
Two Novel Co-Crystals of Naproxen: Comparison of Stability, Solubility and Intermolecular Interaction


 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
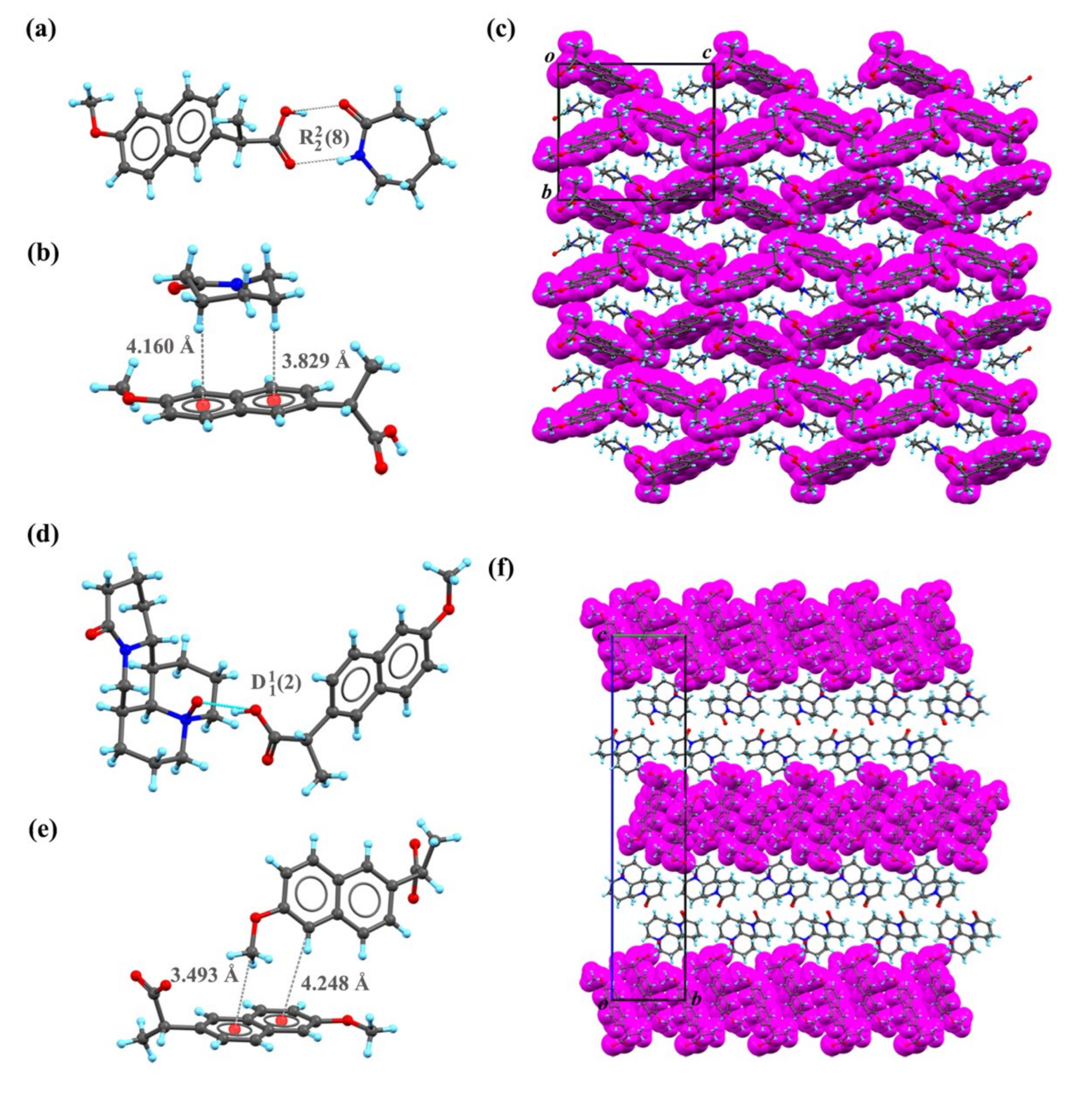
2.1. SC XRD Analysis
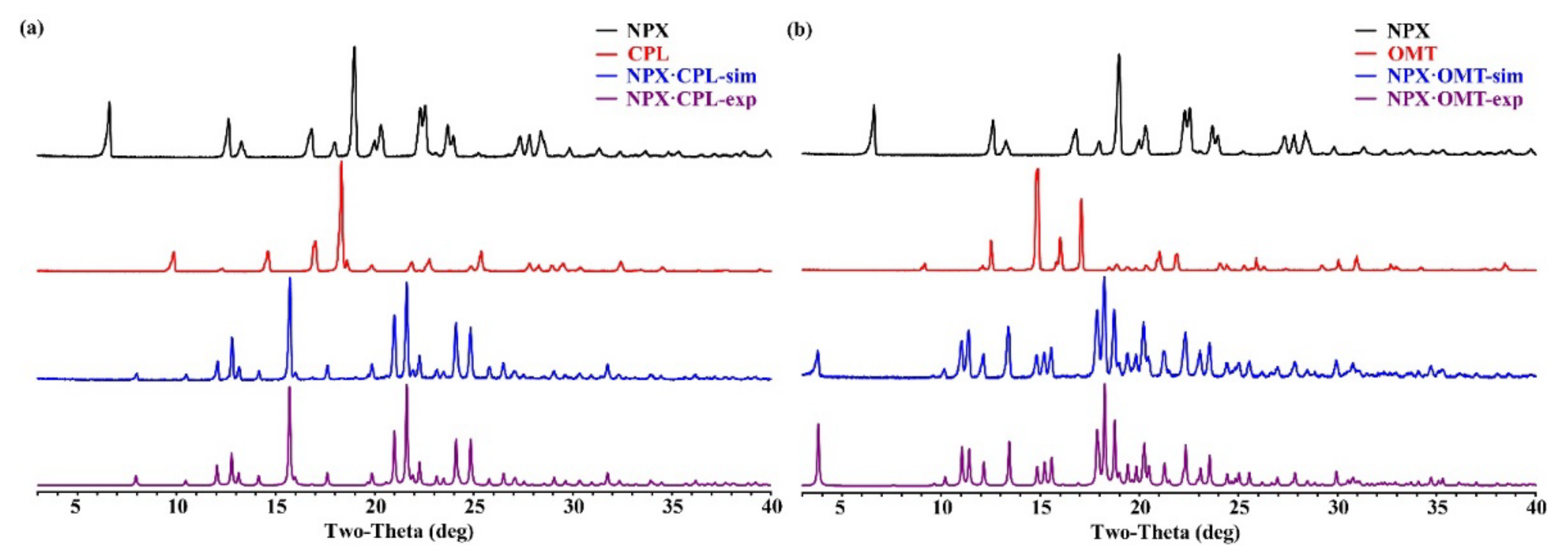
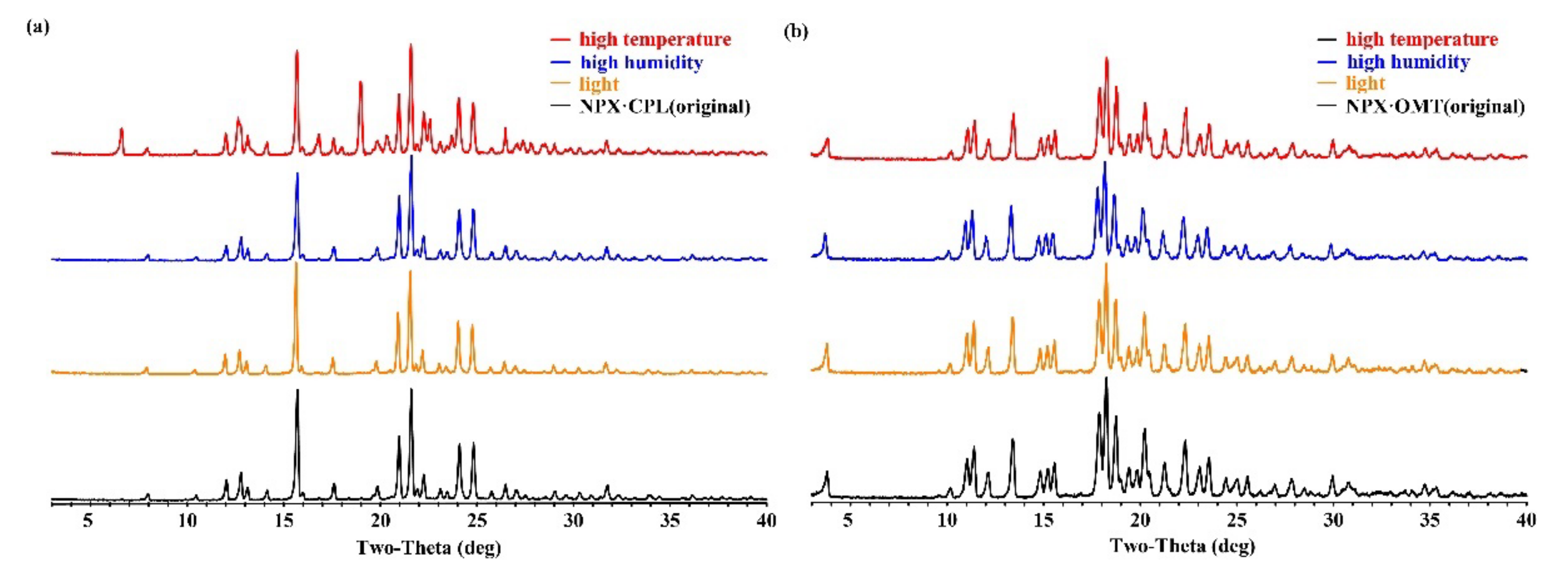
2.2. PXRD Analysis
2.3. DSC Analysis
2.4. IR Analysis
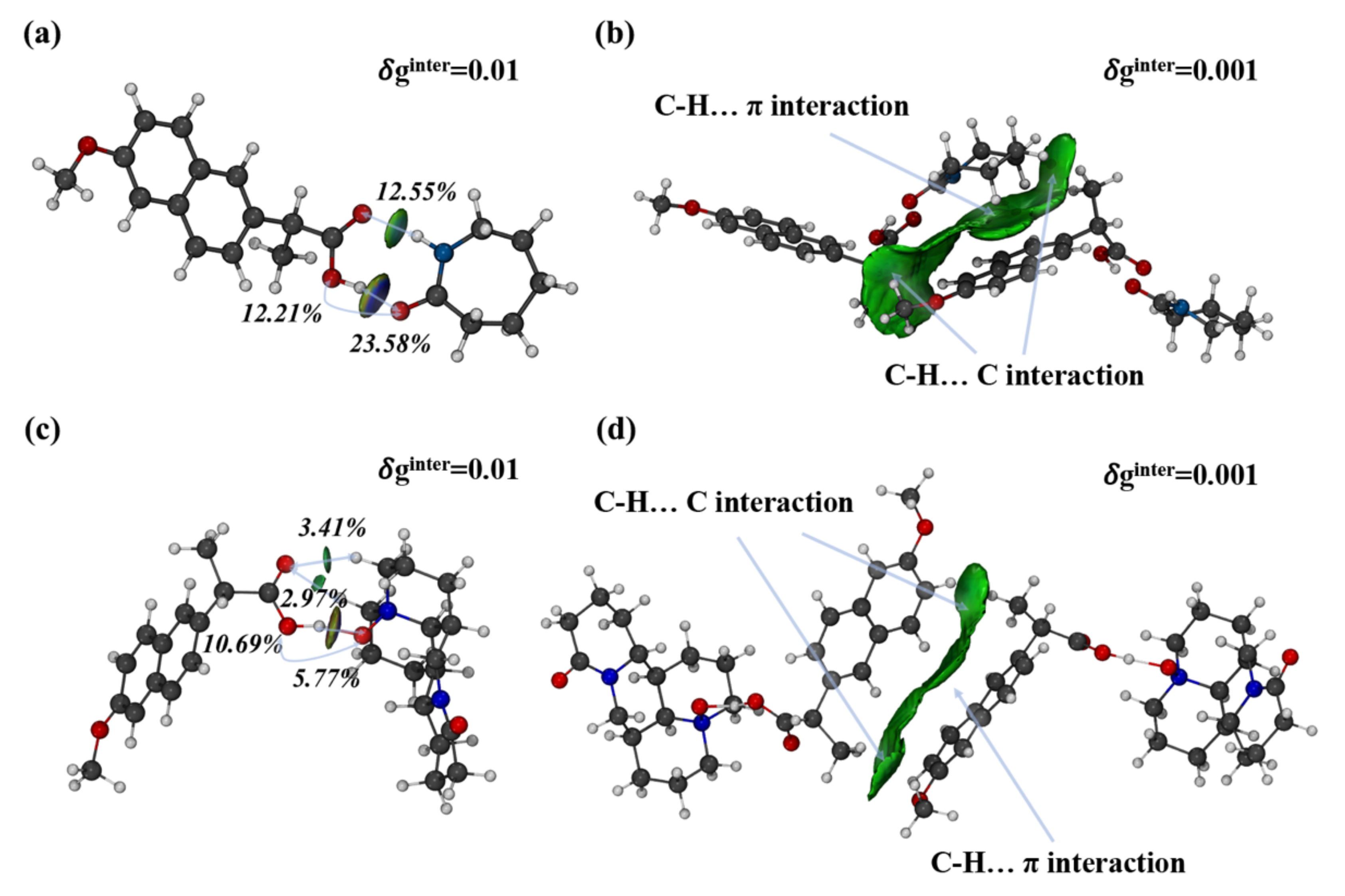
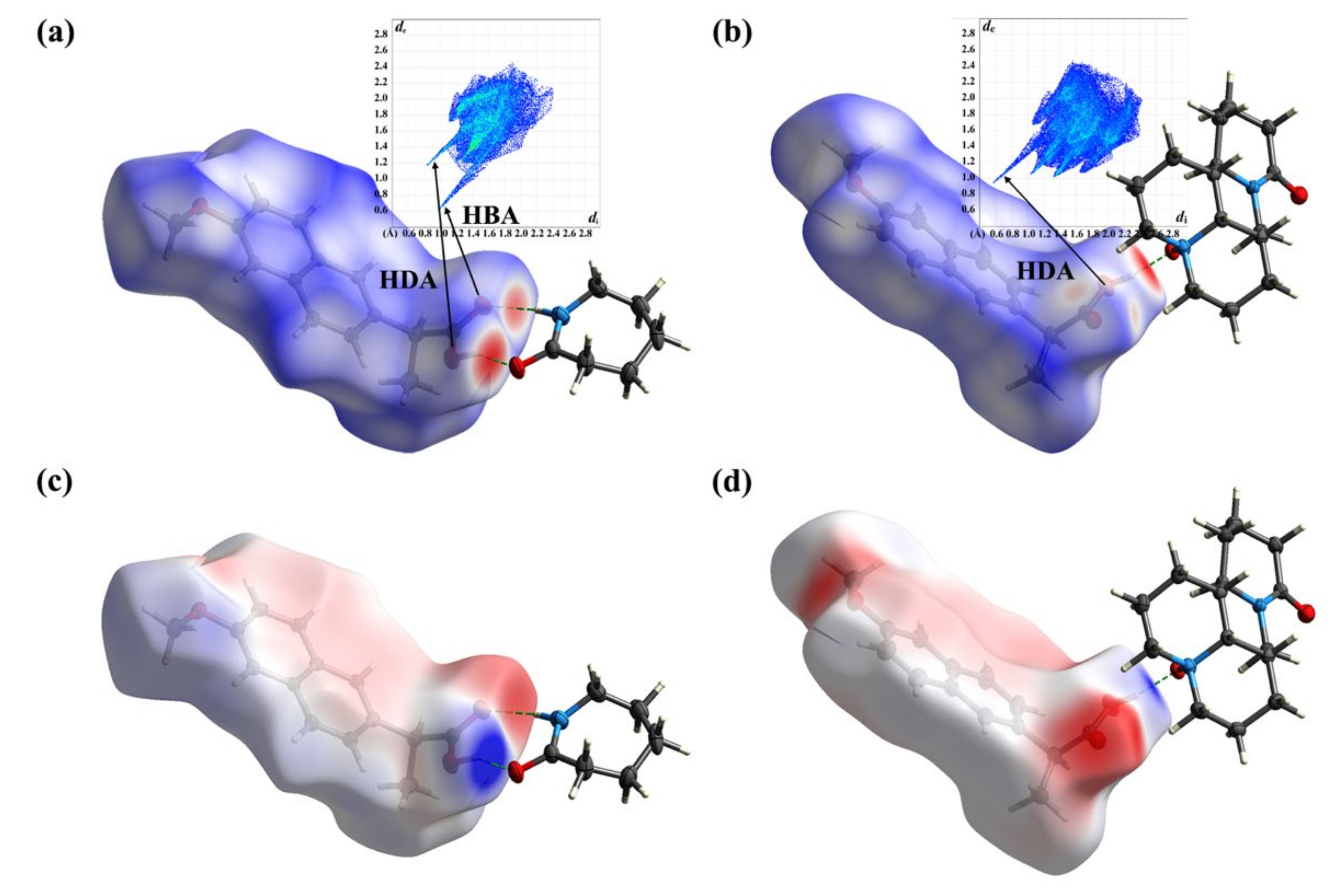
2.5. Computation
2.6. Physical Stability
2.7. Apparent Solubility
3. Materials and Methods
3.1. Materials
3.2. Preparation of NPX–CPL Co-Crystal
3.3. Preparation of NPX–OMT Co-Crystal
3.4. SC XRD
3.5. PXRD
3.6. DSC
3.7. IR Spectrum
3.8. Stability Test
3.9. Apparent Solubility Test
3.10. Theoretical Computation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- He, B.-S.; Wang, J.; Liu, J.; Hu, X.-M. Eco-pharmacovigilance of non-steroidal anti-inflammatory drugs: Necessity and opportunities. Chemosphere 2017, 181, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Bindu, S.; Mazumder, S.; Bandyopadhyay, U. Non-steroidal anti-inflammatory drugs (NSAIDs) and organ damage: A current perspective. Biochem. Pharmacol. 2020, 180, 114147. [Google Scholar] [CrossRef] [PubMed]
- Garg, U.; Azim, Y. Challenges and opportunities of pharmaceutical cocrystals: A focused review on non-steroidal anti-inflammatory drugs. RSC Med. Chem. 2021, 12, 705–721. [Google Scholar] [CrossRef] [PubMed]
- Urban, M.K. COX-2 specific inhibitors offer improved advantages over traditional NSAIDs. Orthopedics 2000, 23, 761–764. [Google Scholar] [CrossRef]
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: The correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, H.E.; Softley, L.K.; Suresh, K.; Hodgkinson, P.; Evans, I.R. Structure and physicochemical characterization of a naproxen–picolinamide cocrystal. Acta Crystallogr. Sect. C Struct. Chem. 2017, 73, 168–175. [Google Scholar] [CrossRef] [Green Version]
- Shaikh, R.; Singh, R.; Walker, G.M.; Croker, D.M. Pharmaceutical Cocrystal Drug Products: An Outlook on Product Development. Trends Pharmacol. Sci. 2018, 39, 1033–1048. [Google Scholar] [CrossRef]
- Berry, D.J.; Steed, J.W. Pharmaceutical cocrystals, salts and multicomponent systems; intermolecular interactions and property based design. Adv. Drug Deliv. Rev. 2017, 117, 3–24. [Google Scholar] [CrossRef] [Green Version]
- Ngilirabanga, J.B.; Samsodien, H. Pharmaceutical co-crystal: An alternative strategy for enhanced physicochemical properties and drug synergy. Nano Select 2021, 2, 512–526. [Google Scholar] [CrossRef]
- Ancheria, R.K.; Jain, S.; Kumar, D.; Soni, S.L.; Sharma, M. An Overview of Pharmaceutical Co-Crystal. Asian J. Pharm. Res. Dev. 2019, 7, 39–46. [Google Scholar] [CrossRef]
- Duggirala, N.K.; Perry, M.L.; Almarsson, O.; Zaworotko, M.J. Pharmaceutical cocrystals: Along the path to improved medicines. Chem. Commun. 2016, 52, 640–655. [Google Scholar] [CrossRef] [PubMed]
- Ando, S.; Kikuchi, J.; Fujimura, Y.; Ida, Y.; Higashi, K.; Moribe, K.; Yamamoto, K. Physicochemical characterization and structural evaluation of a specific 2:1 cocrystal of naproxen-nicotinamide. J. Pharm. Sci. 2012, 101, 3214–3221. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Jang, S.; Kim, I.W. Enhanced Dissolution of Naproxen by Combining Cocrystallization and Eutectic Formation. Pharmaceutics 2021, 13, 618. [Google Scholar] [CrossRef] [PubMed]
- Skorupska, E.; Jeziorna, A.; Potrzebowski, M.J. Thermal Solvent-Free Method of Loading of Pharmaceutical Cocrystals into the Pores of Silica Particles: A Case of Naproxen/Picolinamide Cocrystal. J. Phys. Chem. C 2016, 120, 13169–13180. [Google Scholar] [CrossRef]
- Buschmann, H.H.; Solà, C.; Buchholz, J.B.; Bertran, J.C. Co-crystals of duloxetine and co-crystal formers for the treatment of pain. EP2123626 A1, 25 November 2009. [Google Scholar]
- Latif, S.; Ijaz, Q.A.; Hameed, M.; Fatima, K.; Hussain, A.; Arshad, M.S.; Abbas, N. Improvement of physico-mechanical and pharmacokinetic attributes of naproxen by cocrystallization with L-alanine. J. Drug Deliv. Sci. Technol. 2021, 61, 102236. [Google Scholar] [CrossRef]
- Tumanova, N.; Tumanov, N.; Robeyns, K.; Filinchuk, Y.; Wouters, J.; Leyssens, T. Structural insight into cocrystallization with zwitterionic co-formers: Cocrystals of S-naproxen. CrystEngComm 2014, 16, 8185–8196. [Google Scholar] [CrossRef]
- Kasten, G.; Lobo, L.; Dengale, S.; Grohganz, H.; Rades, T.; Lobmann, K. In vitro and in vivo comparison between crystalline and co-amorphous salts of naproxen-arginine. Eur. J. Pharm. Biopharm. 2018, 132, 192–199. [Google Scholar] [CrossRef]
- Tilborg, A.; Springuel, G.; Norberg, B.; Wouters, J.; Leyssens, T. On the influence of using a zwitterionic coformer for cocrystallization: Structural focus on naproxen–proline cocrystals. CrystEngComm 2013, 15, 3341–3350. [Google Scholar] [CrossRef]
- Heinrich, B.H.; Lluis, S.C.; Jordi, B.B.; Carles, C.B.J.; Ramon, P.; Nicolas, T. Co-crystals of tramadol and NSAIDs. JP5645830B2, 24 December 2014. [Google Scholar]
- Manoj, K.; Tamura, R.; Takahashi, H.; Tsue, H. Crystal engineering of homochiral molecular organization of naproxen in cocrystals and their thermal phase transformation studies. CrystEngComm 2014, 16, 5811–5819. [Google Scholar] [CrossRef]
- Nechipadappu, S.K.; Trivedi, D.R. Structural and physicochemical characterization of pyridine derivative salts of anti-inflammatory drugs. J. Mol. Struct. 2017, 1141, 64–74. [Google Scholar] [CrossRef]
- Rajurkar, V.G.; Gite, R.D.; Ghawate, V.B. Development of Naproxen Co Crystal Formation: An Efficient Approach to Enhance Aqueous Solubility. Anal. Chem. Lett. 2015, 5, 229–238. [Google Scholar] [CrossRef]
- Wang, K.; Zhao, X.; Zhu, W. Application of molecular electrostatic potential surface to predict Supramolecular Synthons for RDX/solvent Cocrystals. Cryst. Res. Technol. 2019, 54, 1900171. [Google Scholar] [CrossRef]
- Rajbongshi, T.; Sarmah, K.K.; Sarkar, A.; Ganduri, R.; Cherukuvada, S.; Thakur, T.S.; Thakuria, R. Preparation of pyrazinamide eutectics versus cocrystals based on supramolecular synthon variations. Cryst. Growth Des. 2018, 18, 6640–6651. [Google Scholar] [CrossRef]
- Wang, Y.; Shou, Z.; Fan, H.; Xu, M.; Chen, Q.; Tang, Q.; Liu, X.; Wu, H.; Zhang, M.; Yu, T. Protective effects of oxymatrine against DSS-induced acute intestinal inflammation in mice via blocking the RhoA/ROCK signaling pathway. Biosci. Rep. 2019, 39, BSR20182297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pindelska, E.; Sokal, A.; Kolodziejski, W. Pharmaceutical cocrystals, salts and polymorphs: Advanced characterization techniques. Adv. Drug Deliv. Rev. 2017, 117, 111–146. [Google Scholar] [CrossRef]
- Hathwar, V.R.; Pal, R.; Guru Row, T.N. Charge Density Analysis of Crystals of Nicotinamide with Salicylic Acid and Oxalic Acid: An Insight into the Salt to Cocrystal Continuum. Cryst. Growth Des. 2010, 10, 3306–3310. [Google Scholar] [CrossRef]
- Jones, C.L.; Skelton, J.M.; Parker, S.C.; Raithby, P.R.; Walsh, A.; Wilson, C.C.; Thomas, L.H. Living in the salt-cocrystal continuum: Indecisive organic complexes with thermochromic behaviour. CrystEngComm 2019, 21, 1626–1634. [Google Scholar] [CrossRef] [Green Version]
- Childs, S.L.; Stahly, G.P.; Park, A. The salt—Cocrystal continuum: The influence of crystal structure on ionization state. Mol. Pharm. 2007, 4, 323–338. [Google Scholar] [CrossRef] [Green Version]
- Sedghiniya, S.; Soleimannejad, J.; Janczak, J. The salt–cocrystal spectrum in salicylic acid–adenine: The influence of crystal structure on proton-transfer balance. Acta Crystallogr. Sect. C Struct. Chem. 2019, 75, 412–421. [Google Scholar] [CrossRef]
- Saganowska, P.; Wesolowski, M. DSC as a screening tool for rapid co-crystal detection in binary mixtures of benzodiazepines with co-formers. J. Therm. Anal. Calorim. 2018, 133, 785–795. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S. Pharmaceutical cocrystals: An overview. Indian J. Pharm. Sci. 2018, 79, 858–871. [Google Scholar] [CrossRef]
- Smith, B. Infrared Spectral Interpretation: A Systematic Approach; CRC Press: Boca Raton, FL, USA, 2018. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Salmon, D.J.; Smith, M.M.; Desper, J. Cyanophenyloximes: Reliable and versatile tools for hydrogen-bond directed supramolecular synthesis of cocrystals. Cryst. Growth Des. 2006, 6, 1033–1042. [Google Scholar] [CrossRef]
- Castro, R.A.E.; Ribeiro, J.D.B.; Maria, T.M.R.; Ramos Silva, M.; Yuste-Vivas, C.; Canotilho, J.; Eusébio, M.E.S. Naproxen Cocrystals with Pyridinecarboxamide Isomers. Cryst. Growth Des. 2011, 11, 5396–5404. [Google Scholar] [CrossRef]
- Liu, L.; Yu, Y.-M.; Bu, F.-Z.; Li, Y.-T.; Yan, C.-W.; Wu, Z.-Y. The First Cocrystallization of Milrinone with Nutraceuticals: The Adjusting Effects of Hydrophilicity/Hydrophobicity in Cavities on the In Vitro/In Vivo Properties of the Cocrystals. Cryst. Growth Des. 2022, 22, 1623–1637. [Google Scholar] [CrossRef]
- Ying, P.; Yu, J.; Su, W. Liquid-Assisted Grinding Mechanochemistry in the Synthesis of Pharmaceuticals. Adv. Synth. Catal. 2021, 363, 1246–1271. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Bourhis, L.J.; Dolomanov, O.V.; Gildea, R.J.; Howard, J.A.; Puschmann, H. The anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment–Olex2 dissected. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 59–75. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Shanthala, H.; Jayaprakash, H.; Radhakrishna, M.; Bh, J.G.; Paul, K.; Shankar, S.; Ahmed, M.G.; Sanjana, A. Enhancement of solubility and dissolution rate of acetylsalicylic acid via co-crystallization technique: A novel ASA-valine cocrystal. Int. J. Appl. Pharm. 2021, 13, 199–205. [Google Scholar] [CrossRef]
- Yang, D.; Wang, H.; Liu, Q.; Yuan, P.; Chen, T.; Zhang, L.; Yang, S.; Zhou, Z.; Lu, Y.; Du, G. Structural landscape on a series of rhein: Berberine cocrystal salt solvates: The formation, dissolution elucidation from experimental and theoretical investigations. Chin. Chem. Lett. 2022, 10, 43. [Google Scholar] [CrossRef]
- Wang, H.; Yang, D.; Zhang, W.; Song, J.; Gong, N.; Yu, M.; Yang, S.; Zhang, B.; Liu, Q.; Du, G. An innovative rhein-matrine cocrystal: Synthesis, characterization, formation mechanism and pharmacokinetic study. Chin. Chem. Lett. 2022. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H. Gaussian 16; Gaussian. Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Yang, D.; Cao, J.; Heng, T.; Xing, C.; Yang, S.; Zhang, L.; Lu, Y.; Du, G. Theoretical Calculation and Structural Analysis of the Cocrystals of Three Flavonols with Praziquantel. Cryst. Growth Des. 2021, 21, 2292–2300. [Google Scholar] [CrossRef]
- Liu, Q.; Yang, D.; Chen, T.; Zhang, B.; Xing, C.; Zhang, L.; Lu, Y.; Du, G. Insights into the Solubility and Structural Features of Four Praziquantel Cocrystals. Cryst. Growth Des. 2021, 21, 6321–6331. [Google Scholar] [CrossRef]
- Nguyen, A.L.P.; Izgorodina, E.I. Behavior of counterpoise correction in many-body molecular clusters of organic compounds: Hartree-Fock interaction energy perspective. J. Comput. Chem. 2022, 43, 568–576. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. Independent gradient model based on Hirshfeld partition: A new method for visual study of interactions in chemical systems. J. Comput. Chem. 2022, 43, 539–555. [Google Scholar] [CrossRef]
- Palatinus, L.; Brázda, P.; Boullay, P.; Perez, O.; Klementová, M.; Petit, S.; Eigner, V.; Zaarour, M.; Mintova, S. Hydrogen positions in single nanocrystals revealed by electron diffraction. Science 2017, 355, 166–169. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Co-Crystal | NPX–CPL (1:1) | NPX–OMT (1:1) |
|---|---|---|
| formula | C14H14O3·C6H11NO | C14H14O3 ·C15H24N2O2 |
| crystal size (mm) | 0.15 × 0.280 × 0.340 | 0.15 × 0.170 × 0.300 |
| molecular weight | 343.17 | 494.27 |
| Temperature (K) | 293(2) | 293(2) |
| crystal system | orthorhombic | orthorhombic |
| space group | P212121 | P212121 |
| α (Å) | 7.565(1) | 5.867(1) |
| b (Å) | 14.642(1) | 9.343(1) |
| c (Å) | 16.833(1) | 46.507(2) |
| a (deg) | 90 | 90 |
| β (deg) | 90 | 90 |
| γ (deg) | 90 | 90 |
| volume (Å3) | 1864.69(6) | 2549.39(16) |
| Z | 4 | 4 |
| density (g/cm³) | 1.223 | 1.289 |
| R1 (I > 2σ(I)) | 0.056 | 0.079 |
| wR2 (I > 3σ(I)) | 0.154 | 0.212 |
| goodness-of-fit on F2 | 1.050 | 1.084 |
| Completeness (%) | 99.9 | 99.5 |
| CCDC deposition no. | 2,172,223 | 2,172,224 |
| Co-Crystal | D–H…A | D…A | ∠DHA |
|---|---|---|---|
| NPX–CPL | a O3NPX–H3NPX…O1CPL | 1.75 | 155.35 |
| b N2CPL-H2CPL…O2NPX | 2.165 | 167.11 | |
| NPX–OMT | O2CPL -H2CPL…O2OMT | 1.697 | 157.72 |
| O2CPL -H2CPL…N2OMT | 2.634 | 165.05 |
| Vibrational Data | Vibrational Assignment | |
|---|---|---|
| NPX–CPL | NPX–OMT | |
| 3270 | – | –NH stretch |
| 3055 | – | –COOH stretch |
| 2978 | 2968 | –CH3 stretch |
| 1451 | 1439 | –CH3 stretch |
| 2924 | 2945 | –CH2– stretch |
| 2850 | – | –CH2– stretch |
| 1673 | 1679 | C=O stretch |
| 1630 | – | –NH bend |
| 1439 | – | –COOH bend |
| 1392 | 1392 | –CH3 bend |
| 1472 | 1462 | –CH2 bend |
| 744 | 667 | Ar-H bend |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xing, C.; Chen, T.; Wang, L.; An, Q.; Jin, Y.; Yang, D.; Zhang, L.; Du, G.; Lu, Y. Two Novel Co-Crystals of Naproxen: Comparison of Stability, Solubility and Intermolecular Interaction. Pharmaceuticals 2022, 15, 807. https://doi.org/10.3390/ph15070807
Xing C, Chen T, Wang L, An Q, Jin Y, Yang D, Zhang L, Du G, Lu Y. Two Novel Co-Crystals of Naproxen: Comparison of Stability, Solubility and Intermolecular Interaction. Pharmaceuticals. 2022; 15(7):807. https://doi.org/10.3390/ph15070807
Chicago/Turabian StyleXing, Cheng, Ting Chen, Li Wang, Qi An, Yali Jin, Dezhi Yang, Li Zhang, Guanhua Du, and Yang Lu. 2022. "Two Novel Co-Crystals of Naproxen: Comparison of Stability, Solubility and Intermolecular Interaction" Pharmaceuticals 15, no. 7: 807. https://doi.org/10.3390/ph15070807
APA StyleXing, C., Chen, T., Wang, L., An, Q., Jin, Y., Yang, D., Zhang, L., Du, G., & Lu, Y. (2022). Two Novel Co-Crystals of Naproxen: Comparison of Stability, Solubility and Intermolecular Interaction. Pharmaceuticals, 15(7), 807. https://doi.org/10.3390/ph15070807