
Chalcones as Potential Ligands for the Treatment of Parkinson’s Disease


Abstract
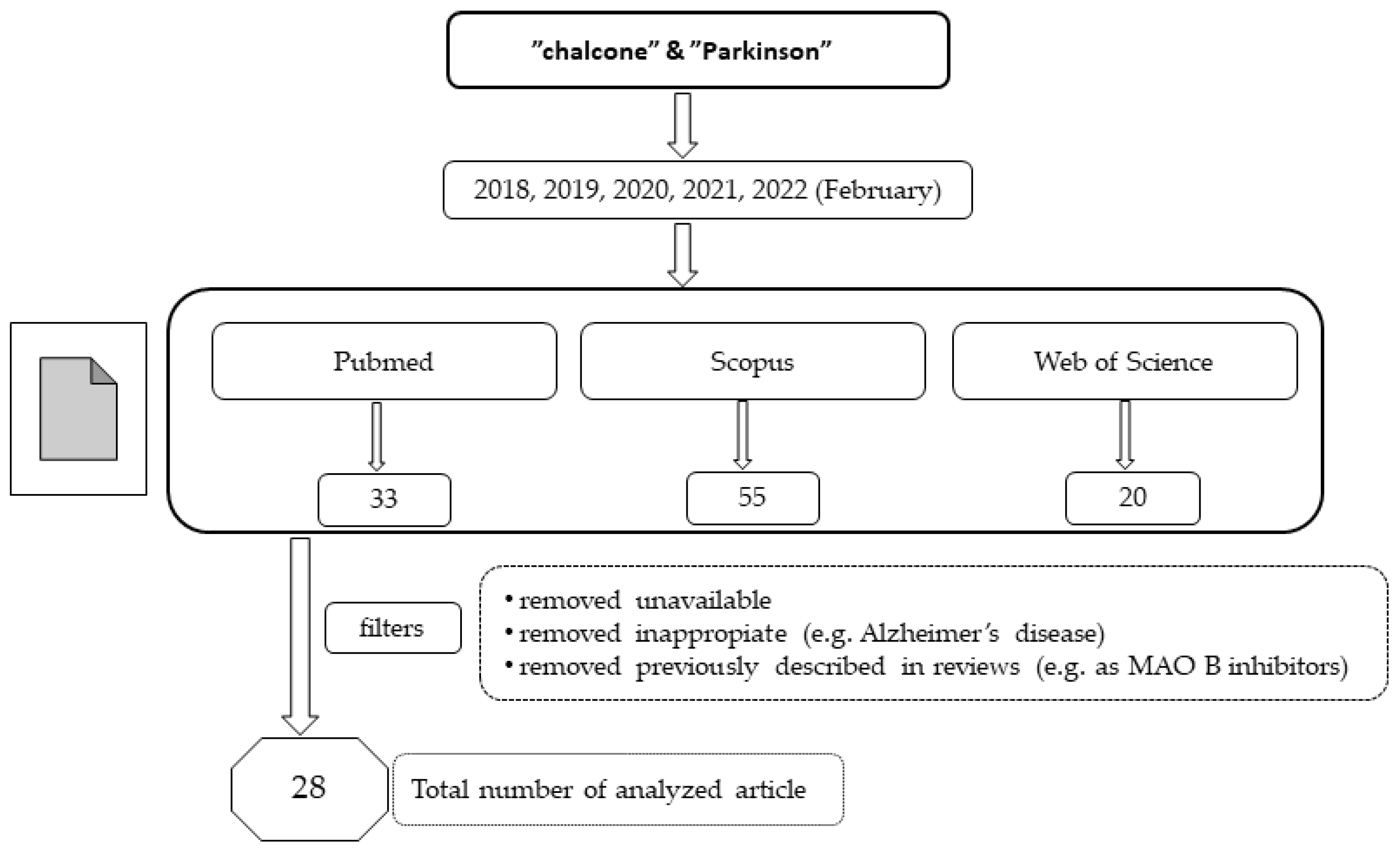
:1. Introduction
- Presymptomatic stages (stage 1–2), in which the pathology of inclusion bodies is confined to the medulla oblongata/pontine. Diagnosis of the disease in these stages is very rare and mainly non-motor symptoms (e.g., loss of smell, depressive symptoms) are present;
- Stages 3–4, in which the black matter and other grey nuclei of the midbrain and forebrain become the focus of pathological changes and symptoms are exacerbated. The lesions in the nervous system no longer respond to administered medication;
- The final stages 5–6, in which the process enters the mature neocortex. The disease manifests itself in all clinical dimensions and the progression of motor difficulties is so marked that the patient ceases to be independent.
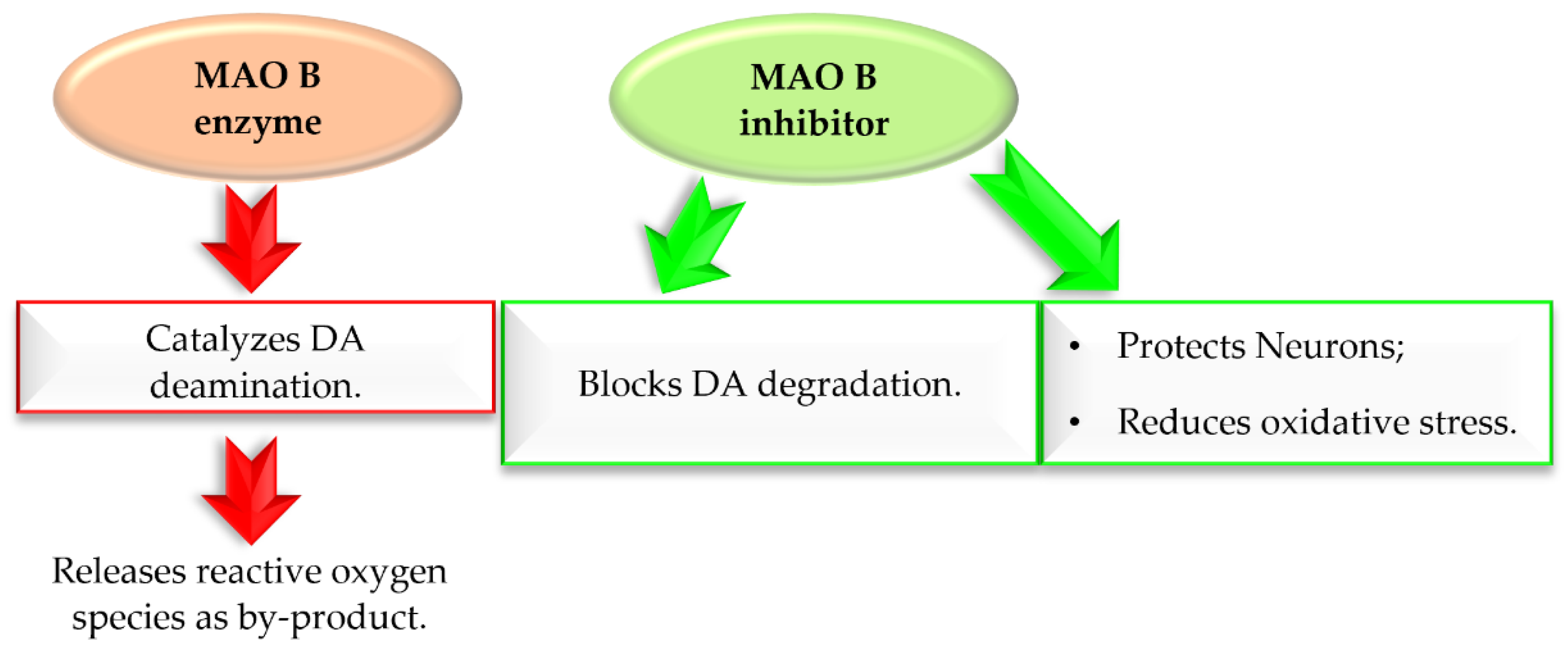
2. Monoamine Oxidase B Inhibitors
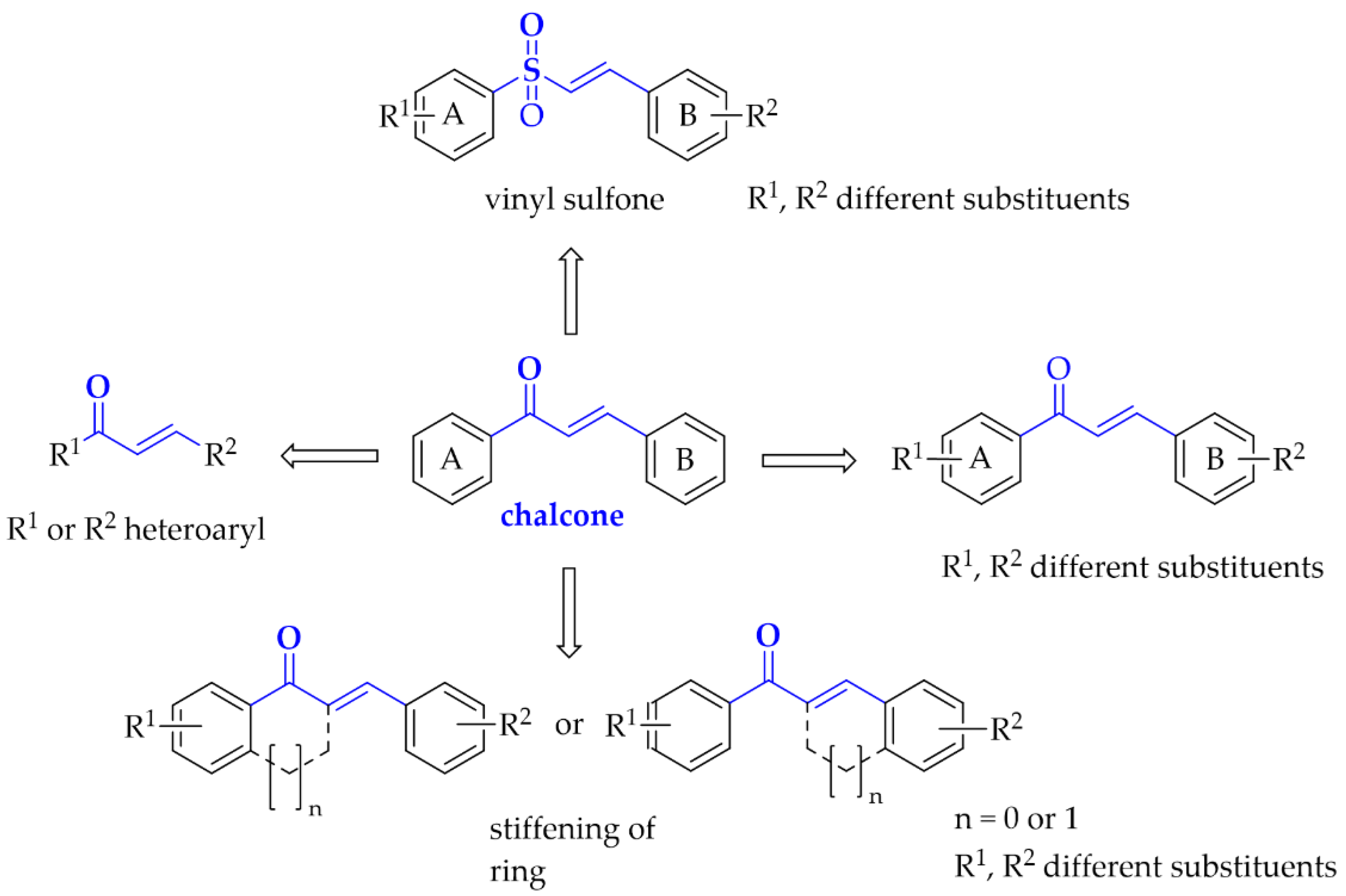
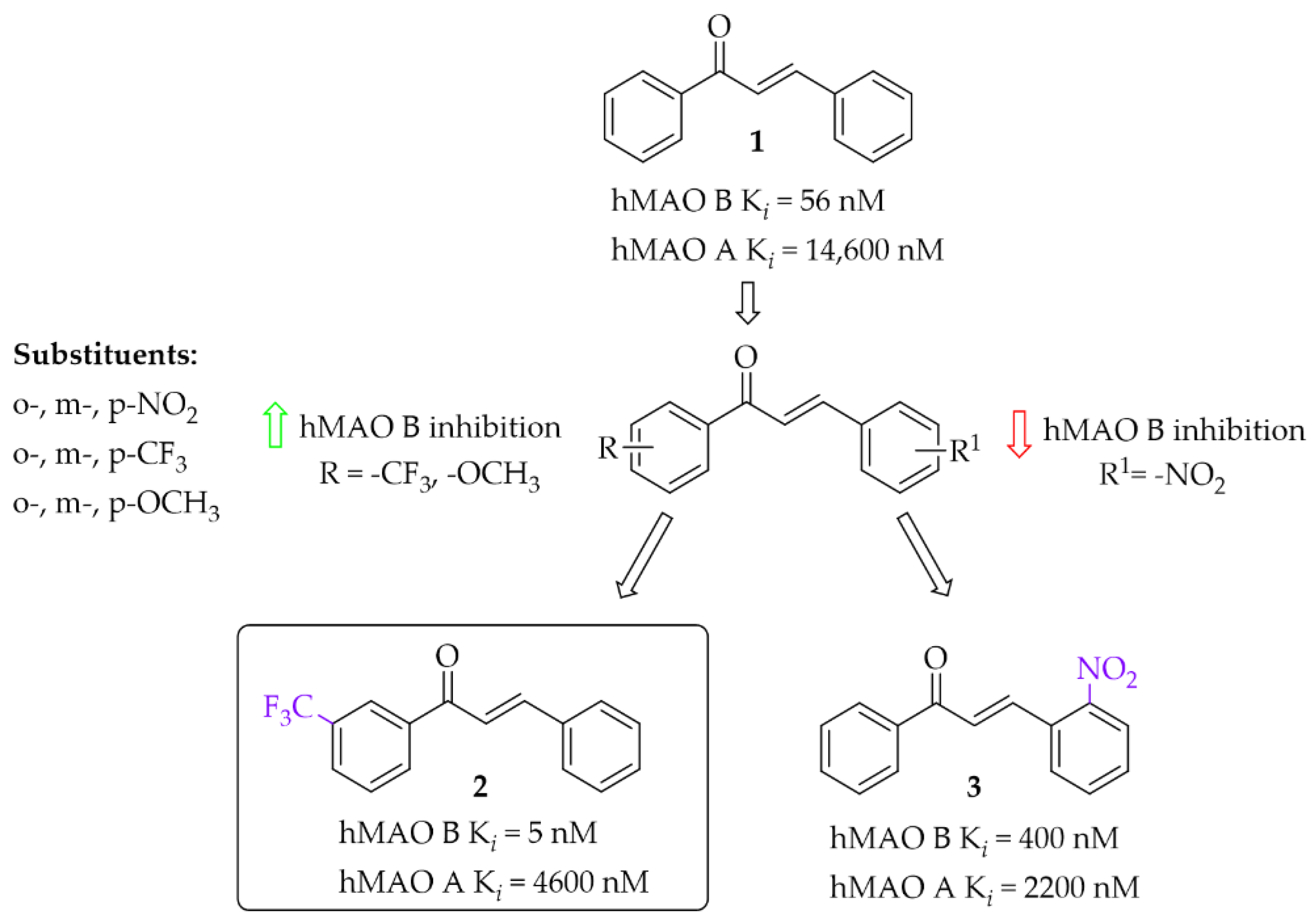
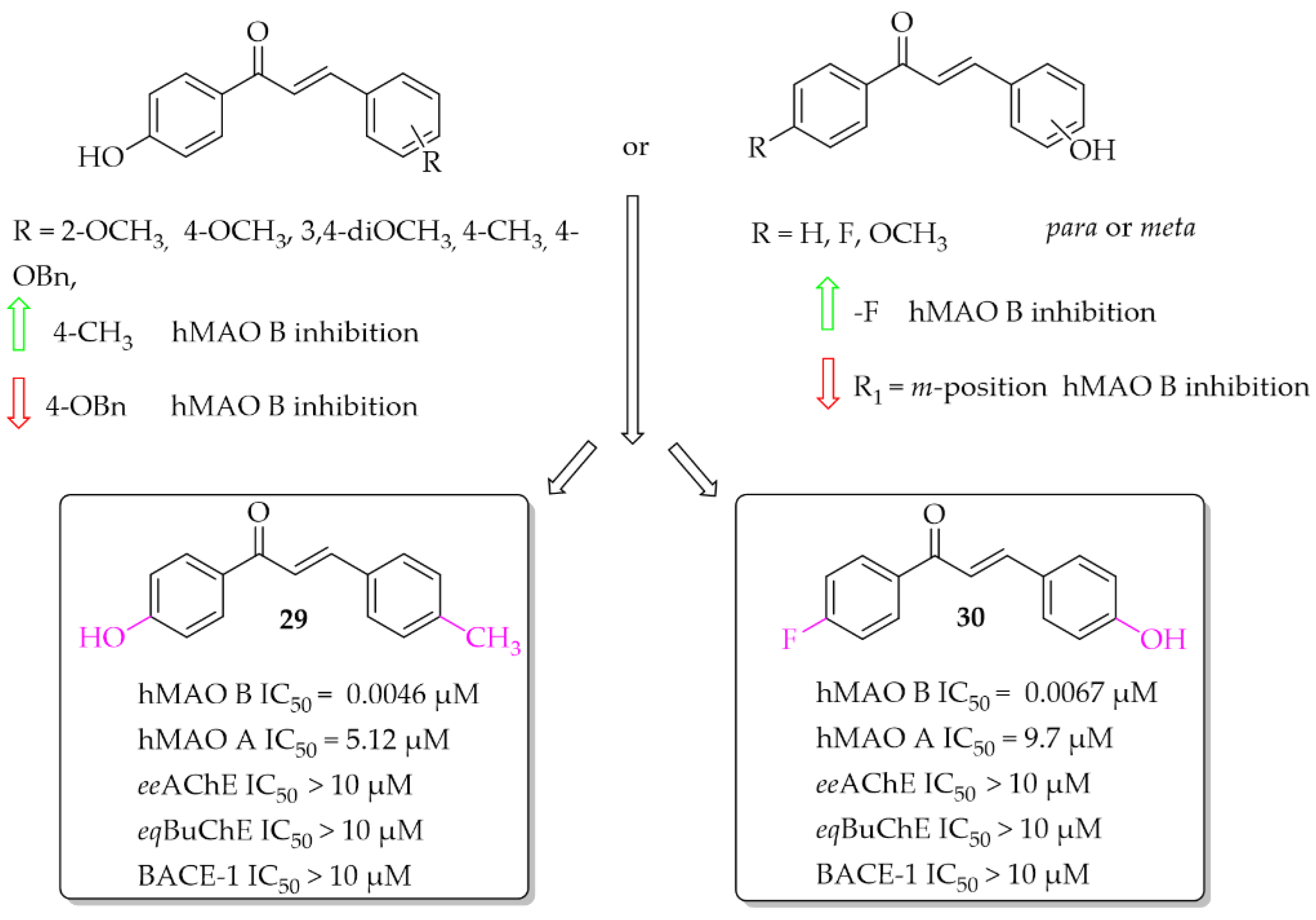
2.1. Monosubstituted Chalcones
2.2. Disubstituted Chalcones
2.2.1. Chalcones Containing a Thioether Group
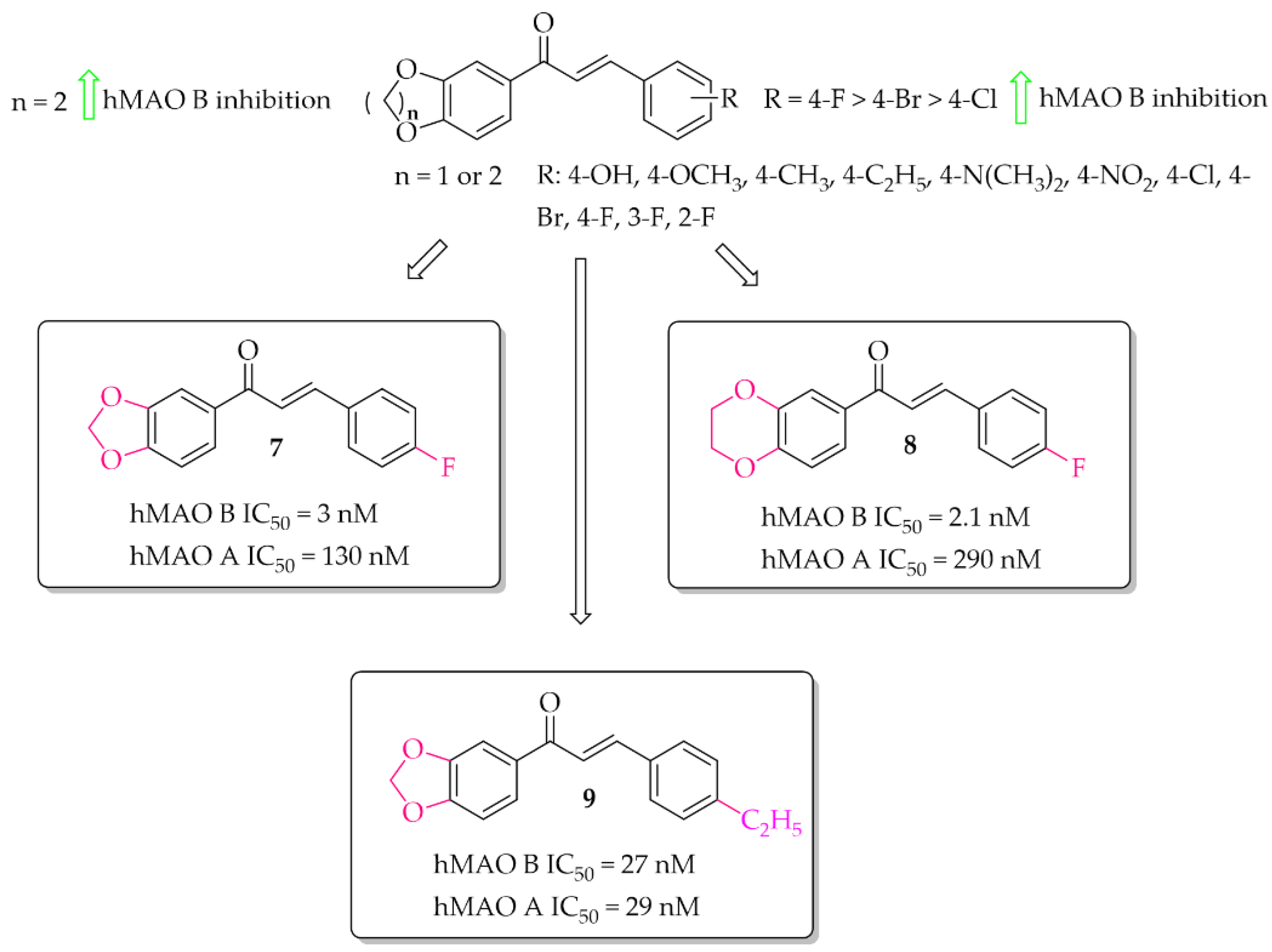
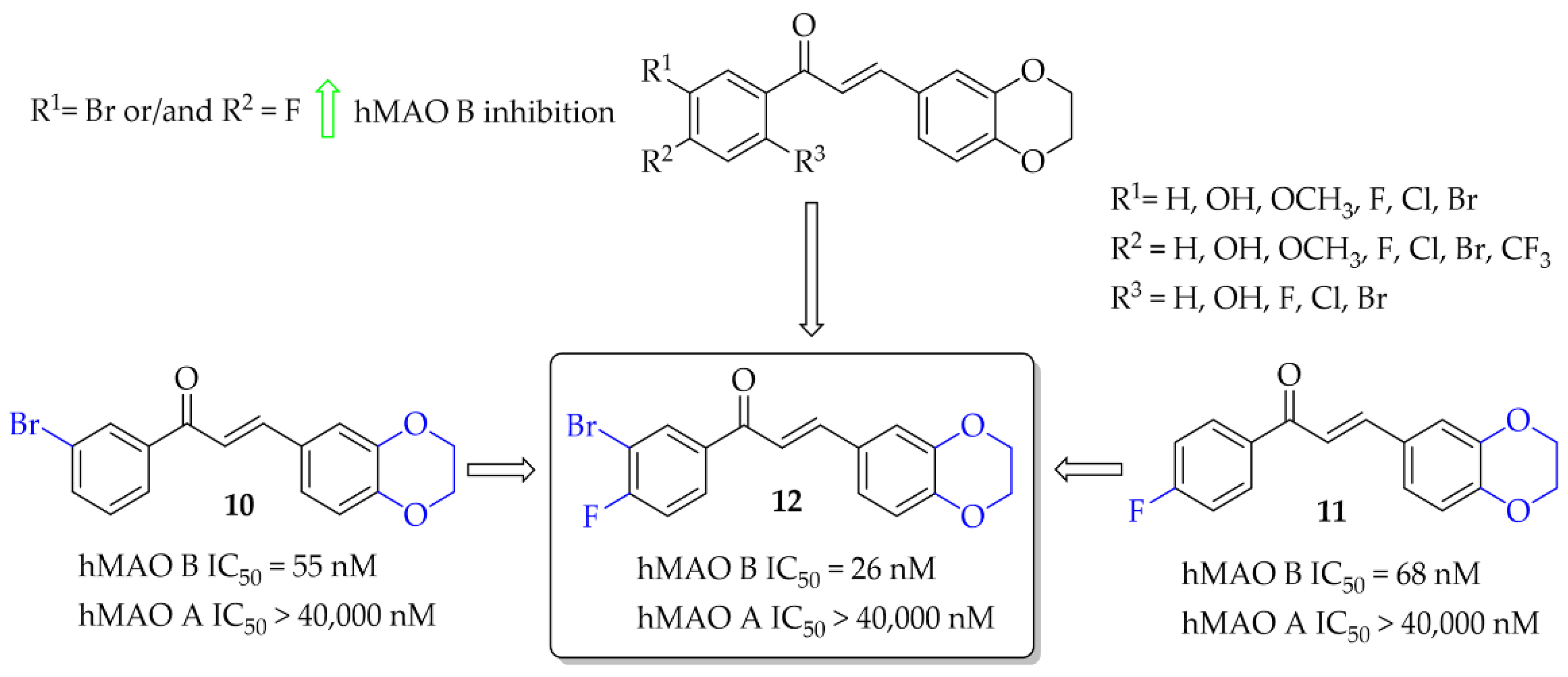
2.2.2. Oxygenated Chalcones
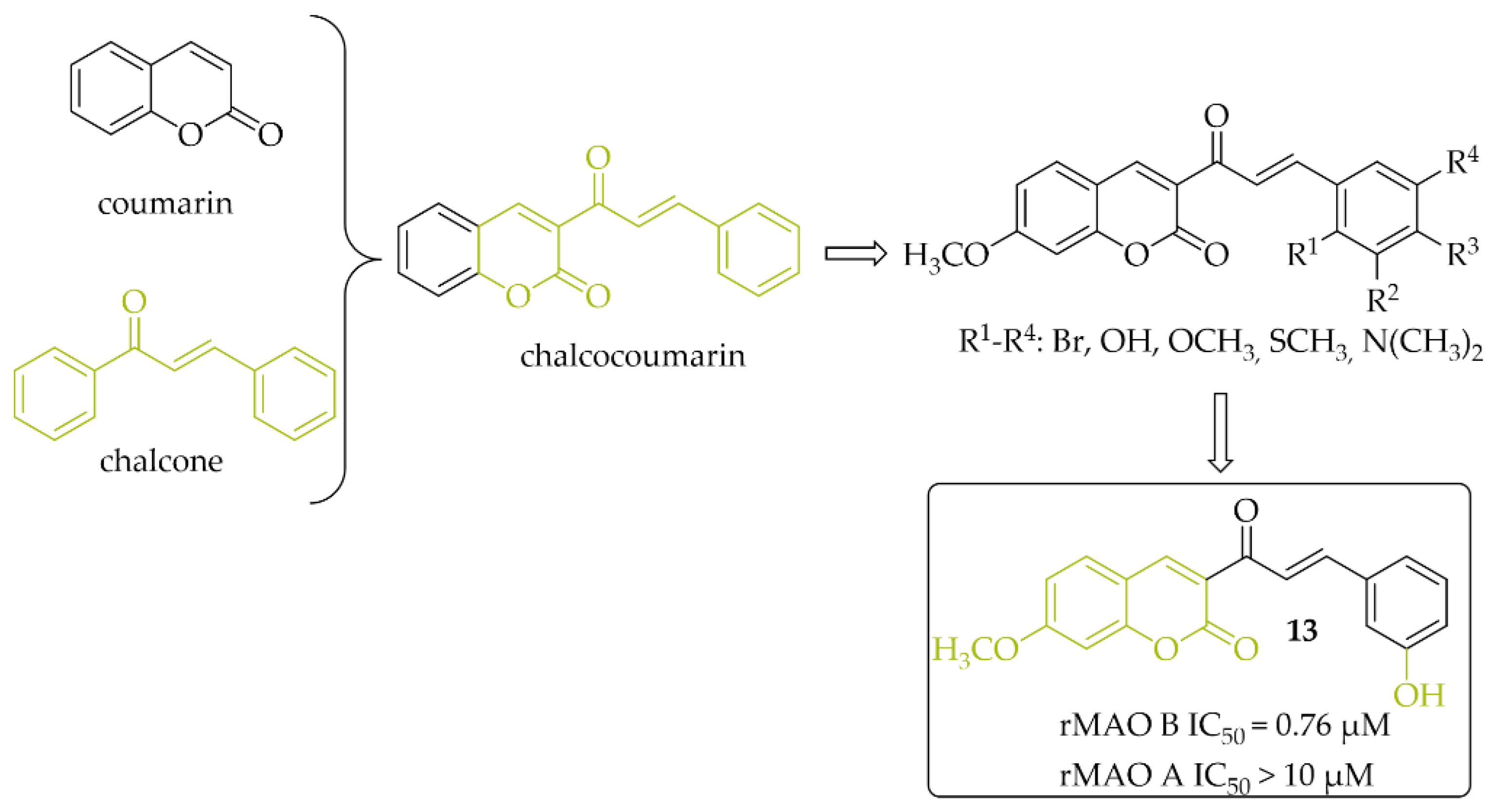
2.2.3. Chalcocoumarin Hybrids
2.3. Dual and Multi-Target Monoamine Oxidase B Inhibitors
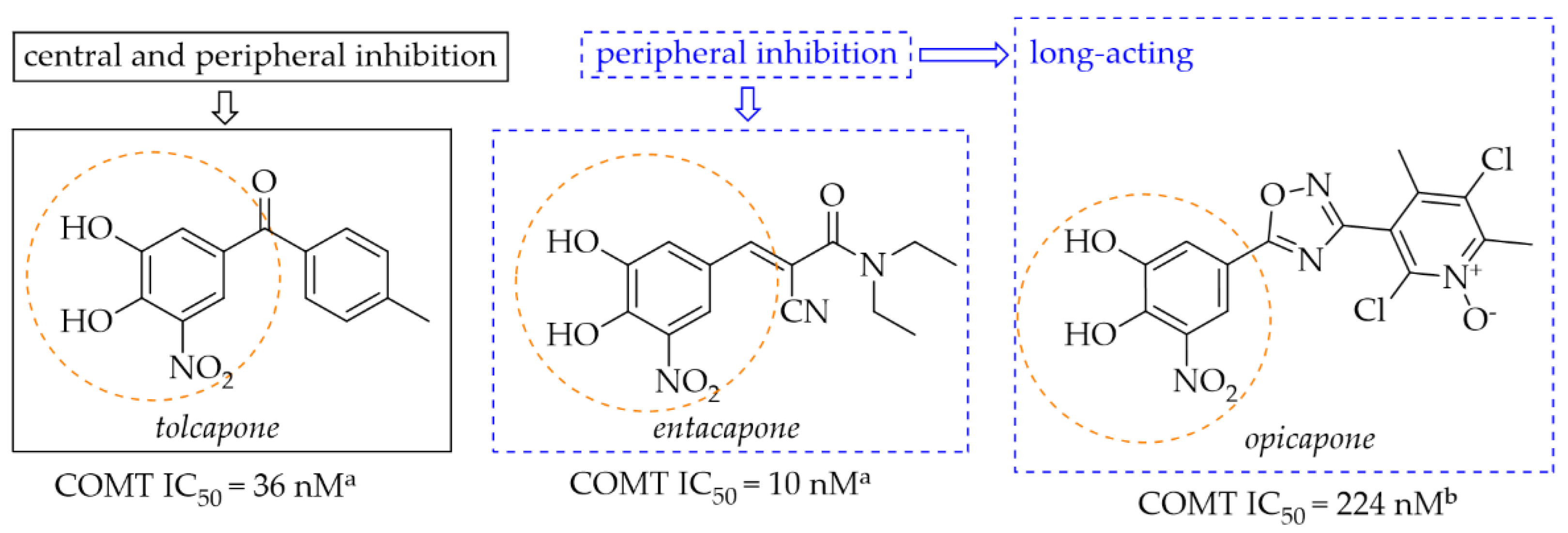
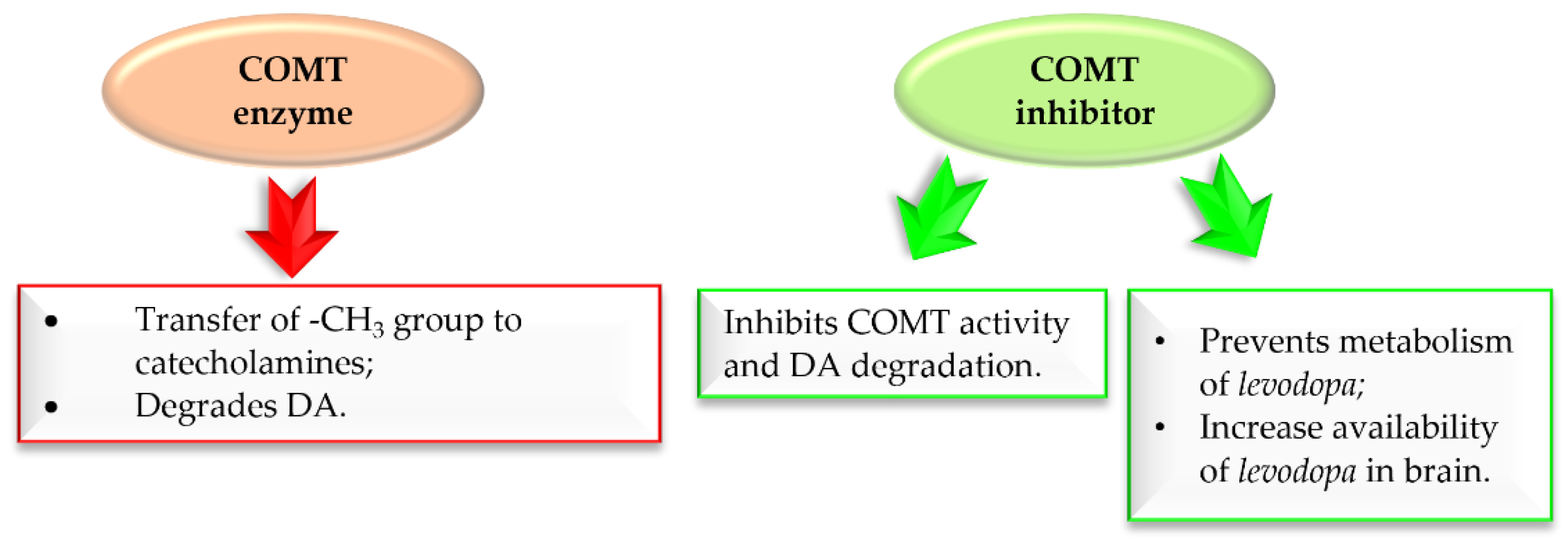
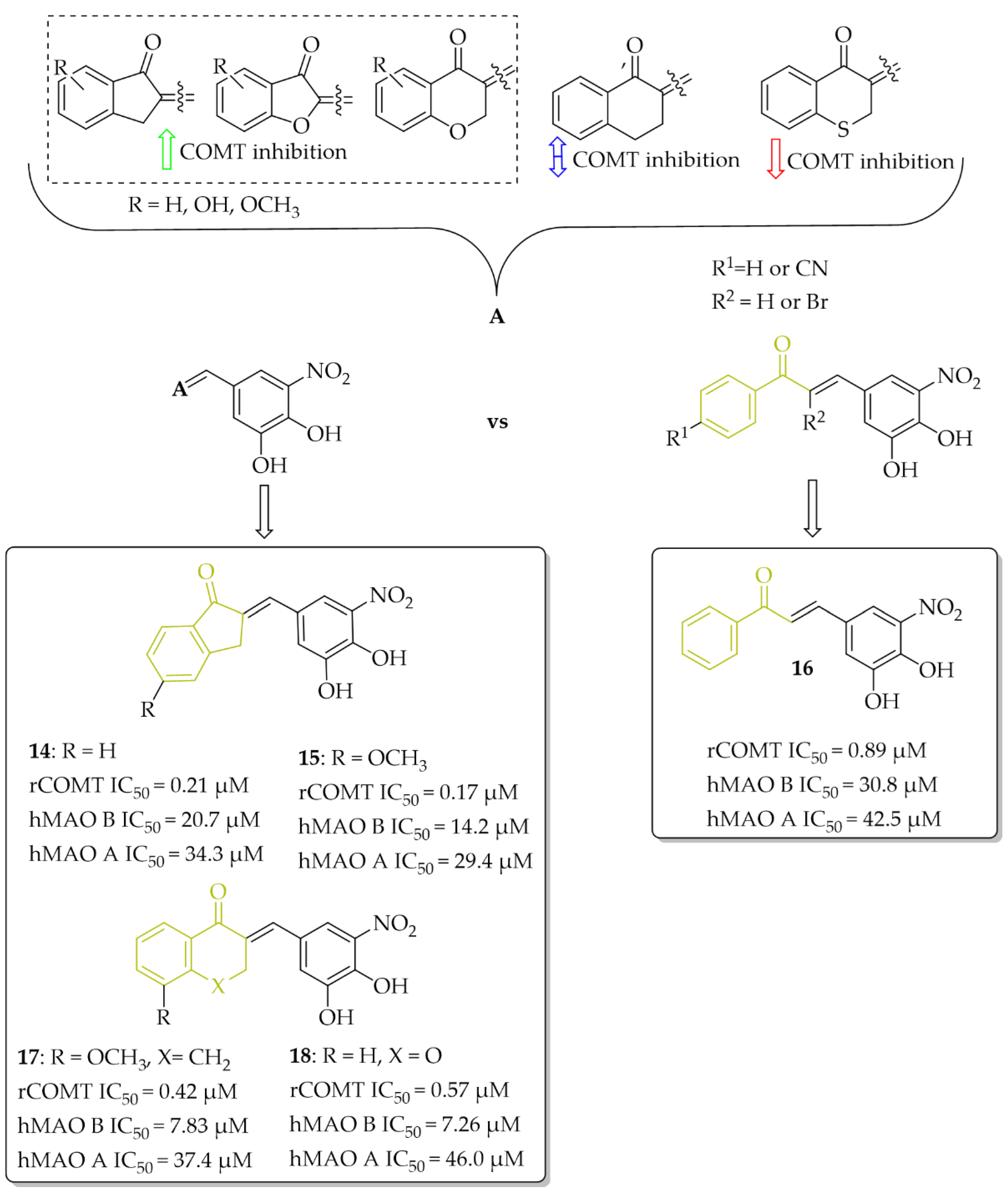
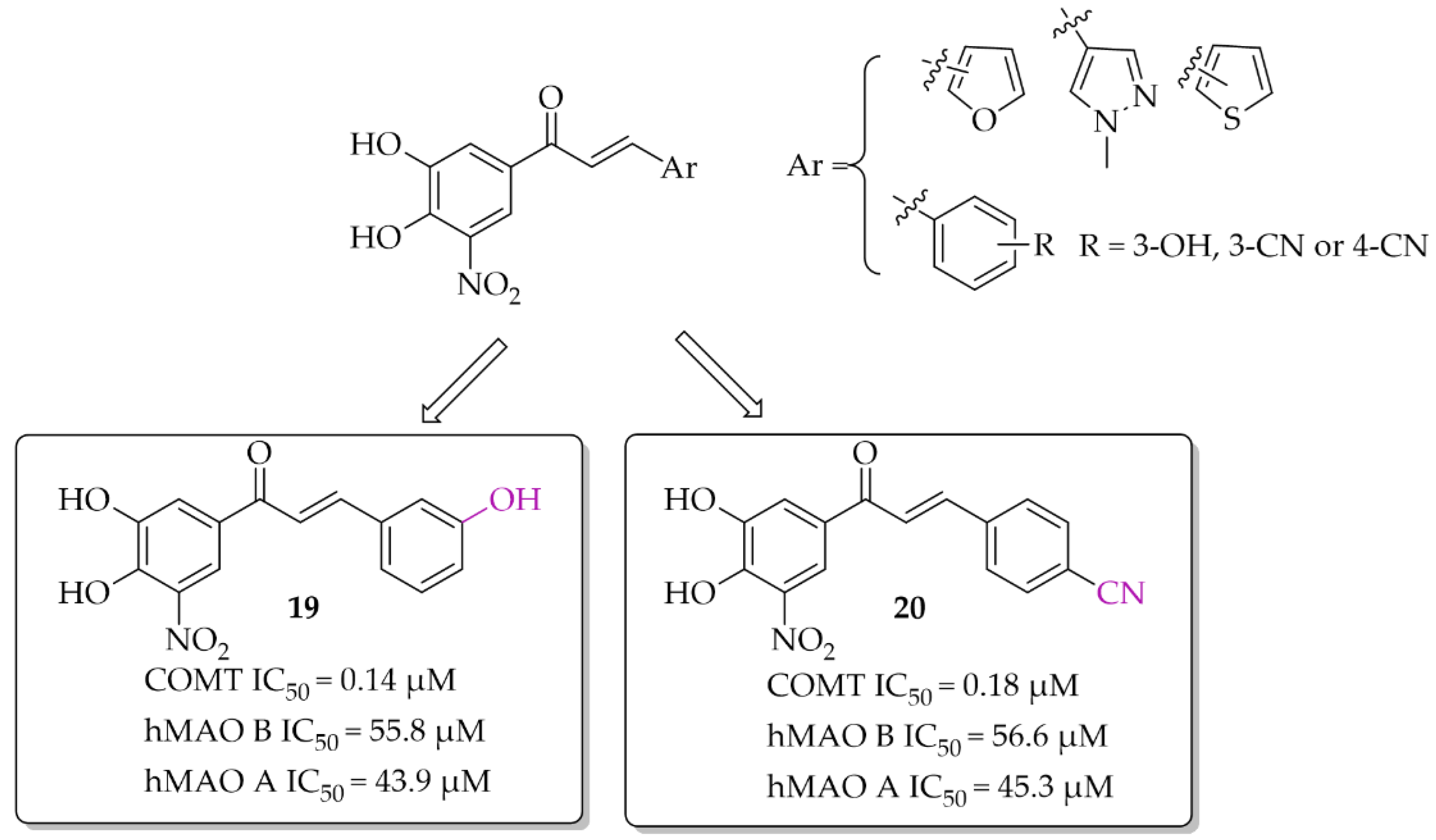
2.3.1. Monoamine Oxidase B and Catechol-O-Methyltransferase Inhibitors—Chalcones with a Nitrocatechol Moiety
2.3.2. Monoamine Oxidase B and Cholinesterase Inhibitors
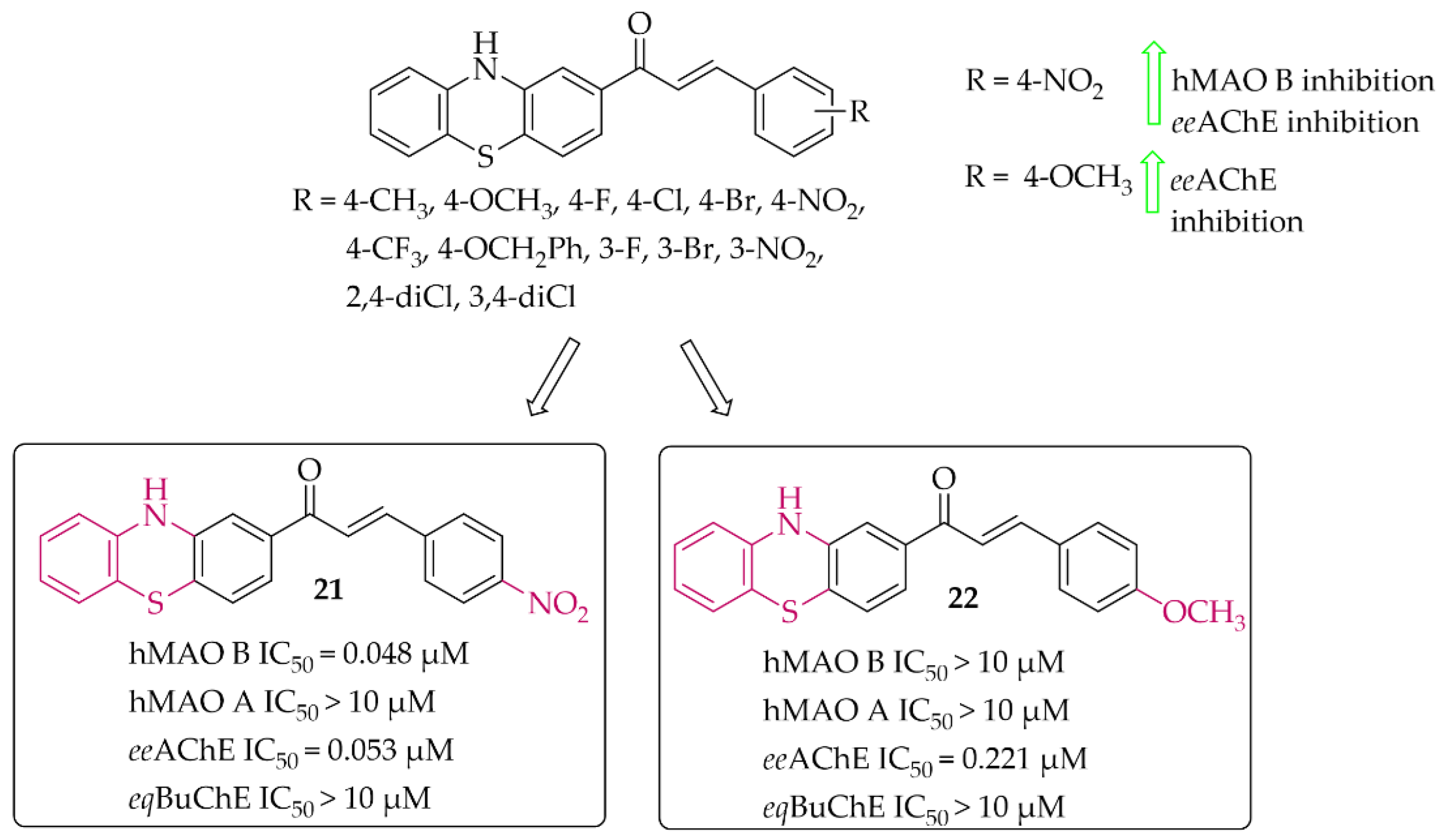
Phenothiazine-Based Chalcones
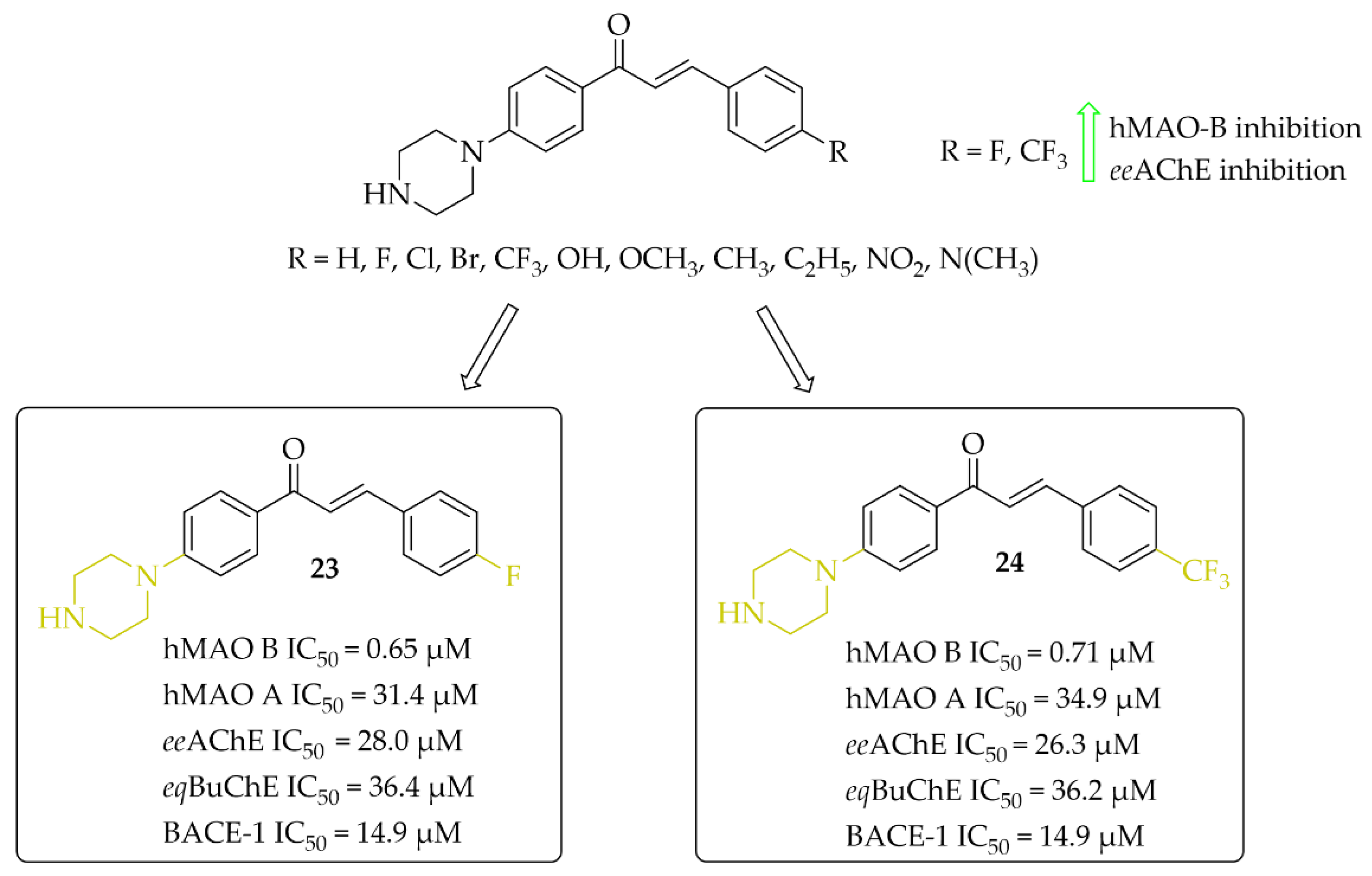
Piperazine-Based Chalcones
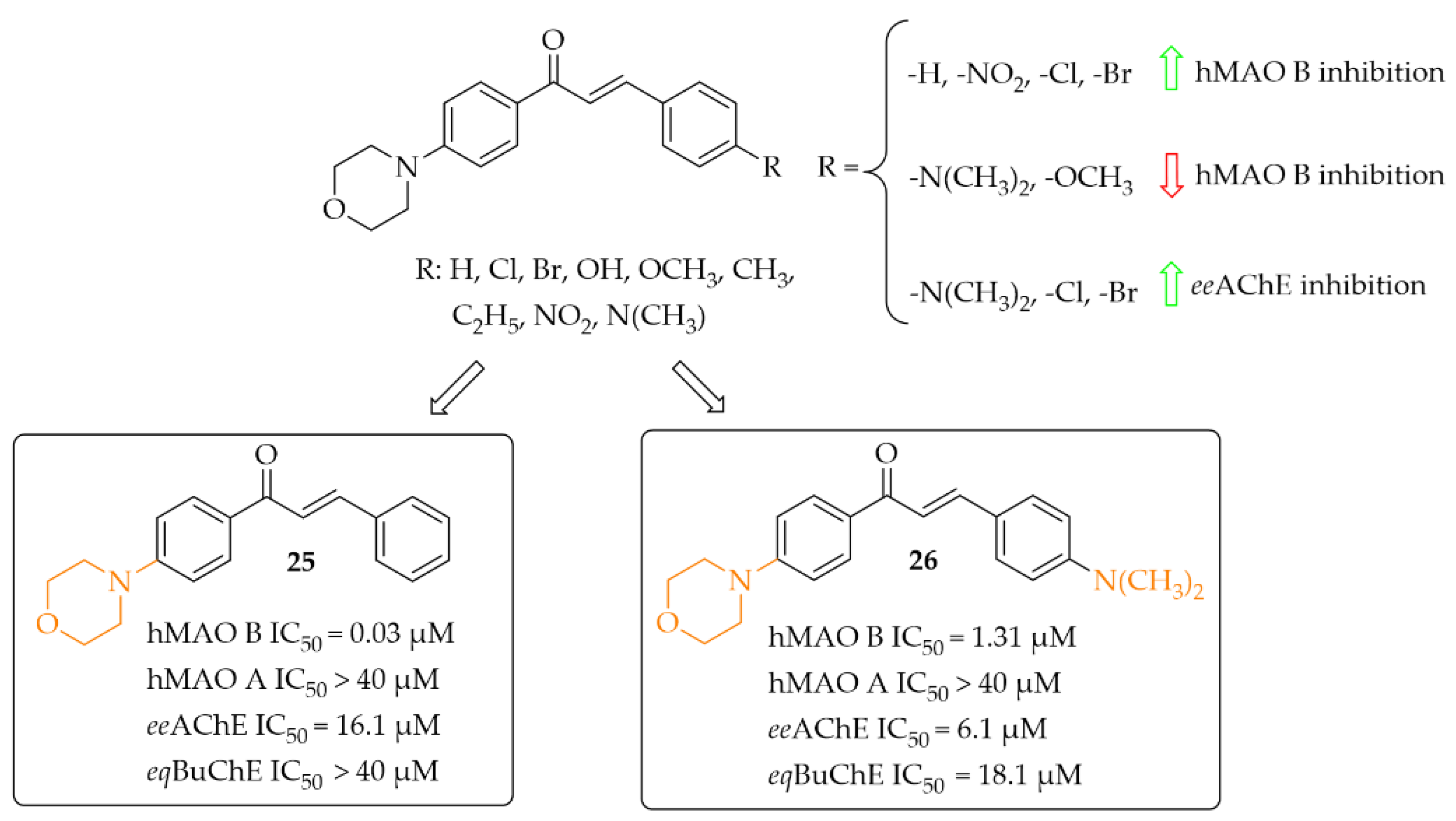
Morpholine-Based Chalcones
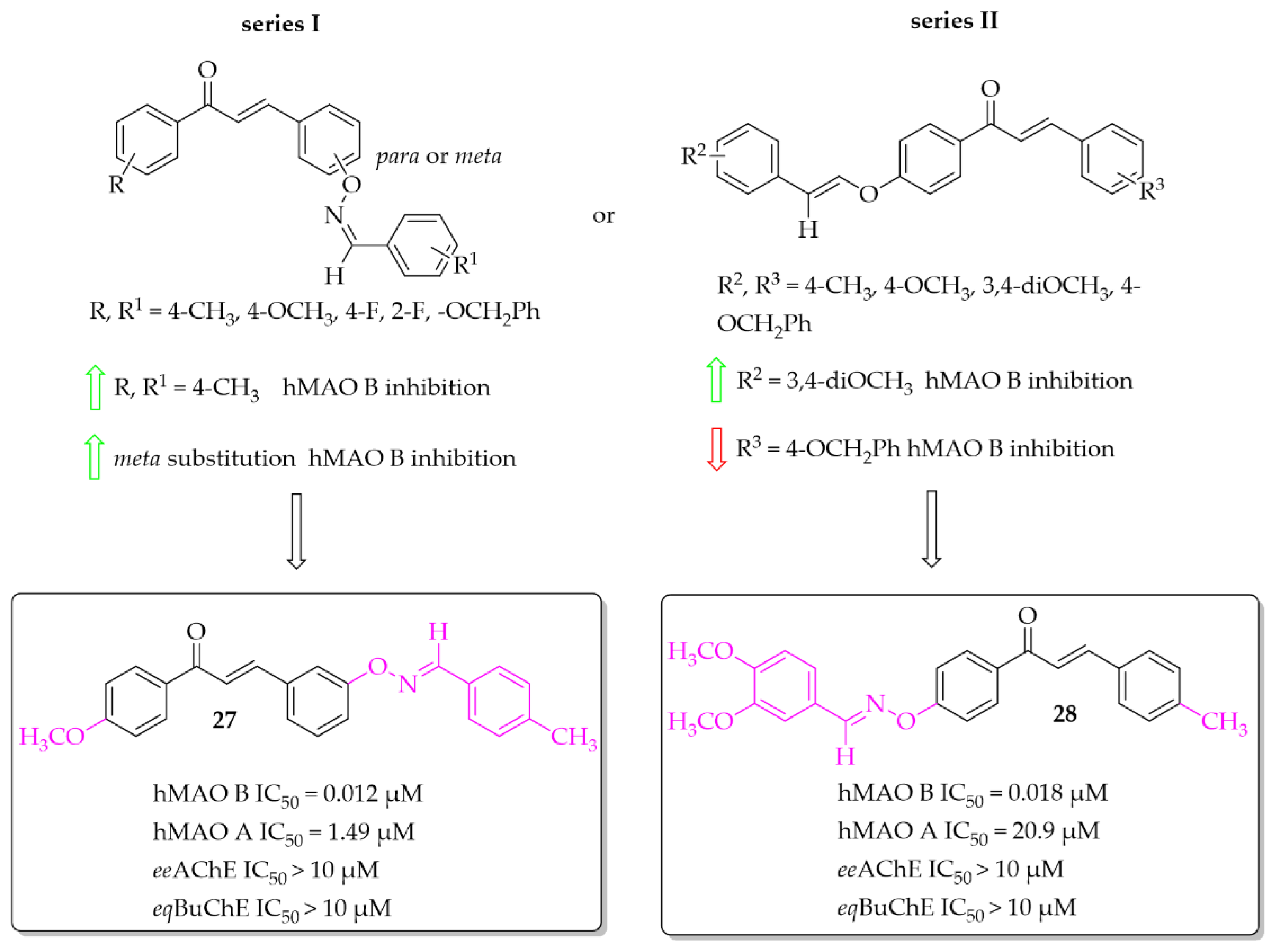
Aldoxime Ethers
Hydroxychalcones
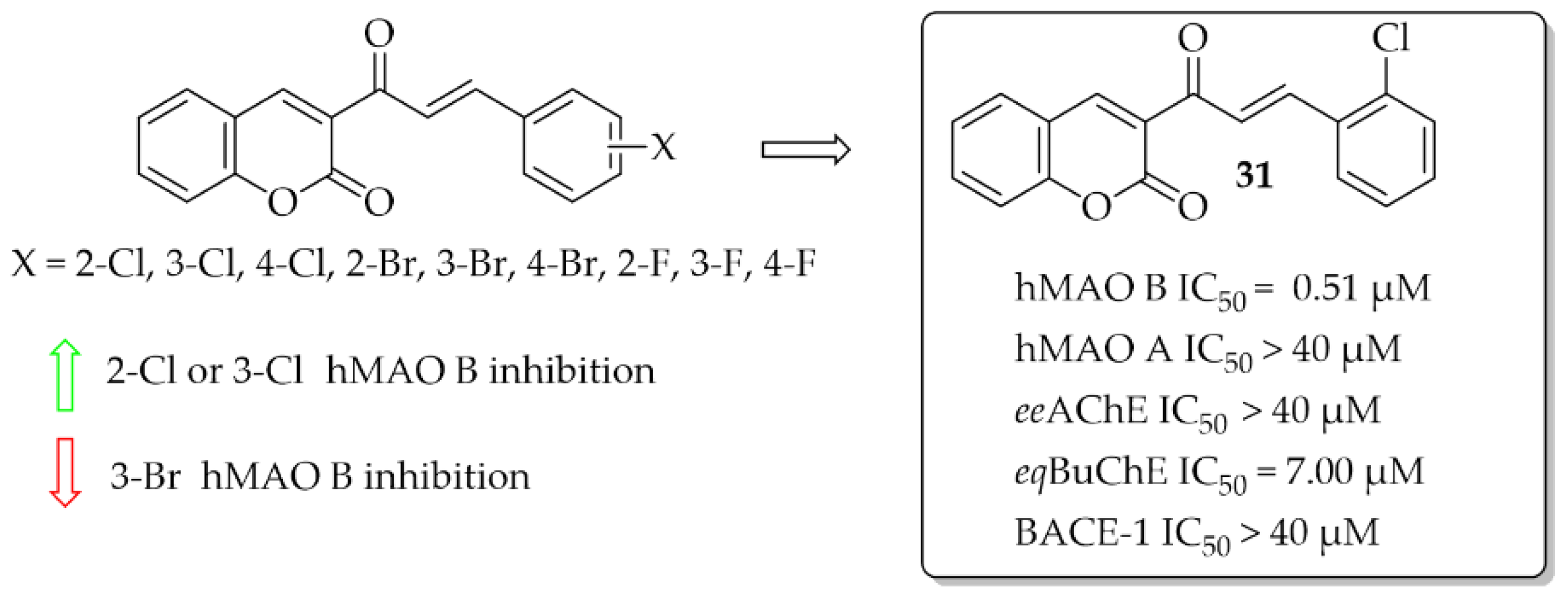
Coumarin-Chalcones
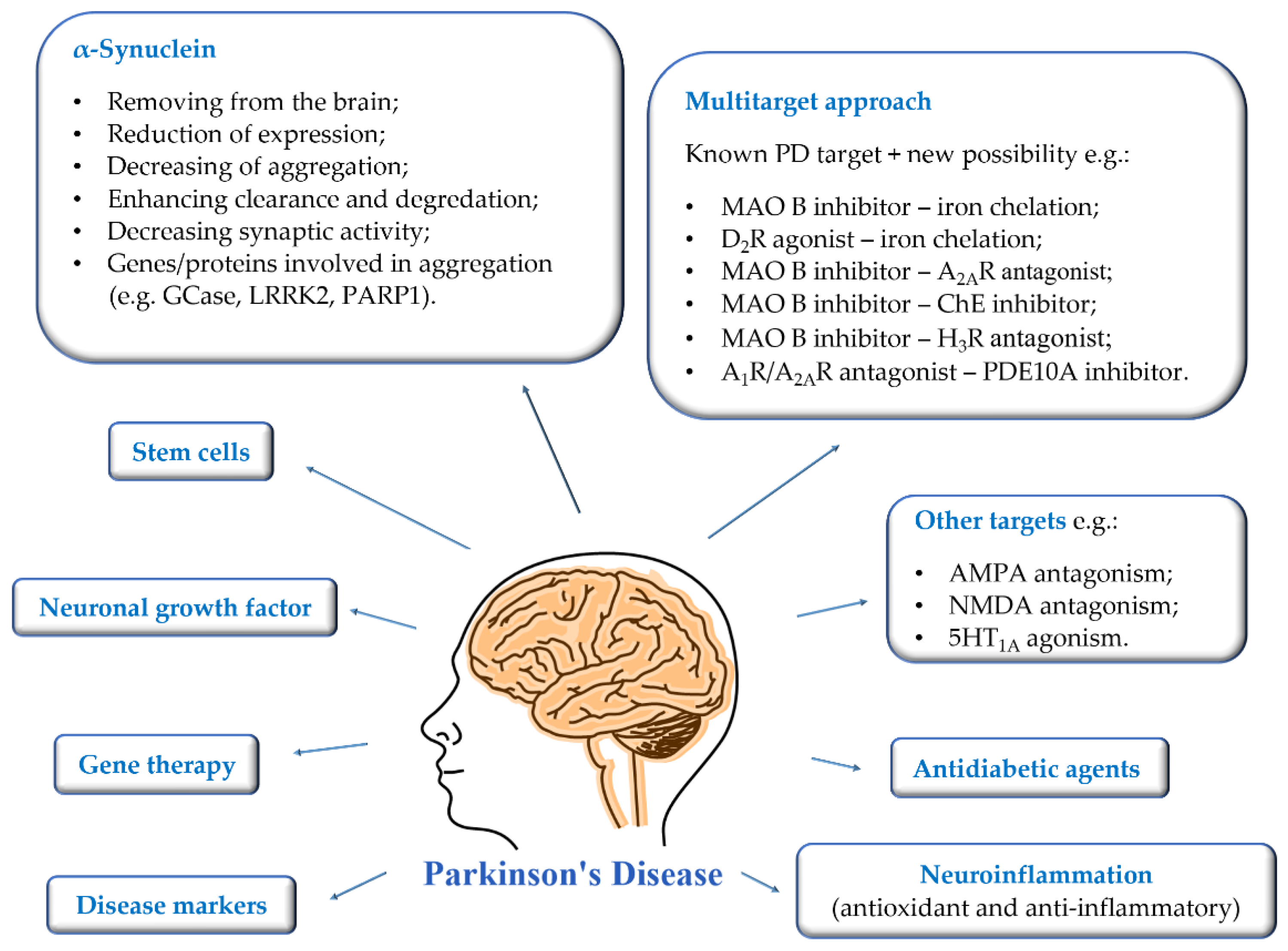
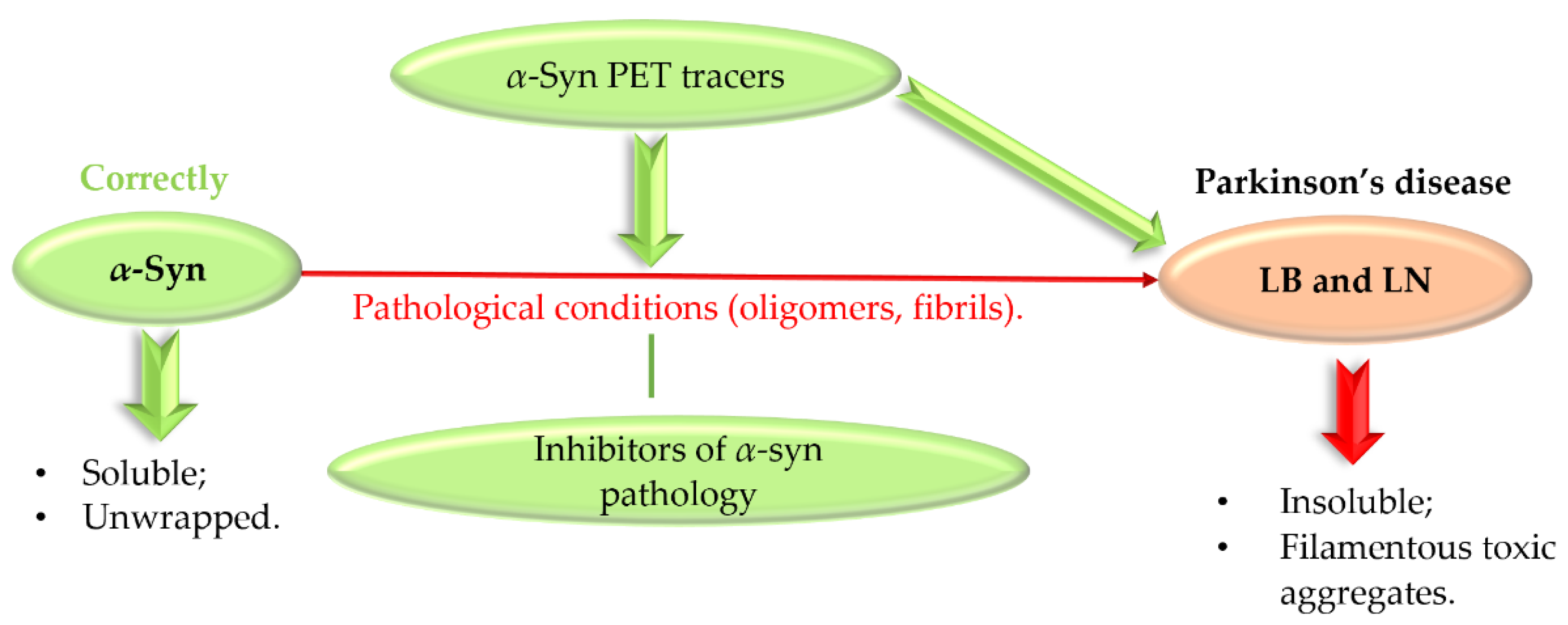
3. Targeting α-Synuclein
- Removing of pathological aggregates from the brain—immunisation bullet;
- Reduction of α-syn expression;
- Inhibition of α-syn aggregation;
- Enhancing clearance and degradation of α-syn;
- Other genes/proteins involved in synucleinopathies;
- Decreasing synaptic activity.
- Very high affinity for α-syn (even in the subnanomolar range);
- High selectivity of imaging α-syn (quantify α-syn in the presence of higher density of Aβ or/and tau proteins);
- Good permeability to cross not only BBB but cell membranes;
- Low binding to off-targets (>500 nM);
- Good metabolic stability.
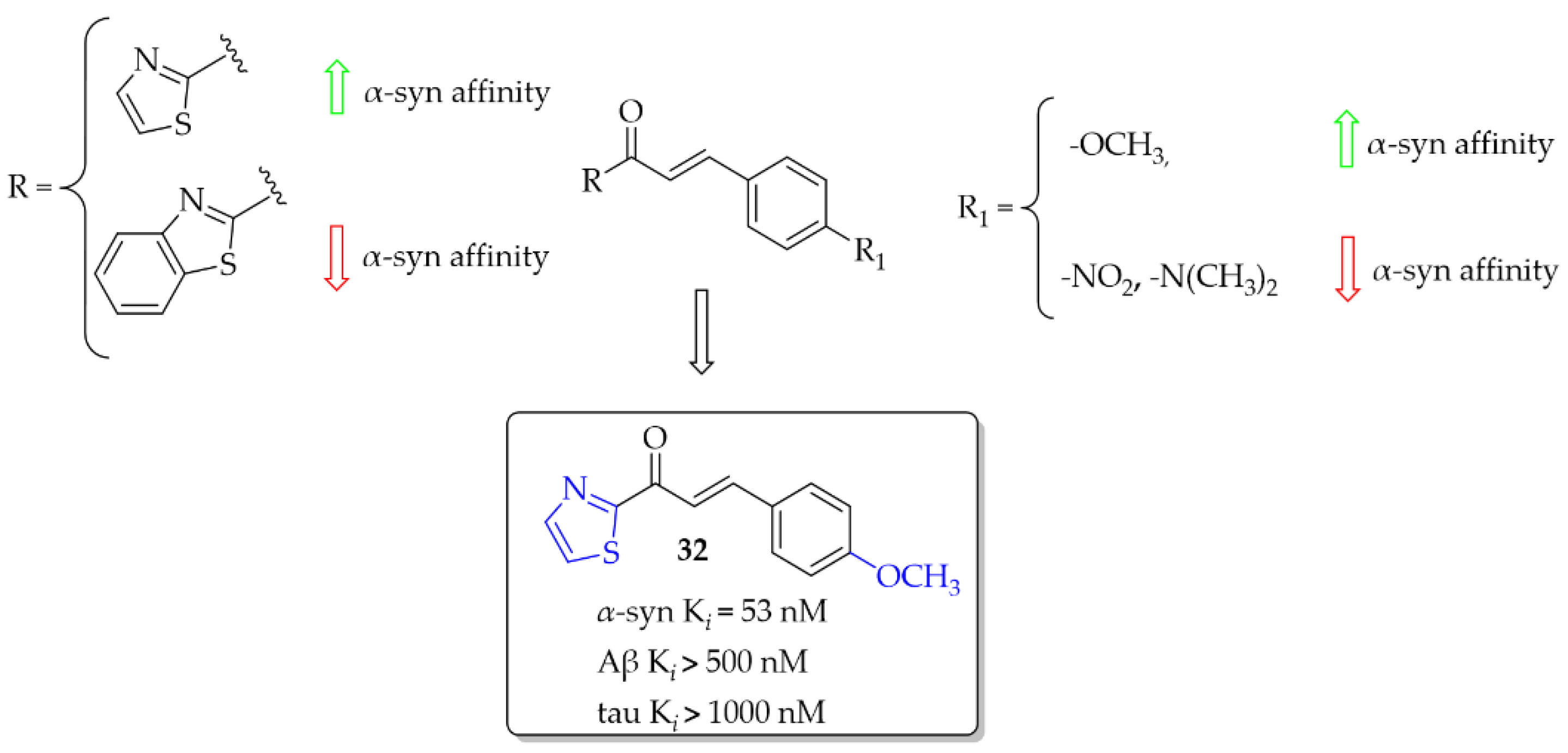
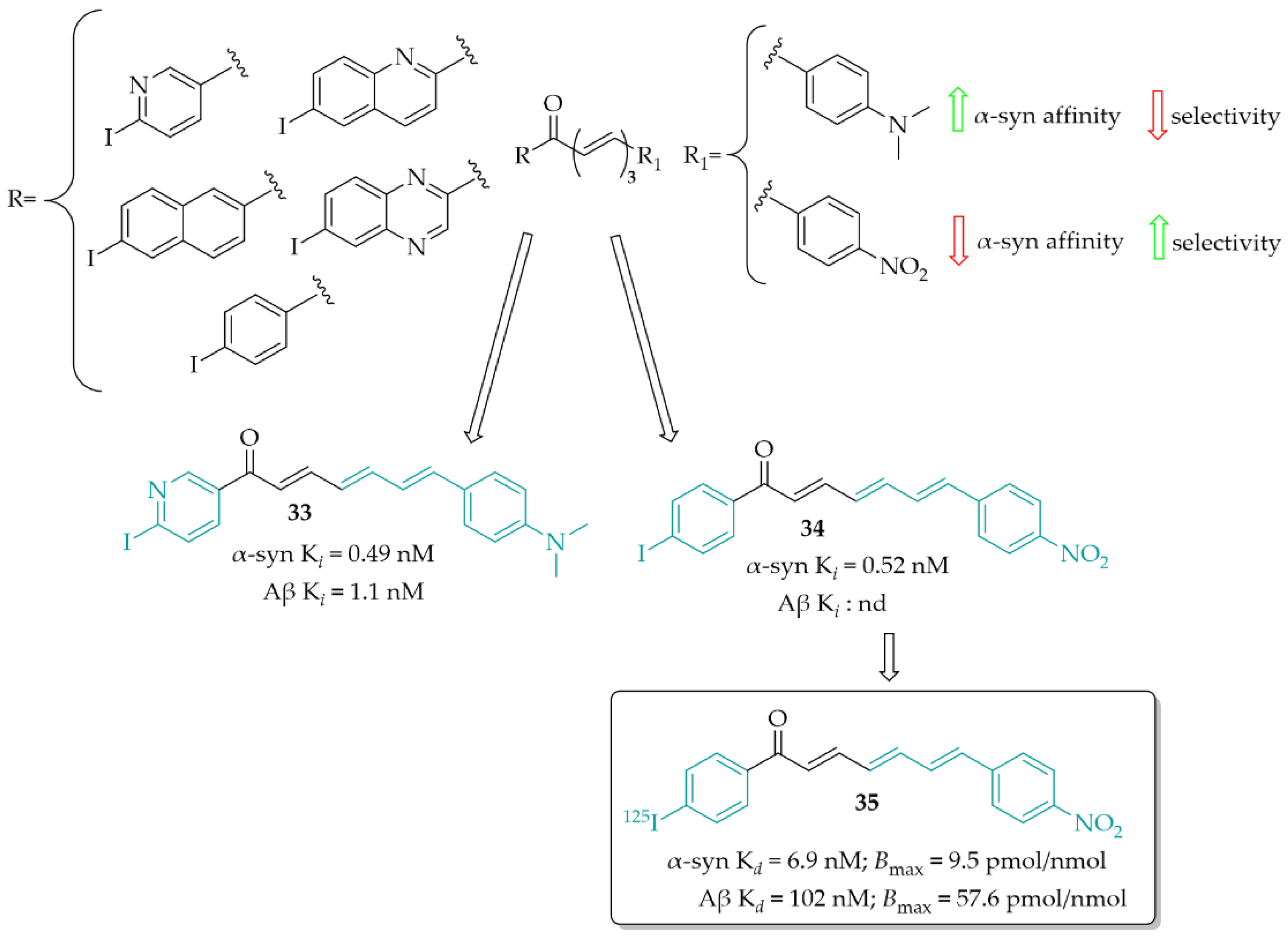
3.1. Imaging Probes of α-Syn Inclusions
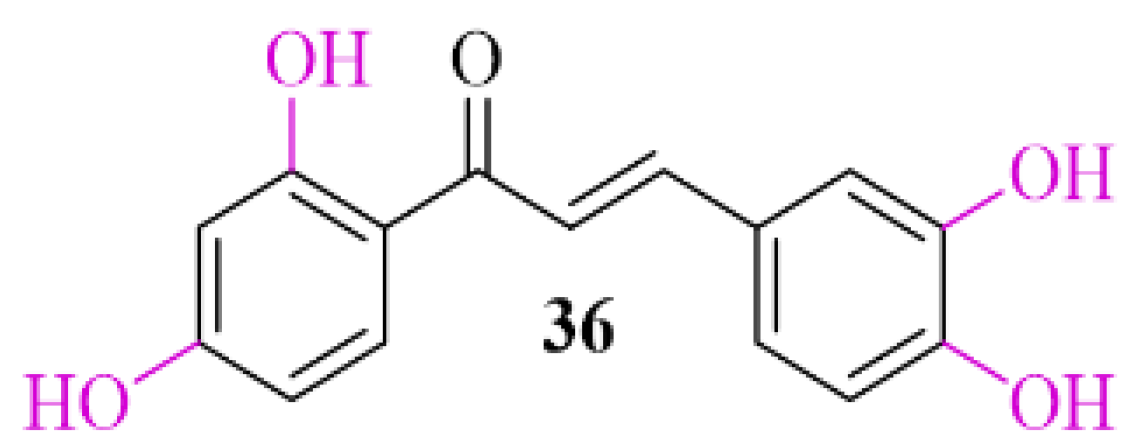
3.2. α-Synuclein Inhibitor—Natural Chalcone Derivative—Butein
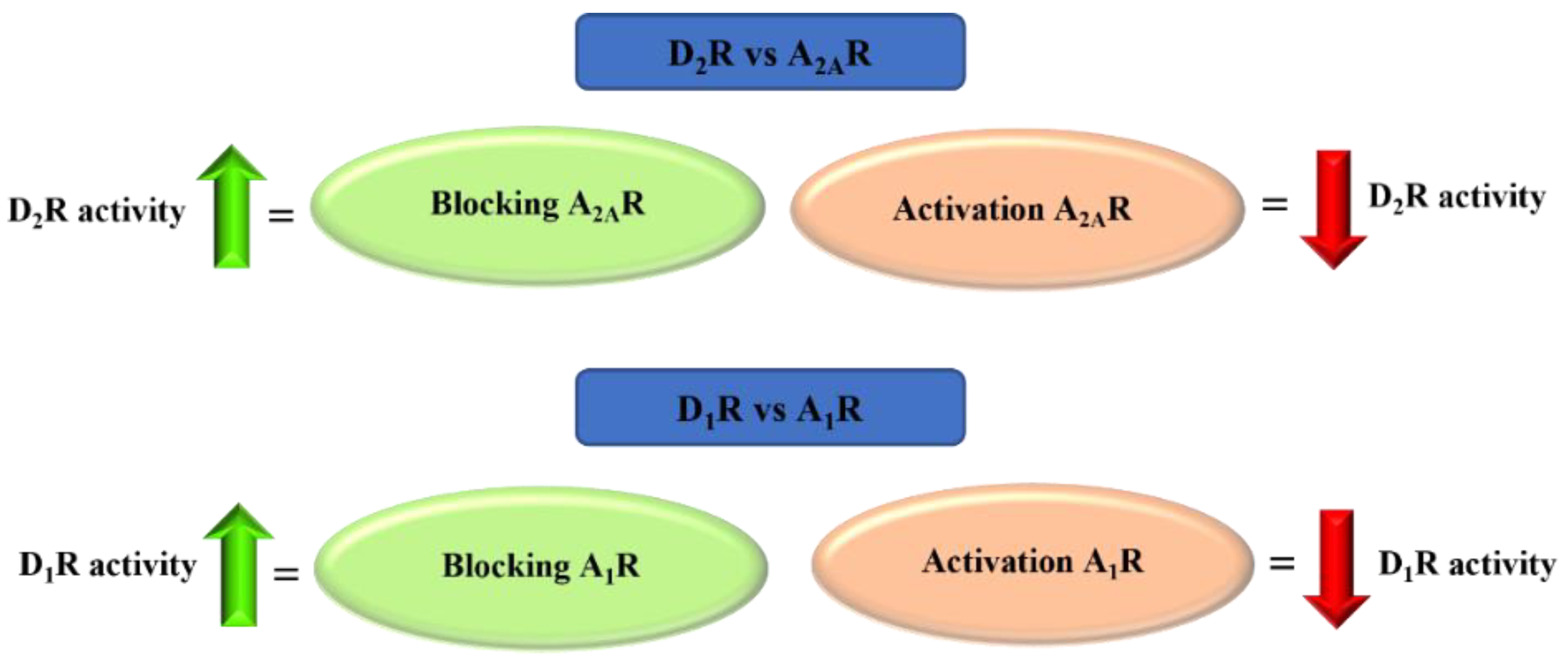
4. Adenosine A2A and A1 Receptor Antagonists
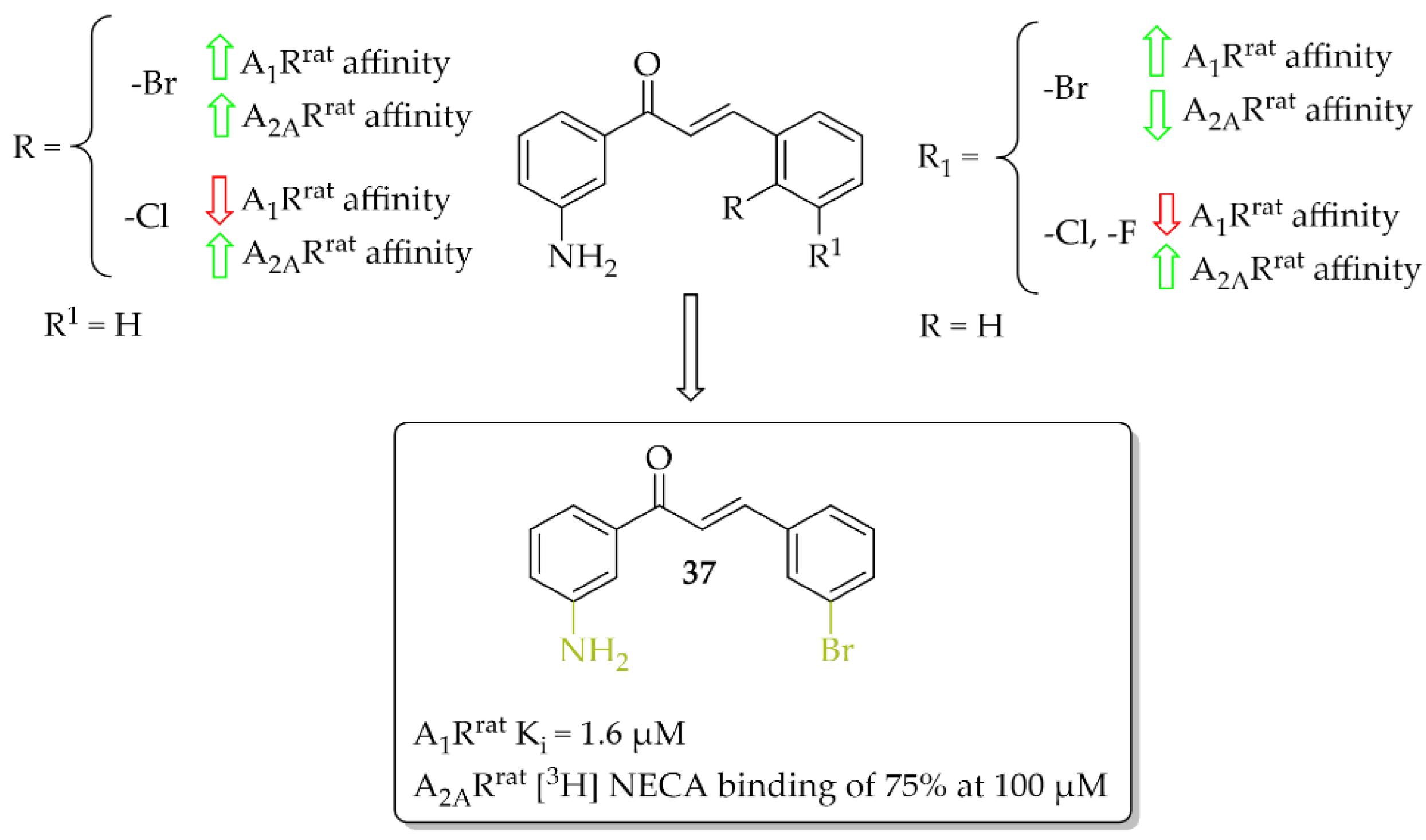
4.1. Aniline-Based Chalcones
4.2. Chalcone Hybrids
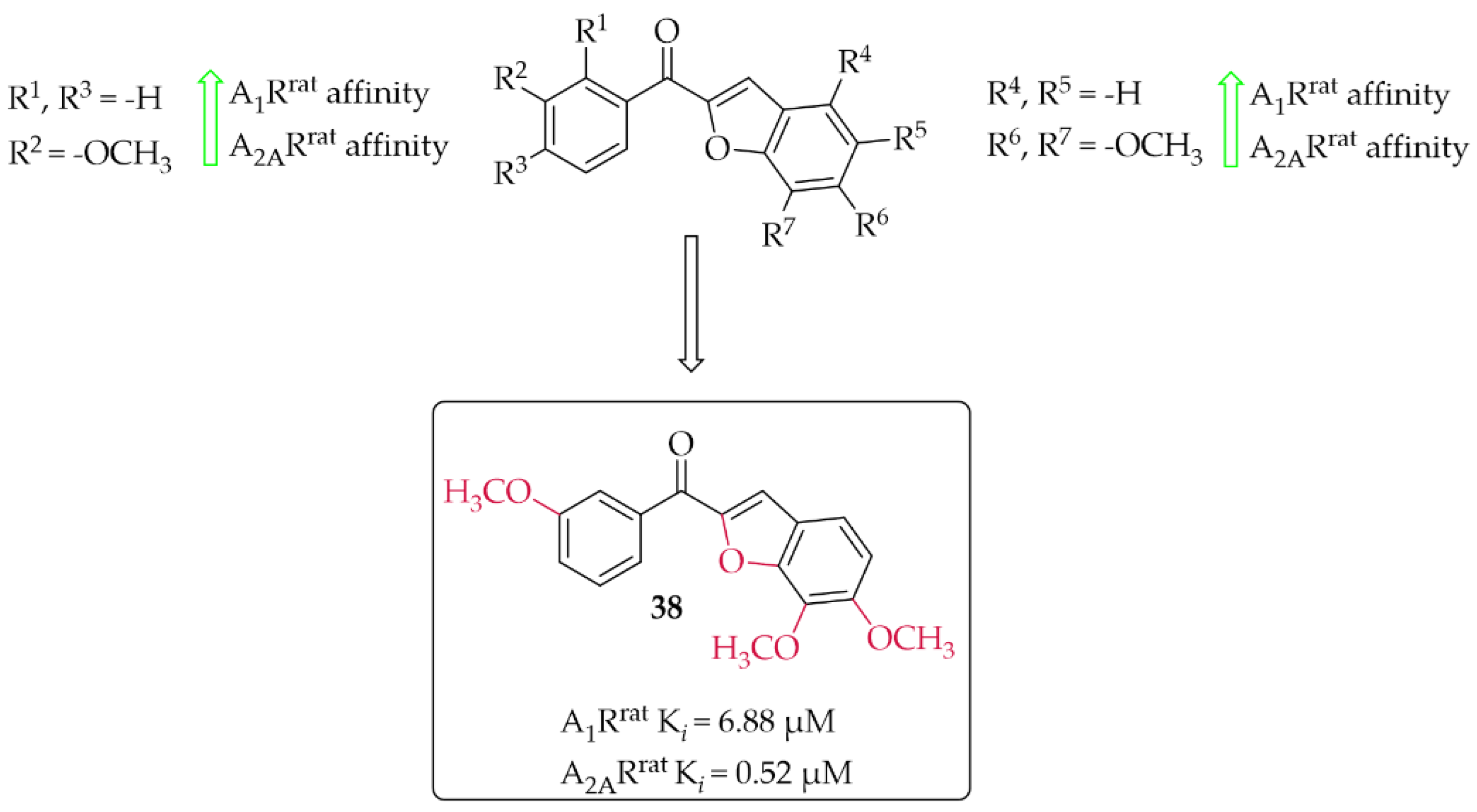
4.2.1. Benzofuran Derivatives
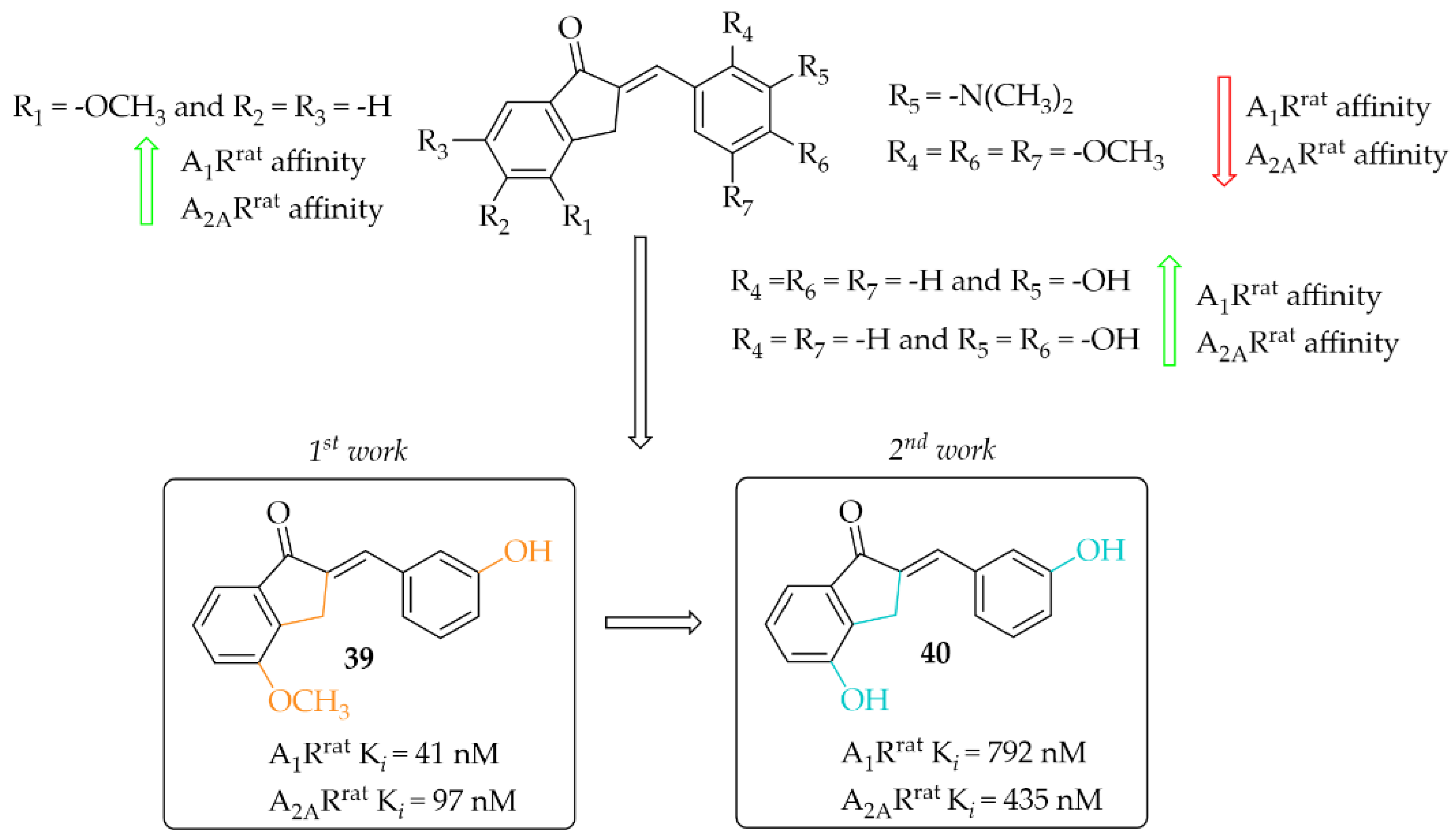
4.2.2. Indanone Derivatives
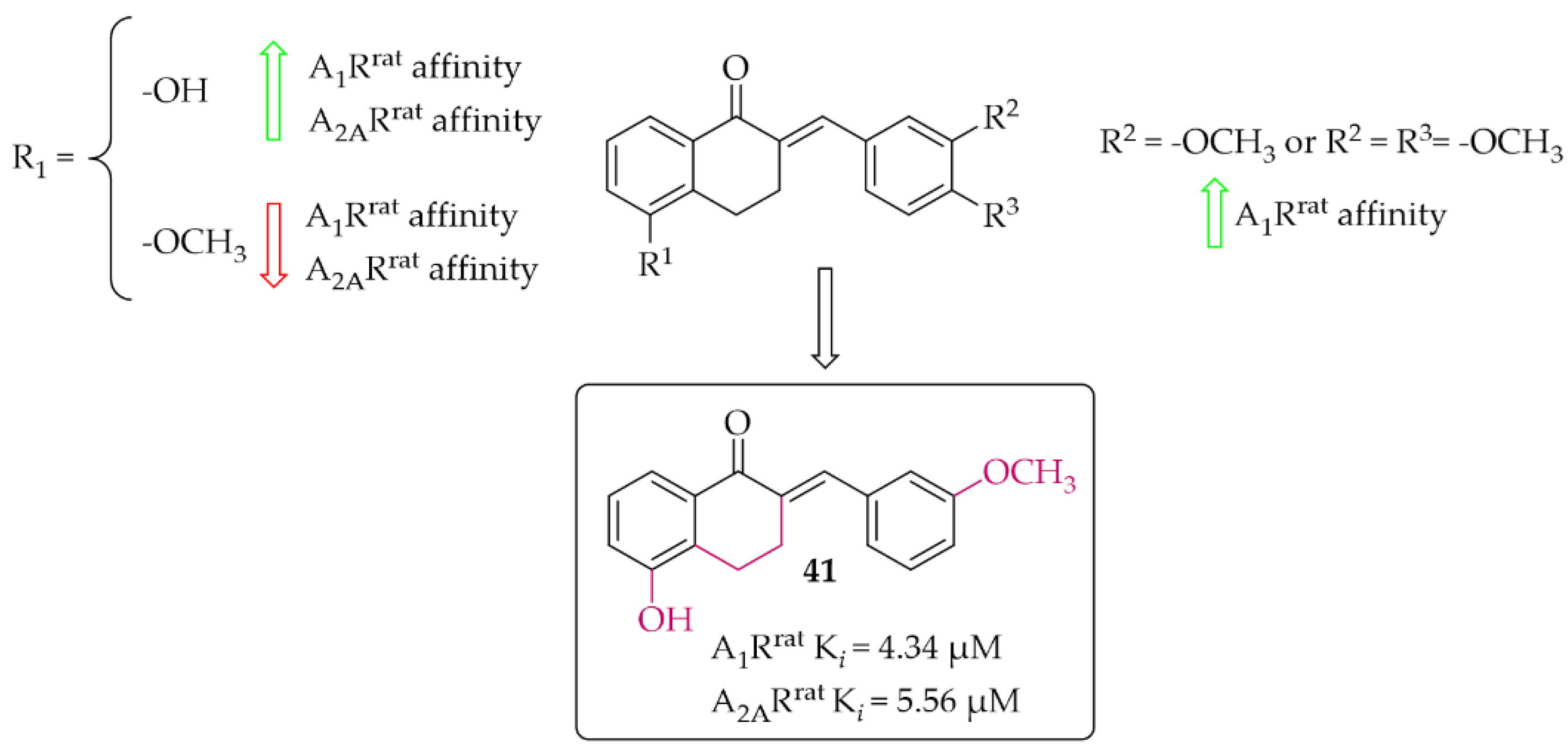
4.2.3. Tetralone Derivatives
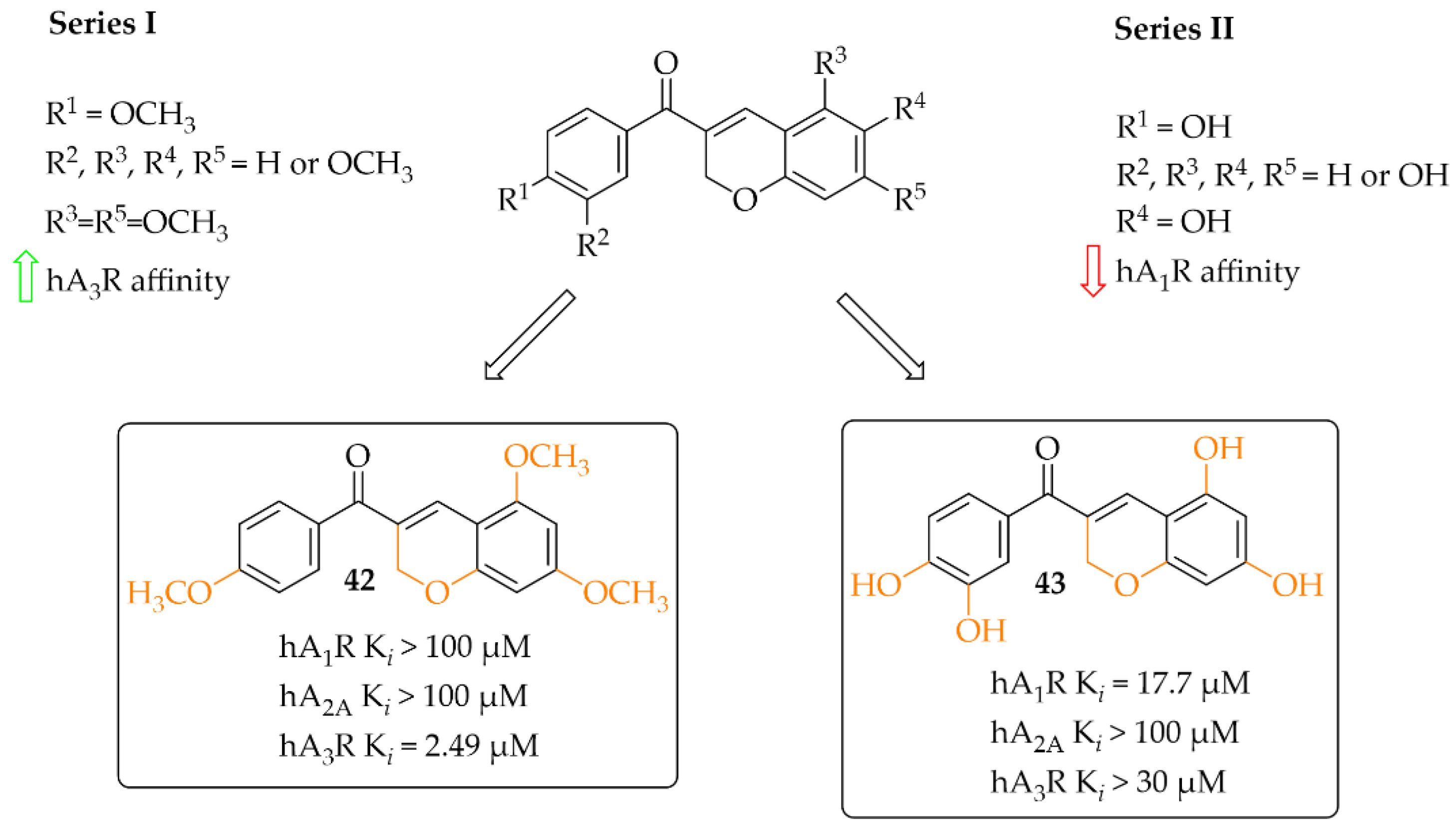
4.2.4. Coumarin Derivatives
5. Targeting Oxidative Stress and Neuroinflammation
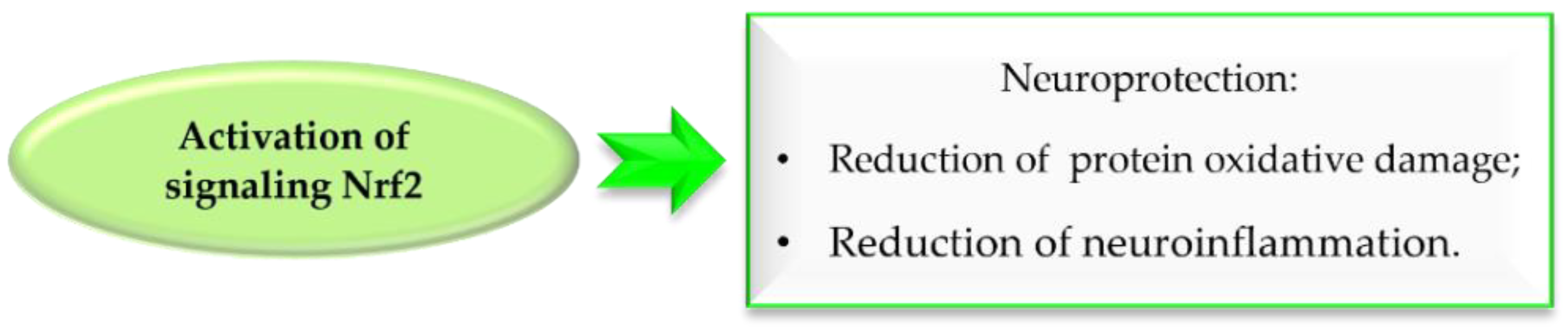
5.1. The Nuclear Factor Erythroid 2–Related Factor 2 Activation
5.1.1. Compound KMS99220
5.1.2. Compound AN07
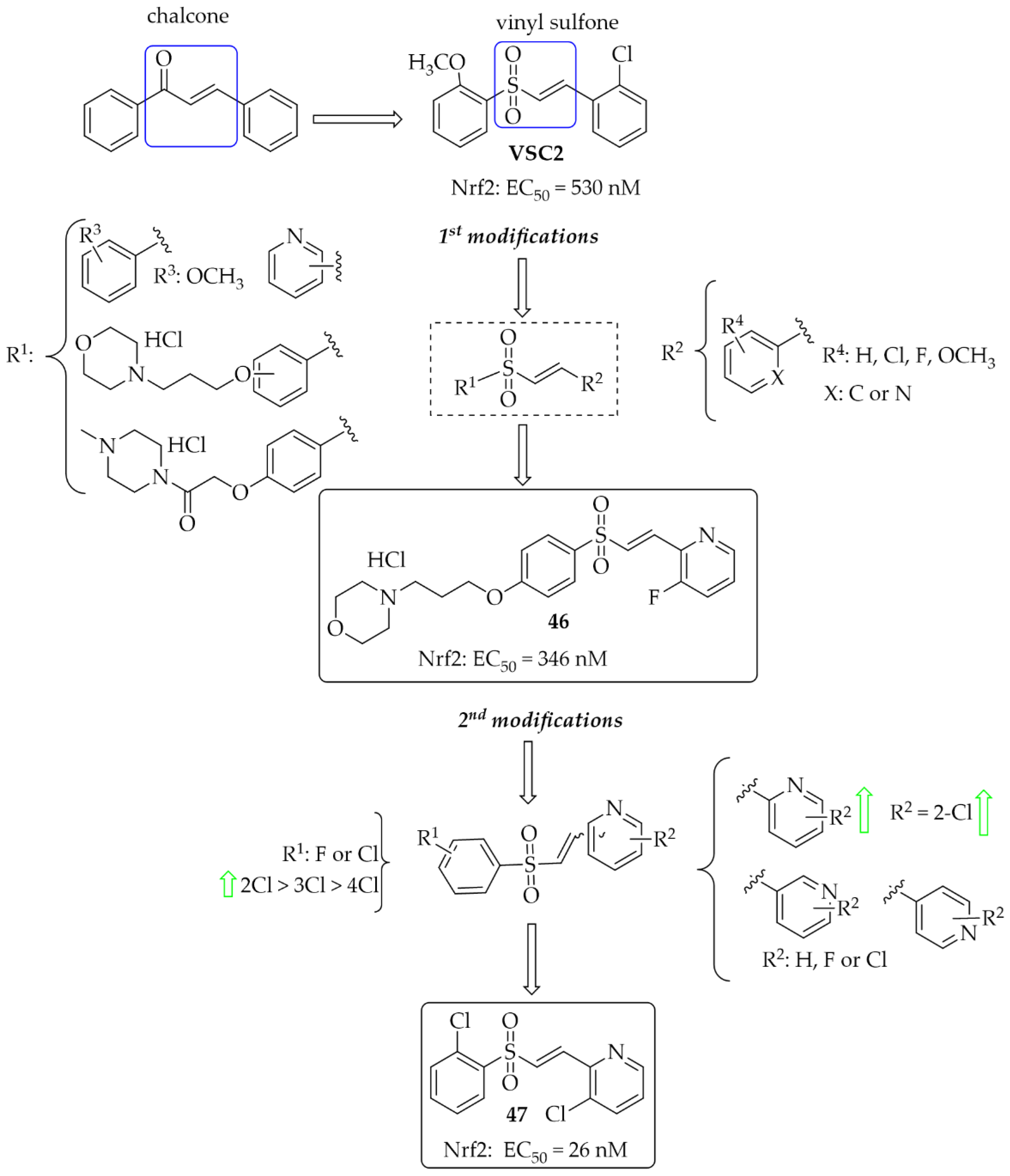
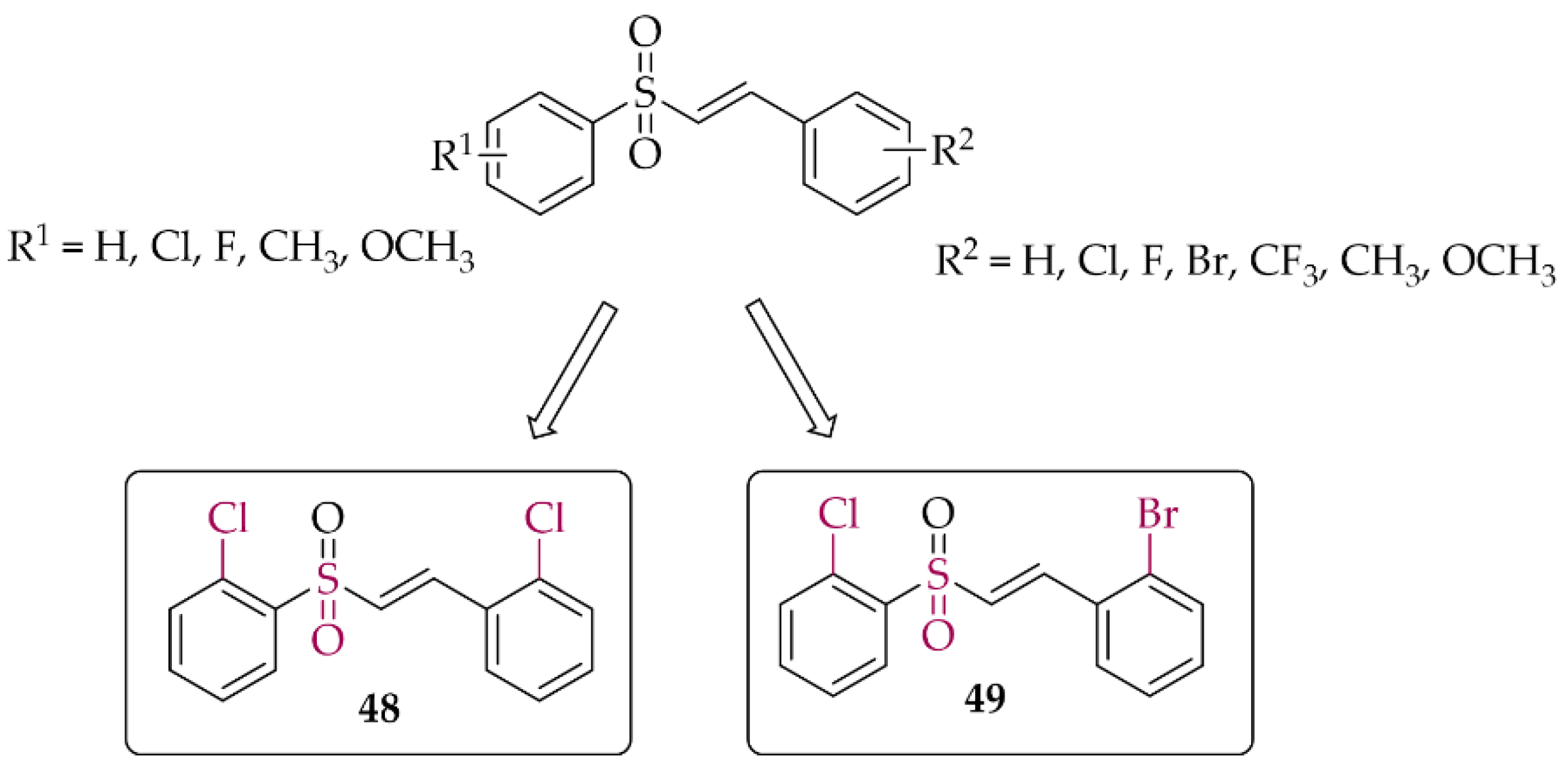
5.1.3. Chalcones Containing a Vinylsulfone Scaffold
5.2. Inducible Nitric Oxide Synthase Inhibitors
6. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| α-syn | alpha-synuclein |
| ACE | aldoxime chalcone ethers |
| ACh | acetylcholine |
| AChE | acetylcholinesterase |
| AMP | adenosine 5′-monophosphate |
| AMPK | AMP protein kinase |
| AR | adenosine receptors |
| ARE | antioxidant response elements |
| Aβ | beta-amyloid |
| A1R | adenosine A1 receptor |
| A1Rrat | adenosine A1 receptor rat whole brain membranes |
| A2AR | adenosine A2A receptor |
| A2ARrat | adenosine A2A receptor rat striatal membranes |
| BACE-1 | β-site amyloid precursor cleaving enzyme 1 |
| BBB | blood-brain barrier |
| BuChE | butyrylcholinesterase |
| eeBuChE | equine serum butyrylcholinesterase |
| CAT | catalase anti-oxidant markers |
| ChE | cholinesterase |
| ChEI | cholinesterase inhibitor |
| CNS | central nervous system |
| COMT | catechol-O-methyltransferase |
| COX-2 | cyclooxygenase-2 |
| CYP | cytochrome |
| DA | dopamine |
| D1R | dopamine D1 receptors |
| D2R | dopamine D2 receptors |
| ELISA | enzyme-linked immunosorbent assay |
| eeAChE | electric eel acetylcholinesterase |
| eNOS | endothelial NOS |
| GCase | glucocerebrosidase |
| GCL | glutamate cysteine ligase |
| GCLC | glutamate cysteine ligase catalytic subunit |
| GCLM | glutamate cysteine ligase modifier subunit |
| GTP | guanosine triphosphate |
| hMAO A | human monoamine oxidase A |
| hMAO B | human monoamine oxidase B |
| HO-1 | heme oxygenase-1 |
| H3R | histamine H3 receptor |
| iNOS | inducible nitric oxide synthases |
| IKK | ikappaB kinase |
| JNK | c-Jun N-terminal kinases |
| LB | Lewy bodies |
| LN | Lewy neurons |
| LPS | lipopolysaccharide |
| LRRK2 | leucine-rich repeat kinase 2 |
| MAO A | monoamine oxidase A |
| MAO B | monoamine oxidase B |
| MB-COMT | membrane-bound catechol-O-methyltransferase |
| MDA | malondialdehyde |
| MG | methylglyoxal |
| MM/GBSA | molecular mechanics with generalized Born and surface area solvation |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| rMAO A | rat brain mitochondria MAO A |
| rMAO B | rat brain mitochondria MAO B |
| ND | neurodegenerative disorders |
| NMDA | N-methyl-D-aspartate |
| NMS | non-motor symptoms |
| nNOS | neuronal nitric oxide synthases |
| NO | nitric oxide |
| NOS | nitric oxide synthases |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| PAMPA assay | parallel artificial membrane permeability assay |
| PARP1 | poly [ADP-ribose] polymerase 1 |
| PD | Parkinson’s disease |
| PDE10A | phosphodiesterase 10A |
| PD-MCI | Parkinson’s disease–mild cognitive impairment |
| PD-D | Parkinson’s disease dementia |
| PET | positron emission tomography |
| p-LIMK1 | phosphorylated LIM kinase 1 |
| PSMA1 | proteasome 20S subunit alpha 1 |
| PSMB5 | proteasome 20S subunit beta 5 |
| PSMB7 | proteasome 20S subunit beta 7 |
| PSMB8 | proteasome 20S subunit beta 8 |
| ROCK2 | pho-associated protein kinase 2 |
| ROS | reactive oxygen species |
| SN | substantia nigra |
| SOD | superoxide dismutase |
| ThT | thioflavin T |
References
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s Disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef]
- Jankovic, J.; Tan, E.K. Parkinson’s Disease: Etiopathogenesis and Treatment. J. Neurol. Neurosurg. Psychiatry 2020, 91, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Ntetsika, T.; Papathoma, P.E.; Markaki, I. Novel Targeted Therapies for Parkinson’s Disease. Mol. Med. 2021, 27, 17. [Google Scholar] [CrossRef]
- Nussbaum, R.L.; Ellis, C.E. Alzheimer’s Disease and Parkinson’s Disease. N. Engl. J. Med. 2003, 348, 1356–1364. [Google Scholar] [CrossRef] [Green Version]
- Sveinbjornsdottir, S. The Clinical Symptoms of Parkinson’s Disease. J. Neurochem. 2016, 139 (Suppl. S1), 318–324. [Google Scholar] [CrossRef] [Green Version]
- Aarsland, D.; Creese, B.; Politis, M.; Chaudhuri, K.R.; Ffytche, D.H.; Weintraub, D.; Ballard, C. Cognitive Decline in Parkinson Disease. Nat. Rev. Neurol. 2017, 13, 217–231. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Ghebremedhin, E.; Rüb, U.; Bratzke, H.; Del Tredici, K. Stages in the Development of Parkinson’s Disease-Related Pathology. Cell Tissue Res. 2004, 318, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA-J. Am. Med. Assoc. 2020, 323, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Ehrt, U.; Broich, K.; Larsen, J.P.; Ballard, C.; Aarsland, D. Use of Drugs with Anticholinergic Effect and Impact on Cognition in Parkinson’s Disease: A Cohort Study. J. Neurol. Neurosurg. Psychiatry 2010, 81, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Dungo, R.; Deeks, E.D. Istradefylline: First Global Approval. Drugs 2013, 73, 875–882. [Google Scholar] [CrossRef]
- Chen, J.F.; Cunha, R.A. The Belated US FDA Approval of the Adenosine A2A Receptor Antagonist Istradefylline for Treatment of Parkinson’s Disease. Purinergic Signal. 2020, 16, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Cheong, S.L.; Federico, S.; Spalluto, G.; Klotz, K.N.; Pastorin, G. The Current Status of Pharmacotherapy for the Treatment of Parkinson’s Disease: Transition from Single-Target to Multitarget Therapy. Drug Discov. Today 2019, 24, 1769–1783. [Google Scholar] [CrossRef] [PubMed]
- Rane, P.; Sarmah, D.; Bhute, S.; Kaur, H.; Goswami, A.; Kalia, K.; Borah, A.; Dave, K.R.; Sharma, N.; Bhattacharya, P. Novel Targets for Parkinson’s Disease: Addressing Different Therapeutic Paradigms and Conundrums. ACS Chem. Neurosci. 2019, 10, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Jasutkar, H.G.; Oh, S.E.; Mouradian, M.M. Therapeutics in the Pipeline Targeting α-Synuclein for Parkinson’s Disease. Pharmacol. Rev. 2022, 74, 207–237. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef]
- Singh, P.; Anand, A.; Kumar, V. Recent Developments in Biological Activities of Chalcones: A Mini Review. Eur. J. Med. Chem. 2014, 85, 758–777. [Google Scholar] [CrossRef]
- Ouyang, Y.; Li, J.; Chen, X.; Fu, X.; Sun, S.; Wu, Q. Chalcone Derivatives: Role in Anticancer Therapy. Biomolecules 2021, 11, 894. [Google Scholar] [CrossRef]
- Jasim, H.A.; Nahar, L.; Jasim, M.A.; Moore, S.A.; Ritchie, K.J.; Sarker, S.D.; Uversky, N. Chalcones: Synthetic Chemistry Follows Where Nature Leads. Biomolecules 2021, 11, 1203. [Google Scholar] [CrossRef]
- Thapa, P.; Upadhyay, S.P.; Suo, W.Z.; Singh, V.; Gurung, P.; Lee, E.S.; Sharma, R.; Sharma, M. Chalcone and Its Analogs: Therapeutic and Diagnostic Applications in Alzheimer’s Disease. Bioorg. Chem. 2021, 108, 104681. [Google Scholar] [CrossRef]
- de Freitas Silva, M.; Pruccoli, L.; Morroni, F.; Sita, G.; Seghetti, F.; Viegas, C.; Tarozzi, A. The Keap1/Nrf2-ARE Pathway as a Pharmacological Target for Chalcones. Molecules 2018, 23, 1803. [Google Scholar] [CrossRef] [Green Version]
- Mathew, B.; Mathew, G.E.; Uçar, G.; Baysal, I.; Suresh, J.; Vilapurathu, J.K.; Prakasan, A.; Suresh, J.K.; Thomas, A. Development of Fluorinated Methoxylated Chalcones as Selective Monoamine Oxidase-B Inhibitors: Synthesis, Biochemistry and Molecular Docking Studies. Bioorg. Chem. 2015, 62, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, H.; Santos, C.; Cavaleiro, J.; Silva, A. Chalcones as Versatile Synthons for the Synthesis of 5- and 6-Membered Nitrogen Heterocycles. Curr. Org. Chem. 2014, 18, 2750–2775. [Google Scholar] [CrossRef]
- Finberg, J.P.M. Inhibitors of MAO-B and COMT: Their Effects on Brain Dopamine Levels and Uses in Parkinson’s Disease. J. Neural Transm. (Vienna) 2019, 126, 433–448. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Arawaka, S.; Sato, H.; Sasaki, A.; Shigekiyo, T.; Takahata, K.; Tsunekawa, H.; Kato, T. Monoamine Oxidase-B Inhibition Facilitates a-Synuclein Secretion In Vitro and Delays Its Aggregation in RAAV-Based Rat Models of Parkinson’s Disease. J. Neurosci. 2021, 41, 7479–7491. [Google Scholar] [CrossRef]
- Sharma, P.; Singh, M.; Mathew, B. An Update of Synthetic Approaches and Structure-Activity Relationships of Various Classes of Human MAO-B Inhibitors. ChemistrySelect 2021, 6, 1404–1429. [Google Scholar] [CrossRef]
- Guglielmi, P.; Mathew, B.; Secci, D.; Carradori, S. Chalcones: Unearthing Their Therapeutic Possibility as Monoamine Oxidase B Inhibitors. Eur. J. Med. Chem. 2020, 205, 112650. [Google Scholar] [CrossRef]
- Iacovino, L.G.; Pinzi, L.; Facchetti, G.; Bortolini, B.; Christodoulou, M.S.; Binda, C.; Rastelli, G.; Rimoldi, I.; Passarella, D.; Di Paolo, M.L.; et al. Promising Non-Cytotoxic Monosubstituted Chalcones to Target Monoamine Oxidase-B. ACS Med. Chem. Lett. 2021, 12, 1151–1158. [Google Scholar] [CrossRef]
- Mathew, B.; Oh, J.M.; Khames, A.; Abdelgawad, M.A.; Rangarajan, T.M.; Nath, L.R.; Agoni, C.; Soliman, M.E.S.; Mathew, G.E.; Kim, H. Replacement of Chalcone-Ethers with Chalcone-Thioethers as Potent and Highly Selective Monoamine Oxidase-b Inhibitors and Their Protein-Ligand Interactions. Pharmaceuticals 2021, 14, 1148. [Google Scholar] [CrossRef]
- Parambi, D.G.T.; Oh, J.M.; Baek, S.C.; Lee, J.P.; Tondo, A.R.; Nicolotti, O.; Kim, H.; Mathew, B. Design, Synthesis and Biological Evaluation of Oxygenated Chalcones as Potent and Selective MAO-B Inhibitors. Bioorg. Chem. 2019, 93, 103335. [Google Scholar] [CrossRef]
- Parambi, D.G.T.; Saleem, U.; Shah, M.A.; Anwar, F.; Ahmad, B.; Manzar, A.; Itzaz, A.; Harilal, S.; Uddin, M.S.; Kim, H.; et al. Exploring the Therapeutic Potentials of Highly Selective Oxygenated Chalcone Based MAO-B Inhibitors in a Haloperidol-Induced Murine Model of Parkinson’s Disease. Neurochem. Res. 2020, 45, 2786–2799. [Google Scholar] [CrossRef]
- Kong, Z.; Sun, D.; Jiang, Y.; Hu, Y. Design, Synthesis, and Evaluation of 1, 4-Benzodioxan-Substituted Chalcones as Selective and Reversible Inhibitors of Human Monoamine Oxidase B. J. Enzyme Inhib. Med. Chem. 2020, 35, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- Moya-Alvarado, G.; Yañez, O.; Morales, N.; González-González, A.; Areche, C.; Núñez, M.T.; Fierro, A.; García-Beltrán, O. Coumarin-Chalcone Hybrids as Inhibitors of MAO-B: Biological Activity and in Silico Studies. Molecules 2021, 26, 2430. [Google Scholar] [CrossRef] [PubMed]
- Silva, T.B.; Borges, F.; Serrão, M.P.; Soares-da-Silva, P. Liver Says No: The Ongoing Search for Safe Catechol O-Methyltransferase Inhibitors to Replace Tolcapone. Drug Discov. Today 2020, 25, 1846–1854. [Google Scholar] [CrossRef] [PubMed]
- Müller, T. Catechol-O-Methyltransferase Inhibitors in Parkinson’s Disease. Drugs 2015, 75, 157–174. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, M.J.; Yar, M.S.; Grover, G.; Nath, R. Neurological and Psychiatric Management Using COMT Inhibitors: A Review. Bioorg. Chem. 2020, 94, 103418. [Google Scholar] [CrossRef] [PubMed]
- Kiss, L.E.; Soares-Da-Silva, P. Medicinal Chemistry of Catechol O-Methyltransferase (COMT) Inhibitors and Their Therapeutic Utility. J. Med. Chem. 2014, 57, 8692–8717. [Google Scholar] [CrossRef] [PubMed]
- de Beer, A.D.; Legoabe, L.J.; Petzer, A.; Petzer, J.P. The Inhibition of Catechol O-Methyltransferase and Monoamine Oxidase by Tetralone and Indanone Derivatives Substituted with the Nitrocatechol Moiety. Bioorg. Chem. 2021, 114, 105130. [Google Scholar] [CrossRef]
- Kitajima, T.; Mizote, S.; Bonifácio, M.J.; Umemura, T.; Yoneda, K.; Moser, P.; Soares-da-Silva, P.; Tanaka, M. Inhibition of Catechol-O-Methyltransferase in the Cynomolgus Monkey by Opicapone after Acute and Repeated Administration. Neuropharmacology 2018, 143, 282–288. [Google Scholar] [CrossRef]
- Hitge, R.; Smit, S.; Petzer, A.; Petzer, J.P. Evaluation of Nitrocatechol Chalcone and Pyrazoline Derivatives as Inhibitors of Catechol-O-Methyltransferase and Monoamine Oxidase. Bioorg. Med. Chem. Lett. 2020, 30, 127188. [Google Scholar] [CrossRef]
- Calabresi, P.; Picconi, B.; Parnetti, L.; Di Filippo, M. A Convergent Model for Cognitive Dysfunctions in Parkinson’s Disease: The Critical Dopamine–Acetylcholine Synaptic Balance. Lancet Neurol. 2006, 5, 974–983. [Google Scholar] [CrossRef]
- Sun, C.; Armstrong, M.J. Treatment of Parkinson’s Disease with Cognitive Impairment: Current Approaches and Future Directions. Behav. Sci. 2021, 11, 54. [Google Scholar] [CrossRef] [PubMed]
- Neumann, S.; Taylor, J.; Bamford, A.; Metcalfe, C.; Gaunt, D.M.; Whone, A.; Steeds, D.; Emmett, S.R.; Hollingworth, W.; Ben-Shlomo, Y.; et al. Cholinesterase Inhibitor to Prevent Falls in Parkinson’s Disease (CHIEF-PD) Trial: A Phase 3 Randomised, Double-Blind Placebo-Controlled Trial of Rivastigmine to Prevent Falls in Parkinson’s Disease. BMC Neurol. 2021, 21, 422. [Google Scholar] [CrossRef] [PubMed]
- Yamali, C.; Engin, F.S.; Bilginer, S.; Tugrak, M.; Ozmen Ozgun, D.; Ozli, G.; Levent, S.; Saglik, B.N.; Ozkay, Y.; Gul, H.I. Phenothiazine-Based Chalcones as Potential Dual-Target Inhibitors toward Cholinesterases (AChE, BuChE) and Monoamine Oxidases (MAO-A, MAO-B). J. Heterocycl. Chem. 2021, 58, 161–171. [Google Scholar] [CrossRef]
- Mathew, B.; Oh, J.M.; Baty, R.S.; Batiha, G.E.S.; Parambi, D.G.T.; Gambacorta, N.; Nicolotti, O.; Kim, H. Piperazine-Substituted Chalcones: A New Class of MAO-B, AChE, and BACE-1 Inhibitors for the Treatment of Neurological Disorders. Environ. Sci. Pollut. Res. Int. 2021, 28, 38855–38866. [Google Scholar] [CrossRef] [PubMed]
- Sasidharan, R.; Eom, B.H.; Heo, J.H.; Park, J.E.; Abdelgawad, M.A.; Musa, A.; Gambacorta, N.; Nicolotti, O.; Manju, S.L.; Mathew, B.; et al. Morpholine-Based Chalcones as Dual-Acting Monoamine Oxidase-B and Acetylcholinesterase Inhibitors: Synthesis and Biochemical Investigations. J. Enzyme Inhib. Med. Chem. 2021, 36, 188–197. [Google Scholar] [CrossRef]
- Oh, J.M.; Rangarajan, T.M.; Chaudhary, R.; Gambacorta, N.; Nicolotti, O.; Kumar, S.; Mathew, B.; Kim, H. Aldoxime- and Hydroxy-Functionalized Chalcones as Highly Potent and Selective Monoamine Oxidase-B Inhibitors. J. Mol. Struct. 2022, 1250, 131817. [Google Scholar] [CrossRef]
- Oh, J.M.; Rangarajan, T.M.; Chaudhary, R.; Singh, R.P.; Singh, M.; Singh, R.P.; Tondo, A.R.; Gambacorta, N.; Nicolotti, O.; Mathew, B.; et al. Novel Class of Chalcone Oxime Ethers as Potent Monoamine Oxidase-B and Acetylcholinesterase Inhibitors. Molecules 2020, 25, 2356. [Google Scholar] [CrossRef]
- Rehuman, N.A.; Oh, J.M.; Nath, L.R.; Khames, A.; Abdelgawad, M.A.; Gambacorta, N.; Nicolotti, O.; Jat, R.K.; Kim, H.; Mathew, B. Halogenated Coumarin-Chalcones as Multifunctional Monoamine Oxidase-B and Butyrylcholinesterase Inhibitors. ACS Omega 2021, 6, 28182–28193. [Google Scholar] [CrossRef]
- Freyssin, A.; Page, G.; Fauconneau, B.; Rioux Bilan, A. Natural Polyphenols Effects on Protein Aggregates in Alzheimer’s and Parkinson’s Prion-like Diseases. Neural Regen. Res. 2018, 13, 955–961. [Google Scholar] [CrossRef]
- Rocha, E.M.; De Miranda, B.; Sanders, L.H. Alpha-Synuclein: Pathology, Mitochondrial Dysfunction and Neuroinflammation in Parkinson’s Disease. Neurobiol. Dis. 2018, 109, 249–257. [Google Scholar] [CrossRef]
- Mehra, S.; Sahay, S.; Maji, S.K. α-Synuclein Misfolding and Aggregation: Implications in Parkinson’s Disease Pathogenesis. Biochim. Biophys. Acta-Proteins Proteom. 2019, 1867, 890–908. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.-M.; Ryan, P.; Rudrawar, S.; Quinn, R.J.; Zhang, H.-Y.; Mellick, G.D. Advances in the Development of Imaging Probes and Aggregation Inhibitors for Alpha-Synuclein. Acta Pharmacol. Sin. 2020, 41, 483–498. [Google Scholar] [CrossRef] [PubMed]
- Korat, Š.; Bidesi, N.S.R.; Bonanno, F.; Di Nanni, A.; Hoàng, A.N.N.; Herfert, K.; Maurer, A.; Battisti, U.M.; Bowden, G.D.; Thonon, D.; et al. Alpha-Synuclein PET Tracer Development-an Overview about Current Efforts. Pharmaceuticals 2021, 14, 847. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.-J.; Xu, K.; Lee, I.; Graham, T.J.A.; Tu, Z.; Dhavale, D.; Kotzbauer, P.; Mach, R.H. Chalcones and Five-Membered Heterocyclic Isosteres Bind to Alpha Synuclein Fibrils in Vitro. ACS Omega 2018, 3, 4486–4493. [Google Scholar] [CrossRef] [Green Version]
- Kaide, S.; Watanabe, H.; Iikuni, S.; Hasegawa, M.; Itoh, K.; Ono, M. Chalcone Analogue as New Candidate for Selective Detection of α-Synuclein Pathology. ACS Chem. Neurosci. 2022, 13, 16–26. [Google Scholar] [CrossRef]
- Ono, M.; Hori, M.; Haratake, M.; Tomiyama, T.; Mori, H.; Nakayama, M. Structure-Activity Relationship of Chalcones and Related Derivatives as Ligands for Detecting of β-Amyloid Plaques in the Brain. Bioorg. Med. Chem. 2007, 15, 6388–6396. [Google Scholar] [CrossRef]
- Caruana, M.; Högen, T.; Levin, J.; Hillmer, A.; Giese, A.; Vassallo, N. Inhibition and Disaggregation of α-Synuclein Oligomers by Natural Polyphenolic Compounds. FEBS Lett. 2011, 585, 1113–1120. [Google Scholar] [CrossRef] [Green Version]
- Padmavathi, G.; Roy, N.K.; Bordoloi, D.; Arfuso, F.; Mishra, S.; Sethi, G.; Bishayee, A.; Kunnumakkara, A.B. Butein in Health and Disease: A Comprehensive Review. Phytomedicine 2017, 25, 118–127. [Google Scholar] [CrossRef]
- Tinku; Paithankar, H.; Rane, A.R.; Hosur, R.V.; Choudhary, S. Title of the Manuscript: Mechanistic Insights into Chalcone Butein-Induced Inhibition of α-Synuclein Fibrillation: Biophysical and Insilico Studies. J. Mol. Liq. 2021, 334, 116105. [Google Scholar] [CrossRef]
- Saini, A.; Patel, R.; Gaba, S.; Singh, G.; Gupta, G.D.; Monga, V. Adenosine Receptor Antagonists: Recent Advances and Therapeutic Perspective. Eur. J. Med. Chem. 2022, 227, 113907. [Google Scholar] [CrossRef]
- Morelli, M.; Di Paolo, T.; Wardas, J.; Calon, F.; Xiao, D.; Schwarzschild, M.A. Role of Adenosine A2A Receptors in Parkinsonian Motor Impairment and L-DOPA-Induced Motor Complications. Prog. Neurobiol. 2007, 83, 293–309. [Google Scholar] [CrossRef] [PubMed]
- Pinna, A.; Serra, M.; Morelli, M.; Simola, N. Role of Adenosine A2A Receptors in Motor Control: Relevance to Parkinson’s Disease and Dyskinesia. J. Neural Transm. (Vienna) 2018, 125, 1273–1286. [Google Scholar] [CrossRef] [PubMed]
- Ferré, S.; Popoli, P.; Giménez-Llort, L.; Rimondini, R.; Müller, C.E.; Strömberg, I.; Ögren, S.O.; Fuxe, K. Adenosine/Dopamine Interaction: Implications for the Treatment of Parkinson’s Disease. Parkinsonism Relat. Disord. 2001, 7, 235–241. [Google Scholar] [CrossRef]
- Morin, N.; Di Paolo, T. Interaction of Adenosine Receptors with Other Receptors from Therapeutic Perspective in Parkinson’s Disease. Int. Rev. Neurobiol. 2014, 119, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Matthee, C.; Terre’Blanche, G.; Legoabe, L.J.; Janse van Rensburg, H.D. Exploration of Chalcones and Related Heterocycle Compounds as Ligands of Adenosine Receptors: Therapeutics Development. Mol. Divers. 2022, 26, 1779–1821. [Google Scholar] [CrossRef]
- Janse van Rensburg, H.D.; Legoabe, L.J.; Terre’Blanche, G. C3 Amino-Substituted Chalcone Derivative with Selective Adenosine RA1 Receptor Affinity in the Micromolar Range. Chem. Pap. 2021, 75, 1581–1605. [Google Scholar] [CrossRef]
- Janse van Rensburg, H.D.; Legoabe, L.J.; Terre’Blanche, G.; Aucamp, J. Synthesis and Evaluation of Methoxy Substituted 2-Benzoyl-1-Benzofuran Derivatives as Lead Compounds for the Development Adenosine A1 and/or A2A Receptor Antagonists. Bioorg. Chem. 2020, 94, 103459. [Google Scholar] [CrossRef]
- Janse Van Rensburg, H.D.; Legoabe, L.J.; Terre’Blanche, G.; Van Der Walt, M.M. Methoxy Substituted 2-Benzylidene-1-Indanone Derivatives as A1 and/or A2A AR Antagonists for the Potential Treatment of Neurological Conditions. Medchemcomm 2019, 10, 300–309. [Google Scholar] [CrossRef]
- Janse Van Rensburg, H.D.; Legoabe, L.J.; Terre’Blanche, G. Synthesis and Structure Activity Relationships of Chalcone Based Benzocycloalkanone Derivatives as Adenosine A1 and/or A2A Receptor Antagonists. Drug Res. (Stuttg) 2020, 70, 243–256. [Google Scholar] [CrossRef]
- Vazquez-Rodriguez, S.; Vilar, S.; Kachler, S.; Klotz, K.N.; Uriarte, E.; Borges, F.; Matos, M.J. Adenosine Receptor Ligands: Coumarin–Chalcone Hybrids as Modulating Agents on the Activity of HARs. Molecules 2020, 25, 4306. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s Disease and Its Potential as Therapeutic Target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Zhu, G.; Wang, G.; Zhang, F. Oxidative Stress and Neuroinflammation Potentiate Each Other to Promote Progression of Dopamine Neurodegeneration. Oxid. Med. Cell. Longev. 2020, 2020, 6137521. [Google Scholar] [CrossRef] [PubMed]
- Cobb, C.A.; Cole, M.P. Oxidative and Nitrative Stress in Neurodegeneration. Neurobiol. Dis. 2015, 84, 4–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dionísio, P.A.; Amaral, J.D.; Rodrigues, C.M.P. Oxidative Stress and Regulated Cell Death in Parkinson’s Disease. Ageing Res. Rev. 2021, 67, 101263. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Kazantsev, A.G. The Role of Nrf2 Signaling in Counteracting Neurodegenerative Diseases. FEBS J. 2018, 285, 3576–3590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magesh, S.; Chen, Y.; Hu, L. Small Molecule Modulators of Keap1-Nrf2-ARE Pathway as Potential Preventive and Therapeutic Agents. Med. Res. Rev. 2012, 32, 687–726. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.A.; Son, H.J.; Choi, J.W.; Kim, J.; Han, S.H.; Shin, N.; Kim, J.H.; Kim, S.J.; Heo, J.Y.; Kim, D.J.; et al. Activation of the Nrf2 Signaling Pathway and Neuroprotection of Nigral Dopaminergic Neurons by a Novel Synthetic Compound KMS99220. Neurochem. Int. 2018, 112, 96–107. [Google Scholar] [CrossRef]
- Lee, J.A.; Kim, H.R.; Kim, J.; Park, K.D.; Kim, D.J.; Hwang, O. The Novel Neuroprotective Compound KMS99220 Has an Early Anti-Neuroinflammatory Effect via AMPK and HO-1, Independent of Nrf2. Exp. Neurobiol. 2018, 27, 408–418. [Google Scholar] [CrossRef]
- Lee, J.A.; Kim, D.J.; Hwang, O. KMS99220 Exerts Anti-Inflammatory Effects, Activates the Nrf2 Signaling and Interferes with IKK, JNK and P38 MAPK via HO-1. Mol. Cells 2019, 42, 702–710. [Google Scholar] [CrossRef]
- Chen, Y.-F.; Wu, S.-N.; Gao, J.-M.; Liao, Z.-Y.; Tseng, Y.-T.; Fülöp, F.; Chang, F.-R.; Lo, Y.-C. The Antioxidant, Anti-Inflammatory, and Neuroprotective Properties of the Synthetic Chalcone Derivative AN07. Molecules 2020, 25, 2907. [Google Scholar] [CrossRef]
- Woo, S.Y.; Kim, J.H.; Moon, M.K.; Han, S.H.; Yeon, S.K.; Choi, J.W.; Jang, B.K.; Song, H.J.; Kang, Y.G.; Kim, J.W.; et al. Discovery of Vinyl Sulfones as a Novel Class of Neuroprotective Agents toward Parkinson’s Disease Therapy. J. Med. Chem. 2014, 57, 1473–1487. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Kim, S.; Park, J.H.; Kim, H.J.; Shin, S.J.; Kim, J.W.; Woo, S.Y.; Lee, C.; Han, S.M.; Lee, J.; et al. Optimization of Vinyl Sulfone Derivatives as Potent Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2) Activators for Parkinson’s Disease Therapy. J. Med. Chem. 2019, 62, 811–830. [Google Scholar] [CrossRef]
- Choi, J.W.; Kim, S.; Yoo, J.S.; Kim, H.J.; Kim, H.J.; Kim, B.E.; Lee, E.H.; Lee, Y.S.; Park, J.H.; Park, K.D. Development and Optimization of Halogenated Vinyl Sulfones as Nrf2 Activators for the Treatment of Parkinson’s Disease. Eur. J. Med. Chem. 2021, 212, 113103. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.L.; Hou, Y.; Bai, F.; Fang, J. Generation of Potent Nrf2 Activators via Tuning the Electrophilicity and Steric Hindrance of Vinyl Sulfones for Neuroprotection. Bioorg. Chem. 2021, 107, 104520. [Google Scholar] [CrossRef] [PubMed]
- Minhas, R.; Bansal, Y.; Bansal, G. Inducible Nitric Oxide Synthase Inhibitors: A Comprehensive Update. Med. Res. Rev. 2020, 40, 823–855. [Google Scholar] [CrossRef] [PubMed]
- Rojas, J.; Payá, M.; Dominguez, J.N.; Luisa Ferrándiz, M. The Synthesis and Effect of Fluorinated Chalcone Derivatives on Nitric Oxide Production. Bioorg. Med. Chem. Lett. 2002, 12, 1951–1954. [Google Scholar] [CrossRef]
- Rojas, J.; Payá, M.; Devesa, I.; Dominguez, J.N.; Ferrándiz, M.L. Therapeutic Administration of 3,4,5-Trimethoxy-4′-Fluorochalcone, a Selective Inhibitor of INOS Expression, Attenuates the Development of Adjuvant-Induced Arthritis in Rats. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2003, 368, 225–233. [Google Scholar] [CrossRef]
- Waku, I.; Magalhães, M.S.; Alves, C.O.; de Oliveira, A.R. Haloperidol-induced catalepsy as an animal model for parkinsonism: A systematic review of experimental studies. Eur. J. Neurosci. 2021, 53, 3743–3767. [Google Scholar] [CrossRef]
- Meredith, G.E.; Rademacher, D.J. MPTP mouse models of Parkinson’s disease: An update. J Parkinsons Dis. 2011, 1, 19–33. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Królicka, E.; Kieć-Kononowicz, K.; Łażewska, D. Chalcones as Potential Ligands for the Treatment of Parkinson’s Disease. Pharmaceuticals 2022, 15, 847. https://doi.org/10.3390/ph15070847
Królicka E, Kieć-Kononowicz K, Łażewska D. Chalcones as Potential Ligands for the Treatment of Parkinson’s Disease. Pharmaceuticals. 2022; 15(7):847. https://doi.org/10.3390/ph15070847
Chicago/Turabian StyleKrólicka, Ewelina, Katarzyna Kieć-Kononowicz, and Dorota Łażewska. 2022. "Chalcones as Potential Ligands for the Treatment of Parkinson’s Disease" Pharmaceuticals 15, no. 7: 847. https://doi.org/10.3390/ph15070847
APA StyleKrólicka, E., Kieć-Kononowicz, K., & Łażewska, D. (2022). Chalcones as Potential Ligands for the Treatment of Parkinson’s Disease. Pharmaceuticals, 15(7), 847. https://doi.org/10.3390/ph15070847