Google Scholar] [CrossRef]
- Zhang, X.; Rao, Q.; Qiu, Z.; Lin, Y.; Zhang, L.; Hu, Q.; Chen, T.; Ma, Z.; Gao, H.; Luo, D.; et al. Using Acetone/Water Binary Solvent to Enhance the Stability and Bioavailability of Spray Dried Enzalutamide/HPMC-AS Solid Dispersions. J. Pharm. Sci. 2021, 110, 1160–1171. [Google Scholar] [CrossRef]
- Faran, S.A.; Asghar, S.; Khalid, S.H.; Khan, I.U.; Asif, M.; Khalid, I.; Gohar, U.F.; Hussain, T. Hepatoprotective and renoprotective properties of lovastatin-loaded ginger and garlic oil nanoemulsomes: Insights into serum biological parameters. Medicina 2019, 55, 579. [Google Scholar] [CrossRef]
- Jahangiri, A.; Barzegar-Jalali, M.; Garjani, A.; Javadzadeh, Y.; Hamishehkar, H.; Asadpour-Zeynali, K.; Adibkia, K. Evaluation of physicochemical properties and in vivo efficiency of atorvastatin calcium/ezetimibe solid dispersions. Eur. J. Pharm. Sci. 2016, 82, 21–30. [Google Scholar] [CrossRef]
- Behiry, E.G.; El Nady, N.M.; AbdEl Haie, O.M.; Mattar, M.K.; Magdy, A. Evaluation of TG-HDL Ratio Instead of HOMA Ratio as Insulin Resistance Marker in Overweight and Children with Obesity. Endocrine Metab. Immune Disord.-Drug Targets 2019, 19, 676–682. [Google Scholar] [CrossRef]
- Shukr, M.H.; Ismail, S.; Ahmed, S.M. Development and optimization of ezetimibe nanoparticles with improved antihyperlipidemic activity. J. Drug Deliv. Sci. Technol. 2019, 49, 383–395. [Google Scholar] [CrossRef]
- Rasul, A.; Riaz, A.; Wei, W.; Sarfraz, I.; Hassan, M.; Li, J.; Asif, F.; Adem, Ş.; Bukhari, S.A.; Asrar, M.; et al. Mangifera indica Extracts as Novel PKM2 Inhibitors for Treatment of Triple Negative Breast Cancer. Biomed. Res. Int. 2021, 2021. [Google Scholar] [CrossRef] [PubMed]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation Code | Gibbs Energy | Yield (%) | Drug Content (%) |
|---|---|---|---|
| RoC-1 | −12,327.79 | 90.96 ± 0.42 | 90.01 ± 0.29 |
| RoC-2 | −12,337.81 | 95.91 ± 0.28 | 90.69 ± 0.89 |
| RoC-3 | −12,385.67 | 96.87 ± 0.18 | 91.38 ± 0.83 |
| RoC-4 | −11,949.02 | 95.75 ± 0.21 | 92.88 ± 1.02 |
| RoC-5 | −11,309.71 | 95.21 ± 0.56 | 95.48 ± 1.32 |
| RSE-1 | −13,034.54 | 75.73 ± 0.14 | 96.17 ± 2.13 |
| RSE-2 | −12,627.16 | 79.75 ± 0.35 | 98.50 ± 0.88 |
| RSE-3 | −12,114.25 | 72.27 ± 0.09 | 95.89 ± 1.09 |
| RSE-4 | −11,846.16 | 68.75 ± 0.07 | 96.03 ± 1.04 |
| RSE-5 | −11,528.73 | 63.06 ± 0.84 | 95.62 ± 1.22 |
| Formulation Code | RoC-1 | RoC-2 | RoC-3 | RoC-4 | RoC-5 | RSE-1 | RSE-2 | RSE-3 | RSE-4 | RSE-5 |
|---|---|---|---|---|---|---|---|---|---|---|
| Ratio of drug | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Ratio of EPO | 0.5 | 1 | 2 | 3 | 4 | 0.5 | 1 | 2 | 3 | 4 |
| Groups | Hyperlipidemia Induction Period (0–6 Weeks) | Treatment Period (7–10 Weeks) |
|---|---|---|
| Group NC | SCF | SCF |
| Group HC | DRF | DFR |
| Group I | DRF | DFR + pure RoC suspended in 1.33% of CMC solution |
| Group II | DRF | DRF + RoC-3 (equivalent 20 mg) suspended in 1.33% of CMC solution |
| Group III | DRF | DRF + RSE-2 (equivalent 20 mg) suspended in 1.33% of CMC solution |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inam, S.; Irfan, M.; Lali, N.u.a.; Khalid Syed, H.; Asghar, S.; Khan, I.U.; Khan, S.-U.-D.; Iqbal, M.S.; Zaheer, I.; Khames, A.; et al. Development and Characterization of Eudragit® EPO-Based Solid Dispersion of Rosuvastatin Calcium to Foresee the Impact on Solubility, Dissolution and Antihyperlipidemic Activity. Pharmaceuticals 2022, 15, 492. https://doi.org/10.3390/ph15040492
Inam S, Irfan M, Lali Nua, Khalid Syed H, Asghar S, Khan IU, Khan S-U-D, Iqbal MS, Zaheer I, Khames A, et al. Development and Characterization of Eudragit® EPO-Based Solid Dispersion of Rosuvastatin Calcium to Foresee the Impact on Solubility, Dissolution and Antihyperlipidemic Activity. Pharmaceuticals. 2022; 15(4):492. https://doi.org/10.3390/ph15040492
Chicago/Turabian StyleInam, Sana, Muhammad Irfan, Noor ul ain Lali, Haroon Khalid Syed, Sajid Asghar, Ikram Ullah Khan, Salah-Ud-Din Khan, Muhammad Shahid Iqbal, Imran Zaheer, Ahmed Khames, and et al. 2022. "Development and Characterization of Eudragit® EPO-Based Solid Dispersion of Rosuvastatin Calcium to Foresee the Impact on Solubility, Dissolution and Antihyperlipidemic Activity" Pharmaceuticals 15, no. 4: 492. https://doi.org/10.3390/ph15040492
APA StyleInam, S., Irfan, M., Lali, N. u. a., Khalid Syed, H., Asghar, S., Khan, I. U., Khan, S.-U.-D., Iqbal, M. S., Zaheer, I., Khames, A., Abou-Taleb, H. A., & Abourehab, M. A. S. (2022). Development and Characterization of Eudragit® EPO-Based Solid Dispersion of Rosuvastatin Calcium to Foresee the Impact on Solubility, Dissolution and Antihyperlipidemic Activity. Pharmaceuticals, 15(4), 492. https://doi.org/10.3390/ph15040492