
Na+/H+ Exchanger 1, a Potential Therapeutic Drug Target for Cardiac Hypertrophy and Heart Failure

 , and
, and 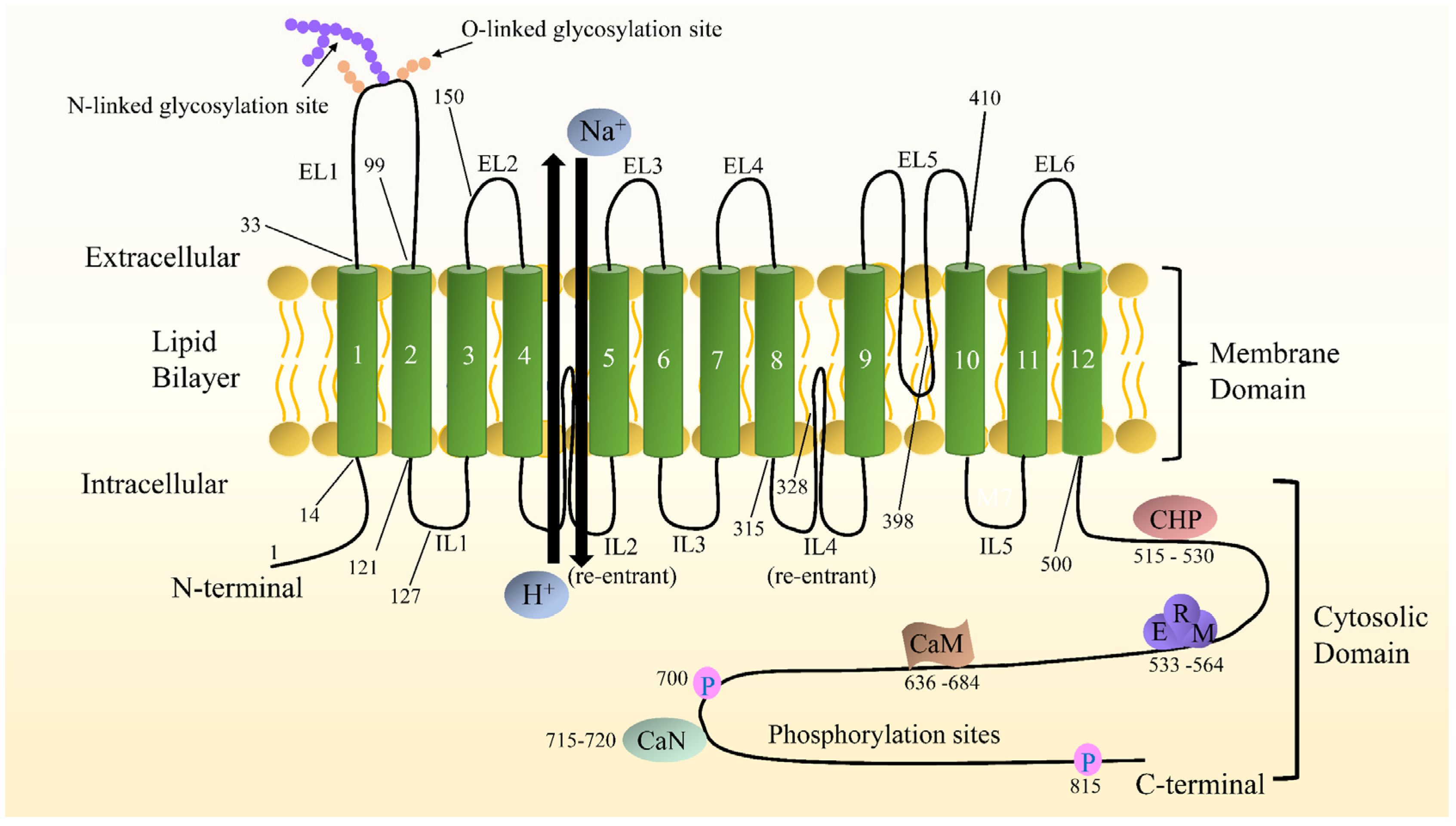{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Distribution and Structure of NHE1
2.1. NHE1 Distribution
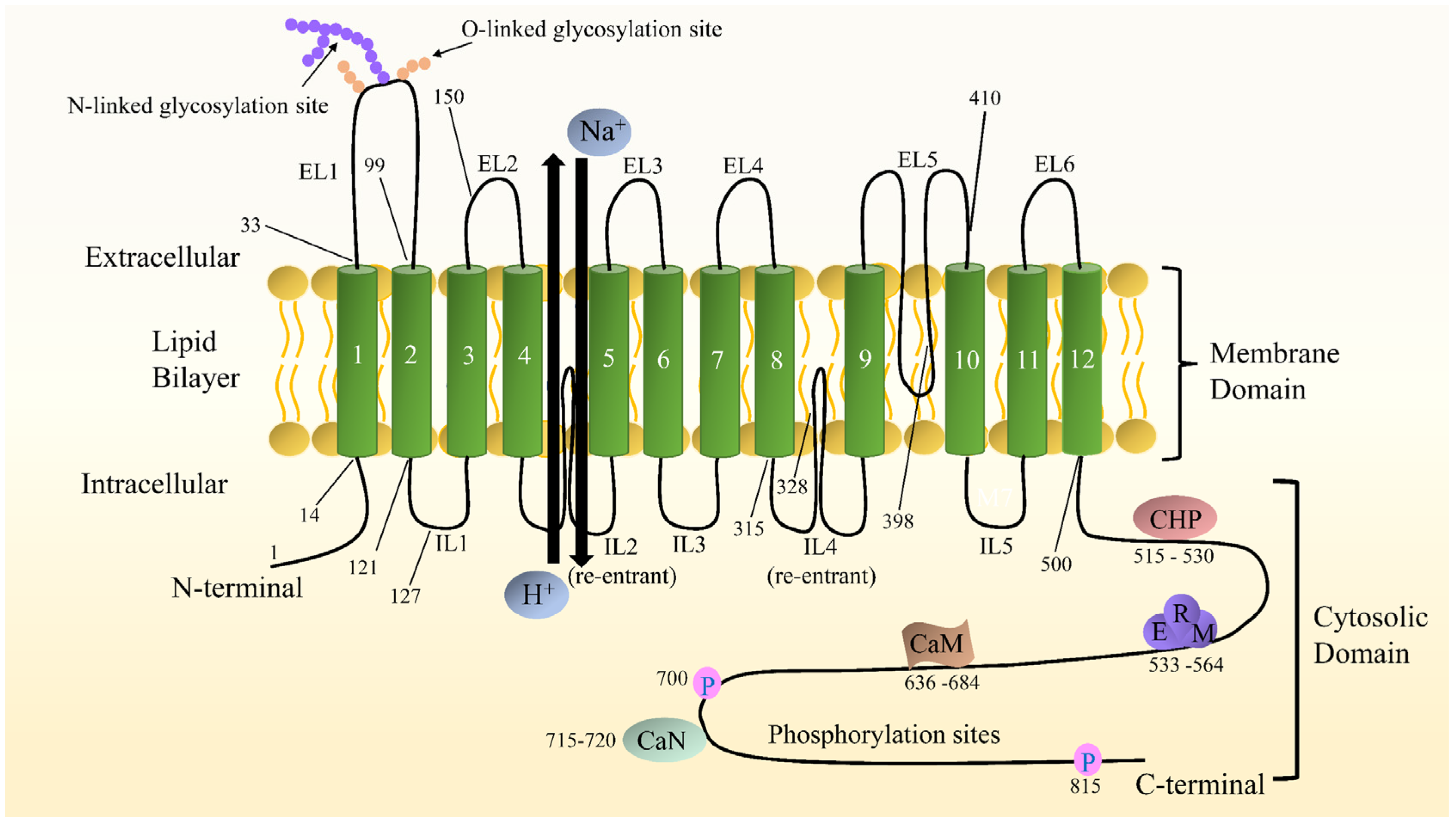
2.2. NHE1 Structure
3. NHE1 in Cardiac Physiology Regulation
4. NHE1 and Myocardial Ischemia-Reperfusion Injury (MIRI)
4.1. Mechanisms of MIRI
4.2. Role of NHE1 in MIRI
5. NHE1 in Cardiac Hypertrophy and Heart Failure
5.1. Experimental Evidence for NHE1 Involvement in Cardiac Hypertrophy
5.2. Experimental Evidence for NHE1 Involvement in Heart Failure
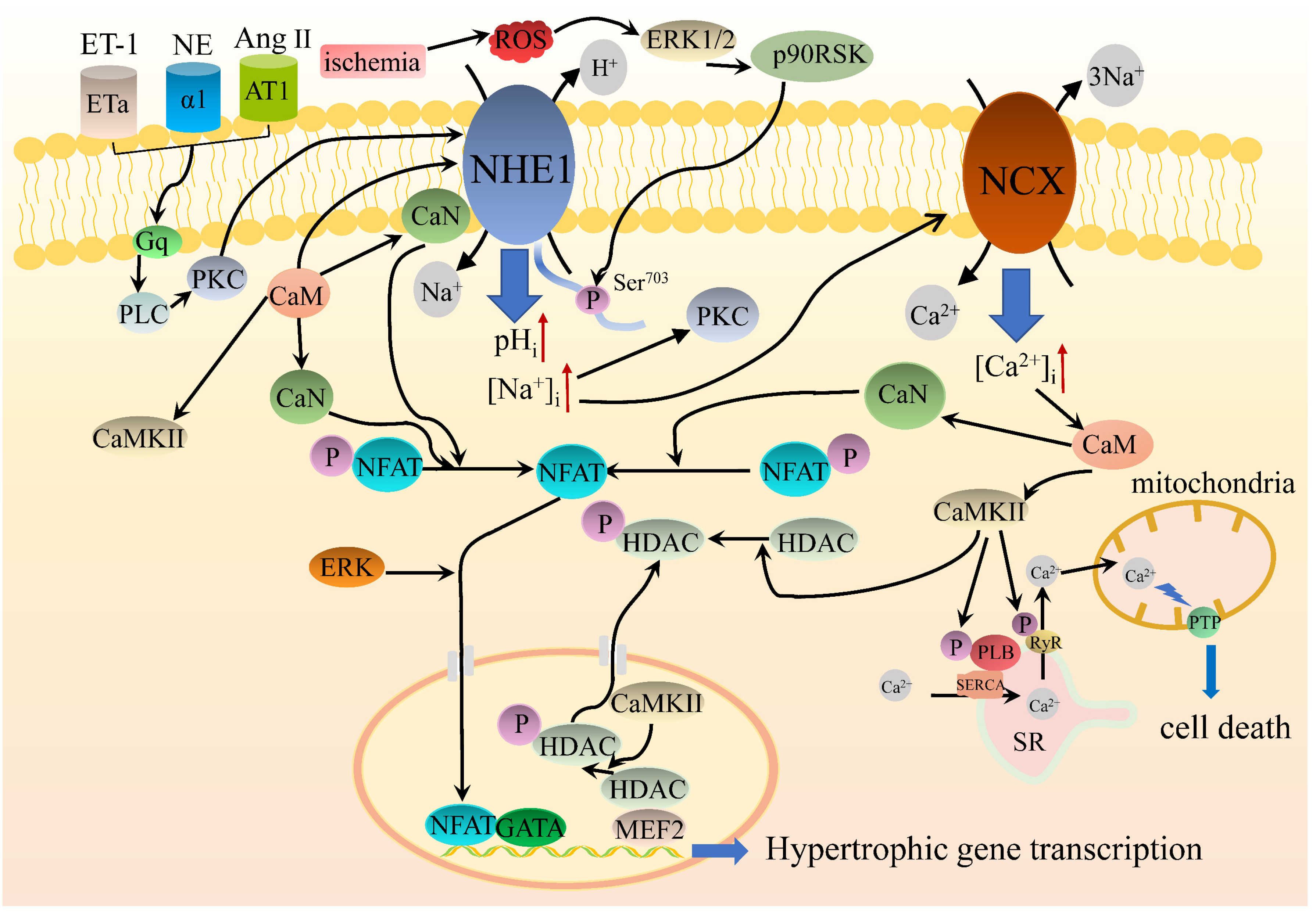
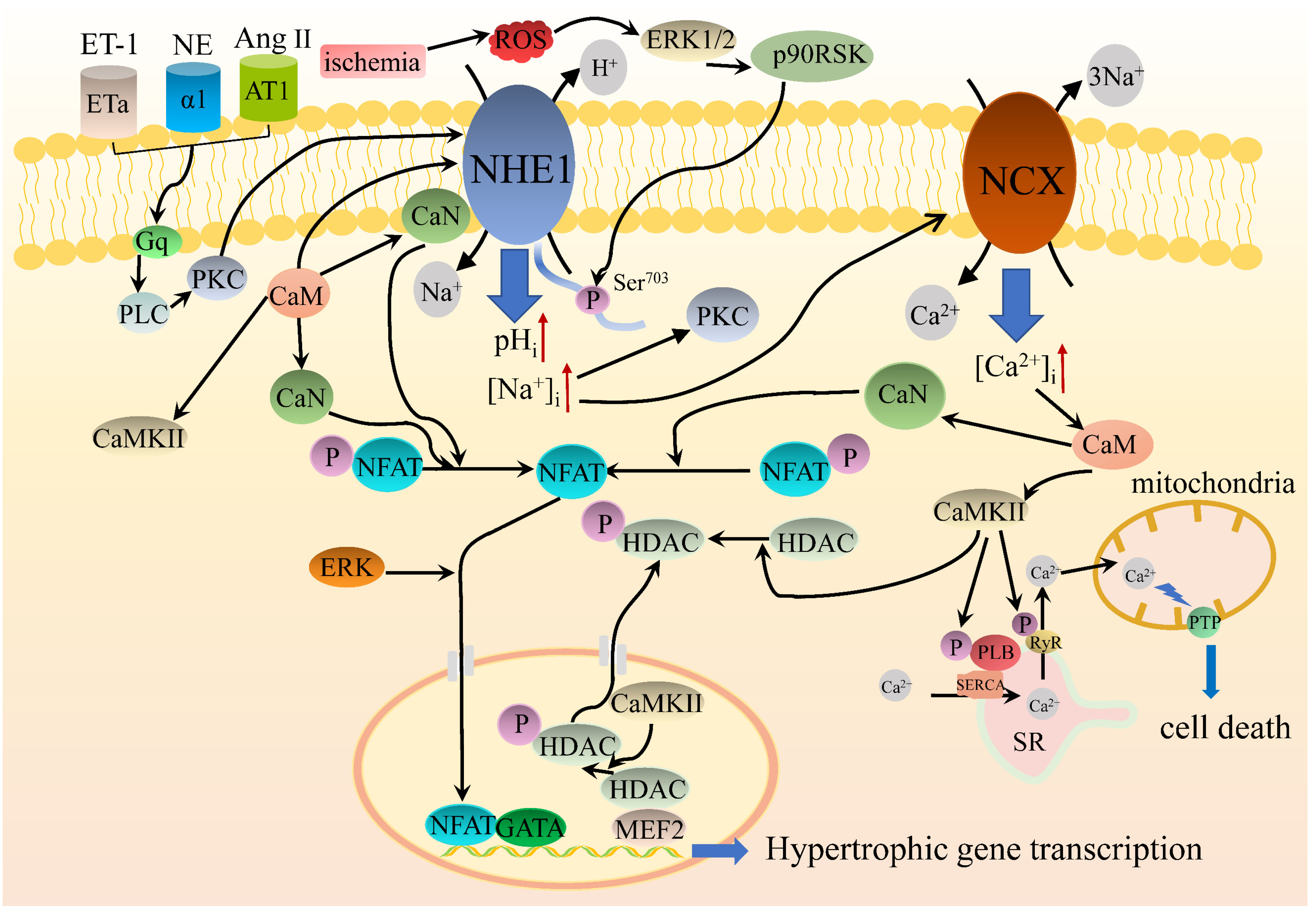
5.3. NHE1 Intracellular Signaling Regulates Cardiac Hypertrophy
6. Left Ventricular Hypertrophy (LVH) Leaves the Myocardium Susceptible to ISCHEMIA-Reperfusion Injury
7. Clinical Perspectives
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tham, Y.K.; Bernardo, B.C.; Ooi, J.Y.; Weeks, K.L.; McMullen, J.R. Pathophysiology of cardiac hypertrophy and heart failure: Signaling pathways and novel therapeutic targets. Arch. Toxicol. 2015, 89, 1401–1438. [Google Scholar] [CrossRef] [PubMed]
- Yeves, A.M.; Ennis, I.L. Na+/H+ exchanger and cardiac hypertrophy. Hipertens. Riesgo. Vasc. 2020, 37, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.M.; Park, M.S.; Youn, J.C. Update on heart failure management and future directions. Korean J. Intern. Med. 2019, 34, 11–43. [Google Scholar] [CrossRef] [PubMed]
- Di Palo, K.E.; Barone, N.J. Hypertension and Heart Failure. Heart Fail. Clin. 2020, 16, 99–106. [Google Scholar] [CrossRef]
- Hammond, G.; Rich, M.W. Hypertensive Heart Failure in the Very Old. Heart. Fail. Clin. 2019, 15, 477–485. [Google Scholar] [CrossRef]
- Agra Bermejo, R.M.; Gonzalez Ferreiro, R.; Varela Roman, A.; Gomez Otero, I.; Kreidieh, O.; Conde Sabaris, P.; Rodriguez-Manero, M.; Moure Gonzalez, M.; Seoane Blanco, A.; Virgos Lamela, A.; et al. Nutritional status is related to heart failure severity and hospital readmissions in acute heart failure. Int. J. Cardiol. 2017, 230, 108–114. [Google Scholar] [CrossRef]
- Yasuhara, S.; Maekawa, M.; Bamba, S.; Kurihara, M.; Nakanishi, N.; Yamamoto, T.; Sakai, H.; Yagi, N.; Nakagawa, Y.; Sasaki, M. Energy Metabolism and Nutritional Status in Hospitalized Patients with Chronic Heart Failure. Ann. Nutr. Metab. 2020, 76, 129–139. [Google Scholar] [CrossRef]
- von Haehling, S.; Doehner, W.; Anker, S.D. Nutrition, metabolism, and the complex pathophysiology of cachexia in chronic heart failure. Cardiovasc. Res. 2007, 73, 298–309. [Google Scholar] [CrossRef]
- Giovannini, S.; Onder, G.; Lattanzio, F.; Bustacchini, S.; Di Stefano, G.; Moresi, R.; Russo, A.; Bernabei, R.; Landi, F. Selenium concentrations and mortality among community-dwelling older adults: Results from ilSIRENTE. J. Nutr. Health Aging 2018, 22, 608–612. [Google Scholar] [CrossRef]
- Krishnaswami, A.; Steinman, M.A.; Goyal, P.; Zullo, A.R.; Anderson, T.S.; Birtcher, K.K.; Goodlin, S.J.; Maurer, M.S.; Alexander, K.P.; Rich, M.W.; et al. Deprescribing in Older Adults With Cardiovascular Disease. J. Am. Coll. Cardiol. 2019, 73, 2584–2595. [Google Scholar] [CrossRef]
- Georgiev, K.D.; Hvarchanova, N.; Georgieva, M.; Kanazirev, B. The role of the clinical pharmacist in the prevention of potential drug interactions in geriatric heart failure patients. Int. J. Clin. Pharm. 2019, 41, 1555–1561. [Google Scholar] [CrossRef] [PubMed]
- Tanai, E.; Frantz, S. Pathophysiology of Heart Failure. Compr. Physiol. 2015, 6, 187–214. [Google Scholar] [CrossRef] [PubMed]
- Basso, C.; Thiene, G. The pathophysiology of myocardial reperfusion: A pathologist’s perspective. Heart 2006, 92, 1559–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Ischemia/Reperfusion. Compr. Physiol. 2016, 7, 113–170. [Google Scholar] [CrossRef] [Green Version]
- Wan Ab Naim, W.N.; Mohamed Mokhtarudin, M.J.; Chan, B.T.; Lim, E.; Ahmad Bakir, A.; Nik Mohamed, N.A. The study of myocardial ischemia-reperfusion treatment through computational modelling. J. Theor. Biol. 2021, 509, 110527. [Google Scholar] [CrossRef]
- Avkiran, M.; Marber, M.S. Na+/H+ exchange inhibitors for cardioprotective therapy: Progress, problems and prospects. J. Am. Coll. Cardiol. 2002, 39, 747–753. [Google Scholar] [CrossRef]
- Gurney, M.A.; Laubitz, D.; Ghishan, F.K.; Kiela, P.R. Pathophysiology of intestinal Na+/H+ exchange. Cell Mol. Gastroenterol. Hepatol. 2017, 3, 27–40. [Google Scholar] [CrossRef] [Green Version]
- Escudero, D.S.; Perez, N.G.; Diaz, R.G. Myocardial Impact of NHE1 Regulation by Sildenafil. Front. Cardiovasc. Med. 2021, 8, 617519. [Google Scholar] [CrossRef]
- Mithell, P.; Moyle, J. Acid-Base titration across the membrane system of rat-liver mitochondria. Biochem. J. 1967, 104, 588–600. [Google Scholar] [CrossRef] [Green Version]
- Parker, M.D.; Myers, E.J.; Schelling, J.R. Na+-H+ exchanger-1 (NHE1) regulation in kidney proximal tubule. Cell Mol. Life Sci. 2015, 72, 2061–2074. [Google Scholar] [CrossRef] [Green Version]
- Donowitz, M.; Ming Tse, C.; Fuster, D. SLC9/NHE gene family, a plasma membrane and organellar family of Na+/H+ exchangers. Mol. Asp. Med. 2013, 34, 236–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendus-Altenburger, R.; Kragelund, B.B.; Pedersen, S.F. Structural dynamics and regulation of the mammalian SLC9A family of Na+/H+ exchangers. Curr. Top. Membr. 2014, 73, 69–148. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kim, T.; Park, E.S.; Yang, S.; Jeong, D.; Choi, Y.; Rho, J. NHE10, an osteoclast-specific member of the Na+/H+ exchanger family, regulates osteoclast differentiation and survival. Biochem. Biophys. Res. Commun. 2008, 369, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Fliegel, L. Structural and functional changes in the Na+/H+ exchanger isoform 1, induced by Erk1/2 phosphorylation. Int. J. Mol. Sci. 2019, 20, 2378. [Google Scholar] [CrossRef] [Green Version]
- Sardet, C.; Franchi, A.; Pouysségur, J. Molecular cloning, primary structure, and expression of the human growth factor-activatable Na+/H+ antiporter. Cell 1989, 56, 271–280. [Google Scholar] [CrossRef]
- Shrode, L.D.; Gan, B.S.; D’Souza, S.J.; Orlowski, J.; Grinstein, S. Topological analysis of NHE1, the ubiquitous Na+/H+ exchanger using chymotryptic cleavage. Am. J. Physiol. Cell Physiol. 1998, 275, C431–C439. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, S.; Pang, T.; Su, X.; Shigekawa, M. A novel topology model of the human Na+/H+ exchanger isoform 1. J. Biol. Chem. 2000, 275, 7942–7949. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Tuo, B. Pathophysiology of hepatic Na+/H+ exchange (Review). Exp. Ther. Med. 2020, 20, 1220–1229. [Google Scholar] [CrossRef]
- Dutta, D.; Fliegel, L. Molecular modeling and inhibitor docking analysis of the Na+/H+ exchanger isoform one. Biochem. Cell Biol. 2019, 97, 333–343. [Google Scholar] [CrossRef] [Green Version]
- Landau, M.; Herz, K.; Padan, E.; Ben-Tal, N. Model structure of the Na+/H+ exchanger 1 (NHE1): Functional and clinical implications. J. Biol. Chem. 2007, 282, 37854–37863. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.L.; Sykes, B.D.; Fliegel, L. Structural analysis of the Na+/H+ exchanger isoform 1 (NHE1) using the divide and conquer approach. Biochem. Cell Biol. 2011, 89, 189–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Basu, A.; Li, X.; Fliegel, L. Topological analysis of the Na+/H+ exchanger. Biochim. Biophys. Acta 2015, 1848, 2385–2393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fliegel, L. Role of genetic mutations of the Na+/H+ exchanger isoform 1, in human isease and protein targeting and activity. Mol. Cell Biochem. 2021, 476, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, J.; Grinstein, S. Diversity of the mammalian sodium/proton exchanger SLC9 gene family. Pflug. Arch. 2004, 447, 549–565. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, J.; Grinstein, S. Na+/H+ exchangers. Compr. Physiol. 2011, 1, 2083–2100. [Google Scholar] [CrossRef]
- Alexander, R.T.; Grinstein, S. Na+/H+ exchangers and the regulation of volume. Acta Physiol. 2006, 187, 159–167. [Google Scholar] [CrossRef]
- Hoffmann, E.K.; Pedersen, S.F. Cell volume homeostatic mechanisms: Effectors and signalling pathways. Acta Physiol. 2011, 202, 465–485. [Google Scholar] [CrossRef]
- Putney, L.K.; Barber, D.L. Expression profile of genes regulated by activity of the Na-H exchanger NHE1. BMC Genom. 2004, 5, 46. [Google Scholar] [CrossRef] [Green Version]
- Putney, L.K.; Denker, S.P.; Barber, D.L. The changing face of the Na+/H+ exchanger, NHE1: Structure, regulation, and cell actions. Annu. Rev. Pharmacol. Toxicol. 1999, 42, 527–552. [Google Scholar] [CrossRef]
- Fliegel, L. The Na+/H+ exchanger isoform 1. Int. J. Biochem. Cell Biol. 2005, 37, 33–37. [Google Scholar] [CrossRef]
- Pedersen, S.F. The Na+/H+ exchanger NHE1 in stress-induced signal transduction: Implications for cell proliferation and cell death. Pflug. Arch. 2006, 452, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Schelling, J.R.; Abu Jawdeh, B.G. Regulation of cell survival by Na+/H+ exchanger-1. Am. J. Physiol. Ren. Physiol. 2008, 295, F625–F632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, S.F.; Counillon, L. The SLC9A-C mammalian Na+/H+ exchanger family: Molecules, mechanisms, and physiology. Physiol. Rev. 2019, 99, 2015–2113. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Karki, P.; Lei, L.; Wang, H.; Fliegel, L. Na+/H+ exchanger isoform 1 facilitates cardiomyocyte embryonic stem cell differentiation. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H159–H170. [Google Scholar] [CrossRef] [PubMed]
- Malo, M.E.; Fliegel, L. Physiological role and regulation of the Na+/H+ exchanger. Can. J. Physiol. Pharmacol. 2006, 84, 1081–1095. [Google Scholar] [CrossRef] [Green Version]
- Martins, G.F.; Martins, G. Role of trimetazidine in coronary artery bypass graft surgery. World J. Cardiovasc. Surg. 2017, 07, 87–102. [Google Scholar] [CrossRef] [Green Version]
- Frank, A.; Bonney, M.; Bonney, S.; Weitzel, L.; Koeppen, M.; Eckle, T. Myocardial ischemia reperfusion injury: From basic science to clinical bedside. Semin. Cardiothorac. Vasc. Anesth. 2012, 16, 123–132. [Google Scholar] [CrossRef]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar] [CrossRef] [Green Version]
- Rout, A.; Tantry, U.S.; Novakovic, M.; Sukhi, A.; Gurbel, P.A. Targeted pharmacotherapy for ischemia reperfusion injury in acute myocardial infarction. Expert Opin. Pharm. 2020, 21, 1851–1865. [Google Scholar] [CrossRef]
- Wu, M.Y.; Yiang, G.T.; Liao, W.T.; Tsai, A.P.; Cheng, Y.L.; Cheng, P.W.; Li, C.Y.; Li, C.J. Current mechanistic concepts in ischemia and reperfusion injury. Cell Physiol. Biochem. 2018, 46, 1650–1667. [Google Scholar] [CrossRef]
- Fischesser, D.M.; Bo, B.; Benton, R.P.; Su, H.; Jahanpanah, N.; Haworth, K.J. Controlling reperfusion injury with controlled reperfusion: Historical perspectives and new paradigms. J. Cardiovasc. Pharmacol. Ther. 2021, 26, 504–523. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Jia, M.; Wang, Q.; Guo, Y.; Li, C.; Ren, B.; Qian, F.; Wu, J. The electrogenic sodium bicarbonate cotransporter and its roles in the myocardial ischemia-reperfusion induced cardiac diseases. Life Sci. 2021, 270, 119153. [Google Scholar] [CrossRef] [PubMed]
- Fliegel, L. Regulation of the Na+/H+ exchanger in the healthy and diseased myocardium. Expert Opin. Ther. Targets 2009, 13, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Karmazyn, M. Pharmacology and clinical assessment of cariporide for the treatment coronary artery diseases. Exp. Opin. Investig. Drugs 2000, 9, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Rabkin, D.G.; Cabreriza, S.E.; Cheema, F.H.; Hill, A.A.; Curtis, L.J.; Sciacca, R.R.; Mosca, R.S.; Spotnitz, H.M. Cariporide is cardioprotective after iatrogenic ventricular fibrillation in the intact swine heart. Ann. Thorac. Surg. 2003, 76, 1264–1269. [Google Scholar] [CrossRef]
- Wang, Y.; Meyer, J.W.; Ashraf, M.; Shull, G.E. Mice with a null mutation in the NHE1 Na+-H+ exchanger are resistant to cardiac ischemia-reperfusion injury. Circ. Res. 2003, 93, 776–782. [Google Scholar] [CrossRef] [Green Version]
- Murphy, E.; Allen, D.G. Why did the NHE inhibitor clinical trials fail? J. Mol. Cell. Cardiol. 2009, 46, 137–141. [Google Scholar] [CrossRef] [Green Version]
- Gazmuri, R.J.; Radhakrishnan, J.; Ayoub, I.M. Sodium-hydrogen exchanger isoform-1 inhibition: A promising pharmacological intervention for resuscitation from cardiac arrest. Molecules 2019, 24, 1765. [Google Scholar] [CrossRef] [Green Version]
- Klein, H.H.; Pich, S.; Bohle, R.M.; Lindert-Heimberg, S.; Nebendahl, K. Na+/H+ exchange inhibitor cariporide attenuates cell injury predominantly during ischemia and not at onset of reperfusion in porcine hearts with low residual blood flow. Circulation 2000, 102, 1977–1982. [Google Scholar] [CrossRef] [Green Version]
- Rupprecht, H.J.; Dahl, J.V.; Terres, W.; Seyfarth, K.M.; Richardt, G.; Schultheiβ, H.P.; Sheehan, F.H.; Drexler, H. Cardioprotective effects of the Na+/H+ exchange inhibitor cariporide in patients with acute anterior myocardial infarction undergoing direct PTCA. Circulation 2000, 101, 2902–2908. [Google Scholar] [CrossRef] [Green Version]
- Boyce, S.W.; Bartels, C.; Bolli, R.; Chaitman, B.; Chen, J.C.; Chi, E.; Jessel, A.; Kereiakes, D.; Knight, J.; Thulin, L.; et al. Impact of sodium-hydrogen exchange inhibition by cariporide on death or myocardial infarction in high-risk CABG surgery patients: Results of the CABG surgery cohort of the GUARDIAN study. J. Thorac. Cardiov. Surg. 2003, 126, 420–427. [Google Scholar] [CrossRef] [Green Version]
- Mentzer, R.M., Jr.; Bartels, C.; Bolli, R.; Boyce, S.; Buckberg, G.D.; Chaitman, B.; Haverich, A.; Knight, J.; Menasche, P.; Myers, M.L.; et al. Sodium-hydrogen exchange inhibition by cariporide to reduce the risk of ischemic cardiac events in patients undergoing coronary artery bypass grafting: Results of the EXPEDITION study. Ann. Thorac. Surg. 2008, 85, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Catalucci, D.; Latronico, M.V.G.; Ellingsen, O.; Condorelli, G. Physiological myocardial hypertrophy: How and why? Front. Biosci. 2008, 13, 312–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weeks, K.L.; McMullen, J.R. The athlete’s heart vs. the failing heart: Can signaling explain the two distinct outcomes? Physiology 2011, 26, 97–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, I.; Minamino, T. Physiological and pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 2016, 97, 245–262. [Google Scholar] [CrossRef]
- Nolly, M.B.; Pinilla, A.O.; Ennis, I.L.; Cingolani, H.E.; Morgan, P.E. Cardiac hypertrophy reduction in SHR by specific silencing of myocardial Na+/H+ exchanger. J. Appl. Physiol. 2015, 118, 1154–1160. [Google Scholar] [CrossRef] [Green Version]
- Fliegel, L.; Karmazyn, M. The cardiac Na-H exchanger: A key downstream mediator for the cellular hypertrophic effects of paracrine, autocrine and hormonal factors. Biochem. Cell Biol. 2004, 82, 626–635. [Google Scholar] [CrossRef]
- Karmazyn, M. Therapeutic potential of Na-H exchange inhibitors for the treatment of heart failure. Expert Opin. Investig. Drugs 2001, 10, 835–843. [Google Scholar] [CrossRef]
- Suleiman, M.; Abdulrahman, N.; Yalcin, H.; Mraiche, F. The role of CD44, hyaluronan and NHE1 in cardiac remodeling. Life Sci. 2018, 209, 197–201. [Google Scholar] [CrossRef]
- Cingolani, H.E.; Alvarez, B.V.; Ennis, I.L.; Camilión de Hurtado, M.C. Stretch-induced alkalinization of feline papillary muscle an autocrine-paracrine system. Circ. Res. 1998, 83, 775–780. [Google Scholar] [CrossRef] [Green Version]
- Karmazyn, M.; Liu, Q.; Gan, X.T.; Brix, B.J.; Fliegel, L. Aldosterone increases NHE-1 expression and induces NHE-1-dependent hypertrophy in neonatal rat ventricular myocytes. Hypertension 2003, 42, 1171–1176. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, S.; Nakano, M.; Taniguchi, Y.; Imai, S.; Murata, K.; Suzuki, T. Effects of Na+-H+ exchange blocker amiloride on left ventricular remodeling after anterior myocardial infarction in rats. Cardiovasc. Drug Ther. 1995, 9, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Mraiche, F.; Oka, T.; Gan, X.T.; Karmazyn, M.; Fliegel, L. Activated NHE1 is required to induce early cardiac hypertrophy in mice. Basic Res. Cardiol. 2011, 106, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.Y.; Iwata, Y.; Arai, Y.; Komamura, K.; Wakabayashi, S. Activation of Na+/H+ exchanger 1 is sufficient to generate Ca2+ signals that induce cardiac hypertrophy and heart failure. Circ. Res. 2008, 103, 891–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosca, M.G.; Tandler, B.; Hoppel, C.L. Mitochondria in cardiac hypertrophy and heart failure. J. Mol. Cell. Cardiol. 2013, 55, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Baartscheer, A.; Hardziyenka, M.; Schumacher, C.A.; Belterman, C.N.; van Borren, M.M.; Verkerk, A.O.; Coronel, R.; Fiolet, J.W. Chronic inhibition of the Na+/H+ exchanger causes regression of hypertrophy, heart failure, and ionic and electrophysiological remodelling. Br. J. Pharmacol. 2008, 154, 1266–1275. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Chen, X.C.; Gan, X.T.; Beier, N.; Scholz, W.; Karmazyn, M. Inhibition and reversal of myocardial infarctioninduced hypertrophy and heart failure by NHE-1 inhibition. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H381–H387. [Google Scholar] [CrossRef]
- Kilić, A.; Huang, C.X.; Rajapurohitam, V.; Madwed, J.B.; Karmazyn, M. Early and transient sodium-hydrogen exchanger isoform 1 inhibition attenuates subsequent cardiac hypertrophy and heart failure following coronary artery ligation. J. Pharmacol. Exp. Ther. 2014, 351, 492–499. [Google Scholar] [CrossRef] [Green Version]
- Kusumoto, K.; Haist, J.V.; Karmazyn, K. Na+/H+ exchange inhibition reduces hypertrophy and heart failure after myocardial infarction in rats. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H738–H745. [Google Scholar] [CrossRef]
- Packer, M. Activation and inhibition of sodium-hydrogen exchanger is a mechanism that links the pathophysiology and treatment of diabetes Mmellitus with that of heart failure. Circulation 2017, 136, 1548–1559. [Google Scholar] [CrossRef]
- Despa, S. Myocyte [Na+]i dysregulation in heart failure and diabetic cardiomyopathy. Front. Physiol. 2018, 9, 1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippatos, T.D.; Liontos, A.; Papakitsou, I.; Elisaf, M.S. SGLT2 inhibitors and cardioprotection: A matter of debate and multiple hypotheses. Postgrad. Med. 2019, 131, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.W.; Que, J.Q.; Liu, S.; Huang, K.Y.; Qian, L.; Weng, Y.B.; Rong, F.N.; Wang, L.; Zhou, Y.Y.; Xue, Y.J.; et al. Sodium-Glucose Co-transporter-2 Inhibitor of Dapagliflozin Attenuates Myocardial Ischemia/Reperfusion Injury by Limiting NLRP3 Inflammasome Activation and Modulating Autophagy. Front. Cardiovasc. Med. 2021, 8, 768214. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Romer, G.; Kerindongo, R.P.; Hermanides, J.; Albrecht, M.; Hollmann, M.W.; Zuurbier, C.J.; Preckel, B.; Weber, N.C. Sodium Glucose Co-Transporter 2 Inhibitors Ameliorate Endothelium Barrier Dysfunction Induced by Cyclic Stretch through Inhibition of Reactive Oxygen Species. Int. J. Mol. Sci. 2021, 22, 6044. [Google Scholar] [CrossRef] [PubMed]
- Linz, W.J.; Busch, A.E. NHE-1 inhibition: From protection during acute ischaemia/reperfusion to prevention/reversal of myocardial remodelling. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2003, 368, 239–246. [Google Scholar] [CrossRef]
- Karmazyn, M.; Sawyer, M.; Fliegel, L. The Na+/H+ exchanger: A target for cardiac therapeutic intervention. Curr. Drug Targets-Cardiovasc. Hematol. Disord. 2005, 5, 323–335. [Google Scholar] [CrossRef]
- Mohamed, I.A.; Mraiche, F. Targeting osteopontin, the silent partner of Na+/H+ exchanger isoform 1 in cardiac remodeling. J. Cell Physiol. 2015, 230, 2006–2018. [Google Scholar] [CrossRef]
- Wakabayashi, S.; Hisamitsu, T.; Nakamura, T.Y. Regulation of the cardiac Na+/H+ exchanger in health and disease. J. Mol. Cell. Cardiol. 2013, 61, 68–76. [Google Scholar] [CrossRef]
- Backs, J.; Song, K.; Bezprozvannaya, S.; Chang, S.; Olson, E.N. CaM kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J. Clin. Investig. 2006, 116, 1853–1864. [Google Scholar] [CrossRef]
- Anderson, M.E.; Brown, J.H.; Bers, D.M. CaMKII in myocardial hypertrophy and heart failure. J. Mol. Cell. Cardiol. 2011, 51, 468–473. [Google Scholar] [CrossRef] [Green Version]
- Backs, J.; Backs, T.; Neef, S.; Kreusser, M.; Lehmann, L.H.; Patrick, D.M.; Grueter, C.E.; Qi, X.; Richardson, J.A.; Hill, J.A.; et al. The δ isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc. Natl. Acad. Sci. USA 2009, 106, 2342–2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samak, M.; Fatullayev, J.; Sabashnikov, A.; Zeriouh, M.; Schmack, B.; Farag, M.; Popov, A.F.; Dohmen, P.M.; Choi, Y.H.; Wahlers, T.; et al. Cardiac hypertrophy: An introduction to molecular and cellular basis. Med. Sci. Monit. Basic Res. 2016, 22, 75–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hisamitsu, T.; Nakamura, T.Y.; Wakabayashi, S. Na+/H+ exchanger 1 directly binds to calcineurin A and activates downstream NFAT signaling, leading to cardiomyocyte hypertrophy. Mol. Cell Biol. 2012, 32, 3265–3280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popov, S.; Venetsanou, K.; Chedrese, P.J.; Pinto, V.; Takemori, H.; Franco-Cereceda, A.; Eriksson, P.; Mochizuki, N.; Soares-da-Silva, P.; Bertorello, A.M. Increases in intracellular sodium activate transcription and gene expression via the salt-inducible kinase 1 network in an atrial myocyte cell line. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H57–H65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amirak, E.; Fuller, S.J.; Sugden, P.H.; Clerk, A. p90 ribosomal S6 kinases play a significant role in early gene regulation in the cardiomyocyte response to Gq-protein-coupled receptor stimuli, endothelin-1 and α1-adrenergic receptor agonists. Biochem. J. 2013, 450, 351–363. [Google Scholar] [CrossRef] [Green Version]
- Kilić, A.; Velic, A.; De Windt, L.J.; Fabritz, L.; Voss, M.; Mitko, D.; Zwiener, M.; Baba, H.A.; van Eickels, M.; Schlatter, E.; et al. Enhanced activity of the myocardial Na+/H+ exchanger NHE-1 contributes to cardiac remodeling in atrial natriuretic peptide receptor-deficient mice. Circulation 2005, 112, 2307–2317. [Google Scholar] [CrossRef] [Green Version]
- Abdulrahman, N.; Jaspard-Vinassa, B.; Fliegel, L.; Jabeen, A.; Riaz, S.; Gadeau, A.P.; Mraiche, F. Na+/H+ exchanger isoform 1-induced osteopontin expression facilitates cardiac hypertrophy through p90 ribosomal S6 kinase. Physiol. Genom. 2018, 50, 332–342. [Google Scholar] [CrossRef] [Green Version]
- Euler, G.; Locquet, F.; Kociszewska, J.; Osygus, Y.; Heger, J.; Schreckenberg, R.; Schlüter, K.D.; Kenyeres, É.; Szabados, T.; Bencsik, P.; et al. Matrix metalloproteinases repress hypertrophic growth in cardiac myocytes. Cardiovasc. Drug Ther. 2021, 35, 353–365. [Google Scholar] [CrossRef]
- Riaz, S.; Abdulrahman, N.; Uddin, S.; Jabeen, A.; Gadeau, A.P.; Fliegel, L.; Mraiche, F. Anti-hypertrophic effect of Na+/H+ exchanger-1 inhibition is mediated by reduced cathepsin B. Eur. J. Pharmacol. 2020, 888, 173420. [Google Scholar] [CrossRef]
- Dellsperger, K.C.; Clothier, J.L.; Hartnett, J.A.; Haun, L.M.; Marcus, M.L. Acceleration of the wavefront of myocardial necrosis by chronic hypertension and left ventricular hypertrophy in dogs. Circ. Res. 1988, 63, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.L.; Li, Y.; Yin, P.P.; Kong, F.J.; Guo, J.J.; Shi, H.T.; Zhu, J.B.; Zou, Y.Z.; Ge, J.B. Hypertrophied myocardium is vulnerable to ischemia/reperfusion injury and refractory to rapamycin-induced protection due to increased oxidative/nitrative stress. Clin. Sci. 2018, 132, 93–110. [Google Scholar] [CrossRef] [PubMed]
- Pagliaro, P.; Penna, C. Hypertension, hypertrophy, and reperfusion injury. J. Cardiovasc. Med. 2017, 18, 131–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yano, T.; Miki, T.; Tanno, M.; Kuno, A.; Itoh, T.; Takada, A.; Sato, T.; Kouzu, H.; Shimamoto, K.; Miura, T. Hypertensive hypertrophied myocardium is vulnerable to infarction and refractory to erythropoietin-induced protection. Hypertension 2011, 57, 110–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molgaard, S.; Faricelli, B.; Salomonsson, M.; Engstrom, T.; Treiman, M. Increased myocardial vulnerability to ischemia-reperfusion injury in the presence of left ventricular hypertrophy. J. Hypertens. 2016, 34, 513–523; discussion 523. [Google Scholar] [CrossRef]
- Madonna, R.; De Caterina, R. Sodium-hydrogen exchangers (NHE) in human cardiovascular diseases: Interfering strategies and their therapeutic applications. Vasc. Pharmacol. 2013, 59, 127–130. [Google Scholar] [CrossRef]
- Wu, D.; Kraut, J.A. Potential role of NHE1 (sodium-hydrogen exchanger 1) in the cellular dysfunction of lactic acidosis: Implications for treatment. Am. J. Kidney Dis. 2011, 57, 781–787. [Google Scholar] [CrossRef]
- Karmazyn, M. NHE-1: Still a viable therapeutic target. J. Mol. Cell. Cardiol. 2013, 61, 77–82. [Google Scholar] [CrossRef]
- Zeymer, U.; Suryapranata, H.; Monassier, J.P.; Opolski, G.; Davies, J.; Rasmanis, G.; Linssen, G.; Tebble, U.; Schröder, R.; Tiemann, R.; et al. The Na+/H+ exchange inhibitor eniporide as an adjunct to early reperfusion therapy for acute myocardial infarction. J. Am. Coll. Cardiol. 2001, 38, E1644–E1650. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, J.C.; Chouinard, L.; Kerlin, R.L.; Groom, S.N.; Botts, S.; Arezzo, J.C.; Boucher, M.A.; Frazier, D.E.; Buchholz, A.R. Neurotoxic effects of zoniporide: A selective inhibitor of the Na+/H+ exchanger isoform 1. Toxicol. Pathol. 2008, 36, 608–619. [Google Scholar] [CrossRef]
- Thomé, F.P.; Su, J.; Barthelemy, I.; Blanchard-Gutton, N.; Bkaily, G.; Yu, Q.; Nagaraju, K.; Galeh, B.; Blot, S. Translational development of rimeporide, a sodium-hydrogen exchanger (NHE-1) inhibitor, for patients with Duchenne muscular dystrophy. Neuromuscul. Disord. 2016, 26, S155. [Google Scholar] [CrossRef]
- Previtali, S.C.; Gidaro, T.; Diaz-Manera, J.; Zambon, A.; Carnesecchi, S.; Roux-Lombard, P.; Spitali, P.; Signorelli, M.; Szigyarto, C.A.; Johansson, C.; et al. Rimeporide as a first-in-class NHE-1 inhibitor: Results of a phase Ib trial in young patients with Duchenne Muscular Dystrophy. Pharmacol. Res. 2020, 159, 104999. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, H.; Zahra, A.; Jia, M.; Wang, Q.; Wang, Y.; Campbell, S.L.; Wu, J. Na+/H+ Exchanger 1, a Potential Therapeutic Drug Target for Cardiac Hypertrophy and Heart Failure. Pharmaceuticals 2022, 15, 875. https://doi.org/10.3390/ph15070875
Xia H, Zahra A, Jia M, Wang Q, Wang Y, Campbell SL, Wu J. Na+/H+ Exchanger 1, a Potential Therapeutic Drug Target for Cardiac Hypertrophy and Heart Failure. Pharmaceuticals. 2022; 15(7):875. https://doi.org/10.3390/ph15070875
Chicago/Turabian StyleXia, Huiting, Aqeela Zahra, Meng Jia, Qun Wang, Yunfu Wang, Susan L. Campbell, and Jianping Wu. 2022. "Na+/H+ Exchanger 1, a Potential Therapeutic Drug Target for Cardiac Hypertrophy and Heart Failure" Pharmaceuticals 15, no. 7: 875. https://doi.org/10.3390/ph15070875
APA StyleXia, H., Zahra, A., Jia, M., Wang, Q., Wang, Y., Campbell, S. L., & Wu, J. (2022). Na+/H+ Exchanger 1, a Potential Therapeutic Drug Target for Cardiac Hypertrophy and Heart Failure. Pharmaceuticals, 15(7), 875. https://doi.org/10.3390/ph15070875