
Pharmacological Implications of Adjusting Abnormal Fear Memory: Towards the Treatment of Post-Traumatic Stress Disorder


{kind=link}
{kind=link}
Abstract
:1. Current Treatment Regime of PTSD
2. Current Challenges of Managing PTSD
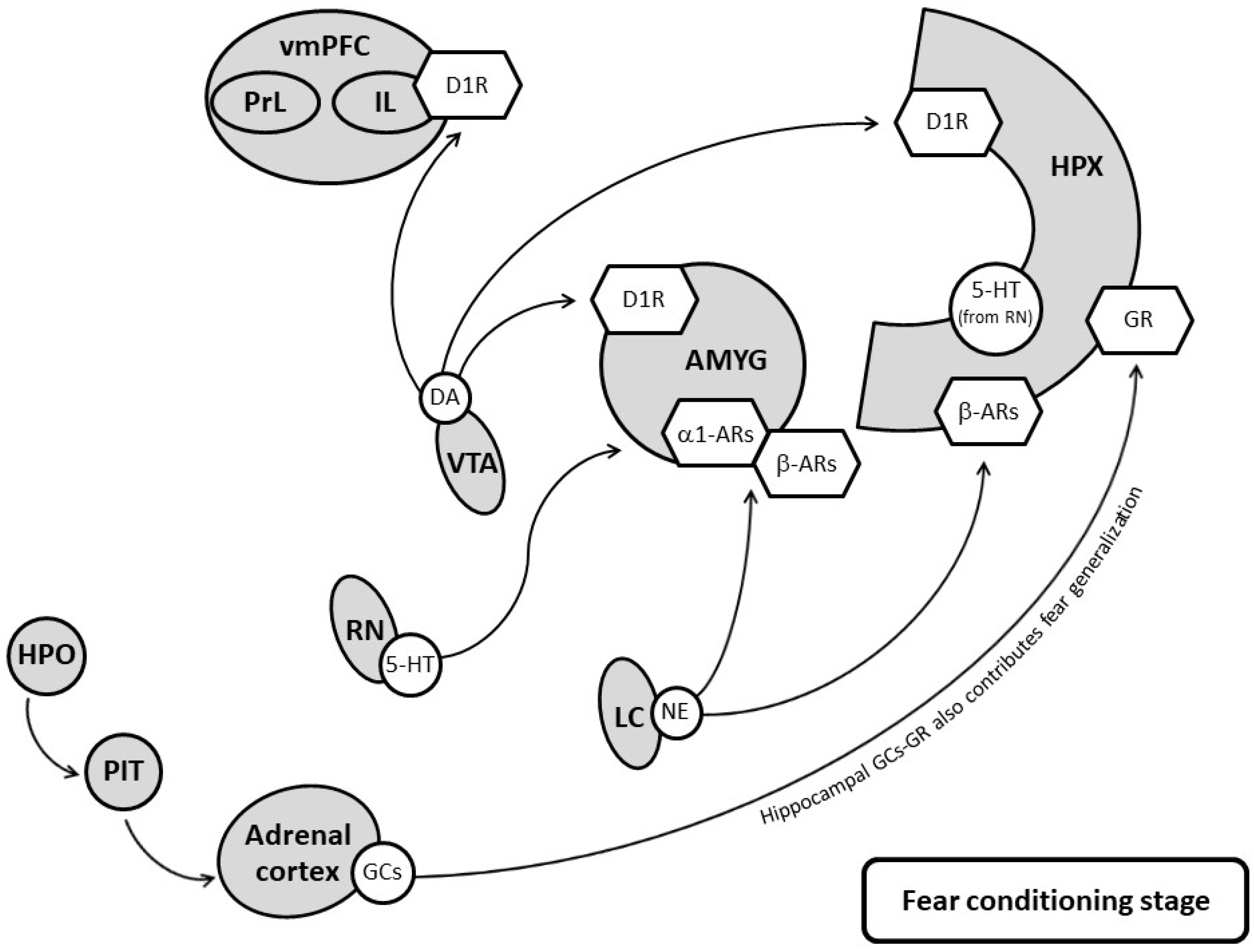
3. Therapeutic Mechanisms of Pharmacological Treatments
3.1. Targeting Serotonergic Systems
3.2. Targeting Noradrenergic Systems
3.3. Targeting DA Systems
3.4. Targeting the Hypothalamic-Pituitary-Adrenal Axis and Glucocorticoid Receptors
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Scott, W.J. PTSD in DSM-III: A Case in the Politics of Diagnosis and Disease. Soc. Probl. 1990, 37, 294–310. [Google Scholar] [CrossRef]
- Hatch, R.; Young, D.; Barber, V.; Griffiths, J.; Harrison, D.A.; Watkinson, P. Anxiety, Depression and Post Traumatic Stress Disorder after critical illness: A UK-wide prospective cohort study. Crit. Care 2018, 22, 310. [Google Scholar] [CrossRef] [Green Version]
- Yehuda, R.; Hoge, C.W.; McFarlane, A.C.; Vermetten, E.; Lanius, R.A.; Nievergelt, C.M.; Hobfoll, S.E.; Koenen, K.C.; Neylan, T.C.; Hyman, S.E. Post-traumatic stress disorder. Nature reviews. Nat. Rev. Dis. Primers 2015, 1, 15057. [Google Scholar] [CrossRef]
- Johnson, L.R.; McGuire, J.; Lazarus, R.; Palmer, A.A. Pavlovian fear memory circuits and phenotype models of PTSD. Neuropharmacology 2012, 62, 638–646. [Google Scholar] [CrossRef]
- Van Marle, H. PTSD as a memory disorder. Eur. J. Psychotraumatol. 2015, 6, 27633. [Google Scholar] [CrossRef]
- American Psychological Association. Clinical Practice Guideline for the Treatment of Posttraumatic Stress Disorder (PTSD) in Adults; American Psychological Association: Washington, DC, USA, 2017. [Google Scholar]
- Huang, Z.-D.; Zhao, Y.-F.; Li, S.; Gu, H.-Y.; Lin, L.-L.; Yang, Z.-Y.; Niu, Y.-M.; Zhang, C.; Luo, J. Comparative Efficacy and Acceptability of Pharmaceutical Management for Adults with Post-Traumatic Stress Disorder: A Systematic Review and Meta-Analysis. Front. Pharmacol. 2020, 11, 559. [Google Scholar] [CrossRef]
- Dubey, V.K.; Ansari, F.; Vohora, D.; Khanam, R. Possible involvement of corticosterone and serotonin in antidepressant and antianxiety effects of chromium picolinate in chronic unpredictable mild stress induced depression and anxiety in rats. J. Trace Elem. Med. Biol. 2015, 29, 222–226. [Google Scholar] [CrossRef]
- Zangrossi, H.; Graeff, F.G. Serotonin in anxiety and panic: Contributions of the elevated T-maze. Neurosci. Biobehav. Rev. 2014, 46, 397–406. [Google Scholar] [CrossRef]
- Gros, D.F.; Price, M.; Magruder, K.M.; Frueh, B.C. Symptom overlap in posttraumatic stress disorder and major depression. Psychiatry Res. 2012, 196, 267–270. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-C.; Tung, C.-S.; Liu, Y.-P. Escitalopram reversed the traumatic stress-induced depressed and anxiety-like symptoms but not the deficits of fear memory. Psychopharmacology 2016, 233, 1135–1146. [Google Scholar] [CrossRef]
- Flory, J.D.; Yehuda, R. Comorbidity between post-traumatic stress disorder and major depressive disorder: Alternative explanations and treatment considerations. Dialogues Clin. Neurosci. 2015, 17, 141–150. [Google Scholar] [CrossRef]
- Harvey, B.H.; Naciti, C.; Brand, L.; Stein, D.J. Serotonin and stress: Protective or malevolent actions in the biobehavioral response to repeated trauma? Ann. N. Y. Acad. Sci. 2004, 1032, 267–272. [Google Scholar] [CrossRef]
- Ravindran, L.N.; Stein, M.B. Pharmacotherapy of PTSD: Premises, principles, and priorities. Brain Res. 2009, 1293, 24–39. [Google Scholar] [CrossRef] [Green Version]
- Sharp, T.; Cowen, P.J. 5-HT and depression: Is the glass half-full? Curr. Opin. Pharmacol. 2011, 11, 45–51. [Google Scholar] [CrossRef]
- Bocchio, M.; McHugh, S.B.; Bannerman, D.; Sharp, T.; Capogna, M. Serotonin, Amygdala and Fear: Assembling the Puzzle. Front. Neural Circuits 2016, 10, 24. [Google Scholar] [CrossRef]
- Murrough, J.W.; Huang, Y.; Hu, J.; Henry, S.; Williams, W.; Gallezot, J.-D.; Bailey, C.R.; Krystal, J.H.; Carson, R.E.; Neumeister, A. Reduced Amygdala Serotonin Transporter Binding in Posttraumatic Stress Disorder. Biol. Psychiatry 2011, 70, 1033–1038. [Google Scholar] [CrossRef] [Green Version]
- Murrough, J.W.; Czermak, C.; Henry, S.; Nabulsi, N.; Gallezot, J.-D.; Gueorguieva, R.; Planeta-Wilson, B.; Krystal, J.H.; Neumaier, J.F.; Huang, Y.; et al. The Effect of Early Trauma Exposure on Serotonin Type 1B Receptor Expression Revealed by Reduced Selective Radioligand Binding. Arch. Gen. Psychiatry 2011, 68, 892–900. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.X.; Han, H.L.; Tian, M.; Cao, J.; Xiu, J.B.; Song, N.N.; Huang, Y.; Xu, T.L.; Ding, Y.Q.; Xu, L. Enhanced contextual fear memory in central serotonin-deficient mice. Proc. Natl. Acad. Sci. USA 2008, 105, 11981–11986. [Google Scholar] [CrossRef] [Green Version]
- Waider, J.; Popp, S.; Mlinar, B.; Montalbano, A.; Bonfiglio, F.; Aboagye, B.; Thuy, E.; Kern, R.; Thiel, C.; Araragi, N.; et al. Serotonin Deficiency Increases Context-Dependent Fear Learning Through Modulation of Hippocampal Activity. Front. Neurosci. 2019, 13, 245. [Google Scholar] [CrossRef]
- Kompagne, H.; Bárdos, G.; Szénási, G.; Gacsályi, I.; Hársing, L.G.; Lévay, G. Chronic mild stress generates clear depressive but ambiguous anxiety-like behaviour in rats. Behav. Brain Res. 2008, 193, 311–314. [Google Scholar] [CrossRef]
- Pitkänen, A.; Savander, V.; LeDoux, J.E. Organization of intra-amygdaloid circuitries in the rat: An emerging framework for understanding functions of the amygdala. Trends Neurosci. 1997, 20, 517–523. [Google Scholar] [CrossRef]
- Friedman, M.J.; Marmar, C.R.; Baker, D.G.; Sikes, C.R.; Farfel, G.M. Randomized, Double-Blind Comparison of Sertraline and Placebo for Posttraumatic Stress Disorder in a Department of Veterans Affairs Setting. J. Clin. Psychiatry 2007, 68, 711–720. [Google Scholar] [CrossRef]
- Robert, S.; Hamner, M.B.; Ulmer, H.G.; Lorberbaum, J.P.; Durkalsk, V.L. Open-Label Trial of Escitalopram in the Treatment of Posttraumatic Stress Disorder. J. Clin. Psychiatry 2006, 67, 1522–1526. [Google Scholar] [CrossRef]
- Furmaga, H.; Shah, A.; Frazer, A. Serotonergic and Noradrenergic Pathways Are Required for the Anxiolytic-like and Antidepressant-like Behavioral Effects of Repeated Vagal Nerve Stimulation in Rats. Biol. Psychiatry 2011, 70, 937–945. [Google Scholar] [CrossRef]
- Kosten, T.R.; Mason, J.W.; Giller, E.L.; Ostroff, R.; Harkness, L. Sustained urinary norepinephrine and epinephrine elevation in post-traumatic stress disorder. Psychoneuroendocrinology 1987, 12, 13–20. [Google Scholar] [CrossRef]
- Naegeli, C.; Zeffiro, T.; Piccirelli, M.; Jaillard, A.; Weilenmann, A.; Hassanpour, K.; Schick, M.; Rufer, M.; Orr, S.P.; Mueller-Pfeiffer, C. Locus Coeruleus Activity Mediates Hyperresponsiveness in Posttraumatic Stress Disorder. Biol. Psychiatry 2018, 83, 254–262. [Google Scholar] [CrossRef]
- Strawn, J.R.; Geracioti, T.D. Noradrenergic dysfunction and the psychopharmacology of posttraumatic stress disorder. Depress. Anxiety 2008, 25, 260–271. [Google Scholar] [CrossRef]
- Yehuda, R.; Southwick, S.; Giller, E.L.; Mason, J.W. Urinary Catecholamine Excretion and Severity of PTSD Symptoms in Vietnam Combat Veterans. J. Nerv. Ment. Dis. 1992, 180, 321–325. [Google Scholar] [CrossRef]
- Geracioti, T.D.; Baker, D.G.; Ekhator, N.N.; West, S.A.; Hill, K.K.; Bruce, A.B.; Schmidt, D.; Rounds-Kugler, B.; Yehuda, R.; Keck, P.E.; et al. CSF Norepinephrine Concentrations in Posttraumatic Stress Disorder. Am. J. Psychiatry 2001, 158, 1227–1230. [Google Scholar] [CrossRef]
- Liberzon, I.; López, J.F.; Flagel, S.B.; Vázquez, D.M.; Young, E.A. Differential regulation of hippocampal glucocorticoid receptors mRNA and fast feedback: Relevance to post-traumatic stress disorder. J. Neuroendocrinol. 1999, 11, 11–17. [Google Scholar] [CrossRef]
- Orr, S.P.; Metzger, L.J.; Lasko, N.B.; Macklin, M.L.; Hu, F.B.; Shalev, A.Y.; Pitman, R.K. Physiologic responses to sudden, loud tones in monozygotic twins discordant for combat exposure: Association with posttraumatic stress disorder. Arch. Gen. Psychiatry 2003, 60, 283–288. [Google Scholar] [CrossRef] [Green Version]
- Pitman, R.K.; Orr, S.P. Twenty-four hour urinary cortisol and catecholamine excretion in combat-related posttraumatic stress disorder. Biol. Psychiatry 1990, 27, 245–247. [Google Scholar] [CrossRef]
- Hendrickson, R.; Raskind, M.A. Noradrenergic dysregulation in the pathophysiology of PTSD. Exp. Neurol. 2016, 284, 181–195. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, P.J.; Giustino, T.F.; Seemann, J.R.; Maren, S. Noradrenergic blockade stabilizes prefrontal activity and enables fear extinction under stress. Proc. Natl. Acad. Sci. USA 2015, 112, E3729–E3737. [Google Scholar] [CrossRef] [Green Version]
- Uematsu, A.; Tan, B.Z.; Ycu, E.A.; Cuevas, J.S.; Koivumaa, J.; Junyent, F.; Kremer, E.; Witten, I.B.; Deisseroth, K.; Johansen, J.P. Modular organization of the brainstem noradrenaline system coordinates opposing learning states. Nat. Neurosci. 2017, 20, 1602–1611. [Google Scholar] [CrossRef]
- Giustino, T.F.; Ramanathan, K.R.; Totty, M.S.; Miles, O.W.; Maren, S. Locus Coeruleus Norepinephrine Drives Stress-Induced Increases in Basolateral Amygdala Firing and Impairs Extinction Learning. J. Neurosci. 2019, 40, 907–916. [Google Scholar] [CrossRef]
- Hauser, M.A.; Garrett, M.E.; Liu, Y.; Dennis, M.F.; Kimbrel, N.A.; Beckham, J.C.; Ashley-Koch, A. Further evidence for a role of the ADRB2 gene in risk for posttraumatic stress disorder. J. Psychiatry Res. 2017, 84, 59–61. [Google Scholar] [CrossRef]
- Giustino, T.F.; Maren, S. Noradrenergic Modulation of Fear Conditioning and Extinction. Front. Behav. Neurosci. 2018, 12, 43. [Google Scholar] [CrossRef]
- Ji, J.-Z.; Wang, X.-M.; Li, B.-M. Deficit in long-term contextual fear memory induced by blockade of beta-adrenoceptors in hippocampal CA1 region. Eur. J. Neurosci. 2003, 17, 1947–1952. [Google Scholar] [CrossRef]
- Schiff, H.C.; Johansen, J.P.; Hou, M.; Bush, D.E.; Smith, E.K.; Klein, J.E.; LeDoux, J.E.; Sears, R.M. β-Adrenergic Receptors Regulate the Acquisition and Consolidation Phases of Aversive Memory Formation Through Distinct, Temporally Regulated Signaling Pathways. Neuropsychopharmacology 2017, 42, 895–903. [Google Scholar] [CrossRef] [Green Version]
- Lopresto, D.; Schipper, P.; Homberg, J.R. Neural circuits and mechanisms involved in fear generalization: Implications for the pathophysiology and treatment of posttraumatic stress disorder. Neurosci. Biobehav. Rev. 2016, 60, 31–42. [Google Scholar] [CrossRef]
- Seo, D.-O.; Zhang, E.T.; Piantadosi, S.C.; Marcus, D.J.; Motard, L.E.; Kan, B.K.; Gomez, A.M.; Nguyen, T.K.; Xia, L.; Bruchas, M.R. A locus coeruleus to dentate gyrus noradrenergic circuit modulates aversive contextual processing. Neuron 2021, 109, 2116–2130.E6. [Google Scholar] [CrossRef]
- Martinho, R.; Oliveira, A.; Correia, G.; Marques, M.; Seixas, R.; Serrão, P.; Moreira-Rodrigues, M. Epinephrine May Contribute to the Persistence of Traumatic Memories in a Post-traumatic Stress Disorder Animal Model. Front. Mol. Neurosci. 2020, 13, 588802. [Google Scholar] [CrossRef]
- Steenen, S.A.; van Wijk, A.J.; van der Heijden, G.M.J.; van Westrhenen, R.; de Lange, J.; de Jongh, A. Propranolol for the treatment of anxiety disorders: Systematic review and meta-analysis. J. Psychopharmacol. 2015, 30, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Young, C.; Butcher, R. Propranolol for post-traumatic stress disorder: A review of clinical effectiveness [Internet]. In CADTH Rapid Response Reports; Canadian Agency for Drugs and Technologies in Health: Ottawa, ON, Canada, 2020. [Google Scholar]
- Brunet, A.; Saumier, D.; Liu, A.; Streiner, D.L.; Tremblay, J.; Pitman, R.K. Reduction of PTSD Symptoms with Pre-Reactivation Propranolol Therapy: A Randomized Controlled Trial. Am. J. Psychiatry 2018, 175, 427–433. [Google Scholar] [CrossRef]
- Vetere, G.; Piserchia, V.; Borreca, A.; Novembre, G.; Aceti, M.; Ammassari-Teule, M. Reactivating fear memory under propranolol resets pre-trauma levels of dendritic spines in basolateral amygdala but not dorsal hippocampus neurons. Front. Behav. Neurosci. 2013, 7, 211. [Google Scholar] [CrossRef] [Green Version]
- Roozendaal, B.; Hui, G.; Hui, I.; Berlau, D.; McGaugh, J.; Weinberger, N. Basolateral amygdala noradrenergic activity mediates corticosterone-induced enhancement of auditory fear conditioning. Neurobiol. Learn. Mem. 2006, 86, 249–255. [Google Scholar] [CrossRef]
- Costanzo, M.; Jovanovic, T.; Norrholm, S.D.; Ndiongue, R.; Reinhardt, B.; Roy, C.M.J. Psychophysiological Investigation of Combat Veterans with Subthreshold Post-traumatic Stress Disorder Symptoms. Mil. Med. 2016, 181, 793–802. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Deng, M.; Wu, J.; Lan, Q.; Yang, H.; Zhang, C. Ventral Tegmental Area Dysfunction and Disruption of Dopaminergic Homeostasis: Implications for Post-traumatic Stress Disorder. Mol. Neurobiol. 2021, 58, 2423–2434. [Google Scholar] [CrossRef]
- Fadok, J.P.; Dickerson, T.M.; Palmiter, R.D. Dopamine is necessary for cue-dependent fear conditioning. J. Neurosci. 2009, 29, 11089–11097. [Google Scholar] [CrossRef]
- Lin, C.-C.; Tung, C.-S.; Lin, P.-H.; Huang, C.-L.; Liu, Y.-P. Traumatic stress causes distinctive effects on fear circuit catecholamines and the fear extinction profile in a rodent model of posttraumatic stress disorder. Eur. Neuropsychopharmacol. 2016, 26, 1484–1495. [Google Scholar] [CrossRef]
- Espejo, E.F. Prefrontocortical Dopamine Loss in Rats Delays Long-Term Extinction of Contextual Conditioned Fear, and Reduces Social Interaction Without Affecting Short-Term Social Interaction Memory. Neuropsychopharmacology 2002, 28, 490–498. [Google Scholar] [CrossRef] [Green Version]
- Abraham, A.D.; Neve, K.; Lattal, K.M. Dopamine and extinction: A convergence of theory with fear and reward circuitry. Neurobiol. Learn. Mem. 2013, 108, 65–77. [Google Scholar] [CrossRef] [Green Version]
- Inglis, F.M.; Moghaddam, B. Dopaminergic Innervation of the Amygdala Is Highly Responsive to Stress. J. Neurochem. 2008, 72, 1088–1094. [Google Scholar] [CrossRef]
- Mueller, D.; Bravo-Rivera, C.; Quirk, G.J. Infralimbic D2 Receptors Are Necessary for Fear Extinction and Extinction-Related Tone Responses. Biol. Psychiatry 2010, 68, 1055–1060. [Google Scholar] [CrossRef] [Green Version]
- Gerlicher, A.M.V.; Tuscher, O.; Kalisch, R. Dopamine-dependent prefrontal reactivations explain long-term benefit of fear extinction. Nat. Commun. 2018, 9, 4294. [Google Scholar] [CrossRef] [Green Version]
- Cabib, S.; Puglisi-Allegra, S. The mesoaccumbens dopamine in coping with stress. Neurosci. Biobehav. Rev. 2012, 36, 79–89. [Google Scholar] [CrossRef]
- Friedman, A.K.; Walsh, J.J.; Juarez, B.; Ku, S.M.; Chaudhury, D.; Wang, J.; Li, X.; Dietz, D.M.; Pan, N.; Vialou, V.F.; et al. Enhancing Depression Mechanisms in Midbrain Dopamine Neurons Achieves Homeostatic Resilience. Science 2014, 344, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Levis, D.J. The prolonged CS exposure techniques of implosive (flooding) therapy. In General Principles and Empirically Supported Techniques of Cognitive Behavior Therapy; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009; pp. 370–380. [Google Scholar]
- Zalta, A.K. Psychological Mechanisms of Effective Cognitive–Behavioral Treatments for PTSD. Curr. Psychiatry Rep. 2015, 17, 23. [Google Scholar] [CrossRef] [Green Version]
- Hitora-Imamura, N.; Miura, Y.; Teshirogi, C.; Ikegaya, Y.; Matsuki, N.; Nomura, H. Prefrontal dopamine regulates fear reinstatement through the downregulation of extinction circuits. eLife 2015, 30, 08274. [Google Scholar] [CrossRef]
- Dadkhah, M.; Abdullahi, P.R.; Rashidy-Pour, A.; Sameni, H.R.; Vafaei, A.A. Infralimbic dopamine D2 receptors mediate glucocorticoid-induced facilitation of auditory fear memory extinction in rats. Brain Res. 2018, 1682, 84–92. [Google Scholar] [CrossRef]
- Heath, F.C.; Jurkus, R.; Bast, T.; Pezze, M.A.; Lee, J.L.C.; Voigt, J.P.; Stevenson, C.W. Dopamine D1-like receptor signalling in the hippocampus and amygdala modulates the acquisition of contextual fear conditioning. Psychopharmacology 2015, 232, 2619–2629. [Google Scholar] [CrossRef] [Green Version]
- Madsen, H.B.; Guerin, A.; Kim, J.H. Investigating the role of dopamine receptor- and parvalbumin-expressing cells in extinction of conditioned fear. Neurobiol. Learn. Mem. 2017, 145, 7–17. [Google Scholar] [CrossRef]
- Lonsdorf, T.B.; Weike, A.I.; Nikamo, P.; Schalling, M.; Hamm, A.O.; Ohman, A. Genetic gating of human fear learning and extinction: Possible implications for gene-environment interaction in anxiety disorder. Psychol. Sci. 2009, 20, 198–206. [Google Scholar] [CrossRef]
- Lonsdorf, T.B.; Rück, C.; Bergström, J.; Andersson, G.; Ohman, A.; Lindefors, N.; Schalling, M. The COMTval158met polymorphism is associated with symptom relief during exposure-based cognitive-behavioral treatment in panic disorder. BMC Psychiatry 2010, 10, 99. [Google Scholar] [CrossRef] [Green Version]
- Farrell, S.M.; Tunbridge, E.M.; Braeutigam, S.; Harrison, P.J. COMT Val158Met Genotype Determines the Direction of Cognitive Effects Produced by Catechol-O-Methyltransferase Inhibition. Biol. Psychiatry 2012, 71, 538–544. [Google Scholar] [CrossRef] [Green Version]
- Wood, M.; Reavill, C. Aripiprazole acts as a selective dopamine D2receptor partial agonist. Expert Opin. Investig. Drugs 2007, 16, 771–775. [Google Scholar] [CrossRef]
- Ganella, D.E.; Lee-Kardashyan, L.; Luikinga, S.J.; Nguyen, D.L.D.; Madsen, H.B.; Zbukvic, I.C.; Coulthard, R.; Lawrence, A.J.; Kim, J.H. Aripiprazole Facilitates Extinction of Conditioned Fear in Adolescent Rats. Front. Behav. Neurosci. 2017, 11, 76. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-C.; Chang, H.-A.; Tai, Y.-M.; Chen, T.-Y.; Wan, F.-J.; Chang, C.-C.; Tung, C.-S.; Liu, Y.-P. Subchronic administration of aripiprazole improves fear extinction retrieval of Pavlovian conditioning paradigm in rats experiencing psychological trauma. Behav. Brain Res. 2019, 362, 181–187. [Google Scholar] [CrossRef]
- Sriram, K.; Rodriguez-Fernandez, M.; Doyle, F.J. 3rd Modeling cortisol dynamics in the neuro-endocrine axis distinguishes normal, depression, and post-traumatic stress disorder (PTSD) in humans. PLoS Comput. Biol. 2012, 8, e1002379. [Google Scholar] [CrossRef]
- Yehuda, R. Post-traumatic stress disorder. N. Engl. J. Med. 2002, 346, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Yehuda, R.; Bierer, L.M.; Pratchett, L.C.; Lehrner, A.; Koch, E.C.; Van Manen, J.A.; Flory, J.D.; Makotkine, I.; Hildebrandt, T. Cortisol augmentation of a psychological treatment for warfighters with posttraumatic stress disorder: Randomized trial showing improved treatment retention and outcome. Psychoneuroendocrinology 2015, 51, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Von Känel, R.; Schmid, J.-P.; Abbas, C.C.; Gander, M.-L.; Saner, H.; Begré, S. Stress hormones in patients with posttraumatic stress disorder caused by myocardial infarction and role of comorbid depression. J. Affect. Disord. 2010, 121, 73–79. [Google Scholar] [CrossRef] [PubMed]
- De Kloet, E.R.; Vreugdenhil, E.; Oitzl, M.S.; Joëls, M. Brain Corticosteroid Receptor Balance in Health and Disease. Endocr. Rev. 1998, 19, 269–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finsterwald, C.; Alberini, C.M. Stress and glucocorticoid receptor-dependent mechanisms in long-term memory: From adaptive responses to psychopathologies. Neurobiol. Learn. Mem. 2013, 112, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Reul, J.M.; de Kloet, E.R. Two receptor systems for corticosterone in rat brain: Microdistribution and differential occupation. Endocrinology 1985, 117, 2505–2511. [Google Scholar] [CrossRef]
- Geuze, E.; van Wingen, G.A.; van Zuiden, M.; Rademaker, A.R.; Vermetten, E.; Kavelaars, A.; Fernández, G.; Heijnen, C.J. Glucocorticoid receptor number predicts increase in amygdala activity after severe stress. Psychoneuroendocrinology 2012, 37, 1837–1844. [Google Scholar] [CrossRef] [Green Version]
- Pitman, R.K.; Rasmusson, A.M.; Koenen, K.; Shin, L.M.; Orr, S.P.; Gilbertson, M.W.; Milad, M.R.; Liberzon, I. Biological studies of post-traumatic stress disorder. Nat. Rev. Neurosci. 2012, 13, 769–787. [Google Scholar] [CrossRef]
- Catalani, A.; Alemà, G.S.; Cinque, C.; Zuena, A.R.; Casolini, P. Maternal corticosterone effects on hypothalamus–pituitary–adrenal axis regulation and behavior of the offspring in rodents. Neurosci. Biobehav. Rev. 2011, 35, 1502–1517. [Google Scholar] [CrossRef]
- Engeland, W.C.; Arnhold, M.M. Neural Circuitry in the Regulation of Adrenal Corticosterone Rhythmicity. Endocrine 2005, 28, 325–332. [Google Scholar] [CrossRef]
- Ding, J.; Chen, X.; da Silva, M.S.; Lingeman, J.; Han, F.; Meijer, O.C. Effects of RU486 treatment after single prolonged stress depend on the post-stress interval. Mol. Cell. Neurosci. 2020, 108, 103541. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; da Silva, M.S.; Lingeman, J.; Chen, X.; Shi, Y.; Han, F.; Meijer, O.C. Late glucocorticoid receptor antagonism changes the outcome of adult life stress. Psychoneuroendocrinology 2019, 107, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Golier, J.A.; Caramanica, K.; DeMaria, R.; Yehuda, R. A Pilot Study of Mifepristone in Combat-Related PTSD. Depress. Res. Treat. 2012, 2012, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.-C.; Cheng, P.-Y.; Hsiao, M.; Liu, Y.-P. Effects of RU486 in Treatment of Traumatic Stress-Induced Glucocorticoid Dysregulation and Fear-Related Abnormalities: Early versus Late Intervention. Int. J. Mol. Sci. 2022, 23, 5494. [Google Scholar] [CrossRef] [PubMed]
- Gutièrrez-Mecinas, M.; Trollope, A.F.; Collins, A.; Morfett, H.; Hesketh, S.A.; Kersanté, F.; Reul, J.M. Long-lasting behavioral responses to stress involve a direct interaction of glucocorticoid receptors with ERK1/2-MSK1-Elk-1 signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 13806–13811. [Google Scholar] [CrossRef] [Green Version]
- Revest, J.-M.; Kaouane, N.; Mondin, M.; le Roux, A.; Rougé-Pont, F.; Vallée, M.; Barik, J.; Tronche, F.; Desmedt, A.; Piazza, P.V. The enhancement of stress-related memory by glucocorticoids depends on synapsin-Ia/Ib. Mol. Psychiatry 2010, 15, 1140–1151. [Google Scholar] [CrossRef] [Green Version]
- Revest, J.M.; Le Roux, A.; Roullot-Lacarriere, V.; Kaouane, N.; Vallee, M.; Kasanetz, F.; Rouge-Pont, F.; Tronche, F.; Desmedt, A.; Piazza, P.V. BDNF-TrkB signaling through Erk1/2 MAPK phosphorylation mediates the enhancement of fear memory induced by glucocorticoids. Mol. Psychiatry 2014, 19, 1001–1009. [Google Scholar] [CrossRef] [Green Version]
- Schelling, G.; Stoll, C.; Kapfhammer, H.P.; Rothenhäusler, H.B.; Krauseneck, T.; Durst, K.; Haller, M.; Briegel, J. The effect of stress doses of hydrocortisone during septic shock on posttraumatic stress disorder and health-related quality of life in survivors. Crit. Care Med. 1999, 27, 2678–2683. [Google Scholar] [CrossRef]
- Schelling, G.; Briegel, J.; Roozendaal, B.; Stoll, C.; Rothenhäusler, H.B.; Kapfhammer, H.P. The effect of stress doses of hydrocortisone during septic shock on posttraumatic stress disorder in survivors. Biol. Psychiatry 2001, 50, 978–985. [Google Scholar] [CrossRef]
- Fani, N.; Gutman, D.; Tone, E.B.; Almli, L.; Mercer, K.B.; Davis, J.; Glover, E.; Jovanovic, T.; Bradley, B.; Dinov, I.D.; et al. FKBP5 and attention bias for threat: Associations with hippocampal function and shape. JAMA Psychiatry 2013, 70, 392–400. [Google Scholar] [CrossRef] [Green Version]
- Sawamura, T.; Klengel, T.; Armario, A.; Jovanovic, T.; Norrholm, S.D.; Ressler, K.J.; Andero, R. Dexamethasone Treatment Leads to Enhanced Fear Extinction and Dynamic Fkbp5 Regulation in Amygdala. Neuropsychopharmacology 2015, 41, 832–846. [Google Scholar] [CrossRef] [PubMed]
- Michopoulos, V.; Norrholm, S.D.; Stevens, J.S.; Glover, E.M.; Rothbaum, B.O.; Gillespie, C.; Schwartz, A.C.; Ressler, K.J.; Jovanovic, T. Dexamethasone facilitates fear extinction and safety discrimination in PTSD: A placebo-controlled, double-blind study. Psychoneuroendocrinology 2017, 83, 65–71. [Google Scholar] [CrossRef]
- Surís, A.; Holliday, R.; Adinoff, B.; Holder, N.; North, C.S. Facilitating Fear-Based Memory Extinction with Dexamethasone: A Randomized Controlled Trial in Male Veterans with Combat-Related PTSD. Psychiatry 2017, 80, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Binder, E.B. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology 2009, 34 (Suppl. S1), S186–S195. [Google Scholar] [CrossRef] [PubMed]
- Menke, A.; Arloth, J.; Pütz, B.; Weber, P.; Klengel, T.; Mehta, D.; Gonik, M.; Rex-Haffner, M.; Rubel, J.; Uhr, M.; et al. Dexamethasone stimulated gene expression in peripheral blood is a sensitive marker for glucocorticoid receptor resistance in depressed patients. Neuropsychopharmacology 2012, 37, 1455–1464. [Google Scholar] [CrossRef]
- Lesuis, S.L.; Brosens, N.; Immerzeel, N.; van der Loo, R.J.; Mitrić, M.; Bielefeld, P.; Fitzsimons, C.P.; Lucassen, P.J.; Kushner, S.A.; Oever, M.C.V.D.; et al. Glucocorticoids Promote Fear Generalization by Increasing the Size of a Dentate Gyrus Engram Cell Population. Biol. Psychiatry 2021, 90, 494–504. [Google Scholar] [CrossRef]
- Roozendaal, B.; Mirone, G. Opposite effects of noradrenergic and glucocorticoid activation on accuracy of an episodic-like memory. Psychoneuroendocrinology 2020, 114, 104588. [Google Scholar] [CrossRef]
- Merz, C.J.; Hamacher-Dang, T.C.; Stark, R.; Wolf, O.T.; Hermann, A. Neural Underpinnings of Cortisol Effects on Fear Extinction. Neuropsychopharmacology 2018, 43, 384–392. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, C.-C.; Liu, Y.-P. Pharmacological Implications of Adjusting Abnormal Fear Memory: Towards the Treatment of Post-Traumatic Stress Disorder. Pharmaceuticals 2022, 15, 788. https://doi.org/10.3390/ph15070788
Lin C-C, Liu Y-P. Pharmacological Implications of Adjusting Abnormal Fear Memory: Towards the Treatment of Post-Traumatic Stress Disorder. Pharmaceuticals. 2022; 15(7):788. https://doi.org/10.3390/ph15070788
Chicago/Turabian StyleLin, Chen-Cheng, and Yia-Ping Liu. 2022. "Pharmacological Implications of Adjusting Abnormal Fear Memory: Towards the Treatment of Post-Traumatic Stress Disorder" Pharmaceuticals 15, no. 7: 788. https://doi.org/10.3390/ph15070788
APA StyleLin, C.-C., & Liu, Y.-P. (2022). Pharmacological Implications of Adjusting Abnormal Fear Memory: Towards the Treatment of Post-Traumatic Stress Disorder. Pharmaceuticals, 15(7), 788. https://doi.org/10.3390/ph15070788