
Inflammation Induced by Lipopolysaccharide and Palmitic Acid Increases Cholesterol Accumulation via Enhancing Myeloid Differentiation Factor 88 Expression in HepG2 Cells

 , ,
, ,
Abstract
:
1. Introduction
2. Results
2.1. LPS and PA Treatments Inhibited HepG2 Cell Viabilities
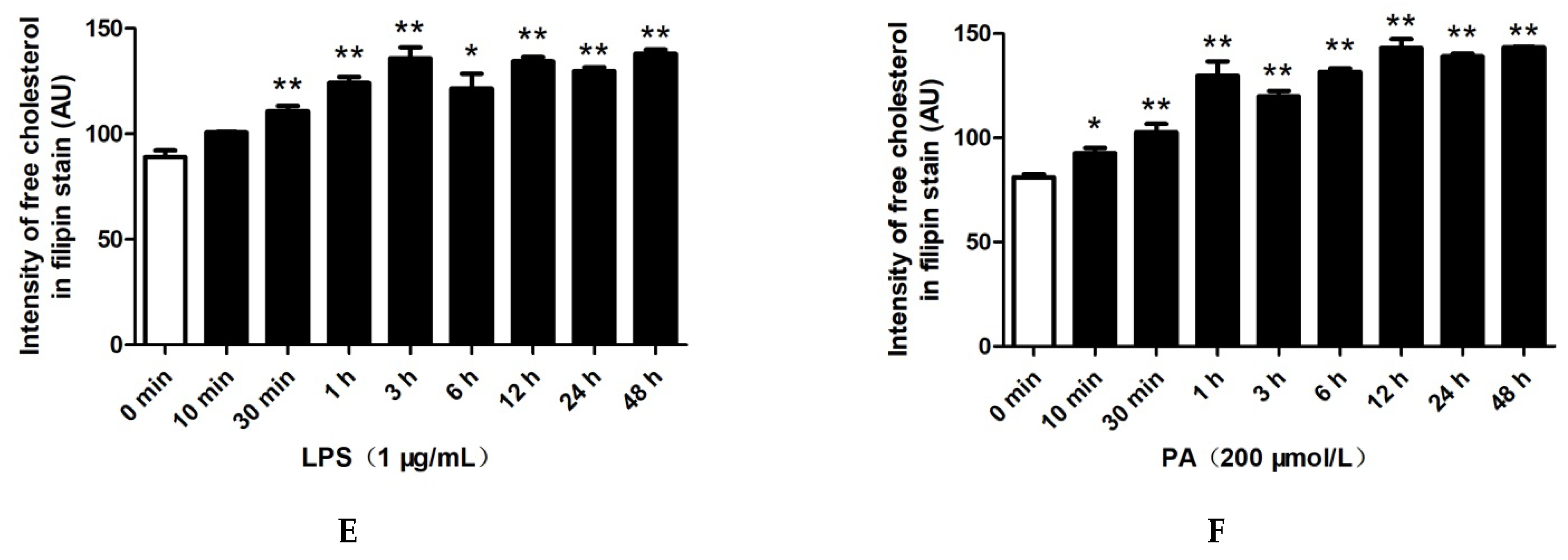
2.2. LPS and PA Treatments Increased Cholesterol Content in HepG2 Cells
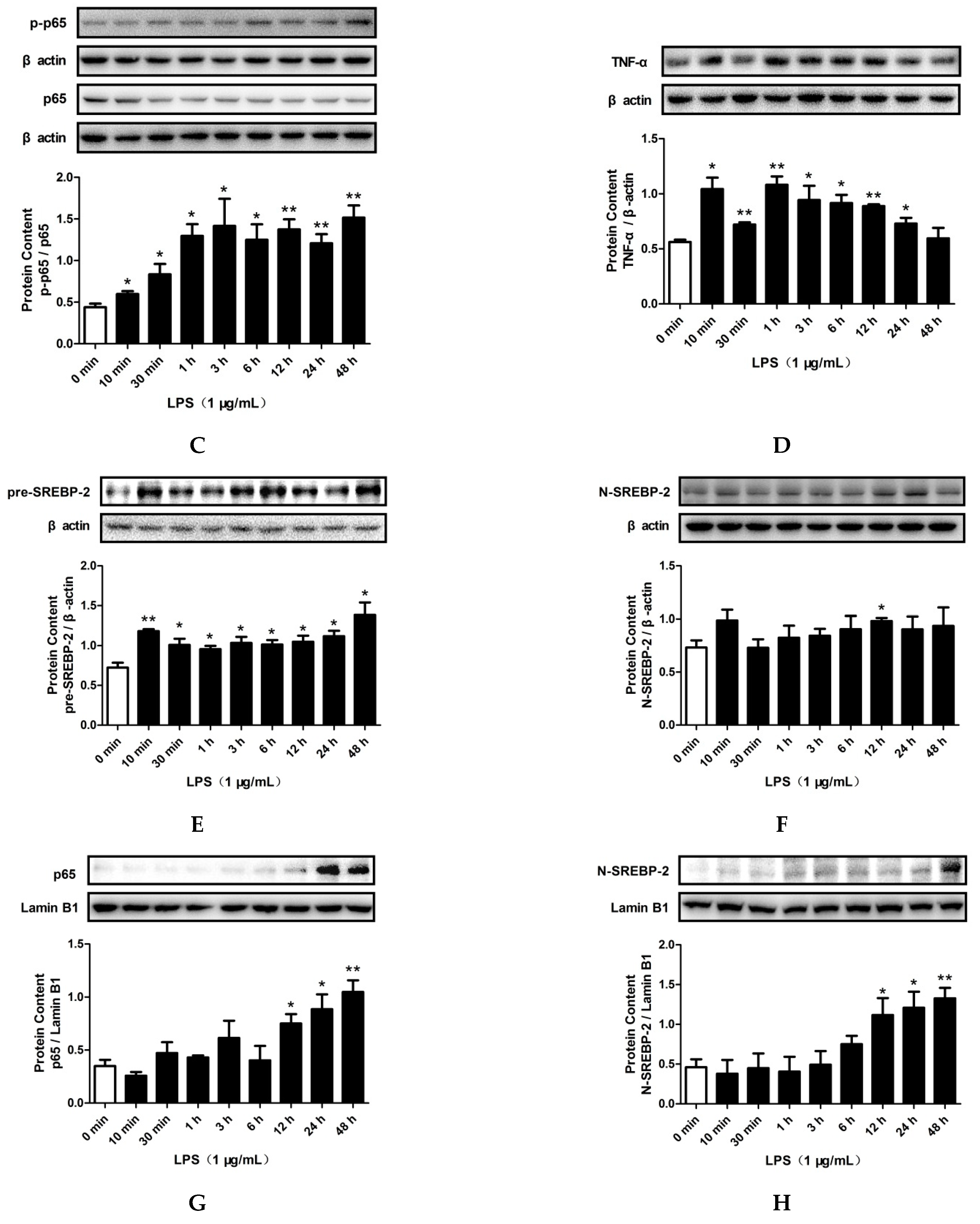
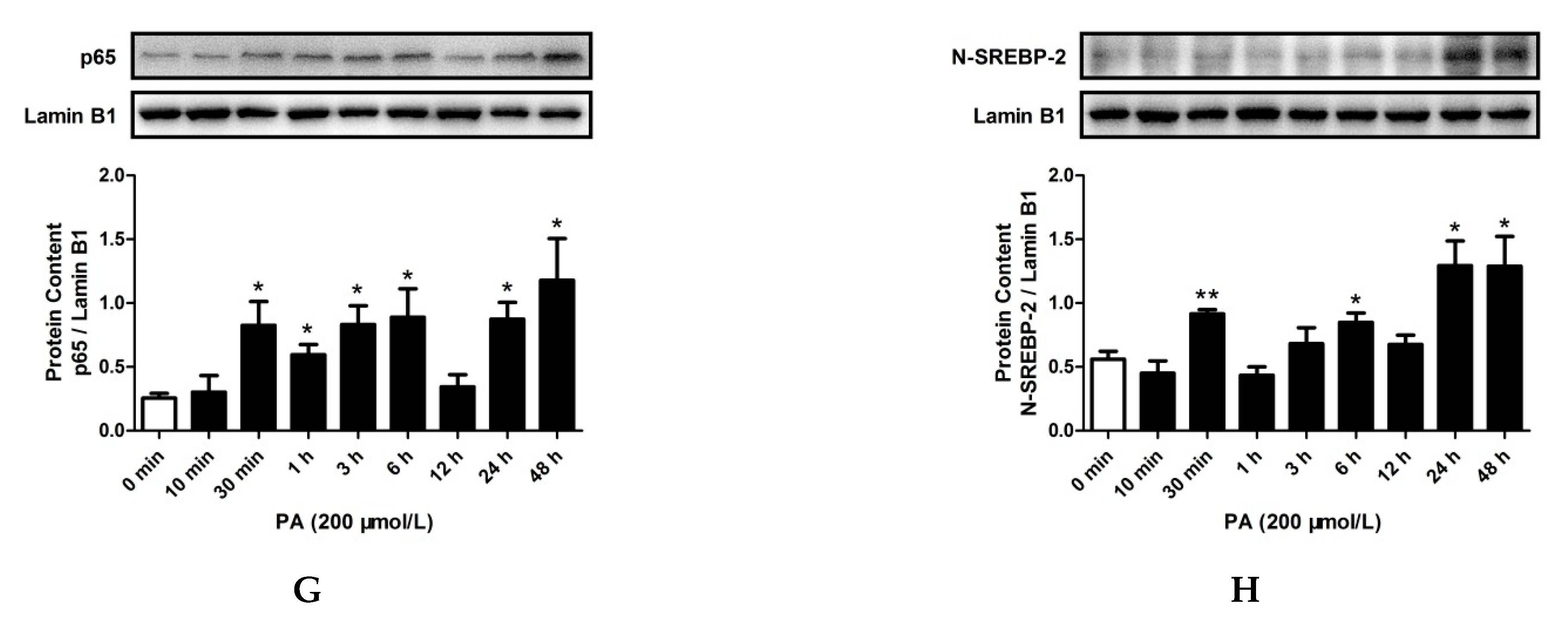
2.3. LPS and PA Treatments Increased TLR4/MyD88/NF-κB Signaling Pathway Activation and SREBP-2 Expression
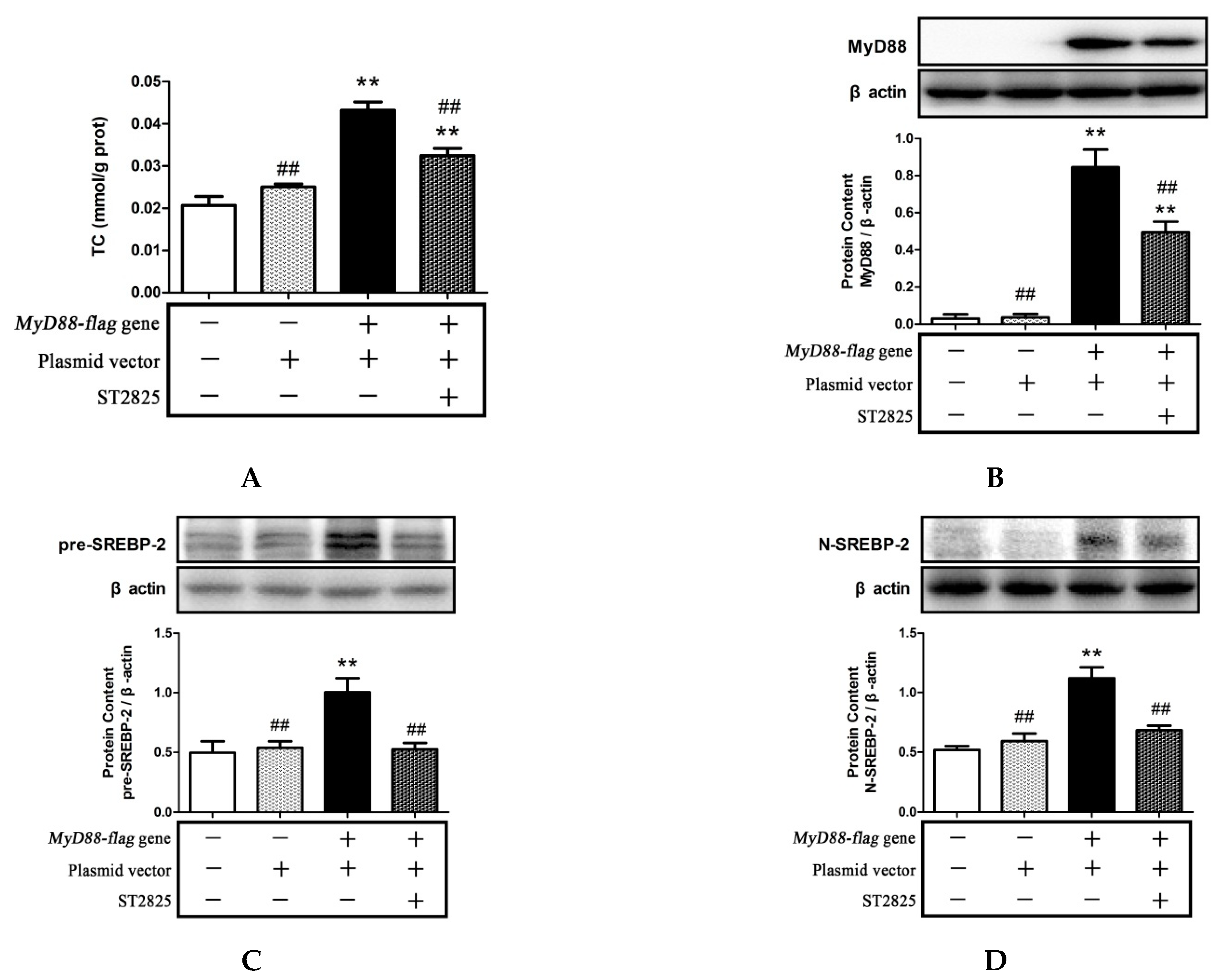
2.4. Inhibition of MyD88 Reduced Cholesterol Content in LPS- and PA-Treated HepG2 Cells
2.5. Inhibition of MyD88 Suppressed TLR4/MyD88/NF-κB Signaling Pathway Activation and SREBP-2 Expression Induced by LPS and PA Treatments
2.6. Role of MyD88 in SREBP-2 Mediated Cholesterol Synthesis
3. Discussion
4. Materials and Methods
4.1. Reagents and Chemicals
4.2. Cell Culture
4.3. MTT Cell Viability Assay
4.4. Transient Transfection
4.5. Measurement of Intracellular Cholesterol
4.6. Filipin Staining
4.7. Western Blotting
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferguson, L.R. Chronic inflammation and mutagenesis. Mutat. Res. 2010, 690, 3–11. [Google Scholar] [CrossRef]
- Magro, D.R.I.; Houben, T.; Gijbels, M.; Lutjohann, D.; Plat, J.; Shiri-Sverdlov, R. Anti-Inflammatory Effects of Dietary Plant Stanol Supplementation Are Largely Dependent on the Intake of Cholesterol in a Mouse Model of Metabolic Inflammation. Biomedicines 2021, 9, 518. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, N.M.; Gucalp, A.; Dannenberg, A.J.; Hudis, C.A. Obesity and Cancer Mechanisms: Tumor Microenvironment and Inflammation. J. Clin. Oncol. 2016, 34, 4270–4276. [Google Scholar] [CrossRef]
- Khovidhunkit, W.; Kim, M.S.; Memon, R.A.; Shigenaga, J.K.; Moser, A.H.; Feingold, K.R.; Grunfeld, C. Effects of infection and inflammation on lipid and lipoprotein metabolism: Mechanisms and consequences to the host. J. Lipid Res. 2004, 45, 1169–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, M.; Zhang, W.; Dong, Y.; Wang, L.; Fang, T.; Tang, W.; Lv, B.; Chen, G.; Yang, B.; Huang, P.; et al. Pro-inflammation NF-kappaB signaling triggers a positive feedback via enhancing cholesterol accumulation in liver cancer cells. J. Exp. Clin. Cancer Res. 2017, 36, 15. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zuo, G.Q.; Zhao, L.; Chen, Y.X.; Ruan, X.Z.; Zuo, D.Y. Effect of inflammatory stress on hepatic cholesterol accumulation and hepatic fibrosis in C57BL/6J mice. Zhonghua Gan Zang Bing Za Zhi 2013, 21, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Westerterp, M.; Gautier, E.L.; Ganda, A.; Molusky, M.M.; Wang, W.; Fotakis, P.; Wang, N.; Randolph, G.J.; D’Agati, V.D.; Yvan-Charvet, L.; et al. Cholesterol Accumulation in Dendritic Cells Links the Inflammasome to Acquired Immunity. Cell Metab. 2017, 25, 1294–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Wang, C.; Lu, J.; Huang, K.; Han, Y.; Chen, J.; Yang, Y.; Liu, B. PPAR delta inhibition protects against palmitic acid-LPS induced lipidosis and injury in cultured hepatocyte L02 cell. J. Med. Sci. 2019, 16, 1593–1603. [Google Scholar] [CrossRef]
- Fang, W.; Xue, H.; Chen, X.; Chen, K.; Ling, W. Supplementation with Sodium Butyrate Modulates the Composition of the Gut Microbiota and Ameliorates High-Fat Diet-Induced Obesity in Mice. J. Nutr. 2019, 149, 747–754. [Google Scholar] [CrossRef]
- Lu, Z.; Li, Y.; Syn, W.K.; Wang, Z.; Lopes-Virella, M.F.; Lyons, T.J.; Huang, Y. Amitriptyline inhibits nonalcoholic steatohepatitis and atherosclerosis induced by high-fat diet and LPS through modulation of sphingolipid metabolism. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E131–E144. [Google Scholar] [CrossRef]
- Rocha, D.M.; Caldas, A.P.; Oliveira, L.L.; Bressan, J.; Hermsdorff, H.H. Saturated fatty acids trigger TLR4-mediated inflammatory response. Atherosclerosis 2016, 244, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Hwangbo, H.; Ji, S.Y.; Kim, M.Y.; Kim, S.Y.; Lee, H.; Kim, G.Y.; Kim, S.; Cheong, J.; Choi, Y.H. Anti-Inflammatory Effect of Auranofin on Palmitic Acid and LPS-Induced Inflammatory Response by Modulating TLR4 and NOX4-Mediated NF-kappaB Signaling Pathway in RAW264.7 Macrophages. Int. J. Mol. Sci. 2021, 22, 5920. [Google Scholar] [CrossRef] [PubMed]
- Lucas, K.; Maes, M. Role of the Toll Like receptor (TLR) radical cycle in chronic inflammation: Possible treatments targeting the TLR4 pathway. Mol. Neurobiol. 2013, 48, 190–204. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Zhou, E.; Wei, Z.; Liang, D.; Wang, W.; Wang, T.; Guo, M.; Zhang, N.; Yang, Z. Glycyrrhizin inhibits the inflammatory response in mouse mammary epithelial cells and a mouse mastitis model. FEBS J. 2014, 281, 2543–2557. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Hu, C.; Yin, L.; Tao, X.; Xu, L.; Qi, Y.; Han, X.; Xu, Y.; Zhao, Y.; Wang, C.; et al. Dioscin reduces lipopolysaccharide-induced inflammatory liver injury via regulating TLR4/MyD88 signal pathway. Int. Immunopharmacol. 2016, 36, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Plociennikowska, A.; Hromada-Judycka, A.; Borzecka, K.; Kwiatkowska, K. Co-operation of TLR4 and raft proteins in LPS-induced pro-inflammatory signaling. Cell. Mol. Life Sci. 2015, 72, 557–581. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.C.; Varghese, Z.; Moorhead, J.F.; Lee, C.T.; Chen, J.B.; Ruan, X.Z. Cross-talk between TLR4-MyD88-NF-kappaB and SCAP-SREBP2 pathways mediates macrophage foam cell formation. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H874–H884. [Google Scholar] [CrossRef]
- Gliozzi, M.; Musolino, V.; Bosco, F.; Scicchitano, M.; Scarano, F.; Nucera, S.; Zito, M.C.; Ruga, S.; Carresi, C.; Macri, R.; et al. Cholesterol homeostasis: Researching a dialogue between the brain and peripheral tissues. Pharmacol. Res. 2021, 163, 105215. [Google Scholar] [CrossRef]
- Haskins, J.W.; Zhang, S.; Means, R.E.; Kelleher, J.K.; Cline, G.W.; Canfran-Duque, A.; Suarez, Y.; Stern, D.F. Neuregulin-activated ERBB4 induces the SREBP-2 cholesterol biosynthetic pathway and increases low-density lipoprotein uptake. Sci. Signal. 2015, 8, ra111. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.J.; Tang, S.L.; Lv, Y.C.; Ouyang, X.P.; He, P.P.; Yao, F.; Tang, Y.Y.; Zhang, M.; Tang, Y.L.; Tang, D.P.; et al. NF-kappaB suppresses the expression of ATP-binding cassette transporter A1/G1 by regulating SREBP-2 and miR-33a in mice. Int. J. Cardiol. 2014, 171, e93–e95. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.L.; Ruan, X.Z.; Powis, S.H.; Chen, Y.; Moorhead, J.F.; Varghese, Z. Inflammatory stress exacerbates lipid accumulation in hepatic cells and fatty livers of apolipoprotein E knockout mice. Hepatology 2008, 48, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Everard, A.; Geurts, L.; Caesar, R.; Van Hul, M.; Matamoros, S.; Duparc, T.; Denis, R.G.; Cochez, P.; Pierard, F.; Castel, J.; et al. Intestinal epithelial MyD88 is a sensor switching host metabolism towards obesity according to nutritional status. Nat. Commun. 2014, 5, 5648. [Google Scholar] [CrossRef] [Green Version]
- Ham, J.R.; Lee, H.I.; Choi, R.Y.; Sim, M.O.; Seo, K.I.; Lee, M.K. Anti-steatotic and anti-inflammatory roles of syringic acid in high-fat diet-induced obese mice. Food Funct. 2016, 7, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jin, Y.; Choi, Y.; Park, T. Resveratrol exerts anti-obesity effects via mechanisms involving down-regulation of adipogenic and inflammatory processes in mice. Biochem. Pharmacol. 2011, 81, 1343–1351. [Google Scholar] [CrossRef]
- Yashiro, T.; Nanmoku, M.; Shimizu, M.; Inoue, J.; Sato, R. Resveratrol increases the expression and activity of the low density lipoprotein receptor in hepatocytes by the proteolytic activation of the sterol regulatory element-binding proteins. Atherosclerosis 2012, 220, 369–374. [Google Scholar] [CrossRef]
- Wu, N.; Sarna, L.K.; Hwang, S.Y.; Zhu, Q.; Wang, P.; Siow, Y.L.; O, K. Activation of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase during high fat diet feeding. Biochim. Biophys. Acta 2013, 1832, 1560–1568. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Ullah, H.; Zheng, Z.; Gu, X.; Su, C.; Xiao, L.; Wu, X.; Xiong, F.; Li, Q.; Zha, L. Soyasaponins reduce inflammation by downregulating MyD88 expression and suppressing the recruitments of TLR4 and MyD88 into lipid rafts. BMC Complementary Med. Ther. 2020, 20, 167. [Google Scholar] [CrossRef]
- Palmer, M.A.; Smart, E.; Haslam, I.S. Localisation and regulation of cholesterol transporters in the human hair follicle: Mapping changes across the hair cycle. Histochem. Cell Biol. 2021, 155, 529–545. [Google Scholar] [CrossRef]
- Xie, Q.; Gu, X.; Chen, J.; Liu, M.; Xiong, F.; Wu, X.; Zhang, Y.; Chen, F.; Chen, H.; Li, M.; et al. Soyasaponins Reduce Inflammation and Improve Serum Lipid Profiles and Glucose Homeostasis in High Fat Diet-Induced Obese Mice. Mol. Nutr. Food Res. 2018, 62, 1800205. [Google Scholar] [CrossRef]
- Ou, T.T.; Kuo, C.Y.; Chyau, C.C.; Lee, H.J.; Peng, J.S.; Wang, C.J. Improvement of lipopolysaccharide-induced hepatic injuries and inflammation with mulberry extracts. J. Sci. Food Agric. 2013, 93, 1880–1886. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yuan, H.; Zhang, C.; Guan, Y.; Wu, Y.; Ling, F.; Niu, Y.; Li, Y. Epigallocatechin gallate improves insulin resistance in HepG2 cells through alleviating inflammation and lipotoxicity. Diabetes Res. Clin. Pract. 2018, 142, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Ricchi, M.; Odoardi, M.R.; Carulli, L.; Anzivino, C.; Ballestri, S.; Pinetti, A.; Fantoni, L.I.; Marra, F.; Bertolotti, M.; Banni, S.; et al. Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. J. Gastroenterol. Hepatol. 2009, 24, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Meng, Z.; Cheng, B.; Liu, M.; Tao, S.; Guan, S. Apigenin reduces the excessive accumulation of lipids induced by palmitic acid via the AMPK signaling pathway in HepG2 cells. Exp. Ther. Med. 2019, 18, 2965–2971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Q.; Zhang, S.; Yang, C.; Du, R.; Zhao, J.; Li, J.; Xu, Y.; Qin, Y.; Gao, Y.; Huang, W. Ginsenoside Rg1 Ameliorates Palmitic Acid-Induced Hepatic Steatosis and Inflammation in HepG2 Cells via the AMPK/NF-kappaB Pathway. Int. J. Endocrinol. 2019, 2019, 7514802. [Google Scholar] [CrossRef] [Green Version]
- Fuke, N.; Nagata, N.; Suganuma, H.; Ota, T. Regulation of Gut Microbiota and Metabolic Endotoxemia with Dietary Factors. Nutrients 2019, 11, 2277. [Google Scholar] [CrossRef] [Green Version]
- Fatima, S.; Hu, X.; Gong, R.H.; Huang, C.; Chen, M.; Wong, H.; Bian, Z.; Kwan, H.Y. Palmitic acid is an intracellular signaling molecule involved in disease development. Cell. Mol. Life Sci. 2019, 76, 2547–2557. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, B.; Zhang, H.; Ge, X.; Ying, S.; Hu, M.; Li, W.; Huang, Y.; Wang, L.; Chen, C.; et al. Inhibition of MD2-dependent inflammation attenuates the progression of non-alcoholic fatty liver disease. J. Cell. Mol. Med. 2018, 22, 936–947. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Guan, Q.; Xiao, Y.; Feng, Z.; Yu, G.; Pan, Q. Strontium Alleviates Endoplasmic Reticulum Stress in a Nonalcoholic Fatty Liver Disease Model. J. Med. Food 2018, 21, 1228–1237. [Google Scholar] [CrossRef]
- Zhou, L.; Tang, J.; Yang, X.; Dong, H.; Xiong, X.; Huang, J.; Zhang, L.; Qin, H.; Yan, S. Five Constituents in Psoralea corylifolia L. Attenuate Palmitic Acid-Induced Hepatocyte Injury via Inhibiting the Protein Kinase C-alpha/Nicotinamide-Adenine Dinucleotide Phosphate Oxidase Pathway. Front. Pharmacol. 2019, 10, 1589. [Google Scholar] [CrossRef]
- Joshi-Barve, S.; Barve, S.S.; Amancherla, K.; Gobejishvili, L.; Hill, D.; Cave, M.; Hote, P.; McClain, C.J. Palmitic acid induces production of proinflammatory cytokine interleukin-8 from hepatocytes. Hepatology 2007, 46, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, N.; Gan, H.; He, Q.; Lei, H.; Wang, S.Y.; Liu, Q.; Zhou, C. Dysfunction of cholesterol sensor SCAP promotes inflammation activation in THP-1 macrophages. Exp. Cell Res. 2018, 367, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.R.; Li, L.; Sun, X.C.; Wang, J.; Ma, C.Y.; Zhang, Y.; Qu, H.L.; Xu, R.X.; Li, J.J. Diallyl disulfide improves lipid metabolism by inhibiting PCSK9 expression and increasing LDL uptake via PI3K/Akt-SREBP2 pathway in HepG2 cells. Nutr. Metab. Cardiovasc. Dis. 2021, 31, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, L.; Liu, X.; Luo, R.; Liao, G.; Li, L.; Liu, J.; Cheng, J.; Lu, Y.; Chen, Y. Oleic acid protects saturated fatty acid mediated lipotoxicity in hepatocytes and rat of non-alcoholic steatohepatitis. Life Sci. 2018, 203, 291–304. [Google Scholar] [CrossRef]
- Tarling, E.J.; Ahn, H.; de Aguiar, V.T. The nuclear receptor FXR uncouples the actions of miR-33 from SREBP-2. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 787–795. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Sato, R.; Goldstein, J.L.; Brown, M.S. Sterol-resistant transcription in CHO cells caused by gene rearrangement that truncates SREBP-2. Genes Dev. 1994, 8, 1910–1919. [Google Scholar] [CrossRef] [Green Version]
- Eberle, D.; Hegarty, B.; Bossard, P.; Ferre, P.; Foufelle, F. SREBP transcription factors: Master regulators of lipid homeostasis. Biochimie 2004, 86, 839–848. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; Cassader, M. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Prog. Lipid Res. 2013, 52, 175–191. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Guo, T.; Deng, R.; Liu, L.; Yu, Y. Apigenin Ameliorates Insulin Resistance and Lipid Accumulation by Endoplasmic Reticulum Stress and SREBP-1c/SREBP-2 Pathway in Palmitate-Induced HepG2 Cells and High-Fat Diet-Fed Mice. J. Pharmacol. Exp. Ther. 2021, 377, 146–156. [Google Scholar] [CrossRef]
- Duparc, T.; Plovier, H.; Marrachelli, V.G.; Van Hul, M.; Essaghir, A.; Stahlman, M.; Matamoros, S.; Geurts, L.; Pardo-Tendero, M.M.; Druart, C.; et al. Hepatocyte MyD88 affects bile acids, gut microbiota and metabolome contributing to regulate glucose and lipid metabolism. Gut 2017, 66, 620–632. [Google Scholar] [CrossRef] [Green Version]
- Lefort, C.; Van Hul, M.; Delzenne, N.M.; Everard, A.; Cani, P.D. Hepatic MyD88 regulates liver inflammation by altering synthesis of oxysterols. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E99–E108. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Bensinger, S.J. Reprogramming cholesterol metabolism in macrophages and its role in host defense against cholesterol-dependent cytolysins. Cell. Mol. Immunol. 2022, 19, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Yan, C.; Wan, J.B. Red yeast rice ameliorates non-alcoholic fatty liver disease through inhibiting lipid synthesis and NF-kappaB/NLRP3 inflammasome-mediated hepatic inflammation in mice. Chin. Med. 2022, 17, 17. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wang, H.; Li, X.; Liu, Y.; Mi, Y.; Kong, H.; Xi, D.; Yan, W.; Luo, X.; Ning, Q.; et al. Fibrinogen-like protein 2 aggravates nonalcoholic steatohepatitis via interaction with TLR4, eliciting inflammation in macrophages and inducing hepatic lipid metabolism disorder. Theranostics 2020, 10, 9702–9720. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Cat. No. | Molecular Weight (kDa) | Dilutions |
|---|---|---|---|
| TLR4 | sc-293072 | 95 | 1:1000 |
| GAPDH | G031 | 37 | 1:2000 |
| TRIF | ab139281 | 76 | 1:1000 |
| TRAF6 | ab137452 | 60 | 1:1000 |
| SREBP-2 | 28212-1-AP | 124 (SREBP-2 precursor) and 73 (SREBP-2 mature forms) | 1:1000 |
| Lamin B1 | 12987-1-AP | 66 | 1:1000 |
| MyD88 | 4283S | 33 | 1:1000 |
| TNF-α | 3707S | 17 | 1:1000 |
| NF-κB-p65 | 4764S | 65 | 1:1000 |
| phospho-NF-κB-p65 | 3031S | 65 | 1:1000 |
| β-actin | 4967S | 45 | 1:1000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Liu, Y.; Luo, H.; Chen, G.; Zheng, Z.; Wang, T.; Hu, X.; Zhao, Y.; Tang, J.; Su, C.; et al. Inflammation Induced by Lipopolysaccharide and Palmitic Acid Increases Cholesterol Accumulation via Enhancing Myeloid Differentiation Factor 88 Expression in HepG2 Cells. Pharmaceuticals 2022, 15, 813. https://doi.org/10.3390/ph15070813
Chen J, Liu Y, Luo H, Chen G, Zheng Z, Wang T, Hu X, Zhao Y, Tang J, Su C, et al. Inflammation Induced by Lipopolysaccharide and Palmitic Acid Increases Cholesterol Accumulation via Enhancing Myeloid Differentiation Factor 88 Expression in HepG2 Cells. Pharmaceuticals. 2022; 15(7):813. https://doi.org/10.3390/ph15070813
Chicago/Turabian StyleChen, Junbin, Yuguo Liu, Huiyu Luo, Guoxun Chen, Zhongdaixi Zheng, Tiannan Wang, Xinge Hu, Yue Zhao, Jiaqi Tang, Chuhong Su, and et al. 2022. "Inflammation Induced by Lipopolysaccharide and Palmitic Acid Increases Cholesterol Accumulation via Enhancing Myeloid Differentiation Factor 88 Expression in HepG2 Cells" Pharmaceuticals 15, no. 7: 813. https://doi.org/10.3390/ph15070813