
Targeting the Hematopoietic Stem Cell Niche in β-Thalassemia and Sickle Cell Disease


Abstract
:1. Introduction
2. Hemoglobinopathies
2.1. Thalassemia
2.2. Sickle Cell Disease
2.3. Therapeutic Options for Hemoglobinopathies
3. HSC and the BM Niche
3.1. HSC Regulation by the BM Niche
3.2. HSC Regulation by Stress Signals
3.3. The HSC Niche in Aging and Disease
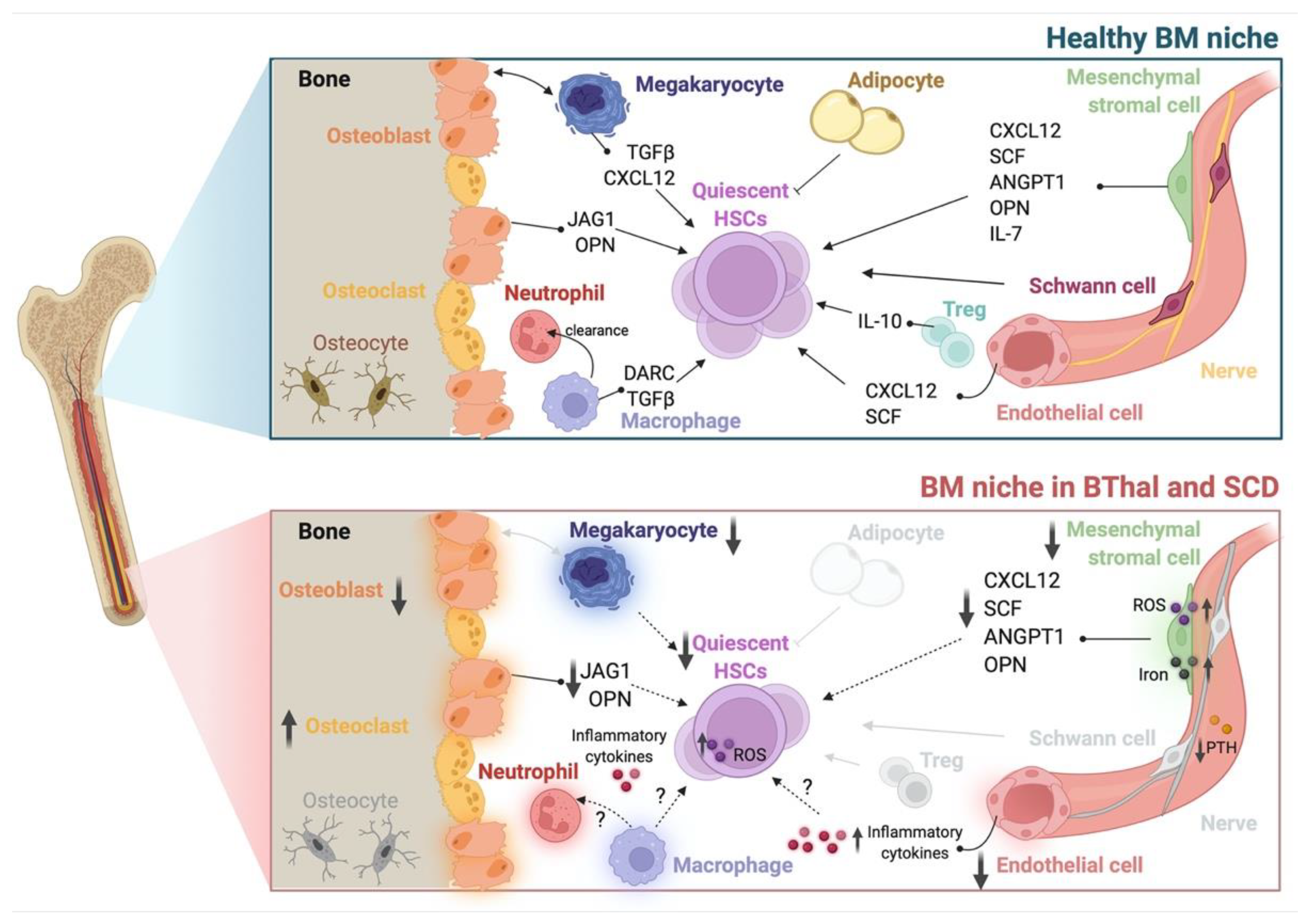
4. The HSC Niche in BThal and SCD
4.1. HSCs
4.2. The Stromal Niche
4.3. Hematopoietic and Soluble Niche Factors
4.4. The Role of IO in the BThal BM Niche
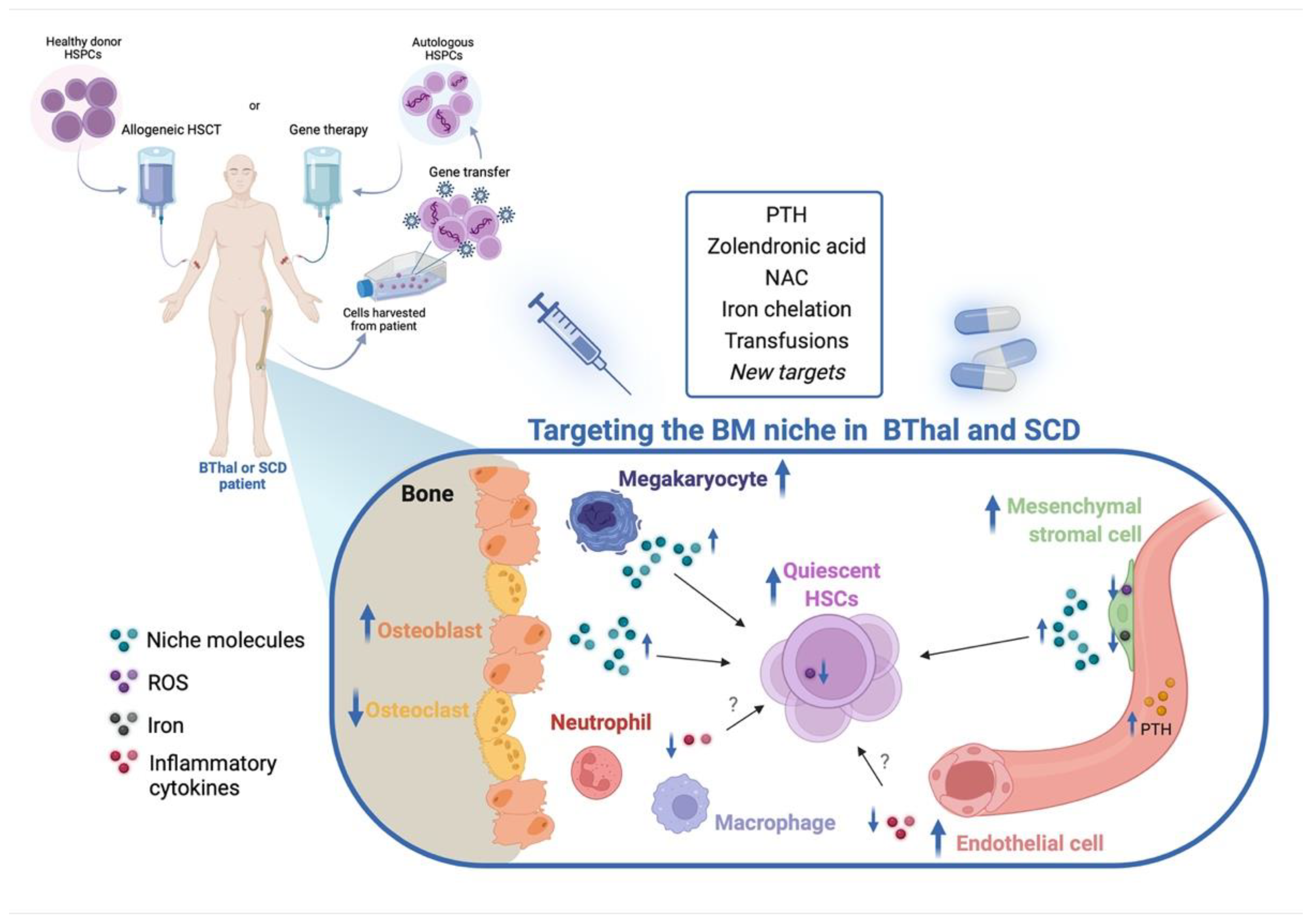
5. Targeting the HSC Niche in BThal and SCD
5.1. Targeting the BM Stromal Niche
5.2. Targeting IO and Other Stress Signals
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Modell, B.; Darlison, M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ 2008, 86, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Musallam, K.M.; Cappellini, M.D. β-Thalassemias. N. Engl. J. Med. 2021, 384, 727–743. [Google Scholar] [CrossRef] [PubMed]
- Ware, R.E.; de Montalembert, M.; Tshilolo, L.; Abboud, M.R. Sickle cell disease. Lancet 2017, 390, 311–323. [Google Scholar] [CrossRef]
- Baronciani, D.; Angelucci, E.; Potschger, U.; Gaziev, J.; Yesilipek, A.; Zecca, M.; Orofino, M.G.; Giardini, C.; Al-Ahmari, A.; Marktel, S.; et al. Hemopoietic stem cell transplantation in thalassemia: A report from the European Society for Blood and Bone Marrow Transplantation Hemoglobinopathy Registry, 2000–2010. Bone Marrow Transplant 2016, 51, 536–541. [Google Scholar] [CrossRef] [Green Version]
- Angelucci, E.; Matthes-Martin, S.; Baronciani, D.; Bernaudin, F.; Bonanomi, S.; Cappellini, M.D.; Dalle, J.H.; Di Bartolomeo, P.; de Heredia, C.D.; Dickerhoff, R.; et al. Hematopoietic stem cell transplantation in thalassemia major and sickle cell disease: Indications and management recommendations from an international expert panel. Haematologica 2014, 99, 811–820. [Google Scholar] [CrossRef] [Green Version]
- Caocci, G.; Orofino, M.G.; Vacca, A.; Piroddi, A.; Piras, E.; Addari, M.C.; Caria, R.; Pilia, M.P.; Origa, R.; Moi, P.; et al. Long-term survival of beta thalassemia major patients treated with hematopoietic stem cell transplantation compared with survival with conventional treatment. Am. J. Hematol. 2017, 92, 1303–1310. [Google Scholar] [CrossRef] [Green Version]
- Gluckman, E.; Cappelli, B.; Bernaudin, F.; Labopin, M.; Volt, F.; Carreras, J.; Pinto Simoes, B.; Ferster, A.; Dupont, S.; de la Fuente, J.; et al. Sickle cell disease: An international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood 2017, 129, 1548–1556. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, G.; Thrasher, A.J.; Aiuti, A. Gene therapy using haematopoietic stem and progenitor cells. Nat. Rev. Genet. 2021, 22, 216–234. [Google Scholar] [CrossRef]
- Thompson, A.A.; Walters, M.C.; Kwiatkowski, J.; Rasko, J.E.J.; Ribeil, J.A.; Hongeng, S.; Magrin, E.; Schiller, G.J.; Payen, E.; Semeraro, M.; et al. Gene Therapy in Patients with Transfusion-Dependent beta-Thalassemia. N. Engl. J. Med. 2018, 378, 1479–1493. [Google Scholar] [CrossRef]
- Marktel, S.; Scaramuzza, S.; Cicalese, M.P.; Giglio, F.; Galimberti, S.; Lidonnici, M.R.; Calbi, V.; Assanelli, A.; Bernardo, M.E.; Rossi, C.; et al. Intrabone hematopoietic stem cell gene therapy for adult and pediatric patients affected by transfusion-dependent β-thalassemia. Nat. Med. 2019, 25, 234–241. [Google Scholar] [CrossRef]
- Locatelli, F.; Thompson, A.A.; Kwiatkowski, J.L.; Porter, J.B.; Thrasher, A.J.; Hongeng, S.; Sauer, M.G.; Thuret, I.; Lal, A.; Algeri, M.; et al. Betibeglogene Autotemcel Gene Therapy for Non-β(0)/β(0) Genotype β-Thalassemia. N. Engl. J. Med. 2022, 386, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Ribeil, J.A.; Hacein-Bey-Abina, S.; Payen, E.; Magnani, A.; Semeraro, M.; Magrin, E.; Caccavelli, L.; Neven, B.; Bourget, P.; El Nemer, W.; et al. Gene Therapy in a Patient with Sickle Cell Disease. N. Engl. J. Med. 2017, 376, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Frangoul, H.; Altshuler, D.; Cappellini, M.D.; Chen, Y.S.; Domm, J.; Eustace, B.K.; Foell, J.; de la Fuente, J.; Grupp, S.; Handgretinger, R.; et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N. Engl. J. Med. 2021, 384, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S.J.; Scadden, D.T. The bone marrow niche for haematopoietic stem cells. Nature 2014, 505, 327–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinho, S.; Frenette, P.S. Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol. 2019, 20, 303–320. [Google Scholar] [CrossRef]
- Mendelson, A.; Frenette, P.S. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat. Med. 2014, 20, 833–846. [Google Scholar] [CrossRef] [Green Version]
- Batsivari, A.; Haltalli, M.L.R.; Passaro, D.; Pospori, C.; Lo Celso, C.; Bonnet, D. Dynamic responses of the haematopoietic stem cell niche to diverse stresses. Nat. Cell Biol. 2020, 22, 7–17. [Google Scholar] [CrossRef]
- Vinchi, F.; Vance, S.Z. Challenging the Erythropoiesis Paradigm in β-Thalassemia. Hemasphere 2020, 4, e475. [Google Scholar] [CrossRef]
- Carlesso, N. Targeting the bone marrow niche in hemoglobinopathies. Blood 2020, 136, 529–531. [Google Scholar] [CrossRef]
- Weatherall, D.J.; Williams, T.N.; Allen, S.J.; O’Donnell, A. The population genetics and dynamics of the thalassemias. Hematol. Oncol. Clin. N. Am. 2010, 24, 1021–1031. [Google Scholar] [CrossRef]
- Taher, A.T.; Cappellini, M.D. How I manage medical complications of beta-thalassemia in adults. Blood 2018, 132, 1781–1791. [Google Scholar] [CrossRef] [PubMed]
- Galanello, R.; Origa, R. Beta-thalassemia. Orphanet J. Rare Dis. 2010, 5, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piel, F.B.; Weatherall, D.J. The α-thalassemias. N. Engl. J. Med. 2014, 371, 1908–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rund, D.; Rachmilewitz, E. β-thalassemia. N. Engl. J. Med. 2005, 353, 1135–1146. [Google Scholar] [CrossRef]
- Higgs, D.R.; Engel, J.D.; Stamatoyannopoulos, G. Thalassaemia. Lancet 2012, 379, 373–383. [Google Scholar] [CrossRef]
- Voskaridou, E.; Terpos, E. New insights into the pathophysiology and management of osteoporosis in patients with beta thalassaemia. Br. J. Haematol. 2004, 127, 127–139. [Google Scholar] [CrossRef]
- Rachmilewitz, E.A.; Giardina, P.J. How I treat thalassemia. Blood 2011, 118, 3479–3488. [Google Scholar] [CrossRef] [Green Version]
- Wong, P.; Fuller, P.J.; Gillespie, M.T.; Milat, F. Bone Disease in Thalassemia: A Molecular and Clinical Overview. Endocr. Rev. 2016, 37, 320–346. [Google Scholar] [CrossRef]
- Morabito, N.; Gaudio, A.; Lasco, A.; Atteritano, M.; Pizzoleo, M.A.; Cincotta, M.; La Rosa, M.; Guarino, R.; Meo, A.; Frisina, N. Osteoprotegerin and RANKL in the pathogenesis of thalassemia-induced osteoporosis: New pieces of the puzzle. J. Bone Miner. Res. 2004, 19, 722–727. [Google Scholar] [CrossRef]
- Bunn, H.F. Pathogenesis and treatment of sickle cell disease. N. Engl. J. Med. 1997, 337, 762–769. [Google Scholar] [CrossRef]
- Kato, G.J.; Piel, F.B.; Reid, C.D.; Gaston, M.H.; Ohene-Frempong, K.; Krishnamurti, L.; Smith, W.R.; Panepinto, J.A.; Weatherall, D.J.; Costa, F.F.; et al. Sickle cell disease. Nat. Rev. Dis. Primers 2018, 4, 18010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rees, D.C.; Williams, T.N.; Gladwin, M.T. Sickle-cell disease. Lancet 2010, 376, 2018–2031. [Google Scholar] [CrossRef]
- Manwani, D.; Frenette, P.S. Vaso-occlusion in sickle cell disease: Pathophysiology and novel targeted therapies. Blood 2013, 122, 3892–3898. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.; Roberts, I. Bone involvement in sickle cell disease. Br. J. Haematol. 2005, 129, 482–490. [Google Scholar] [CrossRef] [PubMed]
- McGann, P.T.; Ware, R.E. Hydroxyurea for sickle cell anemia: What have we learned and what questions still remain? Curr. Opin. Hematol. 2011, 18, 158–165. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Mathews, V.; Kim, S.; George, B.; Hebert, K.; Jiang, H.; Li, C.; Zhu, Y.; Keesler, D.A.; Boelens, J.J.; et al. Related and unrelated donor transplantation for beta-thalassemia major: Results of an international survey. Blood Adv. 2019, 3, 2562–2570. [Google Scholar] [CrossRef] [Green Version]
- Lidonnici, M.R.; Ferrari, G. Gene therapy and gene editing strategies for hemoglobinopathies. Blood Cells Mol. Dis. 2018, 70, 87–101. [Google Scholar] [CrossRef]
- Magrin, E.; Semeraro, M.; Hebert, N.; Joseph, L.; Magnani, A.; Chalumeau, A.; Gabrion, A.; Roudaut, C.; Marouene, J.; Lefrere, F.; et al. Long-term outcomes of lentiviral gene therapy for the β-hemoglobinopathies: The HGB-205 trial. Nat. Med. 2022, 28, 81–88. [Google Scholar] [CrossRef]
- Esrick, E.B.; Lehmann, L.E.; Biffi, A.; Achebe, M.; Brendel, C.; Ciuculescu, M.F.; Daley, H.; MacKinnon, B.; Morris, E.; Federico, A.; et al. Post-Transcriptional Genetic Silencing of BCL11A to Treat Sickle Cell Disease. N. Engl. J. Med. 2021, 384, 205–215. [Google Scholar] [CrossRef]
- Cappellini, M.D.; Viprakasit, V.; Taher, A.T.; Georgiev, P.; Kuo, K.H.M.; Coates, T.; Voskaridou, E.; Liew, H.K.; Pazgal-Kobrowski, I.; Forni, G.L.; et al. A Phase 3 Trial of Luspatercept in Patients with Transfusion-Dependent β-Thalassemia. N. Engl. J. Med. 2020, 382, 1219–1231. [Google Scholar] [CrossRef]
- Casu, C.; Oikonomidou, P.R.; Chen, H.; Nandi, V.; Ginzburg, Y.; Prasad, P.; Fleming, R.E.; Shah, Y.M.; Valore, E.V.; Nemeth, E.; et al. Minihepcidin peptides as disease modifiers in mice affected by β-thalassemia and polycythemia vera. Blood 2016, 128, 265–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nai, A.; Pagani, A.; Mandelli, G.; Lidonnici, M.R.; Silvestri, L.; Ferrari, G.; Camaschella, C. Deletion of TMPRSS6 attenuates the phenotype in a mouse model of β-thalassemia. Blood 2012, 119, 5021–5029. [Google Scholar] [CrossRef] [PubMed]
- Richard, F.; van Lier, J.J.; Roubert, B.; Haboubi, T.; Gohring, U.M.; Durrenberger, F. Oral ferroportin inhibitor VIT-2763: First-in-human, phase 1 study in healthy volunteers. Am. J. Hematol. 2020, 95, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Cavazzana, M.; Ribeil, J.A.; Lagresle-Peyrou, C.; Andre-Schmutz, I. Gene Therapy with Hematopoietic Stem Cells: The Diseased Bone Marrow’s Point of View. Stem Cells Dev. 2017, 26, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Goyal, S.; Tisdale, J.; Schmidt, M.; Kanter, J.; Jaroscak, J.; Whitney, D.; Bitter, H.; Gregory, P.D.; Parsons, G.; Foos, M.; et al. Acute Myeloid Leukemia Case after Gene Therapy for Sickle Cell Disease. N. Engl. J. Med. 2022, 386, 138–147. [Google Scholar] [CrossRef]
- Joseph, C.; Quach, J.M.; Walkley, C.R.; Lane, S.W.; Lo Celso, C.; Purton, L.E. Deciphering hematopoietic stem cells in their niches: A critical appraisal of genetic models, lineage tracing, and imaging strategies. Cell Stem Cell 2013, 13, 520–533. [Google Scholar] [CrossRef] [Green Version]
- Taichman, R.S.; Emerson, S.G. Human osteoblasts support hematopoiesis through the production of granulocyte colony-stimulating factor. J. Exp. Med. 1994, 179, 1677–1682. [Google Scholar] [CrossRef]
- El-Badri, N.S.; Wang, B.Y.; Cherry; Good, R.A. Osteoblasts promote engraftment of allogeneic hematopoietic stem cells. Exp. Hematol. 1998, 26, 110–116. [Google Scholar]
- Calvi, L.M.; Adams, G.B.; Weibrecht, K.W.; Weber, J.M.; Olson, D.P.; Knight, M.C.; Martin, R.P.; Schipani, E.; Divieti, P.; Bringhurst, F.R.; et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003, 425, 841–846. [Google Scholar] [CrossRef]
- Nilsson, S.K.; Johnston, H.M.; Whitty, G.A.; Williams, B.; Webb, R.J.; Denhardt, D.T.; Bertoncello, I.; Bendall, L.J.; Simmons, P.J.; Haylock, D.N. Osteopontin, a key component of the hematopoietic stem cell niche and regulator of primitive hematopoietic progenitor cells. Blood 2005, 106, 1232–1239. [Google Scholar] [CrossRef]
- Ding, L.; Saunders, T.L.; Enikolopov, G.; Morrison, S.J. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 2012, 481, 457–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenbaum, A.; Hsu, Y.M.; Day, R.B.; Schuettpelz, L.G.; Christopher, M.J.; Borgerding, J.N.; Nagasawa, T.; Link, D.C. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 2013, 495, 227–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decker, M.; Leslie, J.; Liu, Q.; Ding, L. Hepatic thrombopoietin is required for bone marrow hematopoietic stem cell maintenance. Science 2018, 360, 106–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.O.; Ding, L.; Morrison, S.J. Hematopoietic stem and progenitor cells regulate the regeneration of their niche by secreting Angiopoietin-1. eLife 2015, 4, e05521. [Google Scholar] [CrossRef] [PubMed]
- Christodoulou, C.; Spencer, J.A.; Yeh, S.A.; Turcotte, R.; Kokkaliaris, K.D.; Panero, R.; Ramos, A.; Guo, G.; Seyedhassantehrani, N.; Esipova, T.V.; et al. Live-animal imaging of native haematopoietic stem and progenitor cells. Nature 2020, 578, 278–283. [Google Scholar] [CrossRef]
- Sacchetti, B.; Funari, A.; Michienzi, S.; Di Cesare, S.; Piersanti, S.; Saggio, I.; Tagliafico, E.; Ferrari, S.; Robey, P.G.; Riminucci, M.; et al. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell 2007, 131, 324–336. [Google Scholar] [CrossRef] [Green Version]
- Bianco, P.; Cao, X.; Frenette, P.S.; Mao, J.J.; Robey, P.G.; Simmons, P.J.; Wang, C.Y. The meaning, the sense and the significance: Translating the science of mesenchymal stem cells into medicine. Nat. Med. 2013, 19, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Mendez-Ferrer, S.; Michurina, T.V.; Ferraro, F.; Mazloom, A.R.; Macarthur, B.D.; Lira, S.A.; Scadden, D.T.; Ma’ayan, A.; Enikolopov, G.N.; Frenette, P.S. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010, 466, 829–834. [Google Scholar] [CrossRef]
- Sugiyama, T.; Kohara, H.; Noda, M.; Nagasawa, T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 2006, 25, 977–988. [Google Scholar] [CrossRef] [Green Version]
- Morikawa, S.; Mabuchi, Y.; Kubota, Y.; Nagai, Y.; Niibe, K.; Hiratsu, E.; Suzuki, S.; Miyauchi-Hara, C.; Nagoshi, N.; Sunabori, T.; et al. Prospective identification, isolation, and systemic transplantation of multipotent mesenchymal stem cells in murine bone marrow. J. Exp. Med. 2009, 206, 2483–2496. [Google Scholar] [CrossRef]
- Wein, F.; Pietsch, L.; Saffrich, R.; Wuchter, P.; Walenda, T.; Bork, S.; Horn, P.; Diehlmann, A.; Eckstein, V.; Ho, A.D.; et al. N-cadherin is expressed on human hematopoietic progenitor cells and mediates interaction with human mesenchymal stromal cells. Stem Cell Res. 2010, 4, 129–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Tao, F.; Venkatraman, A.; Li, Z.; Smith, S.E.; Unruh, J.; Chen, S.; Ward, C.; Qian, P.; Perry, J.M.; et al. N-Cadherin-Expressing Bone and Marrow Stromal Progenitor Cells Maintain Reserve Hematopoietic Stem Cells. Cell Rep. 2019, 26, 652–669.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi Ishikawa, E.; Gonzalez-Nieto, D.; Ghiaur, G.; Dunn, S.K.; Ficker, A.M.; Murali, B.; Madhu, M.; Gutstein, D.E.; Fishman, G.I.; Barrio, L.C.; et al. Connexin-43 prevents hematopoietic stem cell senescence through transfer of reactive oxygen species to bone marrow stromal cells. Proc. Natl. Acad. Sci. USA 2012, 109, 9071–9076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naveiras, O.; Nardi, V.; Wenzel, P.L.; Hauschka, P.V.; Fahey, F.; Daley, G.Q. Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature 2009, 460, 259–263. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.O.; Yu, H.; Yue, R.; Zhao, Z.; Rios, J.J.; Naveiras, O.; Morrison, S.J. Bone marrow adipocytes promote the regeneration of stem cells and haematopoiesis by secreting SCF. Nat. Cell Biol. 2017, 19, 891–903. [Google Scholar] [CrossRef]
- Itkin, T.; Gur-Cohen, S.; Spencer, J.A.; Schajnovitz, A.; Ramasamy, S.K.; Kusumbe, A.P.; Ledergor, G.; Jung, Y.; Milo, I.; Poulos, M.G.; et al. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature 2016, 532, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Winkler, I.G.; Barbier, V.; Nowlan, B.; Jacobsen, R.N.; Forristal, C.E.; Patton, J.T.; Magnani, J.L.; Levesque, J.P. Vascular niche E-selectin regulates hematopoietic stem cell dormancy, self renewal and chemoresistance. Nat. Med. 2012, 18, 1651–1657. [Google Scholar] [CrossRef] [PubMed]
- Doan, P.L.; Russell, J.L.; Himburg, H.A.; Helms, K.; Harris, J.R.; Lucas, J.; Holshausen, K.C.; Meadows, S.K.; Daher, P.; Jeffords, L.B.; et al. Tie2+ bone marrow endothelial cells regulate hematopoietic stem cell regeneration following radiation injury. Stem Cells 2013, 31, 327–337. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Morrison, S.J. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 2013, 495, 231–235. [Google Scholar] [CrossRef]
- Mendez-Ferrer, S.; Lucas, D.; Battista, M.; Frenette, P.S. Haematopoietic stem cell release is regulated by circadian oscillations. Nature 2008, 452, 442–447. [Google Scholar] [CrossRef]
- Park, M.H.; Jin, H.K.; Min, W.K.; Lee, W.W.; Lee, J.E.; Akiyama, H.; Herzog, H.; Enikolopov, G.N.; Schuchman, E.H.; Bae, J.S. Neuropeptide Y regulates the hematopoietic stem cell microenvironment and prevents nerve injury in the bone marrow. EMBO J. 2015, 34, 1648–1660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, S.; Ema, H.; Karlsson, G.; Yamaguchi, T.; Miyoshi, H.; Shioda, S.; Taketo, M.M.; Karlsson, S.; Iwama, A.; Nakauchi, H. Nonmyelinating Schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell 2011, 147, 1146–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruns, I.; Lucas, D.; Pinho, S.; Ahmed, J.; Lambert, M.P.; Kunisaki, Y.; Scheiermann, C.; Schiff, L.; Poncz, M.; Bergman, A.; et al. Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat. Med. 2014, 20, 1315–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Perry, J.M.; Marshall, H.; Venkatraman, A.; Qian, P.; He, X.C.; Ahamed, J.; Li, L. Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat. Med. 2014, 20, 1321–1326. [Google Scholar] [CrossRef]
- Lemieux, J.M.; Horowitz, M.C.; Kacena, M.A. Involvement of integrins α(3)β(1) and α(5)β(1) and glycoprotein IIb in megakaryocyte-induced osteoblast proliferation. J. Cell. Biochem. 2010, 109, 927–932. [Google Scholar] [CrossRef] [Green Version]
- Bord, S.; Frith, E.; Ireland, D.C.; Scott, M.A.; Craig, J.I.; Compston, J.E. Megakaryocytes modulate osteoblast synthesis of type-l collagen, osteoprotegerin, and RANKL. Bone 2005, 36, 812–819. [Google Scholar] [CrossRef]
- Dominici, M.; Rasini, V.; Bussolari, R.; Chen, X.; Hofmann, T.J.; Spano, C.; Bernabei, D.; Veronesi, E.; Bertoni, F.; Paolucci, P.; et al. Restoration and reversible expansion of the osteoblastic hematopoietic stem cell niche after marrow radioablation. Blood 2009, 114, 2333–2343. [Google Scholar] [CrossRef] [Green Version]
- Chow, A.; Lucas, D.; Hidalgo, A.; Mendez-Ferrer, S.; Hashimoto, D.; Scheiermann, C.; Battista, M.; Leboeuf, M.; Prophete, C.; van Rooijen, N.; et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J. Exp. Med. 2011, 208, 261–271. [Google Scholar] [CrossRef]
- Hur, J.; Choi, J.I.; Lee, H.; Nham, P.; Kim, T.W.; Chae, C.W.; Yun, J.Y.; Kang, J.A.; Kang, J.; Lee, S.E.; et al. CD82/KAI1 Maintains the Dormancy of Long-Term Hematopoietic Stem Cells through Interaction with DARC-Expressing Macrophages. Cell Stem Cell 2016, 18, 508–521. [Google Scholar] [CrossRef] [Green Version]
- Ludin, A.; Itkin, T.; Gur-Cohen, S.; Mildner, A.; Shezen, E.; Golan, K.; Kollet, O.; Kalinkovich, A.; Porat, Z.; D’Uva, G.; et al. Monocytes-macrophages that express alpha-smooth muscle actin preserve primitive hematopoietic cells in the bone marrow. Nat. Immunol. 2012, 13, 1072–1082. [Google Scholar] [CrossRef]
- Luo, Y.; Shao, L.; Chang, J.; Feng, W.; Liu, Y.L.; Cottler-Fox, M.H.; Emanuel, P.D.; Hauer-Jensen, M.; Bernstein, I.D.; Liu, L.; et al. M1 and M2 macrophages differentially regulate hematopoietic stem cell self-renewal and ex vivo expansion. Blood Adv. 2018, 2, 859–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casanova-Acebes, M.; Pitaval, C.; Weiss, L.A.; Nombela-Arrieta, C.; Chevre, R.; A-González, N.; Kunisaki, Y.; Zhang, D.; van Rooijen, N.; Silberstein, L.E.; et al. Rhythmic modulation of the hematopoietic niche through neutrophil clearance. Cell 2013, 153, 1025–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawano, Y.; Fukui, C.; Shinohara, M.; Wakahashi, K.; Ishii, S.; Suzuki, T.; Sato, M.; Asada, N.; Kawano, H.; Minagawa, K.; et al. G-CSF-induced sympathetic tone provokes fever and primes antimobilizing functions of neutrophils via PGE2. Blood 2017, 129, 587–597. [Google Scholar] [CrossRef]
- Bowers, E.; Slaughter, A.; Frenette, P.S.; Kuick, R.; Pello, O.M.; Lucas, D. Granulocyte-derived TNFalpha promotes vascular and hematopoietic regeneration in the bone marrow. Nat. Med. 2018, 24, 95–102. [Google Scholar] [CrossRef]
- Fujisaki, J.; Wu, J.; Carlson, A.L.; Silberstein, L.; Putheti, P.; Larocca, R.; Gao, W.; Saito, T.I.; Lo Celso, C.; Tsuyuzaki, H.; et al. In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature 2011, 474, 216–219. [Google Scholar] [CrossRef] [Green Version]
- Hirata, Y.; Furuhashi, K.; Ishii, H.; Li, H.W.; Pinho, S.; Ding, L.; Robson, S.C.; Frenette, P.S.; Fujisaki, J. CD150(high) Bone Marrow Tregs Maintain Hematopoietic Stem Cell Quiescence and Immune Privilege via Adenosine. Cell Stem Cell 2018, 22, 445–453.e5. [Google Scholar] [CrossRef] [Green Version]
- Tan, D.Q.; Suda, T. Reactive Oxygen Species and Mitochondrial Homeostasis as Regulators of Stem Cell Fate and Function. Antioxid. Redox Signal. 2018, 29, 149–168. [Google Scholar] [CrossRef] [PubMed]
- Bigarella, C.L.; Liang, R.; Ghaffari, S. Stem cells and the impact of ROS signaling. Development 2014, 141, 4206–4218. [Google Scholar] [CrossRef] [Green Version]
- Jang, Y.Y.; Sharkis, S.J. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood 2007, 110, 3056–3063. [Google Scholar] [CrossRef] [Green Version]
- Rimmele, P.; Liang, R.; Bigarella, C.L.; Kocabas, F.; Xie, J.; Serasinghe, M.N.; Chipuk, J.; Sadek, H.; Zhang, C.C.; Ghaffari, S. Mitochondrial metabolism in hematopoietic stem cells requires functional FOXO3. EMBO Rep. 2015, 16, 1164–1176. [Google Scholar] [CrossRef] [Green Version]
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Mercurio, S.; Tolosano, E. Heme in pathophysiology: A matter of scavenging, metabolism and trafficking across cell membranes. Front. Pharmacol. 2014, 5, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okabe, H.; Suzuki, T.; Uehara, E.; Ueda, M.; Nagai, T.; Ozawa, K. The bone marrow hematopoietic microenvironment is impaired in iron-overloaded mice. Eur. J. Haematol. 2014, 93, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Muto, Y.; Nishiyama, M.; Nita, A.; Moroishi, T.; Nakayama, K.I. Essential role of FBXL5-mediated cellular iron homeostasis in maintenance of hematopoietic stem cells. Nat. Commun. 2017, 8, 16114. [Google Scholar] [CrossRef] [PubMed]
- Chai, X.; Li, D.; Cao, X.; Zhang, Y.; Mu, J.; Lu, W.; Xiao, X.; Li, C.; Meng, J.; Chen, J.; et al. ROS-mediated iron overload injures the hematopoiesis of bone marrow by damaging hematopoietic stem/progenitor cells in mice. Sci. Rep. 2015, 5, 10181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Gao, X.; Li, H.; Borger, D.K.; Wei, Q.; Yang, E.; Xu, C.; Pinho, S.; Frenette, P.S. The microbiota regulates hematopoietic stem cell fate decisions by controlling iron availability in bone marrow. Cell Stem Cell 2022, 29, 232–247.e7. [Google Scholar] [CrossRef]
- King, K.Y.; Goodell, M.A. Inflammatory modulation of HSCs: Viewing the HSC as a foundation for the immune response. Nat. Rev. Immunol. 2011, 11, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Pietras, E.M.; Mirantes-Barbeito, C.; Fong, S.; Loeffler, D.; Kovtonyuk, L.V.; Zhang, S.; Lakshminarasimhan, R.; Chin, C.P.; Techner, J.M.; Will, B.; et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat. Cell Biol. 2016, 18, 607–618. [Google Scholar] [CrossRef]
- Walter, D.; Lier, A.; Geiselhart, A.; Thalheimer, F.B.; Huntscha, S.; Sobotta, M.C.; Moehrle, B.; Brocks, D.; Bayindir, I.; Kaschutnig, P.; et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature 2015, 520, 549–552. [Google Scholar] [CrossRef]
- Baldridge, M.T.; King, K.Y.; Boles, N.C.; Weksberg, D.C.; Goodell, M.A. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature 2010, 465, 793–797. [Google Scholar] [CrossRef]
- Boettcher, S.; Gerosa, R.C.; Radpour, R.; Bauer, J.; Ampenberger, F.; Heikenwalder, M.; Kopf, M.; Manz, M.G. Endothelial cells translate pathogen signals into G-CSF-driven emergency granulopoiesis. Blood 2014, 124, 1393–1403. [Google Scholar] [CrossRef] [Green Version]
- Schurch, C.M.; Riether, C.; Ochsenbein, A.F. Cytotoxic CD8+ T cells stimulate hematopoietic progenitors by promoting cytokine release from bone marrow mesenchymal stromal cells. Cell Stem Cell 2014, 14, 460–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terashima, A.; Okamoto, K.; Nakashima, T.; Akira, S.; Ikuta, K.; Takayanagi, H. Sepsis-Induced Osteoblast Ablation Causes Immunodeficiency. Immunity 2016, 44, 1434–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geiger, H.; de Haan, G.; Florian, M.C. The ageing haematopoietic stem cell compartment. Nat. Rev. Immunol. 2013, 13, 376–389. [Google Scholar] [CrossRef] [PubMed]
- Sacma, M.; Pospiech, J.; Bogeska, R.; de Back, W.; Mallm, J.P.; Sakk, V.; Soller, K.; Marka, G.; Vollmer, A.; Karns, R.; et al. Haematopoietic stem cells in perisinusoidal niches are protected from ageing. Nat. Cell Biol. 2019, 21, 1309–1320. [Google Scholar] [CrossRef]
- Maryanovich, M.; Zahalka, A.H.; Pierce, H.; Pinho, S.; Nakahara, F.; Asada, N.; Wei, Q.; Wang, X.; Ciero, P.; Xu, J.; et al. Adrenergic nerve degeneration in bone marrow drives aging of the hematopoietic stem cell niche. Nat. Med. 2018, 24, 782–791. [Google Scholar] [CrossRef]
- Mendez-Ferrer, S.; Bonnet, D.; Steensma, D.P.; Hasserjian, R.P.; Ghobrial, I.M.; Gribben, J.G.; Andreeff, M.; Krause, D.S. Bone marrow niches in haematological malignancies. Nat. Rev. Cancer 2020, 20, 285–298. [Google Scholar] [CrossRef]
- Duarte, D.; Hawkins, E.D.; Akinduro, O.; Ang, H.; De Filippo, K.; Kong, I.Y.; Haltalli, M.; Ruivo, N.; Straszkowski, L.; Vervoort, S.J.; et al. Inhibition of Endosteal Vascular Niche Remodeling Rescues Hematopoietic Stem Cell Loss in AML. Cell Stem Cell 2018, 22, 64–77.e6. [Google Scholar] [CrossRef] [Green Version]
- Pitt, L.A.; Tikhonova, A.N.; Hu, H.; Trimarchi, T.; King, B.; Gong, Y.; Sanchez-Martin, M.; Tsirigos, A.; Littman, D.R.; Ferrando, A.A.; et al. CXCL12-Producing Vascular Endothelial Niches Control Acute T Cell Leukemia Maintenance. Cancer Cell 2015, 27, 755–768. [Google Scholar] [CrossRef] [Green Version]
- Baryawno, N.; Przybylski, D.; Kowalczyk, M.S.; Kfoury, Y.; Severe, N.; Gustafsson, K.; Kokkaliaris, K.D.; Mercier, F.; Tabaka, M.; Hofree, M.; et al. A Cellular Taxonomy of the Bone Marrow Stroma in Homeostasis and Leukemia. Cell 2019, 177, 1915–1932.e16. [Google Scholar] [CrossRef]
- Sokolic, R.; Maric, I.; Kesserwan, C.; Garabedian, E.; Hanson, I.C.; Dodds, M.; Buckley, R.; Issekutz, A.C.; Kamani, N.; Shaw, K.; et al. Myeloid dysplasia and bone marrow hypocellularity in adenosine deaminase-deficient severe combined immune deficiency. Blood 2011, 118, 2688–2694. [Google Scholar] [CrossRef] [Green Version]
- Sauer, A.V.; Mrak, E.; Hernandez, R.J.; Zacchi, E.; Cavani, F.; Casiraghi, M.; Grunebaum, E.; Roifman, C.M.; Cervi, M.C.; Ambrosi, A.; et al. ADA-deficient SCID is associated with a specific microenvironment and bone phenotype characterized by RANKL/OPG imbalance and osteoblast insufficiency. Blood 2009, 114, 3216–3226. [Google Scholar] [CrossRef] [PubMed]
- Weisser, M.; Demel, U.M.; Stein, S.; Chen-Wichmann, L.; Touzot, F.; Santilli, G.; Sujer, S.; Brendel, C.; Siler, U.; Cavazzana, M.; et al. Hyperinflammation in patients with chronic granulomatous disease leads to impairment of hematopoietic stem cell functions. J. Allergy Clin. Immunol. 2016, 138, 219–228.e9. [Google Scholar] [CrossRef] [Green Version]
- Lacout, C.; Haddad, E.; Sabri, S.; Svinarchouk, F.; Garcon, L.; Capron, C.; Foudi, A.; Mzali, R.; Snapper, S.B.; Louache, F.; et al. A defect in hematopoietic stem cell migration explains the nonrandom X-chromosome inactivation in carriers of Wiskott-Aldrich syndrome. Blood 2003, 102, 1282–1289. [Google Scholar] [CrossRef] [PubMed]
- Aprile, A.; Gulino, A.; Storto, M.; Villa, I.; Beretta, S.; Merelli, I.; Rubinacci, A.; Ponzoni, M.; Marktel, S.; Tripodo, C.; et al. Hematopoietic stem cell function in beta-thalassemia is impaired and is rescued by targeting the bone marrow niche. Blood 2020, 136, 610–622. [Google Scholar] [CrossRef] [PubMed]
- Tang, A.; Strat, A.N.; Rahman, M.; Zhang, H.; Bao, W.; Liu, Y.; Shi, D.; An, X.; Manwani, D.; Shi, P.; et al. Murine bone marrow mesenchymal stromal cells have reduced hematopoietic maintenance ability in sickle cell disease. Blood 2021, 138, 2570–2582. [Google Scholar] [CrossRef]
- McColl, B.; Vadolas, J. Animal models of beta-hemoglobinopathies: Utility and limitations. J. Blood Med. 2016, 7, 263–274. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Kirby, S.; Lewis, J.; Detloff, P.J.; Maeda, N.; Smithies, O. A mouse model for beta 0-thalassemia. Proc. Natl. Acad. Sci. USA 1995, 92, 11608–11612. [Google Scholar] [CrossRef] [Green Version]
- Trudel, M.; Saadane, N.; Garel, M.C.; Bardakdjian-Michau, J.; Blouquit, Y.; Guerquin-Kern, J.L.; Rouyer-Fessard, P.; Vidaud, D.; Pachnis, A.; Romeo, P.H.; et al. Towards a transgenic mouse model of sickle cell disease: Hemoglobin SAD. EMBO J. 1991, 10, 3157–3165. [Google Scholar] [CrossRef]
- Paszty, C.; Brion, C.M.; Manci, E.; Witkowska, H.E.; Stevens, M.E.; Mohandas, N.; Rubin, E.M. Transgenic knockout mice with exclusively human sickle hemoglobin and sickle cell disease. Science 1997, 278, 876–878. [Google Scholar] [CrossRef] [Green Version]
- Ryan, T.M.; Ciavatta, D.J.; Townes, T.M. Knockout-transgenic mouse model of sickle cell disease. Science 1997, 278, 873–876. [Google Scholar] [CrossRef] [Green Version]
- Hua, P.; Roy, N.; de la Fuente, J.; Wang, G.; Thongjuea, S.; Clark, K.; Roy, A.; Psaila, B.; Ashley, N.; Harrington, Y.; et al. Single-cell analysis of bone marrow-derived CD34+ cells from children with sickle cell disease and thalassemia. Blood 2019, 134, 2111–2115. [Google Scholar] [CrossRef] [PubMed]
- Lidonnici, M.R.; Chianella, G.; Tiboni, F.; Barcella, M.; Merelli, I.; Scaramuzza, S.; Rossi, C.; Crippa, S.; Storto, M.; Bernardo, M.E.; et al. S269 TGF-beta signaling controls the lineage cell fate of hematopoietic stem cells towards erythroid branching in beta-thalassemia. EHA2021 Virtual Congress Abstract Book. HemaSphere 2021, 5, e566. [Google Scholar] [CrossRef]
- Javazon, E.H.; Radhi, M.; Gangadharan, B.; Perry, J.; Archer, D.R. Hematopoietic stem cell function in a murine model of sickle cell disease. Anemia 2012, 2012, 387385. [Google Scholar] [CrossRef] [PubMed]
- Blouin, M.J.; De Paepe, M.E.; Trudel, M. Altered hematopoiesis in murine sickle cell disease. Blood 1999, 94, 1451–1459. [Google Scholar] [CrossRef]
- Tolu, S.S.; Wang, K.; Yan, Z.; Zhang, S.; Roberts, K.; Crouch, A.S.; Sebastian, G.; Chaitowitz, M.; Fornari, E.D.; Schwechter, E.M.; et al. Characterization of Hematopoiesis in Sickle Cell Disease by Prospective Isolation of Stem and Progenitor Cells. Cells 2020, 9, 2159. [Google Scholar] [CrossRef]
- Vogiatzi, M.G.; Tsay, J.; Verdelis, K.; Rivella, S.; Grady, R.W.; Doty, S.; Giardina, P.J.; Boskey, A.L. Changes in bone microarchitecture and biomechanical properties in the th3 thalassemia mouse are associated with decreased bone turnover and occur during the period of bone accrual. Calcif. Tissue Int. 2010, 86, 484–494. [Google Scholar] [CrossRef] [Green Version]
- Crippa, S.; Rossella, V.; Aprile, A.; Silvestri, L.; Rivis, S.; Scaramuzza, S.; Pirroni, S.; Avanzini, M.A.; Basso-Ricci, L.; Hernandez, R.J.; et al. Bone marrow stromal cells from β-thalassemia patients have impaired hematopoietic supportive capacity. J. Clin. Investig. 2019, 129, 1566–1580. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, C.; Nolan, V.G.; Wyszynski, D.F.; Ma, Q.L.; Sebastiani, P.; Embury, S.H.; Bisbee, A.; Farrell, J.; Farrer, L.; Steinberg, M.H. Association of klotho, bone morphogenic protein 6, and annexin A2 polymorphisms with sickle cell osteonecrosis. Blood 2005, 106, 372–375. [Google Scholar] [CrossRef]
- Seguin, C.; Kassis, J.; Busque, L.; Bestawros, A.; Theodoropoulos, J.; Alonso, M.L.; Harvey, E.J. Non-traumatic necrosis of bone (osteonecrosis) is associated with endothelial cell activation but not thrombophilia. Rheumatology 2008, 47, 1151–1155. [Google Scholar] [CrossRef] [Green Version]
- Nouraie, M.; Cheng, K.; Niu, X.; Moore-King, E.; Fadojutimi-Akinsi, M.F.; Minniti, C.P.; Sable, C.; Rana, S.; Dham, N.; Campbell, A.; et al. Predictors of osteoclast activity in patients with sickle cell disease. Haematologica 2011, 96, 1092–1098. [Google Scholar] [CrossRef]
- Kobayashi, K.; Takahashi, N.; Jimi, E.; Udagawa, N.; Takami, M.; Kotake, S.; Nakagawa, N.; Kinosaki, M.; Yamaguchi, K.; Shima, N.; et al. Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J. Exp. Med. 2000, 191, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Green, M.; Akinsami, I.; Lin, A.; Banton, S.; Ghosh, S.; Chen, B.; Platt, M.; Osunkwo, I.; Ofori-Acquah, S.; Guldberg, R.; et al. Microarchitectural and mechanical characterization of the sickle bone. J. Mech. Behav. Biomed. Mater. 2015, 48, 220–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalle Carbonare, L.; Matte, A.; Valenti, M.T.; Siciliano, A.; Mori, A.; Schweiger, V.; Zampieri, G.; Perbellini, L.; De Franceschi, L. Hypoxia-reperfusion affects osteogenic lineage and promotes sickle cell bone disease. Blood 2015, 126, 2320–2328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsay, J.; Yang, Z.; Ross, F.P.; Cunningham-Rundles, S.; Lin, H.; Coleman, R.; Mayer-Kuckuk, P.; Doty, S.B.; Grady, R.W.; Giardina, P.J.; et al. Bone loss caused by iron overload in a murine model: Importance of oxidative stress. Blood 2010, 116, 2582–2589. [Google Scholar] [CrossRef]
- Stenger, E.O.; Chinnadurai, R.; Yuan, S.; Garcia, M.; Arafat, D.; Gibson, G.; Krishnamurti, L.; Galipeau, J. Bone Marrow-Derived Mesenchymal Stromal Cells from Patients with Sickle Cell Disease Display Intact Functionality. Biol. Blood Marrow Transplant. 2017, 23, 736–745. [Google Scholar] [CrossRef]
- Park, S.Y.; Matte, A.; Jung, Y.; Ryu, J.; Anand, W.B.; Han, E.Y.; Liu, M.; Carbone, C.; Melisi, D.; Nagasawa, T.; et al. Pathologic angiogenesis in the bone marrow of humanized sickle cell mice is reversed by blood transfusion. Blood 2020, 135, 2071–2084. [Google Scholar] [CrossRef]
- Aprile, A.; Raggi, L.; Storto, M.; Villa, I.; Bolamperti, S.; Marktel, S.; Motta, I.; Cappellini, M.D.; Rubinacci, A.; Ferrari, G. Inhibition of Fibroblast Growth Factor-23 (FGF-23) Rescues Bone and Hematopoietic Stem Cell Niche Defects in Beta-Thalassemia, Uncovering the Missing Link Between Hematopoiesis and Bone. Blood 2021, 138 (Suppl. 1), 572. [Google Scholar] [CrossRef]
- Edmonston, D.; Wolf, M. FGF23 at the crossroads of phosphate, iron economy and erythropoiesis. Nat. Rev. Nephrol. 2020, 16, 7–19. [Google Scholar] [CrossRef]
- Clinkenbeard, E.L.; Hanudel, M.R.; Stayrook, K.R.; Appaiah, H.N.; Farrow, E.G.; Cass, T.A.; Summers, L.J.; Ip, C.S.; Hum, J.M.; Thomas, J.C.; et al. Erythropoietin stimulates murine and human fibroblast growth factor-23, revealing novel roles for bone and bone marrow. Haematologica 2017, 102, e427–e430. [Google Scholar] [CrossRef]
- Aprile, A.; Storto, M.; Malara, A.; Gulino, A.; Raggi, L.; Sighinolfi, S.; Beretta, S.; Merelli, I.; Marktel, S.; Ponzoni, M.; et al. S249 Reduced Levels of thrombopoietin Contribute to Impaired Hematopoietic stem Cell Function and Defective Megakaryopoiesis in Beta-Thalassemia. Available online: https://library.ehaweb.org/eha/2021/eha2021-virtual-congress/324657/annamaria.aprile.reduced.levels.of.thrombopoietin.contribute.to.impaired.html?f=menu%3D6%2Abrowseby%3D8%2Asortby%3D2%2Amedia%3D3%2Ace_id%3D2035%2Aot_id%3D25563 (accessed on 4 May 2022).
- Yoshihara, H.; Arai, F.; Hosokawa, K.; Hagiwara, T.; Takubo, K.; Nakamura, Y.; Gomei, Y.; Iwasaki, H.; Matsuoka, S.; Miyamoto, K.; et al. Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell Stem Cell 2007, 1, 685–697. [Google Scholar] [CrossRef] [Green Version]
- Nakamura-Ishizu, A.; Takubo, K.; Kobayashi, H.; Suzuki-Inoue, K.; Suda, T. CLEC-2 in megakaryocytes is critical for maintenance of hematopoietic stem cells in the bone marrow. J. Exp. Med. 2015, 212, 2133–2146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seyfried, A.N.; Maloney, J.M.; MacNamara, K.C. Macrophages Orchestrate Hematopoietic Programs and Regulate HSC Function During Inflammatory Stress. Front. Immunol. 2020, 11, 1499. [Google Scholar] [CrossRef] [PubMed]
- Siwaponanan, P.; Siegers, J.Y.; Ghazali, R.; Ng, T.; McColl, B.; Ng, G.Z.; Sutton, P.; Wang, N.; Ooi, I.; Thiengtavor, C.; et al. Reduced PU.1 expression underlies aberrant neutrophil maturation and function in beta-thalassemia mice and patients. Blood 2017, 129, 3087–3099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Xu, C.; Manwani, D.; Frenette, P.S. Neutrophils, platelets, and inflammatory pathways at the nexus of sickle cell disease pathophysiology. Blood 2016, 127, 801–809. [Google Scholar] [CrossRef] [Green Version]
- Canalli, A.A.; Franco-Penteado, C.F.; Saad, S.T.; Conran, N.; Costa, F.F. Increased adhesive properties of neutrophils in sickle cell disease may be reversed by pharmacological nitric oxide donation. Haematologica 2008, 93, 605–609. [Google Scholar] [CrossRef] [Green Version]
- Lum, A.F.; Wun, T.; Staunton, D.; Simon, S.I. Inflammatory potential of neutrophils detected in sickle cell disease. Am. J. Hematol. 2004, 76, 126–133. [Google Scholar] [CrossRef]
- Chen, G.; Zhang, D.; Fuchs, T.A.; Manwani, D.; Wagner, D.D.; Frenette, P.S. Heme-induced neutrophil extracellular traps contribute to the pathogenesis of sickle cell disease. Blood 2014, 123, 3818–3827. [Google Scholar] [CrossRef]
- Zhang, D.; Chen, G.; Manwani, D.; Mortha, A.; Xu, C.; Faith, J.J.; Burk, R.D.; Kunisaki, Y.; Jang, J.E.; Scheiermann, C.; et al. Neutrophil ageing is regulated by the microbiome. Nature 2015, 525, 528–532. [Google Scholar] [CrossRef]
- Villagra, J.; Shiva, S.; Hunter, L.A.; Machado, R.F.; Gladwin, M.T.; Kato, G.J. Platelet activation in patients with sickle disease, hemolysis-associated pulmonary hypertension, and nitric oxide scavenging by cell-free hemoglobin. Blood 2007, 110, 2166–2172. [Google Scholar] [CrossRef] [Green Version]
- Belcher, J.D.; Marker, P.H.; Weber, J.P.; Hebbel, R.P.; Vercellotti, G.M. Activated monocytes in sickle cell disease: Potential role in the activation of vascular endothelium and vaso-occlusion. Blood 2000, 96, 2451–2459. [Google Scholar] [CrossRef]
- Mitroulis, I.; Kalafati, L.; Bornhauser, M.; Hajishengallis, G.; Chavakis, T. Regulation of the Bone Marrow Niche by Inflammation. Front. Immunol 2020, 11, 1540. [Google Scholar] [CrossRef] [PubMed]
- Essers, M.A.; Offner, S.; Blanco-Bose, W.E.; Waibler, Z.; Kalinke, U.; Duchosal, M.A.; Trumpp, A. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature 2009, 458, 904–908. [Google Scholar] [CrossRef] [PubMed]
- Vinchi, F.; Costa da Silva, M.; Ingoglia, G.; Petrillo, S.; Brinkman, N.; Zuercher, A.; Cerwenka, A.; Tolosano, E.; Muckenthaler, M.U. Hemopexin therapy reverts heme-induced proinflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease. Blood 2016, 127, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Huang, L.; Wu, J.; Qu, Y.; Jiang, H.; Zhang, J.; Qiu, S.; Liao, C.; Xu, X.; Xia, J.; et al. Impaired bone marrow microenvironment and stem cells in transfusion-dependent beta-thalassemia. Biomed. Pharmacother. 2022, 146, 112548. [Google Scholar] [CrossRef]
- Fouzia, N.A.; Edison, E.S.; Lakshmi, K.M.; Korula, A.; Velayudhan, S.R.; Balasubramanian, P.; Abraham, A.; Viswabandya, A.; George, B.; Mathews, V.; et al. Long-term outcome of mixed chimerism after stem cell transplantation for thalassemia major conditioned with busulfan and cyclophosphamide. Bone Marrow Transplant 2018, 53, 169–174. [Google Scholar] [CrossRef]
- Angelucci, E. Complication free survival long-term after hemopoietic cell transplantation in thalassemia. Haematologica 2018, 103, 1094–1096. [Google Scholar] [CrossRef] [Green Version]
- Adams, G.B.; Martin, R.P.; Alley, I.R.; Chabner, K.T.; Cohen, K.S.; Calvi, L.M.; Kronenberg, H.M.; Scadden, D.T. Therapeutic targeting of a stem cell niche. Nat. Biotechnol. 2007, 25, 238–243. [Google Scholar] [CrossRef]
- Ballen, K.; Mendizabal, A.M.; Cutler, C.; Politikos, I.; Jamieson, K.; Shpall, E.J.; Dey, B.R.; Attar, E.; McAfee, S.; Delaney, C.; et al. Phase II trial of parathyroid hormone after double umbilical cord blood transplantation. Biol. Blood Marrow Transplant 2012, 18, 1851–1858. [Google Scholar] [CrossRef] [Green Version]
- Vinchi, F.; De Franceschi, L.; Ghigo, A.; Townes, T.; Cimino, J.; Silengo, L.; Hirsch, E.; Altruda, F.; Tolosano, E. Hemopexin therapy improves cardiovascular function by preventing heme-induced endothelial toxicity in mouse models of hemolytic diseases. Circulation 2013, 127, 1317–1329. [Google Scholar] [CrossRef] [Green Version]

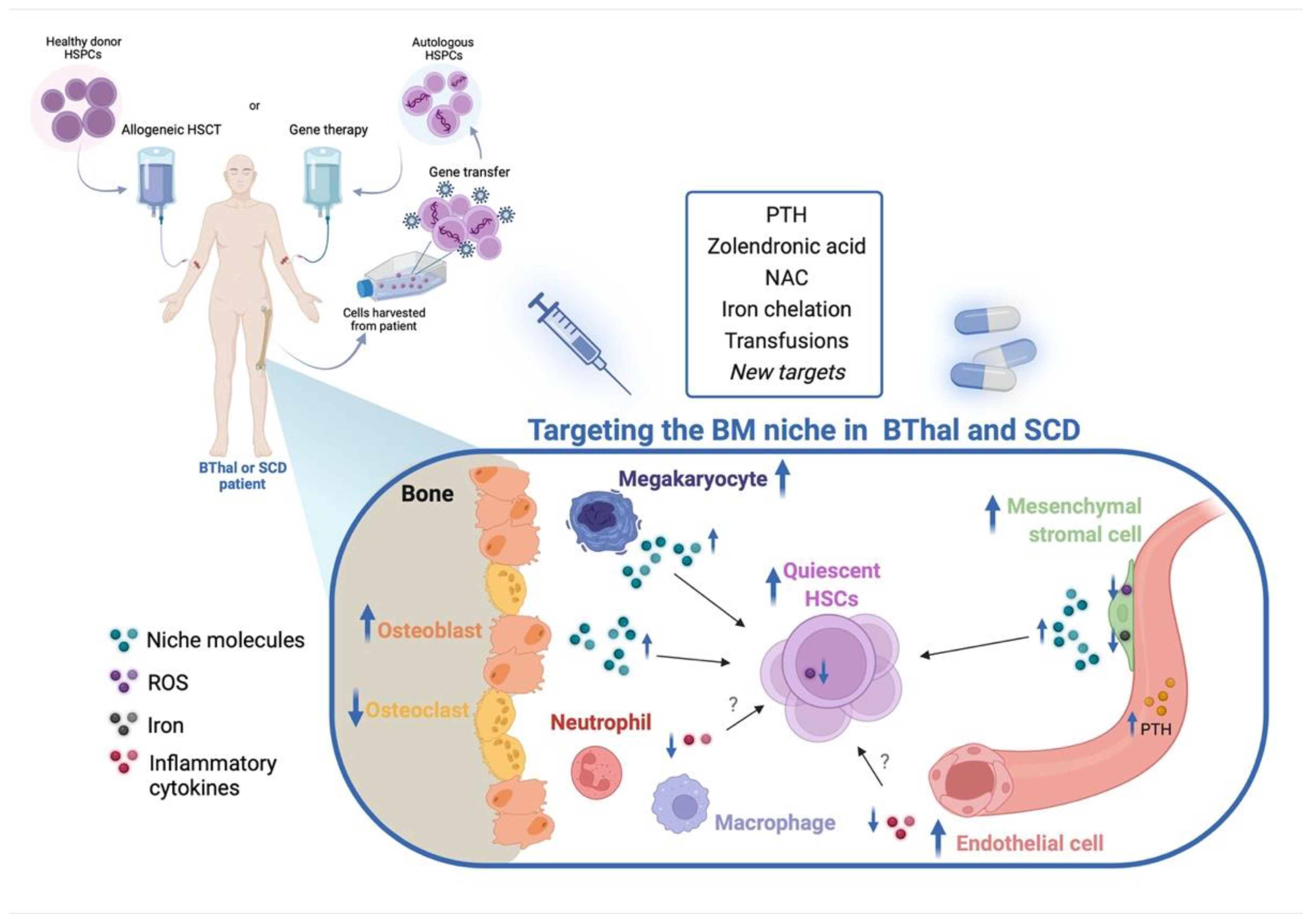

{kind=link}
{kind=link}
| Cell Population | Disease | Species | Alterations | References |
|---|---|---|---|---|
| HSC | BThal | mouse | ⇓ number ⇓ quiescence ⇓ stemness ⇓ reconstitution capacity ⇑ response to stress | [114] |
| human | ⇓ frequency ⇓ quiescence ⇓ stemness ⇑ response to stress (HSPC) | [114,121,122] | ||
| SCD | mouse | ⇓ frequency ⇑ ROS ⇑ DNA damage ⇓ quiescence (HSPC) ⇑ mobilization (HSPC) | [115,123,124] | |
| human | ⇓ frequency ⇑ mobilization | [121,125] | ||
| Osteolineage cell | BThal | mouse | ⇓ BMD ⇓ systemic PTH ⇓ OB activity ⇓ niche molecules ⇑ FGF23 | [114,126,137] |
| human | ⇓ niche molecules | [114] | ||
| SCD | mouse | ⇓ bone microarchitecture | [132] | |
| ⇑ osteoclastogenesis ⇓ osteogenic factors | [133] | |||
| MSC | BThal | mouse | ⇓ frequency ⇓ niche molecules | [114] |
| human | ⇓ frequency ⇓ osteogenic and adipogenic potential ⇑ ROS ⇑ iron content ⇓ niche molecules ⇓ HSPC maintenance | [127] | ||
| SCD | mouse | ⇓ frequency ⇑ ROS ⇓ osteogenic and adipogenic potential ⇓ niche molecules ⇓ HSC maintenance | [115] | |
| human | ⇓ niche molecules | [135] | ||
| EC | SCD | mouse | altered BM vasculature ⇑ inflammatory cytokines | [136] |
| MK | BThal | mouse | ⇓ systemic TPO ⇓ maturation ⇓ niche molecules | [140] |
| Neutrophil | BThal | mouse | altered maturation | [144] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aprile, A.; Sighinolfi, S.; Raggi, L.; Ferrari, G. Targeting the Hematopoietic Stem Cell Niche in β-Thalassemia and Sickle Cell Disease. Pharmaceuticals 2022, 15, 592. https://doi.org/10.3390/ph15050592
Aprile A, Sighinolfi S, Raggi L, Ferrari G. Targeting the Hematopoietic Stem Cell Niche in β-Thalassemia and Sickle Cell Disease. Pharmaceuticals. 2022; 15(5):592. https://doi.org/10.3390/ph15050592
Chicago/Turabian StyleAprile, Annamaria, Silvia Sighinolfi, Laura Raggi, and Giuliana Ferrari. 2022. "Targeting the Hematopoietic Stem Cell Niche in β-Thalassemia and Sickle Cell Disease" Pharmaceuticals 15, no. 5: 592. https://doi.org/10.3390/ph15050592
APA StyleAprile, A., Sighinolfi, S., Raggi, L., & Ferrari, G. (2022). Targeting the Hematopoietic Stem Cell Niche in β-Thalassemia and Sickle Cell Disease. Pharmaceuticals, 15(5), 592. https://doi.org/10.3390/ph15050592