
Pharmacophore-Based Discovery of Viral RNA Conformational Modulators


Abstract
:1. Introduction
2. Results
2.1. Pharmacophore-Based Virtual Screening
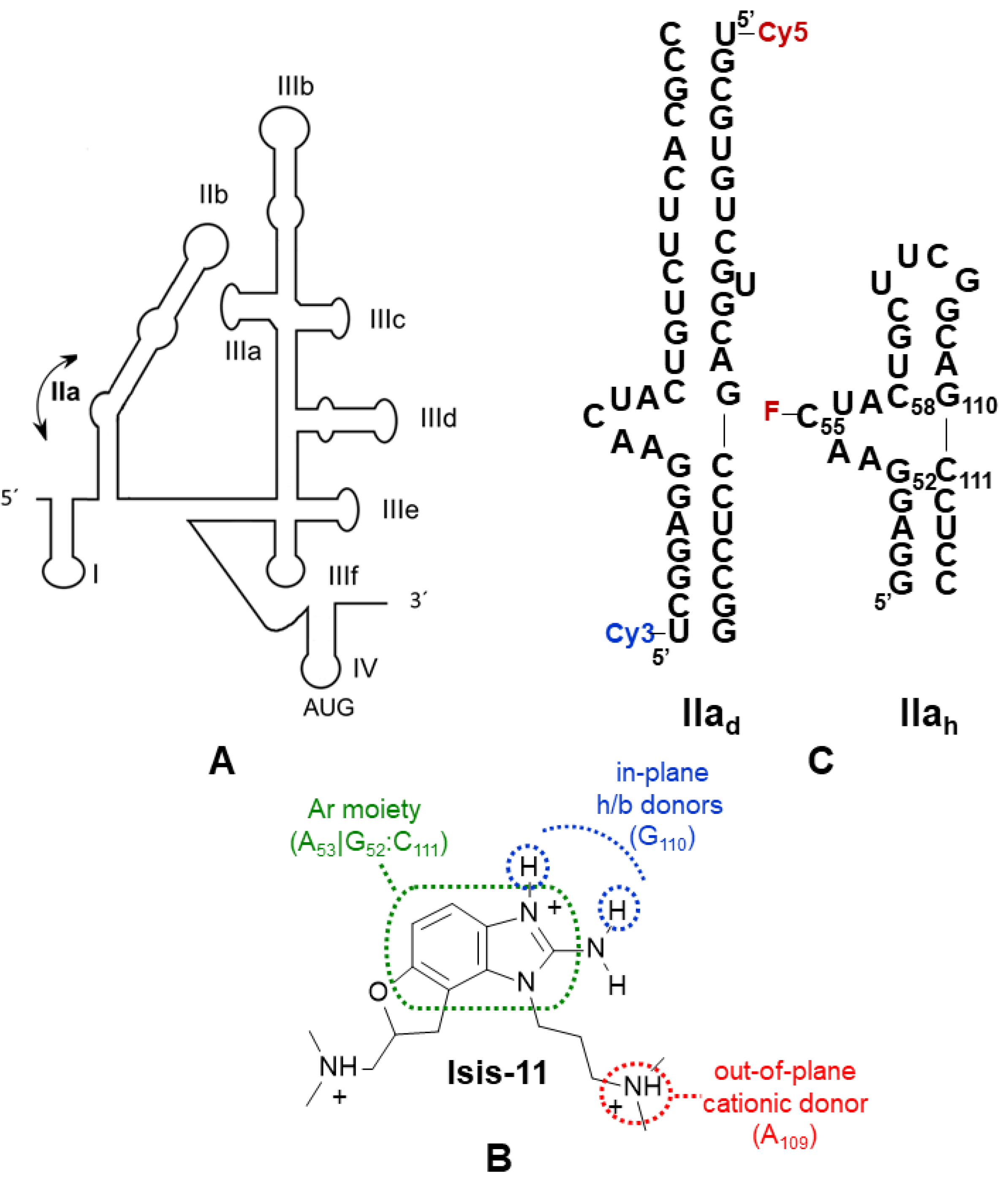
2.2. Conformational Modulation of Subdomain IIa
2.3. Bulge IIa Recognition
2.4. Binding Specificity
3. Discussion
4. Materials and Methods
4.1. Pharmacophore-Based Screening
4.2. Docking Calculations and Compound Selection
4.3. Compounds
4.4. RNA and DNA Samples
4.5. FRET
4.6. Fluorescence Intensity
4.7. NMR Spectroscopy
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gallego, J.; Varani, G. Targeting RNA with small-molecule drugs: Therapeutic promise and chemical challenges. Acc. Chem. Res. 2001, 34, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Warner, K.D.; Hajdin, C.E.; Weeks, K.M. Principles for targeting RNA with drug-like small molecules. Nat. Rev. Drug Discov. 2018, 17, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.M.; Williams, C.C.; Akahori, Y.; Tanaka, T.; Aikawa, H.; Tong, Y.; Childs-Disney, J.L.; Disney, M.D. Small molecule recognition of disease-relevant RNA structures. Chem. Soc. Rev. 2020, 49, 7167–7199. [Google Scholar] [CrossRef] [PubMed]
- Binas, O.; de Jesus, V.; Landgraf, T.; Völklein, A.E.; Martins, J.; Hymon, D.; Kaur Bains, J.; Berg, H.; Biedenbänder, T.; Fürtig, B.; et al. F NMR-Based Fragment Screening for 14 Different Biologically Active RNAs and 10 DNA and Protein Counter-Screens. Chembiochem 2021, 22, 423–433. [Google Scholar] [CrossRef]
- Sreeramulu, S.; Richter, C.; Berg, H.; Wirtz Martin, M.A.; Ceylan, B.; Matzel, T.; Adam, J.; Altincekic, N.; Azzaoui, K.; Bains, J.K.; et al. Exploring the Druggability of Conserved RNA Regulatory Elements in the SARS-CoV-2 Genome. Angew. Chem. Int. Ed. Engl. 2021, 60, 19191–19200. [Google Scholar] [CrossRef]
- Rizvi, N.F.; Santa Maria, J.P.; Nahvi, A.; Klappenbach, J.; Klein, D.J.; Curran, P.J.; Richards, M.P.; Chamberlin, C.; Saradjian, P.; Burchard, J.; et al. Targeting RNA with Small Molecules: Identification of Selective, RNA-Binding Small Molecules Occupying Drug-Like Chemical Space. SLAS Discov. 2020, 25, 384–396. [Google Scholar] [CrossRef]
- Morgan, B.S.; Forte, J.E.; Culver, R.N.; Zhang, Y.; Hargrove, A.E. Discovery of Key Physicochemical, Structural, and Spatial Properties of RNA-Targeted Bioactive Ligands. Angew. Chem. Int. Ed. Engl. 2017, 56, 13498–13502. [Google Scholar] [CrossRef]
- Li, Y.; Shen, J.; Sun, X.; Li, W.; Liu, G.; Tang, Y. Accuracy assessment of protein-based docking programs against RNA targets. J. Chem. Inf. Model. 2010, 50, 1134–1146. [Google Scholar] [CrossRef]
- Lukavsky, P.J.; Kim, I.; Otto, G.A.; Puglisi, J.D. Structure of HCV IRES domain II determined by NMR. Nat. Struct. Biol. 2003, 10, 1033–1038. [Google Scholar] [CrossRef]
- Dibrov, S.M.; Johnston-Cox, H.; Weng, Y.H.; Hermann, T. Functional architecture of HCV IRES domain II stabilized by divalent metal ions in the crystal and in solution. Angew. Chem. Int. Ed. Engl. 2007, 46, 226–229. [Google Scholar] [CrossRef]
- Lukavsky, P.J. Structure and function of HCV IRES domains. Virus Res. 2009, 139, 166–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, J.; Castaldi, M.P.; Dutta, S.; Dibrov, S.M.; Wyles, D.L.; Hermann, T. Conformational inhibition of the hepatitis C virus internal ribosome entry site RNA. Nat. Chem. Biol. 2009, 5, 823–825. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, R.B.; Seth, P.P.; Swayze, E.E.; Griffey, R.H.; Skalicky, J.J.; Cheatham, T.E.; Davis, D.R. Inhibitor-induced structural change in the HCV IRES domain IIa RNA. Proc. Natl. Acad. Sci. USA 2010, 107, 7263–7268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dibrov, S.M.; Ding, K.; Brunn, N.D.; Parker, M.A.; Bergdahl, B.M.; Wyles, D.L.; Hermann, T. Structure of a hepatitis C virus RNA domain in complex with a translation inhibitor reveals a binding mode reminiscent of riboswitches. Proc. Natl. Acad. Sci. USA 2012, 109, 5223–5228. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Nelson, C.A.; Xiao, L.; Lu, L.; Seth, P.P.; Davis, D.R.; Hagedorn, C.H. Measuring antiviral activity of benzimidazole molecules that alter IRES RNA structure with an infectious hepatitis C virus chimera expressing Renilla luciferase. Antivir. Res. 2011, 89, 54–63. [Google Scholar] [CrossRef] [Green Version]
- Seth, P.P.; Miyaji, A.; Jefferson, E.A.; Sannes-Lowery, K.A.; Osgood, S.A.; Propp, S.S.; Ranken, R.; Massire, C.; Sampath, R.; Ecker, D.J.; et al. SAR by MS: Discovery of a new class of RNA-binding small molecules for the hepatitis C virus: Internal ribosome entry site IIA subdomain. J. Med. Chem. 2005, 48, 7099–7102. [Google Scholar] [CrossRef]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A Free Tool to Discover Chemistry for Biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef]
- Bailor, M.H.; Sun, X.; Al-Hashimi, H.M. Topology links RNA secondary structure with global conformation, dynamics, and adaptation. Science 2010, 327, 202–206. [Google Scholar] [CrossRef]
- Murchie, A.I.; Davis, B.; Isel, C.; Afshar, M.; Drysdale, M.J.; Bower, J.; Potter, A.J.; Starkey, I.D.; Swarbrick, T.M.; Mirza, S.; et al. Structure-based drug design targeting an inactive RNA conformation: Exploiting the flexibility of HIV-1 TAR RNA. J. Mol. Biol. 2004, 336, 625–638. [Google Scholar] [CrossRef]
- Stelzer, A.C.; Frank, A.T.; Kratz, J.D.; Swanson, M.D.; Gonzalez-Hernandez, M.J.; Lee, J.; Andricioaei, I.; Markovitz, D.M.; Al-Hashimi, H.M. Discovery of selective bioactive small molecules by targeting an RNA dynamic ensemble. Nat. Chem. Biol. 2011, 7, 553–559. [Google Scholar] [CrossRef] [Green Version]
- Davila-Calderon, J.; Patwardhan, N.N.; Chiu, L.Y.; Sugarman, A.; Cai, Z.; Penutmutchu, S.R.; Li, M.L.; Brewer, G.; Hargrove, A.E.; Tolbert, B.S. IRES-targeting small molecule inhibits enterovirus 71 replication via allosteric stabilization of a ternary complex. Nat. Commun. 2020, 11, 4775. [Google Scholar] [CrossRef] [PubMed]
- Lozano, G.; Trapote, A.; Ramajo, J.; Elduque, X.; Grandas, A.; Robles, J.; Pedroso, E.; Martínez-Salas, E. Local RNA flexibility perturbation of the IRES element induced by a novel ligand inhibits viral RNA translation. RNA Biol. 2015, 12, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Bardaro, M.F.; Shajani, Z.; Patora-Komisarska, K.; Robinson, J.A.; Varani, G. How binding of small molecule and peptide ligands to HIV-1 TAR alters the RNA motional landscape. Nucleic. Acids. Res. 2009, 37, 1529–1540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boerneke, M.A.; Dibrov, S.M.; Gu, J.; Wyles, D.L.; Hermann, T. Functional conservation despite structural divergence in ligand-responsive RNA switches. Proc. Natl. Acad. Sci. USA 2014, 111, 15952–15957. [Google Scholar] [CrossRef] [Green Version]
- Martin, W.J.; Grandi, P.; Marcia, M. Screening strategies for identifying RNA- and ribonucleoprotein-targeted compounds. Trends Pharmacol. Sci. 2021, 42, 758–771. [Google Scholar] [CrossRef]
- Zhou, S.; Rynearson, K.D.; Ding, K.; Brunn, N.D.; Hermann, T. Screening for inhibitors of the hepatitis C virus internal ribosome entry site RNA. Bioorg. Med. Chem. 2013, 21, 6139–6144. [Google Scholar] [CrossRef] [Green Version]
- Angelbello, A.J.; González, À.; Rzuczek, S.G.; Disney, M.D. Development of pharmacophore models for small molecules targeting RNA: Application to the RNA repeat expansion in myotonic dystrophy type 1. Bioorg. Med. Chem. Lett. 2016, 26, 5792–5796. [Google Scholar] [CrossRef] [Green Version]
- Renner, S.; Ludwig, V.; Boden, O.; Scheffer, U.; Göbel, M.; Schneider, G. New inhibitors of the Tat-TAR RNA interaction found with a “fuzzy” pharmacophore model. Chembiochem 2005, 6, 1119–1125. [Google Scholar] [CrossRef]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.; Schlick, T. Candidate RNA structures for domain 3 of the foot-and-mouth-disease virus internal ribosome entry site. Nucleic Acids Res. 2013, 41, 1483–1495. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Kim, N.K.; Peterson, R.D.; Wang, Z.; Feigon, J. Structurally conserved five nucleotide bulge determines the overall topology of the core domain of human telomerase RNA. Proc. Natl. Acad. Sci. USA 2010, 107, 18761–18768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Bradrick, T.D.; Marino, J.P. Ligand-induced changes in 2-aminopurine fluorescence as a probe for small molecule binding to HIV-1 TAR RNA. RNA 2004, 10, 1459–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.; Tonelli, M.; Markley, J.L. NMRFAM-SPARKY: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics 2015, 31, 1325–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd a | IC50 (IIad) (μM) | IC50 (IIad+tRNA) (μM) | IIad-tRNA specb |
|---|---|---|---|
| gn1 | 15.6 (2.67–52.0, 0.6404) | 30.2 (11.2–81.2, 0.7106) | 0.52 |
| qn1 | 10.7 (6.11–17.5, 0.9028) | 27.1 (10.7–68.9, 0.7421) | 0.39 |
| qn2 | 12.3 (5.60–22.6, 0.8358) | 73.8 (13.4–566, 0.7606) | 0.17 |
| qz2 | >50 | n/d | n/d |
| bz1 | >25 | n/d | n/d |
| Cpd a | Kd (μM) | Kd MgCl2 (μM) |
|---|---|---|
| gn1 | 81.5 (44.9–161, 0.8708) | 17.3 (9.21–35.2, 0.8526) |
| qn1 | 44.1 (30.1–66.5, 0.9332) | 60.8 (23.9–178, 0.7612) |
| qn2 | 38.5 (31.7–47.1, 0.9863) | 172 (103–319, 0.9485) |
| qz2 | 91.4 (63.9–135, 0.9522) | 216 (151–326, 0.9743) |
| bz1 | >100 | >100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martín-Villamil, M.; Sanmartín, I.; Moreno, Á.; Gallego, J. Pharmacophore-Based Discovery of Viral RNA Conformational Modulators. Pharmaceuticals 2022, 15, 748. https://doi.org/10.3390/ph15060748
Martín-Villamil M, Sanmartín I, Moreno Á, Gallego J. Pharmacophore-Based Discovery of Viral RNA Conformational Modulators. Pharmaceuticals. 2022; 15(6):748. https://doi.org/10.3390/ph15060748
Chicago/Turabian StyleMartín-Villamil, María, Isaías Sanmartín, Ángela Moreno, and José Gallego. 2022. "Pharmacophore-Based Discovery of Viral RNA Conformational Modulators" Pharmaceuticals 15, no. 6: 748. https://doi.org/10.3390/ph15060748
APA StyleMartín-Villamil, M., Sanmartín, I., Moreno, Á., & Gallego, J. (2022). Pharmacophore-Based Discovery of Viral RNA Conformational Modulators. Pharmaceuticals, 15(6), 748. https://doi.org/10.3390/ph15060748