
QSAR, ADMET In Silico Pharmacokinetics, Molecular Docking and Molecular Dynamics Studies of Novel Bicyclo (Aryl Methyl) Benzamides as Potent GlyT1 Inhibitors for the Treatment of Schizophrenia

 ,
, 
 ,
,  , , ,
, , ,  ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Pricipal Component Analysis
2.2. Statistical Database
2.3. Multiple Linear Regression
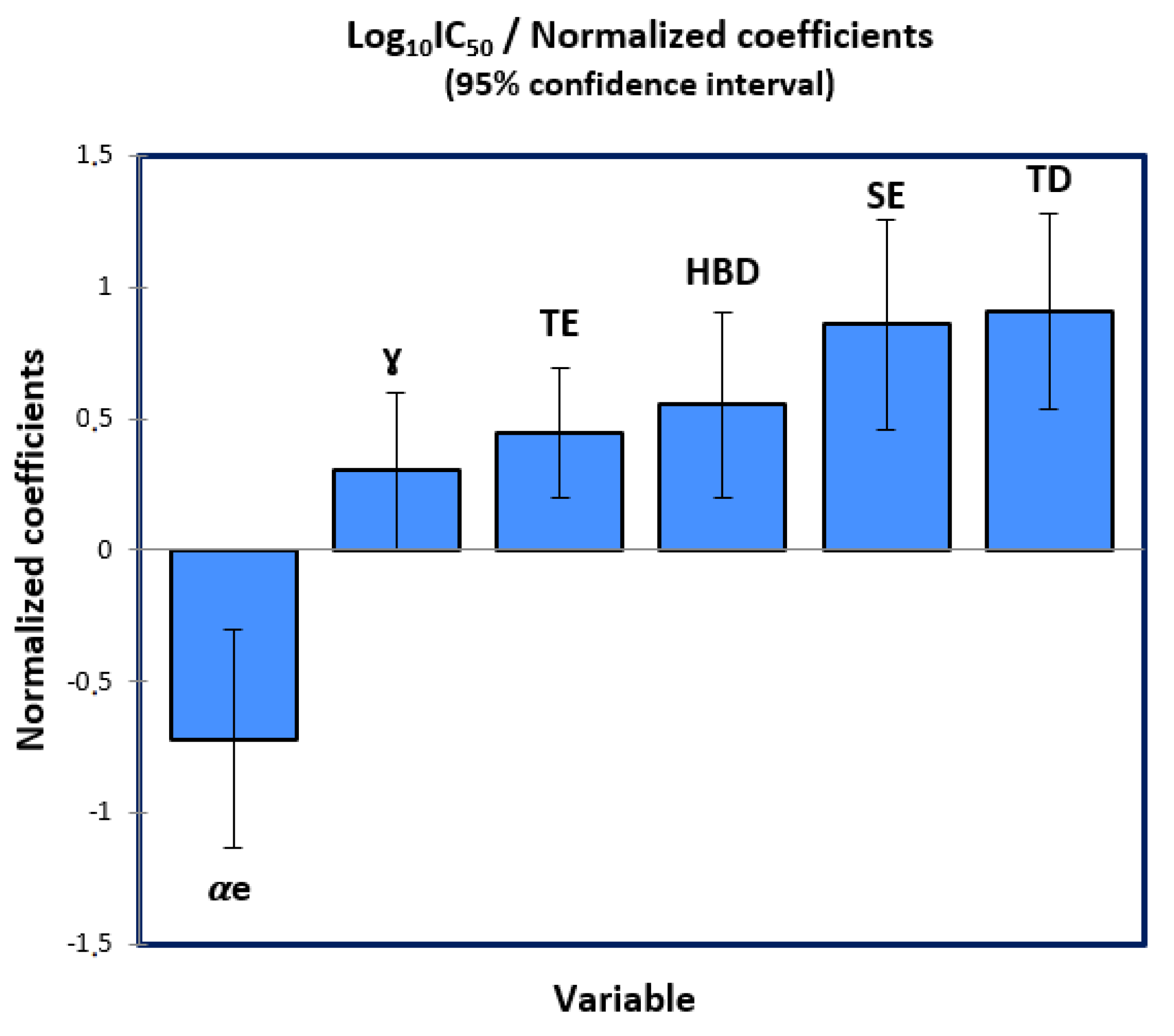
2.4. Multiple Non-Linear Regression
−0.002 × αe^2 + 0.001 × ɣ^2 + 0.013 × TE^2 + 0.148 × SE^2 – 0.07 × TD^2.
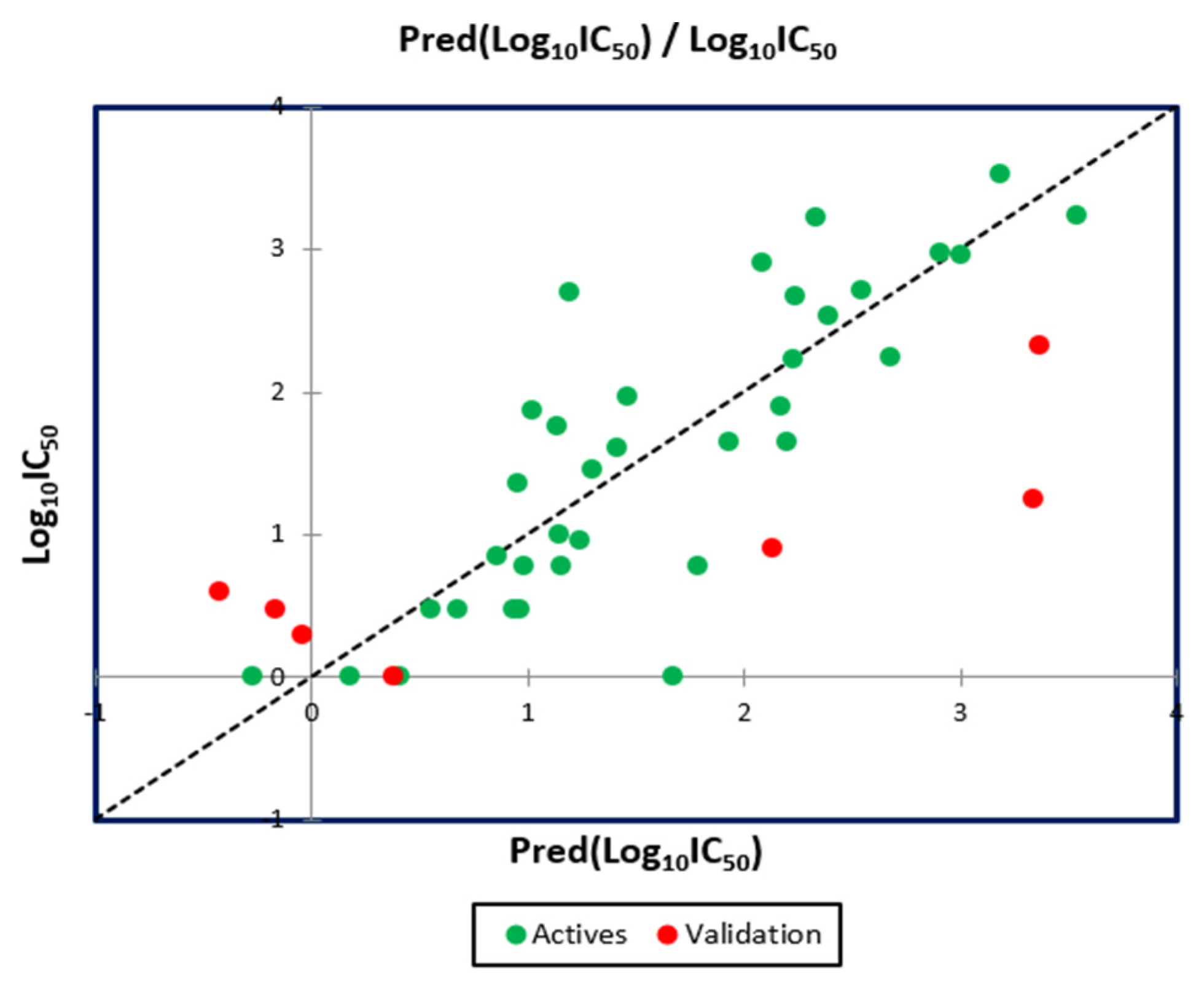
2.5. QSAR Model Validation
2.5.1. Applicability Domain
2.5.2. External Validation
2.5.3. Internal Validation
2.5.4. Validation Using Y-Randomisation Test
2.5.5. Golbreikh and Tropsha Criteria
2.6. In Silico Pharmacokinetics ADMET Prediction
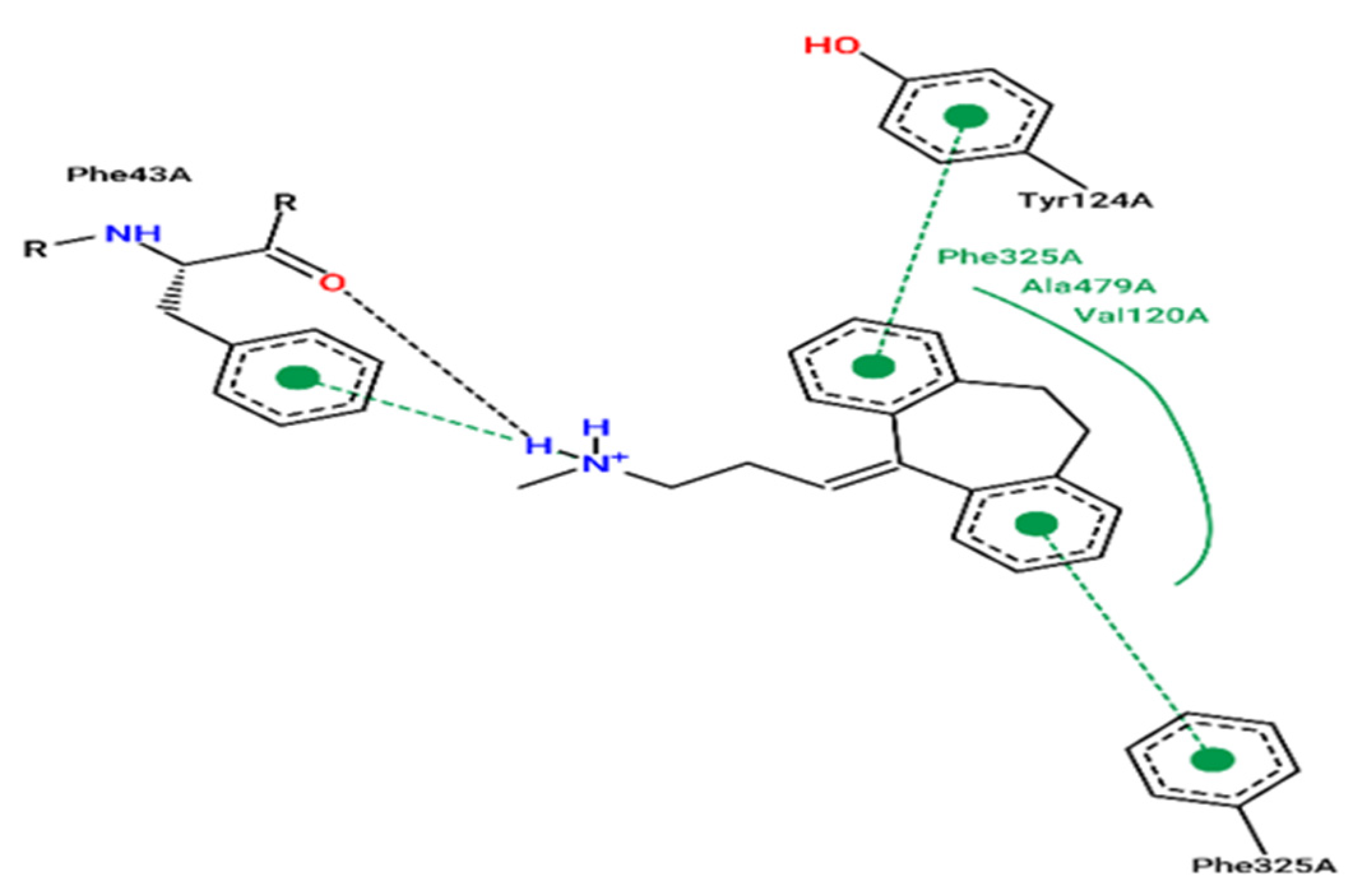
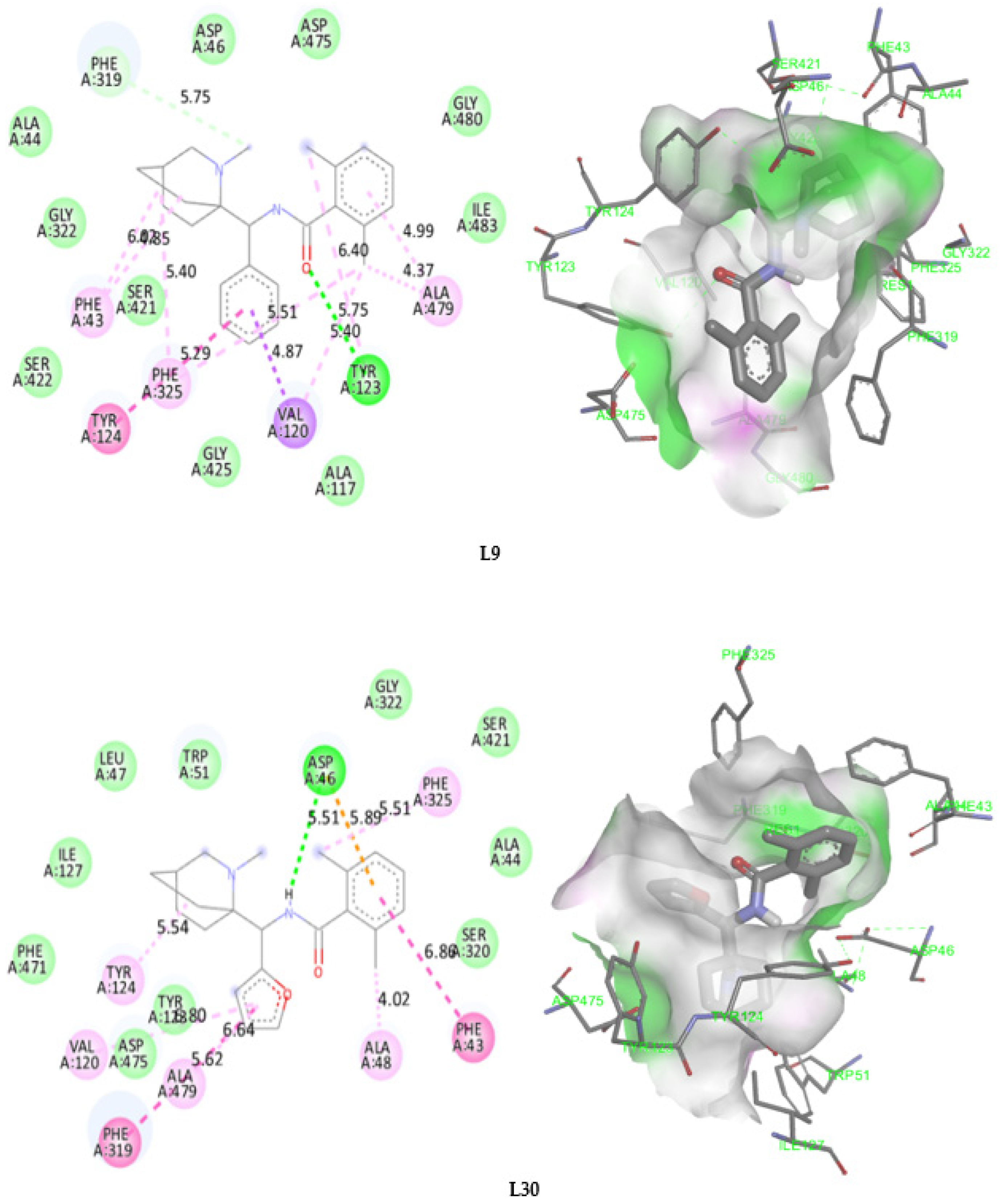
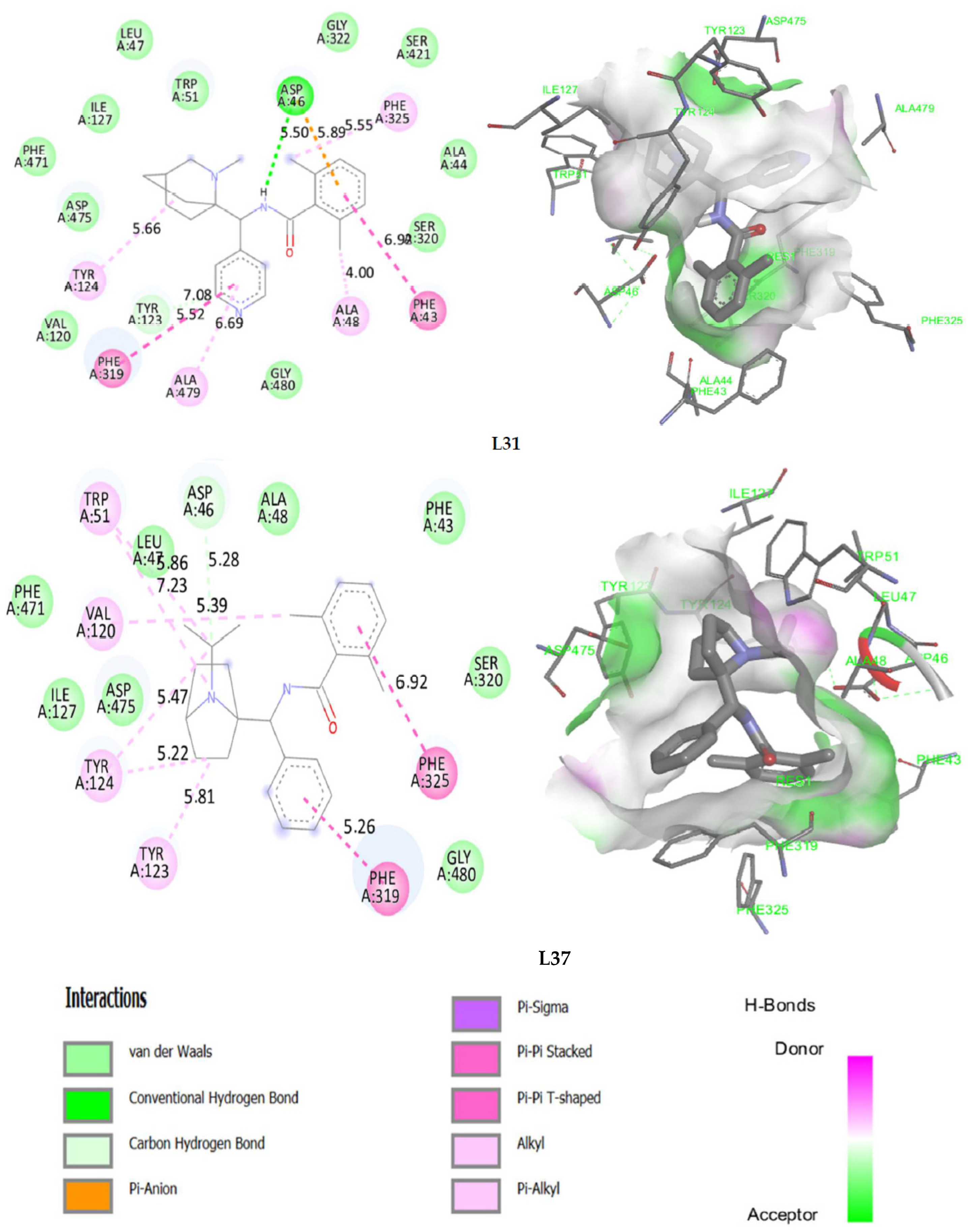
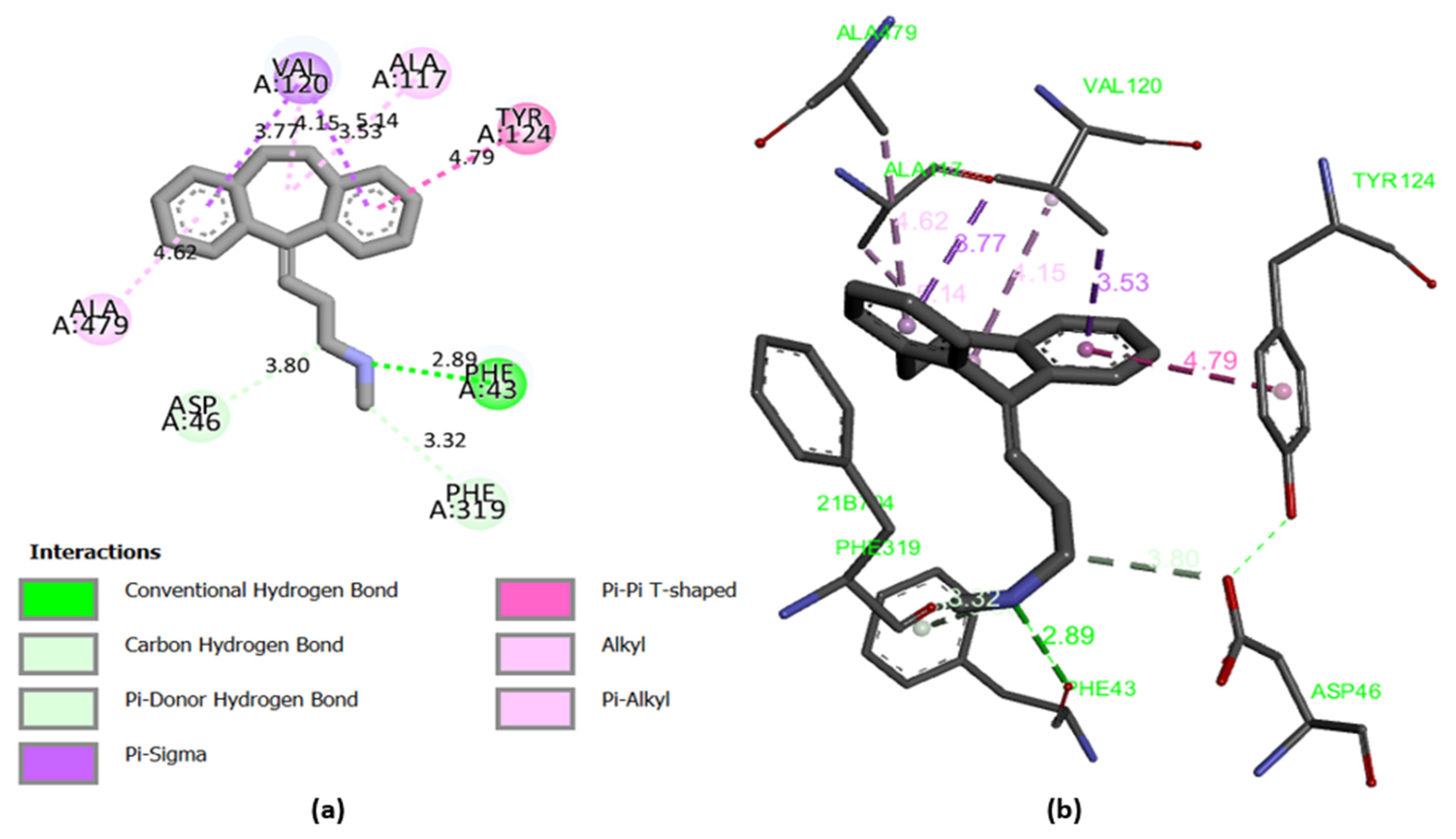
2.7. Molecular Docking
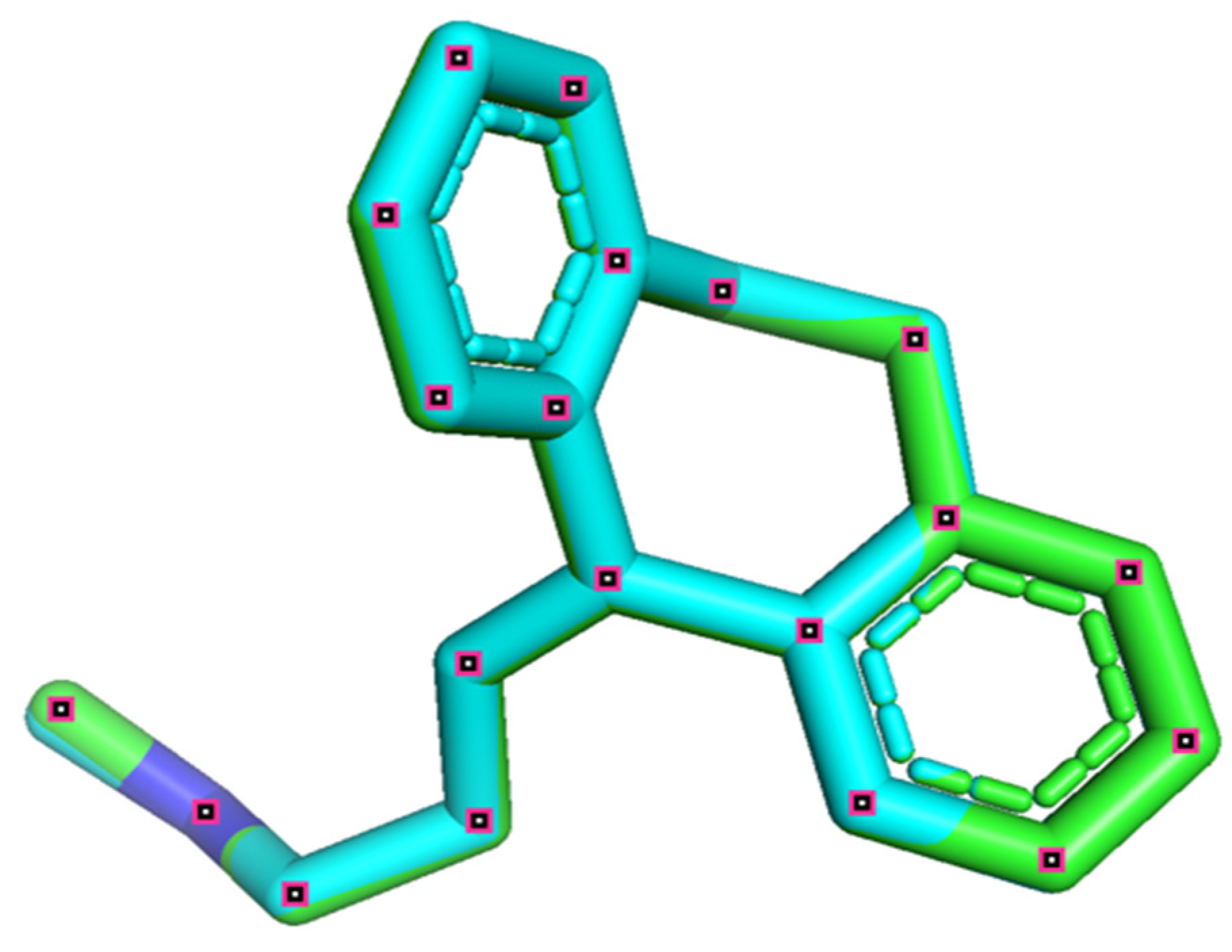
2.8. Docking Validation Protocol
2.9. Molecular Dynamics Simulations
3. Materials and Methods
3.1. Database
3.2. Molecular Descriptors Calculation
3.3. Statistical Methods
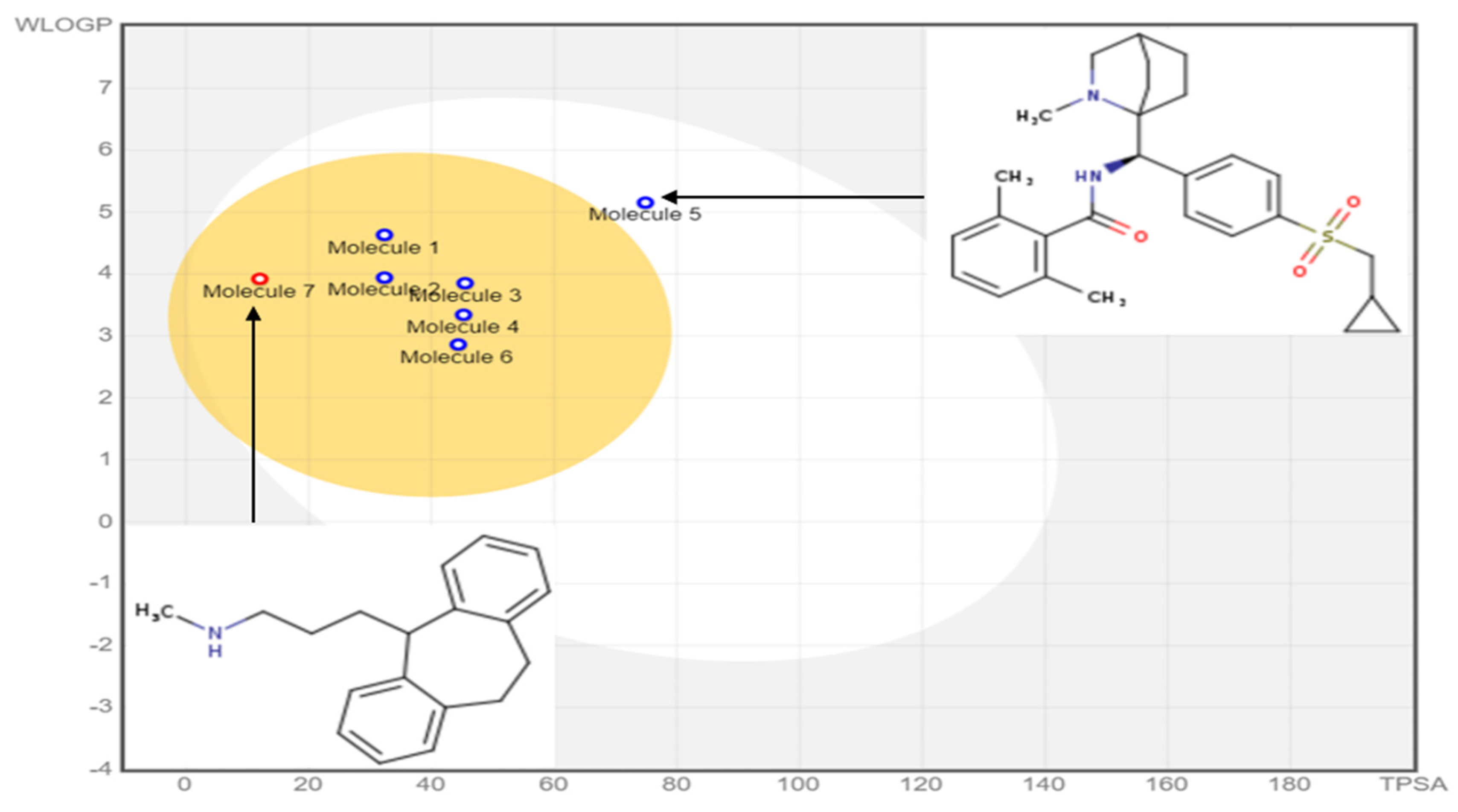
3.4. Drug Likeness and In Silico Pharmacokinetics ADMET Prediction
3.5. Molecular Docking Modeling
3.6. Molecular Dynamics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zheng, X.; Wang, C.; Zhai, N.; Luo, X.; Liu, G.; Ju, X. In Silico Screening of Novel A1-GABAA Receptor PAMs towards Schizophrenia Based on Combined Modeling Studies of Imidazo [1,2-a]-Pyridines. Int. J. Mol. Sci. 2021, 22, 9645. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Hao, C.; Chen, J.; Ma, R.; Zheng, L.; Wu, Q.; Liu, X.; Liu, B.-F.; Zhang, G.; Chen, Y.; et al. Discovery of a New Class of Multi-Target Heterocycle Piperidine Derivatives as Potential Antipsychotics with pro-Cognitive Effect. Bioorg. Med. Chem. Lett. 2021, 40, 127909. [Google Scholar] [CrossRef] [PubMed]
- Shahsavar, A.; Stohler, P.; Bourenkov, G.; Zimmermann, I.; Siegrist, M.; Guba, W.; Pinard, E.; Seeger, M.A.; Schneider, T.R.; Dawson, R.J.P.; et al. Structural Insights into Glycine Reuptake Inhibition; 2020; BioRxiv. Available online: https://www.biorxiv.org/content/10.1101/2020.12.20.110478v1 (accessed on 14 April 2022).
- Varnes, J.G.; Xiong, H.; Forst, J.M.; Holmquist, C.R.; Ernst, G.E.; Frietze, W.; Dembofsky, B.; Andisik, D.W.; Palmer, W.E.; Hinkley, L.; et al. Bicyclo((Aryl)Methyl)Benzamides as Inhibitors of GlyT1. Bioorg. Med. Chem. Lett. 2018, 28, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Cubelos, B.; Giménez, C.; Zafra, F. The Glycine Transporter GLYT1 Interacts with Sec3, a Component of the Exocyst Complex. Neuropharmacology 2005, 49, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Marques, B.L.; Oliveira-Lima, O.C.; Carvalho, G.A.; de Almeida Chiarelli, R.; Ribeiro, R.I.; Parreira, R.C.; da Madeira Freitas, E.M.; Resende, R.R.; Klempin, F.; Ulrich, H.; et al. Neurobiology of Glycine Transporters: From Molecules to Behavior. Neurosci. Biobehav. Rev. 2020, 118, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Lill, M.A. Multi-Dimensional QSAR in Drug Discovery. Drug Discov. Today 2007, 12, 1013–1017. [Google Scholar] [CrossRef]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal Chemistry and the Molecular Operating Environment (MOE): Application of QSAR and Molecular Docking to Drug Discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef]
- Tian, S.; Wang, J.; Li, Y.; Li, D.; Xu, L.; Hou, T. The Application of in Silico Drug-Likeness Predictions in Pharmaceutical Research. Adv. Drug Deliv. Rev. 2015, 86, 2–10. [Google Scholar] [CrossRef]
- Serrano, A.; Imbernón, B.; Pérez-Sánchez, H.; Cecilia, J.M.; Bueno-Crespo, A.; Abellán, J.L. QN-Docking: An Innovative Molecular Docking Methodology Based on Q-Networks. Appl. Soft Comput. 2020, 96, 106678. [Google Scholar] [CrossRef]
- El Khatabi, K.; El-mernissi, R.; Aanouz, I.; Ajana, M.A.; Lakhlifi, T.; Shahinozzaman, M.; Bouachrine, M. Benzimidazole Derivatives in Identifying Novel Acetylcholinesterase Inhibitors: A Combination of 3D-QSAR, Docking and Molecular Dynamics Simulation. Phys. Chem. Res. 2022, 10, 237–249. [Google Scholar] [CrossRef]
- de Oliveira, T.A.; Medaglia, L.R.; Maia, E.H.B.; Assis, L.C.; de Carvalho, P.B.; da Silva, A.M.; Taranto, A.G. Evaluation of Docking Machine Learning and Molecular Dynamics Methodologies for DNA-Ligand Systems. Pharmaceuticals 2022, 15, 132. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jia, Y.; Jing, G.; Wu, X. A Novel Toxicity Prediction Model for Hydrazine Compounds Based on 1D–3D Molecular Descriptors. Comput. Toxicol. 2021, 18, 100169. [Google Scholar] [CrossRef]
- Halder, A.K.; Jha, T. Validated Predictive QSAR Modeling of N-Aryl-Oxazolidinone-5-Carboxamides for Anti-HIV Protease Activity. Bioorg. Med. Chem. Lett. 2010, 20, 6082–6087. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Kar, S.; Das, R.N. Validation of QSAR Models. In Understanding the Basics of QSAR for Applications in Pharmaceutical Sciences and Risk Assessment; Elsevier: Amsterdam, The Netherlands, 2015; pp. 231–289. ISSN 978-0-12-801505-6. [Google Scholar]
- El Mchichi, L.; El Aissouq, A.; Kasmi, R.; Belhassan, A.; El-Mernissi, R.; Ouammou, A.; Lakhlifi, T.; Bouachrine, M. In Silico Design of Novel Pyrazole Derivatives Containing Thiourea Skeleton as Anti-Cancer Agents Using: 3D QSAR, Drug-Likeness Studies, ADMET Prediction and Molecular Docking. Mater. Today Proc. 2021, 45, 7661–7674. [Google Scholar] [CrossRef]
- Mazigh, M.; El’mbarki, C.; Hadni, H.; Elhallaoui, M. QSAR Studies Combined with DFT-Calculations and Molecular Docking of Polyamine-Sensitive Inhibitors of the NMDA Receptor. Mediterr. J. Chem. 2019, 9, 164–174. [Google Scholar] [CrossRef] [Green Version]
- Abdullahi, M.; Shallangwa, G.A.; Uzairu, A. In Silico QSAR and Molecular Docking Simulation of Some Novel Aryl Sulfonamide Derivatives as Inhibitors of H5N1 Influenza A Virus Subtype. Beni-Suef Univ. J. Basic Appl. Sci. 2020, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Krieger, E.; Darden, T.; Nabuurs, S.B.; Finkelstein, A.; Vriend, G. Making Optimal Use of Empirical Energy Functions: Force-Field Parameterization in Crystal Space. Proteins Struct. Funct. Bioinforma. 2004, 57, 678–683. [Google Scholar] [CrossRef]
- Bastianoni, A.; Guastaldi, E.; Barbagli, A.; Bernardinetti, S.; Zirulia, A.; Brancale, M.; Colonna, T. Multivariate Analysis Applied to Aquifer Hydrogeochemical Evaluation: A Case Study in the Coastal Significant Subterranean Water Body between “Cecina River and San Vincenzo”, Tuscany (Italy). Appl. Sci. 2021, 11, 7595. [Google Scholar] [CrossRef]
- Daoui, O.; Elkhattabi, S.; Chtita, S.; Elkhalabi, R.; Zgou, H.; Benjelloun, A.T. QSAR, Molecular Docking and ADMET Properties in Silico Studies of Novel 4,5,6,7-Tetrahydrobenzo[D]-Thiazol-2-Yl Derivatives Derived from Dimedone as Potent Anti-Tumor Agents through Inhibition of C-Met Receptor Tyrosine Kinase. Heliyon 2021, 7, e07463. [Google Scholar] [CrossRef]
- Matsuzaka, Y.; Uesawa, Y. A Deep Learning-Based Quantitative Structure–Activity Relationship System Construct Prediction Model of Agonist and Antagonist with High Performance. Int. J. Mol. Sci. 2022, 23, 2141. [Google Scholar] [CrossRef]
- Uyanık, G.K.; Güler, N. A Study on Multiple Linear Regression Analysis. Procedia Soc. Behav. Sci. 2013, 106, 234–240. [Google Scholar] [CrossRef] [Green Version]
- Kravić, N.; Savosina, J.; Agafonova-Moroz, M.; Babain, V.; Legin, A.; Kirsanov, D. Nonlinear Multivariate Regression Algorithms for Improving Precision of Multisensor Potentiometry in Analysis of Spent Nuclear Fuel Reprocessing Solutions. Chemosensors 2022, 10, 90. [Google Scholar] [CrossRef]
- Ruiz, I.L.; Gómez-Nieto, M.Á. Study of the Applicability Domain of the QSAR Classification Models by Means of the Rivality and Modelability Indexes. Molecules 2018, 23, 2756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pourbasheer, E.; Riahi, S.; Ganjali, M.R.; Norouzi, P. Quantitative Structure–Activity Relationship (QSAR) Study of Interleukin-1 Receptor Associated Kinase 4 (IRAK-4) Inhibitor Activity by the Genetic Algorithm and Multiple Linear Regression (GA-MLR) Method. J. Enzyme Inhib. Med. Chem. 2010, 25, 844–853. [Google Scholar] [CrossRef]
- Chtita, S.; Belhassan, A.; Bakhouch, M.; Taourati, A.I.; Aouidate, A.; Belaidi, S.; Moutaabbid, M.; Belaaouad, S.; Bouachrine, M.; Lakhlifi, T. QSAR Study of Unsymmetrical Aromatic Disulfides as Potent Avian SARS-CoV Main Protease Inhibitors Using Quantum Chemical Descriptors and Statistical Methods. Chemom. Intell. Lab. Syst. 2021, 210, 104266. [Google Scholar] [CrossRef]
- de Castro Rezende, K.B.; da Cunha, A.J.L.; Amim Junior, J.; Bornia, R.G. External Validation of the Fetal Medicine Foundation Algorithm for the Prediction of Preeclampsia in a Brazilian Population. Pregnancy Hypertens. 2019, 17, 64–68. [Google Scholar] [CrossRef]
- Rafało, M. Cross Validation Methods: Analysis Based on Diagnostics of Thyroid Cancer Metastasis. ICT Express 2021, S2405959521000552. [Google Scholar] [CrossRef]
- Chtita, S.; Ghamali, M.; Ousaa, A.; Aouidate, A.; Belhassan, A.; Taourati, A.I.; Masand, V.H.; Bouachrine, M.; Lakhlifi, T. QSAR Study of Anti-Human African Trypanosomiasis Activity for 2-Phenylimidazopyridines Derivatives Using DFT and Lipinski’s Descriptors. Heliyon 2019, 5, e01304. [Google Scholar] [CrossRef] [Green Version]
- Golbraikh, A.; Tropsha, A. Beware of Q2! J. Mol. Graph. Model. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. ILOGP: A Simple, Robust, and Efficient Description of n -Octanol/Water Partition Coefficient for Drug Design Using the GB/SA Approach. J. Chem. Inf. Model. 2014, 54, 3284–3301. [Google Scholar] [CrossRef]
- Daina, A.; Zoete, V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belhassan, A.; Zaki, H.; Benlyas, M.; Lakhlifi, T.; Bouachrine, M. Study of Novel Triazolo-Benzodiazepine Analogues as Antidepressants Targeting by Molecular Docking and ADMET Properties Prediction. Heliyon 2019, 5, e02446. [Google Scholar] [CrossRef] [PubMed]
- Adamski, A.; Kruszka, D.; Dutkiewicz, Z.; Kubicki, M.; Gorczyński, A.; Patroniak, V. Novel Family of Fused Tricyclic [1,4]Diazepines: Design, Synthesis, Crystal Structures and Molecular Docking Studies. Tetrahedron 2017, 73, 3377–3386. [Google Scholar] [CrossRef]
- Penmatsa, A.; Wang, K.H.; Gouaux, E. X-Ray Structure of Dopamine Transporter Elucidates Antidepressant Mechanism. Nature 2013, 503, 85–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zentrum Für Bioinformatik: Universität Hamburg-Proteins Plus Server. Available online: https://proteins.plus/ (accessed on 8 March 2022).
- Spiegel, J.; Senderowitz, H. Evaluation of QSAR Equations for Virtual Screening. Int. J. Mol. Sci. 2020, 21, 7828. [Google Scholar] [CrossRef] [PubMed]
- Österberg, T.; Norinder, U. Prediction of Drug Transport Processes Using Simple Parameters and PLS Statistics The Use of ACD/LogP and ACD/ChemSketch Descriptors. Eur. J. Pharm. Sci. 2001, 12, 327–337. [Google Scholar] [CrossRef]
- Milne, G.W.A. Software Review of ChemBioDraw 12.0. J. Chem. Inf. Model. 2010, 50, 2053. [Google Scholar] [CrossRef]
- Citation Gaussienne 09 | Gaussian.Com. Available online: https://gaussian.com/g09citation/ (accessed on 8 March 2022).
- Parr, R.G.; Weitao, Y. Density-Functional Theory of Atoms and Molecules; International Series of Monographs on Chemistry; Oxford University Press: New York, NY, USA, 1995; ISBN 978-0-19-509276-9. [Google Scholar]
- Introducing the XLSTAT Free Edition. Available online: https://www.xlstat.com/en/news/introducing-the-xlstat-free-edition (accessed on 8 March 2022).
- Guerra Tort, C.; Aguiar Pulido, V.; Suárez Ulloa, V.; Docampo Boedo, F.; López Gestal, J.M.; Pereira Loureiro, J. Electronic Health Records Exploitation Using Artificial Intelligence Techniques. Proceedings 2020, 54, 60. [Google Scholar] [CrossRef]
- Hadni, H.; Elhallaoui, M. 3D-QSAR, Docking and ADMET Properties of Aurone Analogues as Antimalarial Agents. Heliyon 2020, 6, e03580. [Google Scholar] [CrossRef]
- van der Voet, H. Comparing the Predictive Accuracy of Models Using a Simple Randomization Test. Chemom. Intell. Lab. Syst. 1994, 25, 313–323. [Google Scholar] [CrossRef]
- Ebenezer, O.; Damoyi, N.; Jordaan, M.A.; Shapi, M. Unveiling of Pyrimidindinones as Potential Anti-Norovirus Agents—A Pharmacoinformatic-Based Approach. Molecules 2022, 27, 380. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, S. Molecular Docking, Drug Likeness, and ADMET Analyses of Passiflora Compounds as P-Glycoprotein (P-Gp) Inhibitor for the Treatment of Cancer. Curr. Pharmacol. Rep. 2020, 6, 429–440. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Egan, W.J.; Lauri, G. Prediction of Intestinal Permeability. Adv. Drug Deliv. Rev. 2002, 54, 273–289. [Google Scholar] [CrossRef]
- Gomes, D.; Silvestre, S.; Duarte, A.P.; Venuti, A.; Soares, C.P.; Passarinha, L.; Sousa, Â. In Silico Approaches: A Way to Unveil Novel Therapeutic Drugs for Cervical Cancer Management. Pharmaceuticals 2021, 14, 741. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- PkCSM. Available online: http://biosig.unimelb.edu.au/pkcsm/run_example? (accessed on 8 March 2022).
- Bassani, D.; Pavan, M.; Bolcato, G.; Sturlese, M.; Moro, S. Re-Exploring the Ability of Common Docking Programs to Correctly Reproduce the Binding Modes of Non-Covalent Inhibitors of SARS-CoV-2 Protease Mpro. Pharmaceuticals 2022, 15, 180. [Google Scholar] [CrossRef]
- Iqbal, D.; Rehman, M.T.; Bin Dukhyil, A.; Rizvi, S.M.D.; Al Ajmi, M.F.; Alshehri, B.M.; Banawas, S.; Khan, M.S.; Alturaiki, W.; Alsaweed, M. High-Throughput Screening and Molecular Dynamics Simulation of Natural Product-like Compounds against Alzheimer’s Disease through Multitarget Approach. Pharmaceuticals 2021, 14, 937. [Google Scholar] [CrossRef]
- Norgan, A.P.; Coffman, P.K.; Kocher, J.-P.A.; Katzmann, D.J.; Sosa, C.P. Multilevel Parallelization of AutoDock 4.2. J. Cheminformatics 2011, 3, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- BIOVIA Discovery Studio - BIOVIA - Dassault Systèmes®. Available online: https://www.3ds.com/products-services/biovia/products/molecular-modeling-simulation/biovia-discovery-studio/ (accessed on 8 March 2022).
- Desmond | Schrodinger. Available online: https://www.schrodinger.com/products/desmond (accessed on 28 April 2022).
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N° | ae | ɣ | TE | HBD | SE | TD | Log10IC50 |
|---|---|---|---|---|---|---|---|
| 2 | 41.05 | 40.3 | 12.9136 | 1 | 2.609 | 11 | 0.47712126 |
| 3 | 43.9 | 43.9 | 12.833 | 1 | 3.43 | 11 | 1.61278386 |
| 4 | 43.9 | 43.9 | 12.2837 | 1 | 3.7251 | 11 | 0.77815125 |
| 5 | 41.96 | 44.6 | 10.5041 | 1 | 3.3398 | 11 | 1.96378783 |
| 6 | 43.87 | 50.9 | 5.8562 | 1 | 3.5148 | 10 | 0 |
| 7 | 43.11 | 54.3 | 10.9678 | 1 | 3.5012 | 10 | 0.77815125 |
| 8 | 44.29 | 57.7 | 10.6827 | 1 | 3.31 | 10 | 0.47712126 |
| 10 | 43.05 | 49.2 | 13.6092 | 1 | 3.581 | 10 | 2.69372695 |
| 11 | 43.82 | 51.6 | 16.8366 | 1 | 3.7574 | 10 | 2.67577834 |
| 12 | 42.28 | 53.7 | 12.0135 | 1 | 3.2349 | 10 | 1.87506126 |
| 13 | 43.11 | 54.3 | 15.9864 | 1 | 3.5181 | 10 | 2.90794852 |
| 14 | 43.87 | 50.9 | 12.0765 | 1 | 3.6081 | 10 | 1.36172784 |
| 15 | 43.11 | 54.3 | 15.211 | 1 | 3.5393 | 10 | 1.65321251 |
| 17 | 45.28 | 46.3 | 11.4242 | 1 | 4.1245 | 10 | 0.77815125 |
| 18 | 46.55 | 43.3 | 11.1295 | 1 | 4.5556 | 11 | 2.53147892 |
| 19 | 45.81 | 43.2 | 11.5098 | 1 | 4.5198 | 11 | 2.71096312 |
| 20 | 46.95 | 45.7 | 12.3354 | 1 | 3.7527 | 11 | 0.95424251 |
| 21 | 45.81 | 43.2 | 11.5767 | 1 | 3.7965 | 11 | 1.462398 |
| 22 | 46.55 | 43.3 | 12.3649 | 1 | 4.0183 | 12 | 2.24797327 |
| 23 | 45.81 | 43.2 | 11.5647 | 1 | 3.7752 | 12 | 3.23121465 |
| 24 | 46.55 | 43.3 | 12.3272 | 1 | 3.9781 | 13 | 3.24526584 |
| 25 | 41.5 | 38.4 | 18.5378 | 1 | 3.5883 | 11 | 3.52659771 |
| 26 | 40.85 | 43.8 | 14.1068 | 1 | 3.3294 | 11 | 1.8920946 |
| 29 | 43.14 | 46.6 | 17.5352 | 1 | 3.5318 | 11 | 2.95616843 |
| 30 | 42.67 | 45.3 | 11.6496 | 1 | 3.2701 | 10 | 0 |
| 31 | 43.05 | 49.2 | 15.9695 | 1 | 3.5677 | 10 | 0 |
| 33 | 52.92 | 45 | 13.4879 | 1 | 3.9397 | 14 | 2.97589114 |
| 34 | 41.86 | 48.5 | 7.3285 | 2 | 2.3774 | 10 | 0.47712126 |
| 37 | 45.11 | 46 | 10.9824 | 1 | 3.3677 | 10 | 0 |
| 38 | 44.35 | 48.9 | 16.6401 | 1 | 3.3293 | 10 | 1 |
| 40 | 39.53 | 51.1 | 7.6774 | 2 | 2.2337 | 10 | 1.76342799 |
| 41 | 38.77 | 54.5 | 13.3491 | 2 | 2.2124 | 10 | 1.65321251 |
| 42 | 40.15 | 49.3 | 8.3209 | 1 | 3.3316 | 10 | 0.84509804 |
| 43 | 41.98 | 47.7 | 10.8208 | 1 | 3.622 | 10 | 0.47712126 |
| 44 | 38.2 | 52.2 | 5.2114 | 2 | 2.8062 | 10 | 2.22530928 |
| 1 * | 45.25 | 41.3 | 39.2347 | 1 | 2.6809 | 13 | 1.56820172 |
| 9 * | 43.81 | 46.2 | 10.3084 | 1 | 3.6309 | 10 | 0 |
| 16 * | 43.6 | 55.2 | 8.802 | 2 | 3.3159 | 10 | 0.90308999 |
| 27 * | 43.14 | 46.6 | 18.0072 | 1 | 3.6639 | 11 | 1.25527251 |
| 28 * | 43.14 | 46.6 | 18.0105 | 1 | 3.682 | 11 | 2.32428246 |
| 32 * | 53.84 | 50.1 | 58.0893 | 1 | 5.1759 | 14 | 0 |
| 35 * | 41.48 | 47.3 | 9.0526 | 1 | 2.9116 | 10 | 0.47712126 |
| 36 * | 43.31 | 45.9 | 9.4978 | 1 | 3.0768 | 10 | 0.60205999 |
| 39 * | 44.32 | 54.8 | 9.9783 | 1 | 2.9 | 10 | 0.30103 |
| Source | Value | Standard Deviation | t | Pr > |t| | Lower Terminal (95%) | Higher Terminal (95%) |
|---|---|---|---|---|---|---|
| Constante | −10.408 | 3.172 | −3.281 | 0.003 | −16.905 | −3.910 |
| αe | −0.279 | 0.079 | −3.550 | 0.001 | −0.441 | −0.118 |
| ɣ | 0.070 | 0.033 | 2.101 | 0.045 | 0.002 | 0.138 |
| TE | 0.156 | 0.042 | 3.731 | 0.001 | 0.071 | 0.242 |
| HBD | 1.830 | 0.571 | 3.208 | 0.003 | 0.661 | 2.999 |
| SE | 1.716 | 0.387 | 4.429 | 0.000 | 0.922 | 2.510 |
| TD | 1.030 | 0.208 | 4.956 | <0.0001 | 0.604 | 1.455 |
| Source | DDL | Total Square | Mean Square | F | Pr > F |
|---|---|---|---|---|---|
| Model | 6 | 26.753 | 4.459 | 10.325 | <0.0001 |
| Error | 28 | 12.092 | 0.432 | ||
| Adjusted total | 34 | 38.846 |
| Molecule Number | Observed Log10IC50 | Predicted Log10IC50(MLR) | Predicted Log10IC50(MNLR) |
|---|---|---|---|
| 9 * | 0.000 | 0.545 | 0.380 |
| 16 * | 0.903 | 2.285 | 2.134 |
| 27 * | 1.255 | 3.051 | 3.342 |
| 28 * | 2.324 | 3.083 | 3.372 |
| 35 * | 0.477 | −0.158 | −0.164 |
| 36 * | 0.602 | −0.414 | −0.429 |
| 39 * | 0.301 | −0.304 | −0.046 |
| Molecules Number | Observed Log10IC50 | Predicted Log10IC50 (MLR) | Predicted Log10IC50 (MNLR) | Predicted Log10IC50 (CV) |
|---|---|---|---|---|
| 2 | 0.47712126 | 0.588 | 0.679 | 0.664 |
| 3 | 1.61278386 | 1.439 | 1.415 | 1.428 |
| 4 | 0.77815125 | 1.860 | 1.790 | 1.922 |
| 5 | 1.96378783 | 1.511 | 1.464 | 1.441 |
| 6 | 0 | −0.040 | 0.405 | −0.053 |
| 7 | 0.77815125 | 1.186 | 1.157 | 1.245 |
| 8 | 0.47712126 | 0.721 | 0.933 | 0.801 |
| 10 | 2.69372695 | 1.397 | 1.195 | 1.321 |
| 11 | 2.67577834 | 2.157 | 2.240 | 2.057 |
| 12 | 1.87506126 | 1.083 | 1.018 | 0.970 |
| 13 | 2.90794852 | 2.000 | 2.081 | 1.827 |
| 14 | 1.36172784 | 1.093 | 0.954 | 1.075 |
| 15 | 1.65321251 | 1.915 | 1.933 | 1.959 |
| 17 | 0.77815125 | 1.163 | 0.982 | 1.247 |
| 18 | 2.53147892 | 2.322 | 2.392 | 2.250 |
| 19 | 2.71096312 | 2.520 | 2.548 | 2.458 |
| 20 | 0.95424251 | 1.189 | 1.245 | 1.217 |
| 21 | 1.462398 | 1.289 | 1.302 | 1.272 |
| 22 | 2.24797327 | 2.623 | 2.680 | 2.667 |
| 23 | 3.23121465 | 2.281 | 2.338 | 2.174 |
| 24 | 3.24526584 | 3.578 | 3.538 | 3.710 |
| 25 | 3.52659771 | 2.891 | 3.183 | 2.562 |
| 26 | 1.8920946 | 2.311 | 2.171 | 2.396 |
| 29 | 2.95616843 | 2.751 | 3.000 | 2.717 |
| 30 | 0 | 0.391 | 0.176 | 0.443 |
| 31 | 0 | 1.744 | 1.675 | 1.934 |
| 33 | 2.97589114 | 3.062 | 2.907 | 3.204 |
| 34 | 0.47712126 | 0.463 | 0.555 | 0.454 |
| 37 | 0 | −0.178 | −0.276 | −0.228 |
| 38 | 1 | 1.056 | 1.151 | 1.068 |
| 40 | 1.76342799 | 1.104 | 1.136 | 0.867 |
| 41 | 1.65321251 | 2.404 | 2.199 | 2.852 |
| 42 | 0.84509804 | 0.959 | 0.855 | 0.993 |
| 43 | 0.47712126 | 1.226 | 0.961 | 1.294 |
| Model | R | R^2 | Q^2 | Model | R | R^2 | Q^2 |
|---|---|---|---|---|---|---|---|
| Original | 0.829884 | 0.688707 | 0.572045 | Random 51 | 0.206983 | 0.042842 | −0.46272 |
| Random 1 | 0.252331 | 0.063671 | −0.50878 | Random 52 | 0.537396 | 0.288794 | −0.15592 |
| Random 2 | 0.457615 | 0.209411 | −0.17702 | Random 53 | 0.379861 | 0.144294 | −0.43774 |
| Random 3 | 0.47795 | 0.228436 | −0.41001 | Random 54 | 0.367538 | 0.135084 | −0.29876 |
| Random 4 | 0.375518 | 0.141014 | −0.34708 | Random 55 | 0.179251 | 0.032131 | −0.54121 |
| Random 5 | 0.422447 | 0.178462 | −0.39625 | Random 56 | 0.663141 | 0.439756 | 0.029755 |
| Random 6 | 0.480602 | 0.230979 | −0.17775 | Random 57 | 0.36146 | 0.130653 | −0.41471 |
| Random 7 | 0.306791 | 0.09412 | −0.47744 | Random 58 | 0.445943 | 0.198865 | −0.22915 |
| Random 8 | 0.354955 | 0.125993 | −0.40713 | Random 59 | 0.417956 | 0.174687 | −0.19669 |
| Random 9 | 0.209847 | 0.044036 | −0.71484 | Random 60 | 0.204369 | 0.041767 | −0.88175 |
| Random 10 | 0.395267 | 0.156236 | −0.36218 | Random 61 | 0.557804 | 0.311145 | 0.016035 |
| Random 11 | 0.520928 | 0.271366 | −0.1806 | Random 62 | 0.50639 | 0.256431 | −0.3063 |
| Random 12 | 0.510412 | 0.260521 | −0.21009 | Random 63 | 0.37293 | 0.139077 | −0.46899 |
| Random 13 | 0.427634 | 0.182871 | −0.23082 | Random 64 | 0.383643 | 0.147182 | −0.41262 |
| Random 14 | 0.445148 | 0.198156 | −0.41414 | Random 65 | 0.414428 | 0.171751 | −0.30301 |
| Random 15 | 0.21278 | 0.045275 | −0.4451 | Random 66 | 0.292763 | 0.08571 | −0.36258 |
| Random 16 | 0.516892 | 0.267178 | −0.45198 | Random 67 | 0.526141 | 0.276824 | −0.1287 |
| Random 17 | 0.37686 | 0.142024 | −0.55449 | Random 68 | 0.284657 | 0.08103 | −0.54548 |
| Random 18 | 0.154692 | 0.023929 | −0.85659 | Random 69 | 0.456042 | 0.207974 | −0.2171 |
| Random 19 | 0.491084 | 0.241163 | −0.24676 | Random 70 | 0.451139 | 0.203526 | −0.15451 |
| Random 20 | 0.424795 | 0.180451 | −0.30099 | Random 71 | 0.402163 | 0.161735 | −0.15347 |
| Random 21 | 0.513699 | 0.263886 | −0.1961 | Random 72 | 0.480122 | 0.230517 | −0.17729 |
| Random 22 | 0.316251 | 0.100015 | −0.30938 | Random 73 | 0.426294 | 0.181727 | −0.22948 |
| Random 23 | 0.301949 | 0.091173 | −0.63655 | Random 74 | 0.475859 | 0.226442 | −0.23411 |
| Random 24 | 0.332628 | 0.110641 | −0.8224 | Random 75 | 0.462608 | 0.214006 | −0.12839 |
| Random 25 | 0.633727 | 0.401609 | 0.166923 | Random 76 | 0.53816 | 0.289616 | −0.33075 |
| Random 26 | 0.328704 | 0.108046 | −0.48201 | Random 77 | 0.383709 | 0.147233 | −0.30145 |
| Random 27 | 0.46585 | 0.217016 | −0.16011 | Random 78 | 0.38822 | 0.150715 | −0.41903 |
| Random 28 | 0.441731 | 0.195126 | −0.25279 | Random 79 | 0.528782 | 0.279611 | −0.29561 |
| Random 29 | 0.355019 | 0.126039 | −0.31878 | Random 80 | 0.330001 | 0.1089 | −0.41611 |
| Random 30 | 0.329982 | 0.108888 | −0.42698 | Random 81 | 0.413654 | 0.171109 | −0.22613 |
| Random 31 | 0.378435 | 0.143213 | −0.25482 | Random 82 | 0.493491 | 0.243533 | −0.12853 |
| Random 32 | 0.462326 | 0.213746 | −0.13151 | Random 83 | 0.381202 | 0.145315 | −0.49761 |
| Random 33 | 0.343488 | 0.117984 | −0.53921 | Random 84 | 0.323593 | 0.104712 | −0.30559 |
| Random 34 | 0.462673 | 0.214066 | −0.27221 | Random 85 | 0.32106 | 0.103079 | −0.33856 |
| Random 35 | 0.35063 | 0.122941 | −0.3394 | Random 86 | 0.30071 | 0.090427 | −0.55488 |
| Random 36 | 0.522964 | 0.273491 | −0.09258 | Random 87 | 0.518334 | 0.26867 | −0.1494 |
| Random 37 | 0.222631 | 0.049564 | −0.75169 | Random 88 | 0.387695 | 0.150307 | −0.45639 |
| Random 38 | 0.241784 | 0.058459 | −0.47485 | Random 89 | 0.36652 | 0.134337 | −0.30196 |
| Random 39 | 0.339537 | 0.115286 | −0.4132 | Random 90 | 0.279562 | 0.078155 | −0.47573 |
| Random 40 | 0.448316 | 0.200987 | −0.47037 | Random 91 | 0.575806 | 0.331552 | −0.03852 |
| Random 41 | 0.487561 | 0.237716 | −0.34662 | Random 92 | 0.5706 | 0.325585 | 0.021398 |
| Random 42 | 0.369003 | 0.136164 | −0.33599 | Random 93 | 0.381837 | 0.1458 | −0.44739 |
| Random 43 | 0.400756 | 0.160605 | −0.30621 | Random 94 | 0.385236 | 0.148406 | −0.6547 |
| Random 44 | 0.343595 | 0.118058 | −0.42487 | Random 95 | 0.251773 | 0.06339 | −0.49809 |
| Random 45 | 0.390289 | 0.152325 | −0.27962 | Random 96 | 0.446548 | 0.199405 | −0.47359 |
| Random 46 | 0.350185 | 0.12263 | −0.22911 | Random 97 | 0.316743 | 0.100326 | −0.95316 |
| Random 47 | 0.463947 | 0.215247 | −0.27397 | Random 98 | 0.367366 | 0.134958 | −0.3352 |
| Random 48 | 0.37435 | 0.140138 | −0.27999 | Random 99 | 0.631342 | 0.398592 | 0.090509 |
| Random 49 | 0.452168 | 0.204456 | −0.40118 | Random 100 | 0.56162 | 0.315417 | 0.016006 |
| Random 50 | 0.266881 | 0.071225 | −0.47945 |
| Parameter | Equation | Model Score | Threshold | Comment |
|---|---|---|---|---|
| 0.69 | >0.6 | Accepted | ||
| 0.62 | >0.6 | Accepted | ||
| 0.63 | >0.6 | Accepted | ||
| 0.57 | >0.5 | Accepted | ||
| R2 rand | Average of the 100 R2 rand (i) | 0.17 | <R2 | Accepted |
| ‘LOO’ rand | Average of the 100 ‘LOO‘ rand (i) | −0.34 | <Q2cv | Accepted |
| cR2p | cR2p = R* | 0.60 | >0.5 | Accepted |
| Ligands Number | Physico-Chemical Propities | Lipinski Violations | Veber Violations | Egan Violations | Ghose Violations | Synthetic Accessiblity | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Molecular Weight (g/mol) | Molar Refractive Index | Rotatable Bonds | Log p (Octanol/Water) | H-BA | H-BD | ||||||
| Rule | ≤500 | 40 ≤ MR ≤ 130 | <10 | <5 | ≤10 | <5 | ≤1 | Yes/No | Yes/No | Yes/No | 0 < S.A < 10 |
| (1) L6 | 403.34 | 115.02 | 5 | 3.60 | 2 | 1 | 1 | Yes | Yes | Yes | 4.24 |
| (2) L9 | 362.51 | 114.93 | 5 | 3.58 | 2 | 1 | 0 | Yes | Yes | Yes | 4.21 |
| (3) L30 | 366.50 | 112.17 | 5 | 3.75 | 3 | 1 | 0 | Yes | Yes | Yes | 4.97 |
| (4) L31 | 363.50 | 112.73 | 5 | 3.15 | 3 | 1 | 0 | Yes | Yes | Yes | 4.28 |
| (5) L32 | 480.66 | 140.34 | 8 | 3.61 | 4 | 1 | 0 | Yes | Yes | No | 5.19 |
| (6) L37 | 377.52 | 121.65 | 6 | 3.23 | 3 | 2 | 0 | Yes | Yes | Yes | 4.47 |
| (7) nortriptyline | 265.39 | 85.74 | 4 | 3.24 | 1 | 1 | 1 | Yes | Yes | Yes | 3.28 |
| Ligands Number | Absorption | Distribution | Metabolism | Excretion | Toxicity | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Intestinal Absorption (Human) | VDss (Human) | BBB Permeability | CNS Permeability | Substrate | Inhibitor | Total Clearance | AMES Toxicity | ||||||
| CYP | |||||||||||||
| 2D6 | 3A4 | 1A2 | 2C19 | 2C9 | 2D6 | 3A4 | |||||||
| Numeric (% Absorbed) | Numeric (Log L/kg) | Numeric (Log BB) | Numeric (Log PS) | Categorical (Yes/No) | Numeric (Log ml/min/kg) | Categorical (Yes/No) | |||||||
| (1) L6 | 91.194 | 1.431 | 0.199 | −1.06 | Yes | Yes | Yes | Yes | No | Yes | Yes | 1.058 | Not toxic |
| (2) L9 | 93.373 | 1.477 | 0.223 | −1.072 | Yes | Yes | Yes | No | No | Yes | No | 0.978 | Not toxic |
| (3) L30 | 93.344 | 1.242 | 0.176 | −2.055 | Yes | Yes | No | No | No | Yes | No | 0.883 | Not toxic |
| (4) L31 | 95.105 | 1.187 | 0.048 | −1.976 | Yes | Yes | No | No | No | Yes | Yes | 0.948 | Not toxic |
| (5) L32 | 94.331 | 1.038 | −0.378 | −2.005 | No | Yes | Yes | No | No | No | Yes | 0.85 | Not toxic |
| (6) L37 | 92.765 | 1.814 | 0.044 | −0.657 | Yes | Yes | No | No | No | Yes | No | 0.905 | Not toxic |
| (7) nortriptyline | 98.519 | 1.688 | 0.854 | −1.287 | No | Yes | Yes | No | No | Yes | No | 1.077 | Not toxic |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El fadili, M.; Er-Rajy, M.; Kara, M.; Assouguem, A.; Belhassan, A.; Alotaibi, A.; Mrabti, N.N.; Fidan, H.; Ullah, R.; Ercisli, S.; et al. QSAR, ADMET In Silico Pharmacokinetics, Molecular Docking and Molecular Dynamics Studies of Novel Bicyclo (Aryl Methyl) Benzamides as Potent GlyT1 Inhibitors for the Treatment of Schizophrenia. Pharmaceuticals 2022, 15, 670. https://doi.org/10.3390/ph15060670
El fadili M, Er-Rajy M, Kara M, Assouguem A, Belhassan A, Alotaibi A, Mrabti NN, Fidan H, Ullah R, Ercisli S, et al. QSAR, ADMET In Silico Pharmacokinetics, Molecular Docking and Molecular Dynamics Studies of Novel Bicyclo (Aryl Methyl) Benzamides as Potent GlyT1 Inhibitors for the Treatment of Schizophrenia. Pharmaceuticals. 2022; 15(6):670. https://doi.org/10.3390/ph15060670
Chicago/Turabian StyleEl fadili, Mohamed, Mohammed Er-Rajy, Mohammed Kara, Amine Assouguem, Assia Belhassan, Amal Alotaibi, Nidal Naceiri Mrabti, Hafize Fidan, Riaz Ullah, Sezai Ercisli, and et al. 2022. "QSAR, ADMET In Silico Pharmacokinetics, Molecular Docking and Molecular Dynamics Studies of Novel Bicyclo (Aryl Methyl) Benzamides as Potent GlyT1 Inhibitors for the Treatment of Schizophrenia" Pharmaceuticals 15, no. 6: 670. https://doi.org/10.3390/ph15060670