
Antiproliferative Activity of a New Quinazolin-4(3H)-One Derivative via Targeting Aurora Kinase A in Non-Small Cell Lung Cancer

 , ,
, , 
 and
and
Abstract
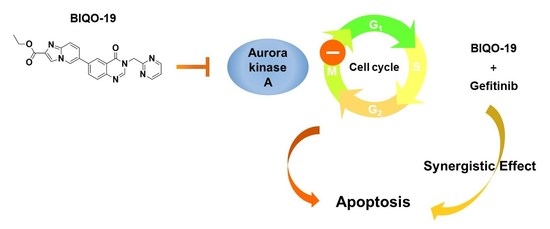
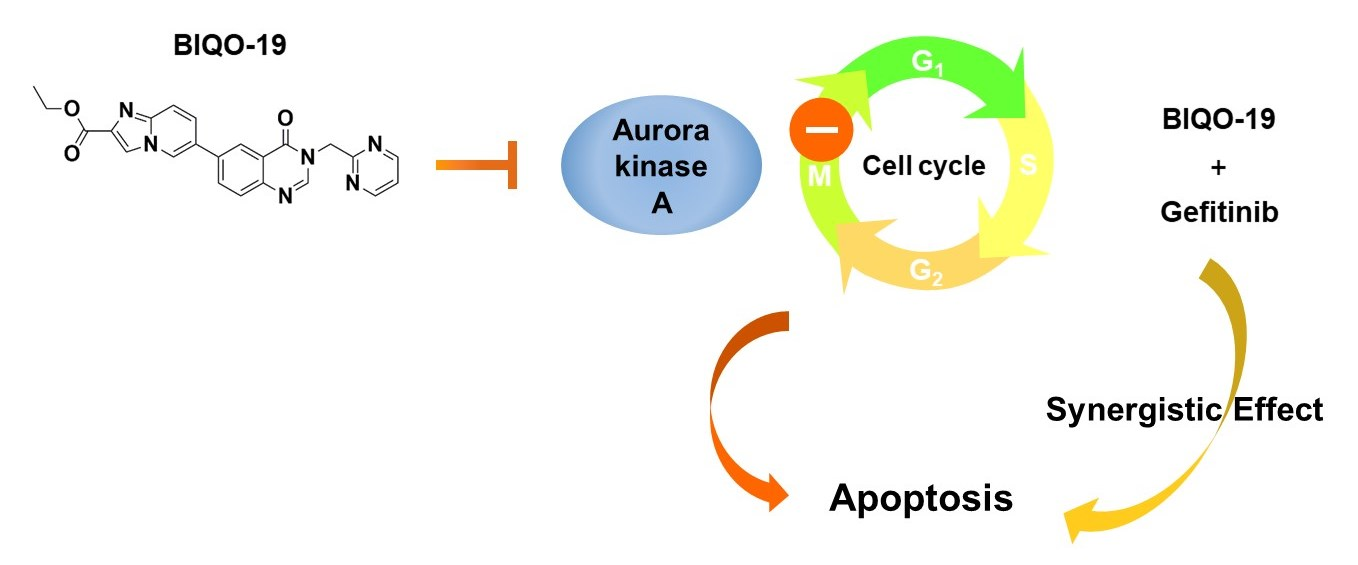
:
1. Introduction
2. Results
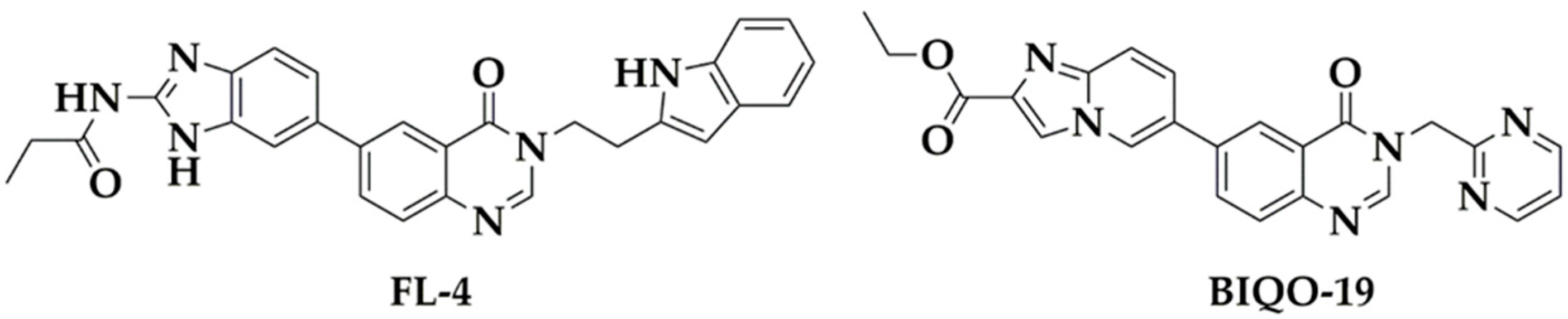
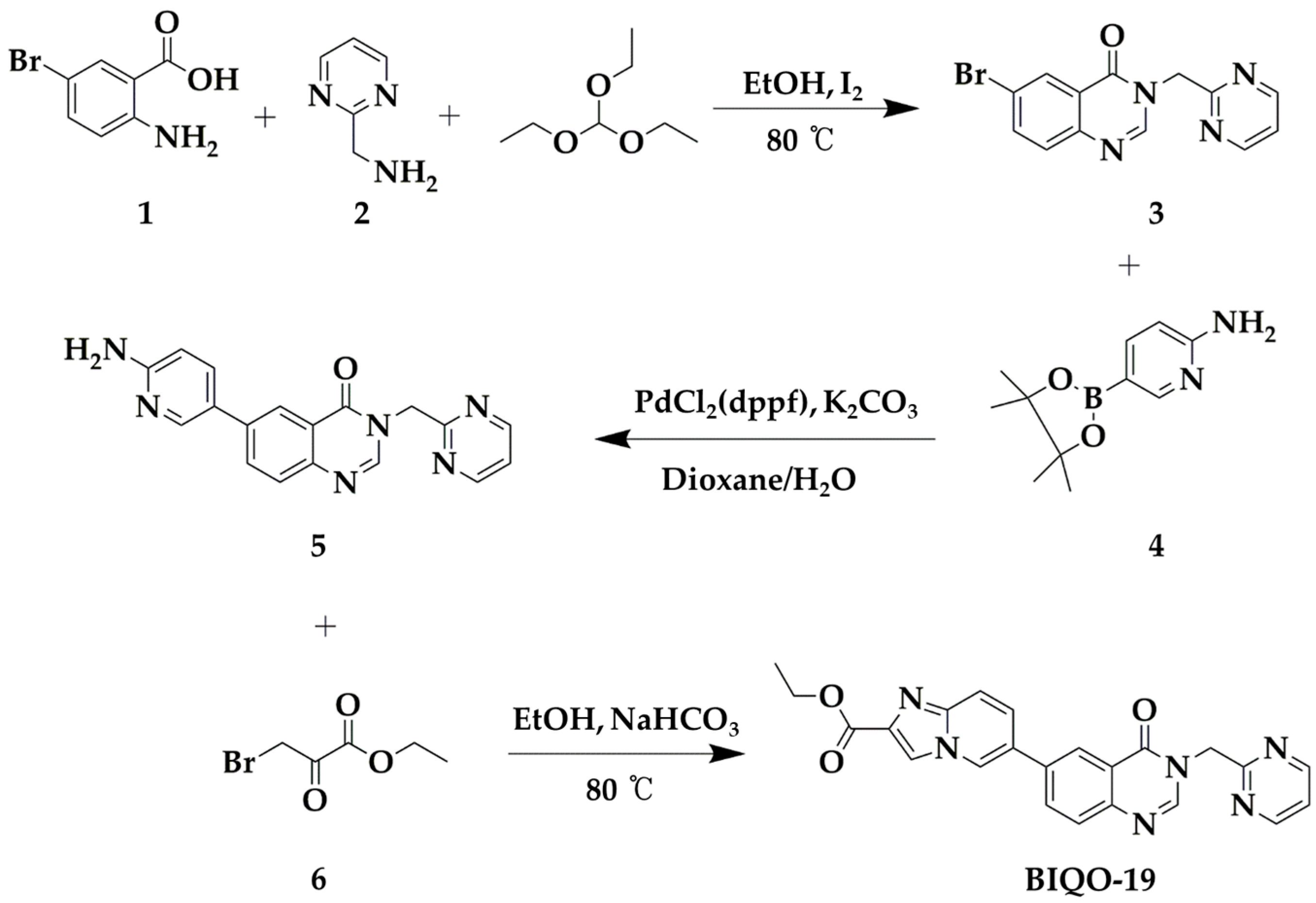
2.1. Synthesis and Pharmacokinetic Profile of the Quinazolin-4(3H)-One Derivative BIQO-19
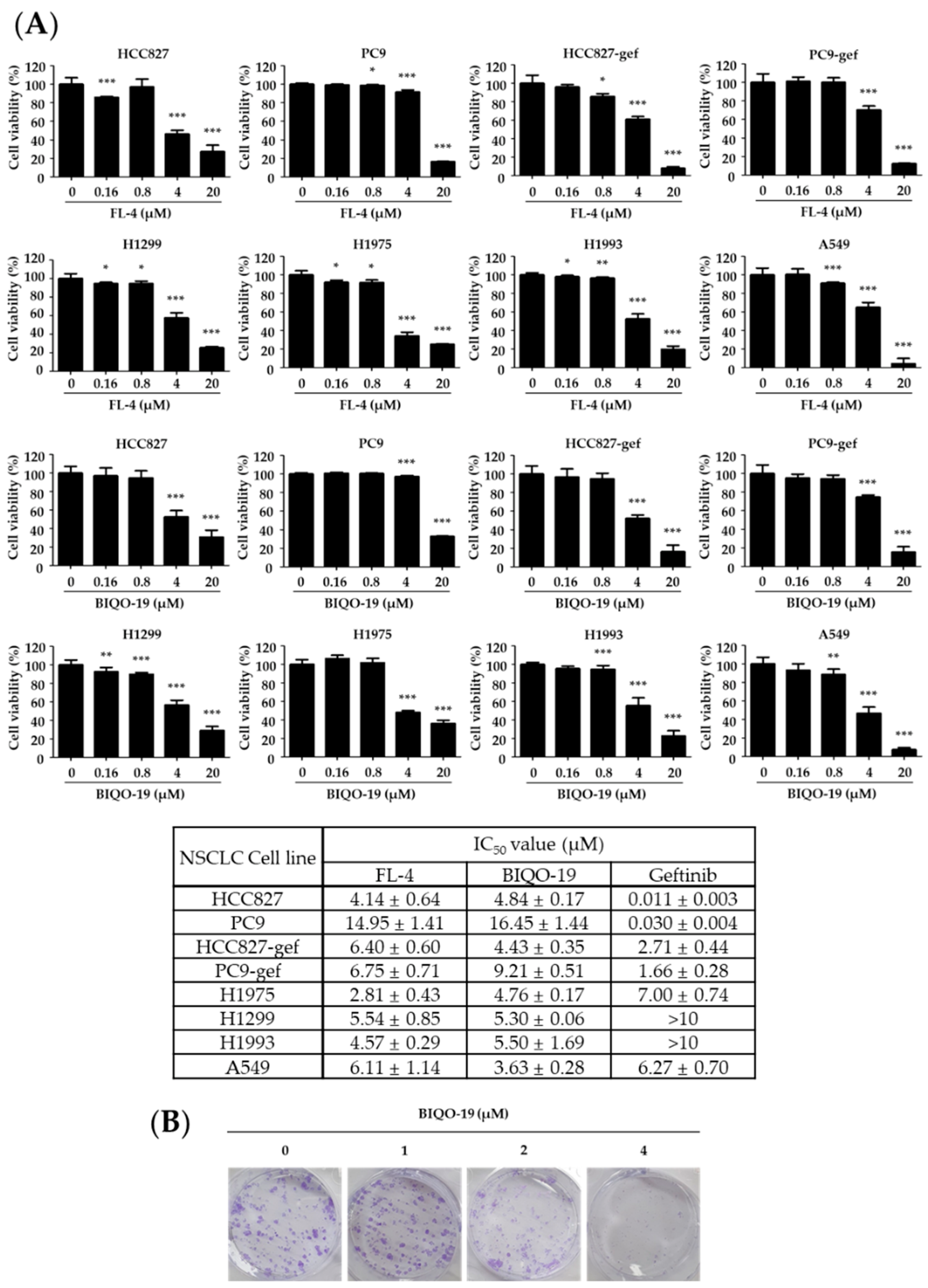
2.2. Antiproliferative Activity of Quinazolin-4(3H)-One Derivative in Human NSCLC Cell Lines
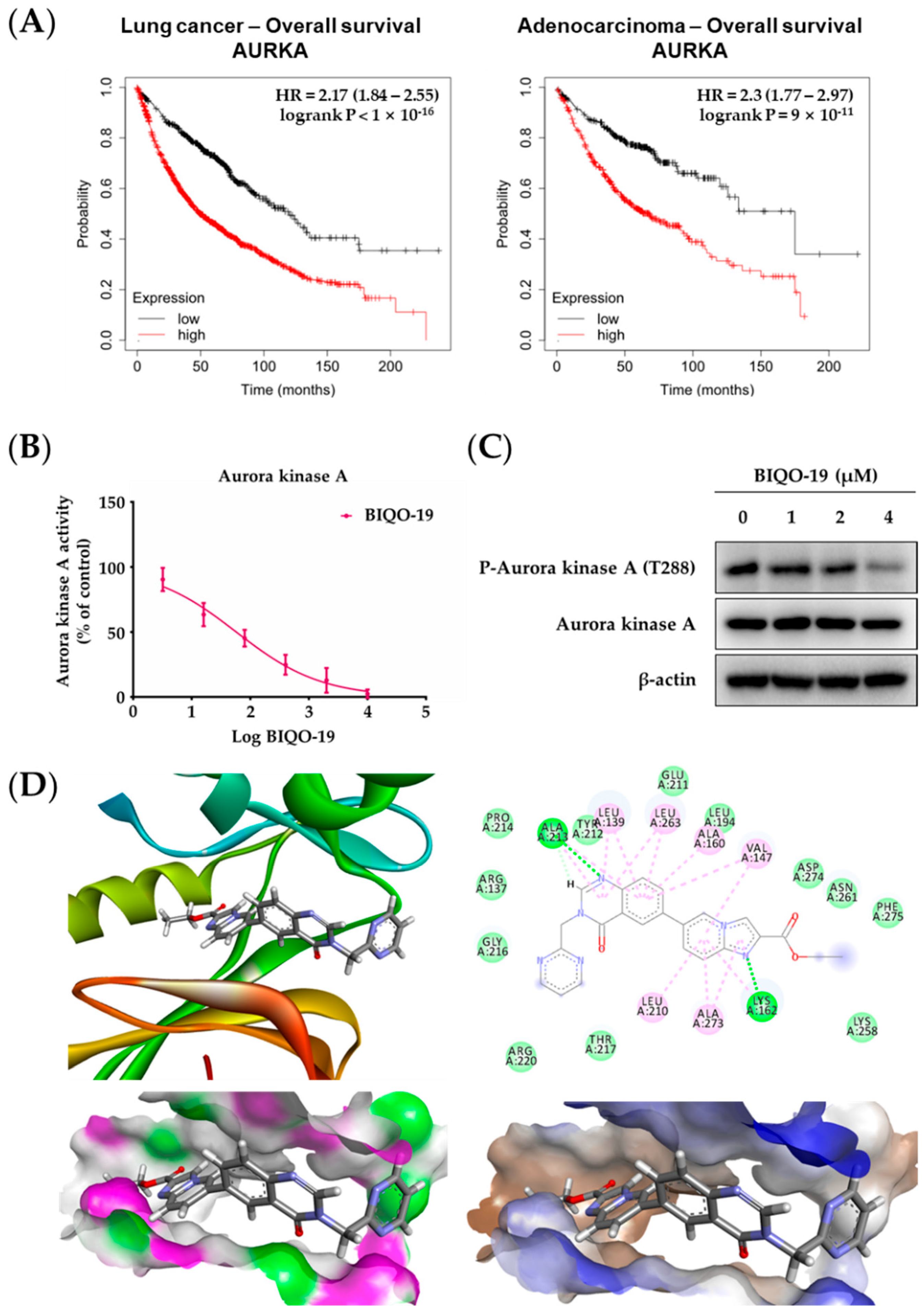
2.3. Effects of BIQO-19 on the Activity and Expression of Aurora Kinase A
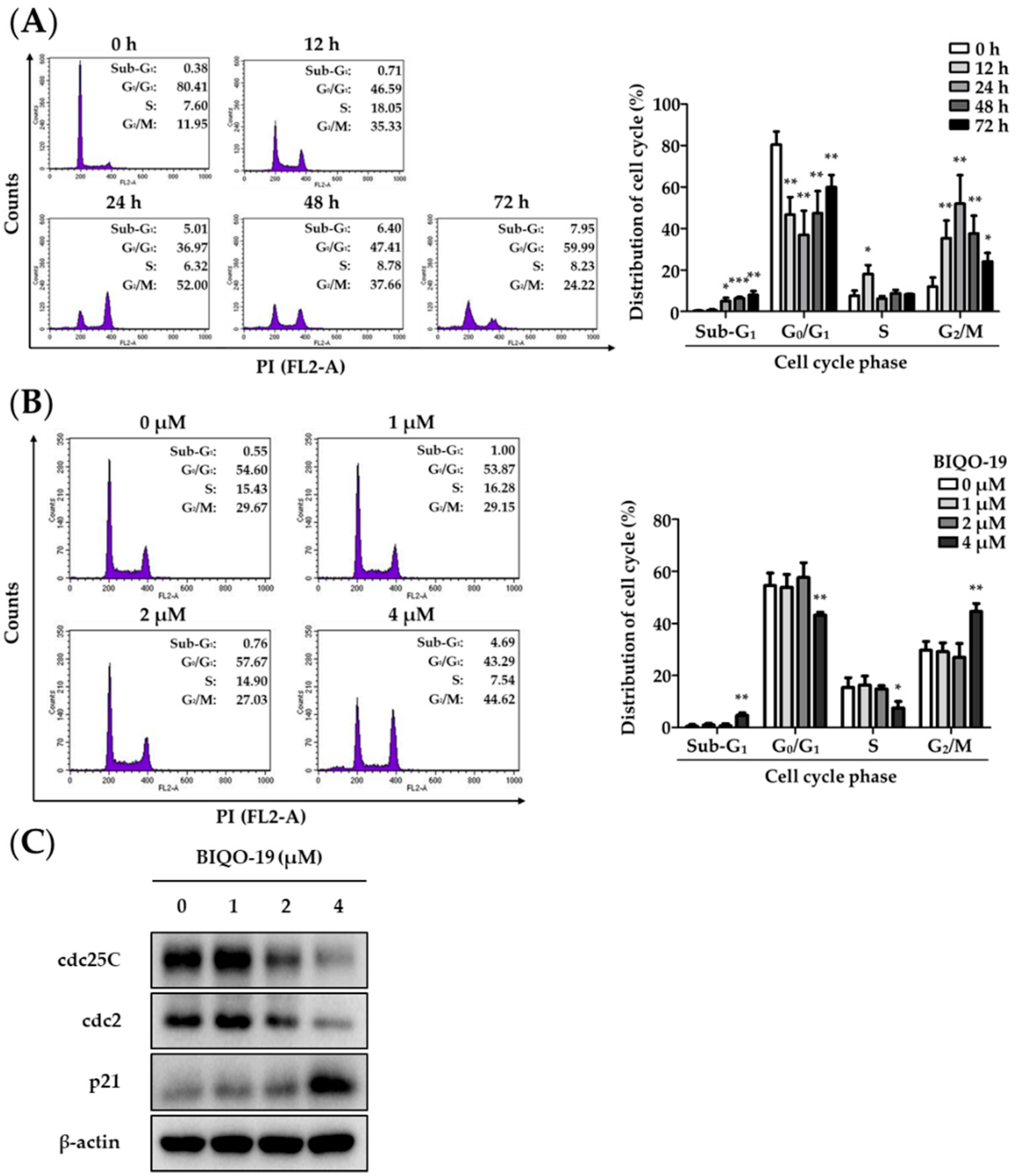
2.4. Effects of BIQO-19 on Cell Cycle Regulation in H1975 Cells
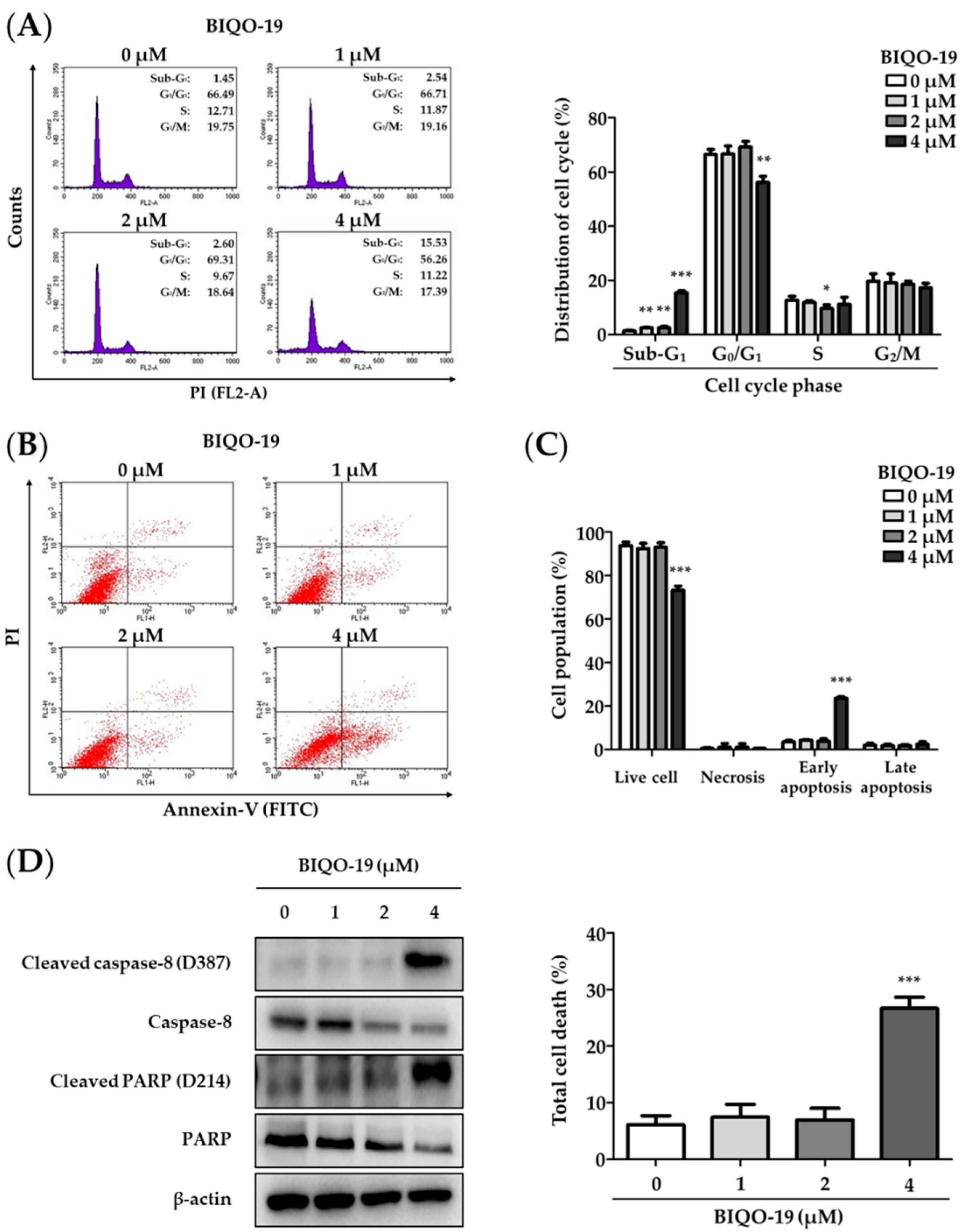
2.5. Effects of BIQO-19 on the Induction of Apoptosis in H1975 Cells
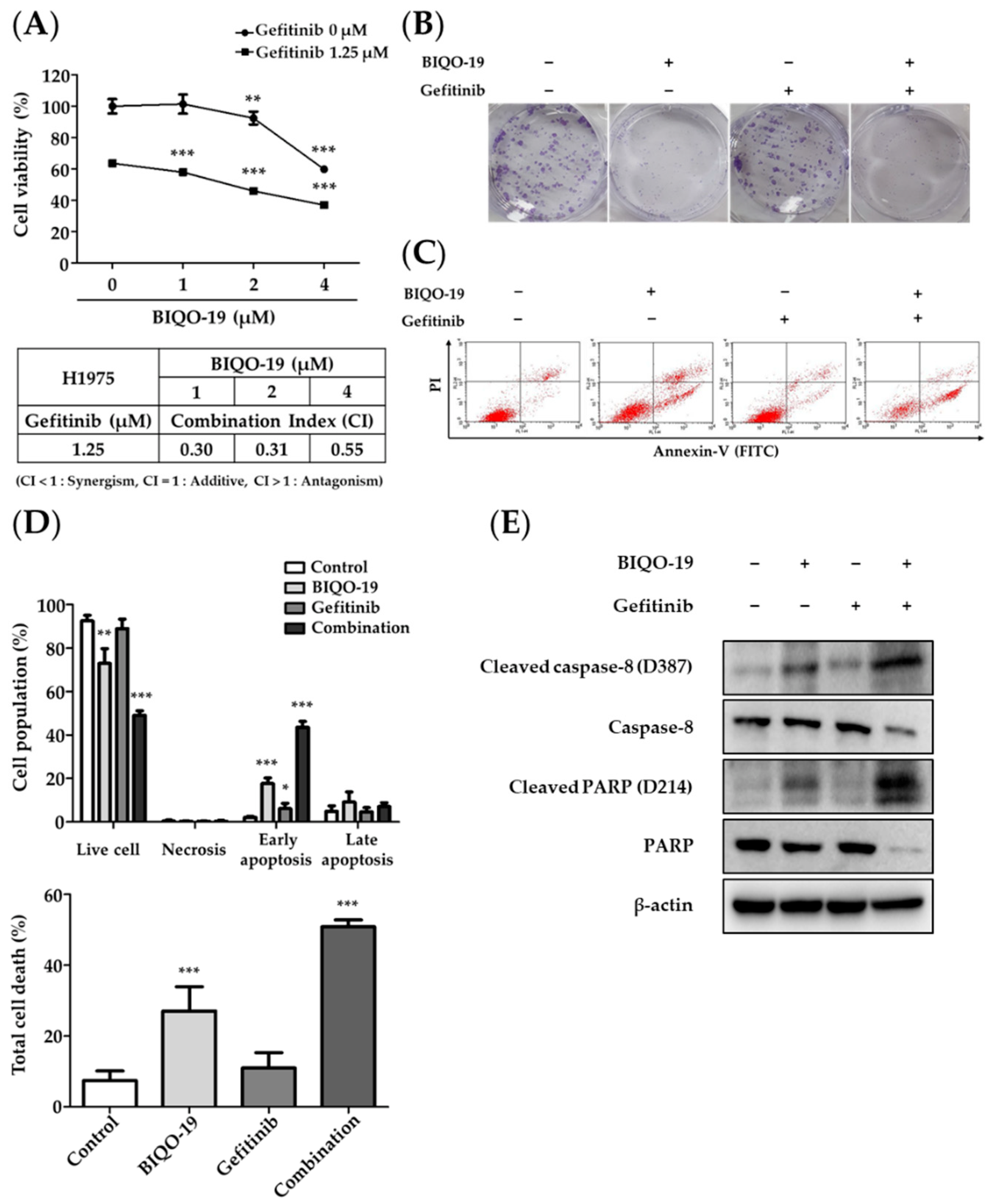
2.6. Combination Effects of BIQO-19 and Gefitinib on the Proliferation of H1975 Cells
3. Discussion
4. Materials and Methods
4.1. Chemistry
General Procedure for the Synthesis of Ethyl 6-(4-Oxo-3-(pyrimidin-2-ylmethyl)-3,4-dihydroquinazolin-6-yl)imidazo [1,2-a]pyridine-2-carboxylate (BIQO-19)
4.2. Reagents
4.3. ADMET Prediction Analysis
4.4. Cell Culture
4.5. Cell Proliferation Assay
4.6. Colony Formation Assay
4.7. Bioinformatics Analysis
4.8. Aurora A Kinase Assay
4.9. Western Blotting Analysis
4.10. Molecular Docking
4.11. Cell Cycle Analysis
4.12. Annexin V-FITC/PI Double Staining
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Heymach, J.V.; Lippman, S.M. Lung Cancer. N. Engl. J. Med. 2008, 359, 1367–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warth, A.; Penzel, R.; Lindenmaier, H.; Brandt, R.; Stenzinger, A.; Herpel, E.; Goeppert, B.; Thomas, M.; Herth, F.; Dienemann, H.; et al. EGFR, KRAS, BRAF and ALK gene alterations in lung adenocarcinomas: Patient outcome, interplay with morphology and immunophenotype. Eur. Respir. J. 2014, 43, 872–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prabhakar, C.N. Epidermal growth factor receptor in non-small cell lung cancer. Transl. Lung Cancer Res. 2015, 4, 110–118. [Google Scholar] [CrossRef]
- Liam, C.-K.; Pang, Y.-K.; Poh, M.-E. EGFR Mutations in Asian Patients with Advanced Lung Adenocarcinoma. J. Thorac. Oncol. 2014, 9, e70–e71. [Google Scholar] [CrossRef] [Green Version]
- Kris, M.G.; Natale, R.B.; Herbst, R.S.; Lynch, J.T., Jr.; Prager, D.; Belani, C.P.; Schiller, J.H.; Kelly, K.; Spiridonidis, H.; Sandler, A.; et al. Efficacy of Gefitinib, an Inhibitor of the Epidermal Growth Factor Receptor Tyrosine Kinase, in Symptomatic Patients with Non–Small Cell Lung Cancer: A randomized trial. JAMA 2003, 290, 2149–2158. [Google Scholar] [CrossRef] [Green Version]
- Costa, D.B.; Nguyen, K.-S.H.; Cho, B.C.; Sequist, L.V.; Jackman, D.M.; Riely, G.J.; Yeap, B.Y.; Halmos, B.; Kim, J.H.; Jänne, P.A.; et al. Effects of Erlotinib in EGFR Mutated Non-Small Cell Lung Cancers with Resistance to Gefitinib. Clin. Cancer Res. 2008, 14, 7060–7067. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of Tumor Specimens at the Time of Acquired Resistance to EGFR-TKI Therapy in 155 Patients with EGFR-Mutant Lung Cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef] [Green Version]
- Mok, T.S.; Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or Platinum–Pemetrexed in EGFR T790M–Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhang, X.; Wang, R.; Qin, J.; Wang, J.; Li, Z.; Song, X. Osimertinib Resistance with a Novel EGFR L858R/A859S/Y891D Triple Mutation in a Patient with Non-Small Cell Lung Cancer: A Case Report. Front. Oncol. 2020, 10, 542277. [Google Scholar] [CrossRef]
- Chen, J.; Lu, H.; Zhou, W.; Yin, H.; Zhu, L.; Liu, C.; Zhang, P.; Hu, H.; Yang, Y.; Han, H. AURKA upregulation plays a role in fibroblast-reduced gefitinib sensitivity in the NSCLC cell line HCC827. Oncol. Rep. 2015, 33, 1860–1866. [Google Scholar] [CrossRef] [Green Version]
- Low, J.-L.; Lau, D.P.; Zhang, X.; Kwang, X.-L.; Rohatgi, N.; Chan, J.V.; Chong, F.-T.; Wong, S.Q.R.; Leong, H.-S.; Thangavelu, M.T.; et al. A chemical genetic screen identifies Aurora kinases as a therapeutic target in EGFR T790M negative, gefitinib-resistant head and neck squamous cell carcinoma (HNSCC). EBioMedicine 2021, 64, 103220. [Google Scholar] [CrossRef]
- Carmena, M.; Earnshaw, W.C. The cellular geography of Aurora kinases. Nat. Rev. Mol. Cell Biol. 2003, 4, 842–854. [Google Scholar] [CrossRef]
- Willems, E.; Dedobbeleer, M.; Digregorio, M.; Lombard, A.; Lumapat, P.N.; Rogister, B. The functional diversity of Aurora kinases: A comprehensive review. Cell Div. 2018, 13, 7. [Google Scholar] [CrossRef] [Green Version]
- Goldenson, B.; Crispino, J.D. The aurora kinases in cell cycle and leukemia. Oncogene 2015, 34, 537–545. [Google Scholar] [CrossRef] [Green Version]
- Vader, G.; Lens, S.M. The Aurora kinase family in cell division and cancer. Biochim. Biophys. Acta 2008, 1786, 60–72. [Google Scholar] [CrossRef]
- Bischoff, J.R.; Anderson, L.; Zhu, Y.; Mossie, K.; Ng, L.; Souza, B.; Schryver, B.; Flanagan, P.; Clairvoyant, F.; Ginther, C.; et al. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J. 1998, 17, 3052–3065. [Google Scholar] [CrossRef]
- Zhang, Z.; Singh, M.; Davidson, S.; Rosen, D.G.; Yang, G.; Liu, J. Activation of BTAK expression in primary ovarian surface epithelial cells of prophylactic ovaries. Mod. Pathol. 2007, 20, 1078–1084. [Google Scholar] [CrossRef] [Green Version]
- Görgün, G.; Calabrese, E.; Hideshima, T.; Ecsedy, J.; Perrone, G.; Mani, M.; Ikeda, H.; Bianchi, G.; Hu, Y.; Cirstea, D.; et al. A novel Aurora-A kinase inhibitor MLN8237 induces cytotoxicity and cell-cycle arrest in multiple myeloma. Blood 2010, 115, 5202–5213. [Google Scholar] [CrossRef] [Green Version]
- Kaestner, P.; Stolz, A.; Bastians, H. Determinants for the efficiency of anticancer drugs targeting either Aurora-A or Aurora-B kinases in human colon carcinoma cells. Mol. Cancer Ther. 2009, 8, 2046–2056. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Yan, L.; Torres, R.; Gong, X.; Bian, H.; Marugán, C.; Boehnke, K.; Baquero, C.; Hui, Y.-H.; Chapman, S.C.; et al. Aurora A–Selective Inhibitor LY3295668 Leads to Dominant Mitotic Arrest, Apoptosis in Cancer Cells, and Shows Potent Preclinical Antitumor Efficacy. Mol. Cancer Ther. 2019, 18, 2207–2219. [Google Scholar] [CrossRef] [Green Version]
- Ndolo, K.M.; Park, K.R.; Lee, H.J.; Yoon, K.B.; Kim, Y.-C.; Han, S.-Y. Characterization of the Indirubin Derivative LDD970 as a Small Molecule Aurora Kinase A Inhibitor in Human Colorectal Cancer Cells. Immune Netw. 2017, 17, 110–115. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.-X.; Wang, J.-D.; Chen, J.-J.; Long, B.; Liu, L.-L.; Tu, X.-X.; Luo, Y.; Hu, Y.; Lin, D.-J.; Lu, G.; et al. Aurora A Kinase Inhibitor AKI603 Induces Cellular Senescence in Chronic Myeloid Leukemia Cells Harboring T315I Mutation. Sci. Rep. 2016, 6, 35533. [Google Scholar] [CrossRef] [Green Version]
- Min, Y.H.; Kim, W.; Kim, J.-E. The Aurora kinase A inhibitor TC-A2317 disrupts mitotic progression and inhibits cancer cell proliferation. Oncotarget 2016, 7, 84718–84735. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos, E.O.; Carneiro-Lobo, T.C.; Aoki, M.N.; Levantini, E.; Bassères, D.S. Aurora kinase targeting in lung cancer reduces KRAS-induced transformation. Mol. Cancer 2016, 15, 12. [Google Scholar] [CrossRef]
- Shah, K.N.; Bhatt, R.; Rotow, J.; Rohrberg, J.; Olivas, V.; Wang, V.E.; Hemmati, G.; Martins, M.M.; Maynard, A.; Kuhn, J.; et al. Aurora kinase A drives the evolution of resistance to third-generation EGFR inhibitors in lung cancer. Nat. Med. 2019, 25, 111–118. [Google Scholar] [CrossRef]
- Blakely, C.M.; Gubens, M.A.; Allen, G.M.; Shah, S.; Jereza, M.; Bacaltos, B.; Bandyopadhyay, S. Phase I study of the aurora kinase A inhibitor alisertib in combination with osimertinib in EGFR-mutant lung cancer. J. Clin. Oncol. 2021, 39, 9074. [Google Scholar] [CrossRef]
- Du, R.; Huang, C.; Liu, K.; Li, X.; Dong, Z. Targeting AURKA in Cancer: Molecular mechanisms and opportunities for Cancer therapy. Mol. Cancer 2021, 20, 15. [Google Scholar] [CrossRef]
- O’Connor, O.A.; Özcan, M.; Jacobsen, E.D.; Roncero, J.M.; Trotman, J.; Demeter, J.; Masszi, T.; Pereira, J.; Ramchandren, R.; Beaven, A.; et al. Randomized Phase III Study of Alisertib or Investigator’s Choice (Selected Single Agent) in Patients with Relapsed or Refractory Peripheral T-Cell Lymphoma. J. Clin. Oncol. 2019, 37, 613–623. [Google Scholar] [CrossRef]
- Yan, V.C.; Butterfield, H.E.; Poral, A.H.; Yan, M.J.; Yang, K.L.; Pham, C.-D.; Muller, F.L. Why Great Mitotic Inhibitors Make Poor Cancer Drugs. Trends Cancer 2020, 6, 924–941. [Google Scholar] [CrossRef]
- Al-Salahi, R.; Abuelizz, H.A.; Ghabbour, H.A.; El-Dib, R.; Marzouk, M. Molecular docking study and antiviral evaluation of 2-thioxo-benzo[g]quinazolin-4(3H)-one derivatives. Chem. Central J. 2016, 10, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanda, A.K.; Ganguli, S.; Chakraborty, R. Antibacterial Activity of Some 3-(Arylideneamino)-2-phenylquinazoline-4(3H)-ones: Synthesis and Preliminary QSAR Studies. Molecules 2007, 12, 2413–2426. [Google Scholar] [CrossRef] [PubMed]
- Zayed, M.F.; Hassan, M. Synthesis and biological evaluation studies of novel quinazolinone derivatives as antibacterial and anti-inflammatory agents. Saudi Pharm. J. 2014, 22, 157–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahdy, H.A.; Ibrahim, M.K.; Metwaly, A.; Belal, A.; Mehany, A.B.; El-Gamal, K.M.; El-Sharkawy, A.; Elhendawy, M.A.; Radwan, M.M.; ElSohly, M.A.; et al. Design, synthesis, molecular modeling, in vivo studies and anticancer evaluation of quinazolin-4(3H)-one derivatives as potential VEGFR-2 inhibitors and apoptosis inducers. Bioorg. Chem. 2020, 94, 103422. [Google Scholar] [CrossRef]
- Fan, Y.-H.; Li, W.; Liu, D.-D.; Bai, M.-X.; Song, H.-R.; Xu, Y.; Lee, S.; Zhou, Z.-P.; Wang, J.; Ding, H.-W. Design, synthesis, and biological evaluation of novel 3-substituted imidazo[1,2- a ]pyridine and quinazolin-4(3H)-one derivatives as PI3Kα inhibitors. Eur. J. Med. Chem. 2017, 139, 95–106. [Google Scholar] [CrossRef]
- Wang, P.-F.; Jensen, A.A.; Bunch, L. From Methaqualone and Beyond: Structure–Activity Relationship of 6-, 7-, and 8-Substituted 2,3-Diphenyl-quinazolin-4(3H)-ones and in Silico Prediction of Putative Binding Modes of Quinazolin-4(3H)-ones as Positive Allosteric Modulators of GABAA Receptors. ACS Chem. Neurosci. 2020, 11, 4362–4375. [Google Scholar] [CrossRef]
- Kumar, D.; Mariappan, G.; Husain, A.; Monga, J.; Kumar, S. Design, synthesis and cytotoxic evaluation of novel imidazolone fused quinazolinone derivatives. Arab. J. Chem. 2017, 10, 344–350. [Google Scholar] [CrossRef] [Green Version]
- Safavi, M.; Ashtari, A.; Khalili, F.; Mirfazli, S.S.; Saeedi, M.; Ardestani, S.K.; Ranjbar, P.R.; Tehrani, M.B.; Larijani, B.; Mahdavi, M. Novel quinazolin-4(3H)-one linked to 1,2,3-triazoles: Synthesis and anticancer activity. Chem. Biol. Drug Des. 2018, 92, 1373–1381. [Google Scholar] [CrossRef]
- Hieu, D.T.; Anh, D.T.; Hai, P.T.; Thuan, N.T.; Huong, L.T.T.; Park, E.J.; Ji, A.Y.; Kang, J.S.; Dung, P.T.P.; Han, S.B.; et al. Quinazolin-4(3H)-one-Based Hydroxamic Acids: Design, Synthesis and Evaluation of Histone Deacetylase Inhibitory Effects and Cytotoxicity. Chem. Biodivers. 2019, 16, e1800502. [Google Scholar] [CrossRef]
- Thakur, A.; Tawa, G.J.; Henderson, M.J.; Danchik, C.; Liu, S.; Shah, P.; Wang, A.Q.; Dunn, G.; Kabir, M.; Padilha, E.C.; et al. Design, Synthesis, and Biological Evaluation of Quinazolin-4-one-Based Hydroxamic Acids as Dual PI3K/HDAC Inhibitors. J. Med. Chem. 2020, 63, 4256–4292. [Google Scholar] [CrossRef]
- Ke, Y.-Y.; Shiao, H.-Y.; Hsu, Y.C.; Chu, C.-Y.; Wang, W.-C.; Lee, Y.-C.; Lin, W.-H.; Chen, C.-H.; Hsu, J.T.A.; Chang, C.-W.; et al. 3D-QSAR-Assisted Drug Design: Identification of a Potent Quinazoline-Based Aurora Kinase Inhibitor. ChemMedChem 2013, 8, 136–148. [Google Scholar] [CrossRef]
- Fan, C.; Zhong, T.; Yang, H.; Yang, Y.; Wang, D.; Yang, X.; Xu, Y.; Fan, Y. Design, synthesis, biological evaluation of 6-(2-amino-1H-benzo[d]imidazole-6-yl)quinazolin-4(3H)-one derivatives as novel anticancer agents with Aurora kinase inhibition. Eur. J. Med. Chem. 2020, 190, 112108. [Google Scholar] [CrossRef]
- Liu, K.; Zheng, M.; Lu, R.; Du, J.; Zhao, Q.; Li, Z.; Li, Y.; Zhang, S. The role of CDC25C in cell cycle regulation and clinical cancer therapy: A systematic review. Cancer Cell Int. 2020, 20, 213. [Google Scholar] [CrossRef]
- Dutto, I.; Tillhon, M.; Cazzalini, O.; Stivala, L.A.; Prosperi, E. Biology of the cell cycle inhibitor p21CDKN1A: Molecular mechanisms and relevance in chemical toxicology. Arch. Toxicol. 2015, 89, 155–178. [Google Scholar] [CrossRef]
- Kim, D.; Bach, D.-H.; Fan, Y.-H.; Luu, T.-T.; Hong, J.-Y.; Park, H.J.; Lee, S.K. AXL degradation in combination with EGFR-TKI can delay and overcome acquired resistance in human non-small cell lung cancer cells. Cell Death Dis. 2019, 10, 361. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.-C.; Chen, C.-H.; Wang, J.-C.; Tsai, A.-C.; Liou, J.-P.; Pan, S.-L.; Teng, C.-M. The HDAC inhibitor, MPT0E028, enhances erlotinib-induced cell death in EGFR-TKI-resistant NSCLC cells. Cell Death Dis. 2013, 4, e810. [Google Scholar] [CrossRef] [Green Version]
- Pai-Scherf, L.; Blumenthal, G.M.; Li, H.; Subramaniam, S.; Mishra-Kalyani, P.S.; He, K.; Zhao, H.; Yu, J.; Paciga, M.; Goldberg, K.B.; et al. FDA Approval Summary: Pembrolizumab for Treatment of Metastatic Non-Small Cell Lung Cancer: First-Line Therapy and Beyond. Oncologist 2017, 22, 1392–1399. [Google Scholar] [CrossRef] [Green Version]
- To, K.K.W.; Fong, W.; Cho, W.C.S. Immunotherapy in Treating EGFR-Mutant Lung Cancer: Current Challenges and New Strategies. Front. Oncol. 2021, 11, 635007. [Google Scholar] [CrossRef]
- Eymin, B.; Gazzeri, S. Role of cell cycle regulators in lung carcinogenesis. Cell Adhes. Migr. 2010, 4, 114–123. [Google Scholar] [CrossRef]
- Kamran, M.; Long, Z.-J.; Xu, D.; Lv, S.-S.; Liu, B.; Wang, C.-L.; Xu, J.; Lam, E.; Liu, Q. Aurora kinase A regulates Survivin stability through targeting FBXL7 in gastric cancer drug resistance and prognosis. Oncogenesis 2017, 6, e298. [Google Scholar] [CrossRef] [Green Version]
- Nikhil, K.; Raza, A.; Haymour, H.S.; Flueckiger, B.V.; Chu, J.; Shah, K. Aurora Kinase A-YBX1 Synergy Fuels Aggressive Oncogenic Phenotypes and Chemoresistance in Castration-Resistant Prostate Cancer. Cancers 2020, 12, 660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Z.; Yuan, Z.; Zhang, Q.; Long, Z.; Chen, J.; Tang, Z.; Zhu, Y.; Chen, S.; Xu, J.; Yan, M.; et al. Aurora kinase A inhibition-induced autophagy triggers drug resistance in breast cancer cells. Autophagy 2012, 8, 1798–1810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Lu, H.; Ma, H.; Feng, F.; Hu, X.; Zhang, Q.; Wang, J.; Xu, Y.; Zhao, Q. Bioactive compounds of Eriocaulon sieboldianum blocking proliferation and inducing apoptosis of HepG2 cells might be involved in Aurora kinase inhibition. Food Funct. 2015, 6, 3746–3759. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Guo, A.; Oler, E.; Wang, F.; Anjum, A.; Peters, H.; Dizon, R.; Sayeeda, Z.; Tian, S.; Lee, B.L.; et al. HMDB 5.0: The Human Metabolome Database for 2022. Nucleic Acids Res. 2022, 50, D622–D631. [Google Scholar] [CrossRef]
- Beltran, H.; Oromendia, C.; Danila, D.C.; Montgomery, B.; Hoimes, C.; Szmulewitz, R.Z.; Vaishampayan, U.; Armstrong, A.J.; Stein, M.; Pinski, J.; et al. A Phase II Trial of the Aurora Kinase A Inhibitor Alisertib for Patients with Castration-resistant and Neuroendocrine Prostate Cancer: Efficacy and Biomarkers. Clin. Cancer Res. 2019, 25, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Chu, Q.S.-C.; Bouganim, N.; Fortier, C.; Zaknoen, S.; Stille, J.R.; Kremer, J.D.; Yuen, E.; Hui, Y.-H.; de la Peña, A.; Lithio, A.; et al. Aurora kinase A inhibitor, LY3295668 erbumine: A phase 1 monotherapy safety study in patients with locally advanced or metastatic solid tumors. Investig. New Drugs 2021, 39, 1001–1010. [Google Scholar] [CrossRef]
- Mou, P.K.; Yang, E.J.; Shi, C.; Ren, G.; Tao, S.; Shim, J.S. Aurora kinase A, a synthetic lethal target for precision cancer medicine. Exp. Mol. Med. 2021, 53, 835–847. [Google Scholar] [CrossRef]
- Jung, C.; Hong, J.-Y.; Bae, S.Y.; Kang, S.S.; Park, H.J.; Lee, S.K. Antitumor Activity of Americanin A Isolated from the Seeds of Phytolacca americana by Regulating the ATM/ATR Signaling Pathway and the Skp2–p27 Axis in Human Colon Cancer Cells. J. Nat. Prod. 2015, 78, 2983–2993. [Google Scholar] [CrossRef]
- Győrffy, B.; Surowiak, P.; Budczies, J.; Lánczky, A. Online Survival Analysis Software to Assess the Prognostic Value of Biomarkers Using Transcriptomic Data in Non-Small-Cell Lung Cancer. PLoS ONE 2013, 8, e82241. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property a | FL-4 | BIQO-19 |
|---|---|---|
| ADMET_Solubility b | −6.224 | −3.318 |
| ADMET_Solubility_Level | 1 (Very low solubility) | 3 (Good solubility) |
| ADMET_PSA_2D c | 105.01 | 97.34 |
| ADMET_AlogP98 d | 4.522 | 1.802 |
| ADMET_BBB e | Undefined | −1.137 |
| ADMET_BBB_Level | 4 (Undefined) | 3 (Low penetration) |
| ADMET_EXT_CYP2D6 f | −9.26 | −7.69 |
| ADMET_EXT_hepatotoxic g | 4.1775 | −3.179 |
| ADMET_EXT_PPB h | 3.54856 | −0.242418 |
| Compound | Aurora Kinase A Inhibition (IC50) a | Aurora Kinase B Inhibition (IC50) a |
|---|---|---|
| BIQO-19 | 68.54 ± 6.45 nM | 581.03 ± 13.77 nM |
| Staurosporine b | 18.09 ± 2.07 nM | 26.13 ± 2.51 nM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.Y.; Yang, H.; Kim, D.; Kyaw, K.Z.; Hu, R.; Fan, Y.; Lee, S.K. Antiproliferative Activity of a New Quinazolin-4(3H)-One Derivative via Targeting Aurora Kinase A in Non-Small Cell Lung Cancer. Pharmaceuticals 2022, 15, 698. https://doi.org/10.3390/ph15060698
Lee JY, Yang H, Kim D, Kyaw KZ, Hu R, Fan Y, Lee SK. Antiproliferative Activity of a New Quinazolin-4(3H)-One Derivative via Targeting Aurora Kinase A in Non-Small Cell Lung Cancer. Pharmaceuticals. 2022; 15(6):698. https://doi.org/10.3390/ph15060698
Chicago/Turabian StyleLee, Ji Yun, Huarong Yang, Donghwa Kim, Kay Zin Kyaw, Ruoci Hu, Yanhua Fan, and Sang Kook Lee. 2022. "Antiproliferative Activity of a New Quinazolin-4(3H)-One Derivative via Targeting Aurora Kinase A in Non-Small Cell Lung Cancer" Pharmaceuticals 15, no. 6: 698. https://doi.org/10.3390/ph15060698
APA StyleLee, J. Y., Yang, H., Kim, D., Kyaw, K. Z., Hu, R., Fan, Y., & Lee, S. K. (2022). Antiproliferative Activity of a New Quinazolin-4(3H)-One Derivative via Targeting Aurora Kinase A in Non-Small Cell Lung Cancer. Pharmaceuticals, 15(6), 698. https://doi.org/10.3390/ph15060698