
Siccanin Is a Dual-Target Inhibitor of Plasmodium falciparum Mitochondrial Complex II and Complex III

 , , ,
, , ,
Abstract
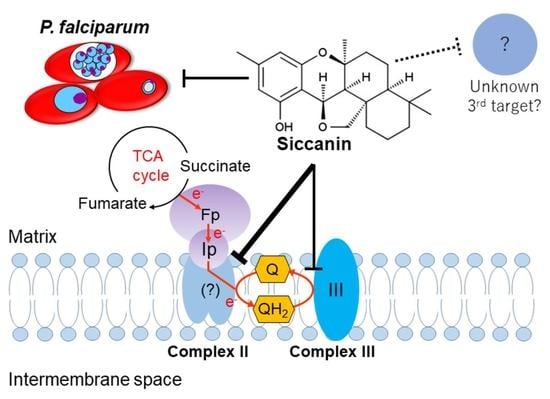
1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Complex II IC50 [μM] | Selectivity a | |
|---|---|---|---|
| Plasmodium spp. | Mammal | ||
| Malonate | 13.2 ± 0.49 b | 3.4 d | 0.26 |
| TTFA | >50 b | 5.4 e | <0.11 |
| Atpenin A5 | 4.6 ± 0.2 c | 0.004 e | 0.00087 |
| Carboxin | 3.6 ± 1.0 c | 1.0 e | 0.28 |
2. Results
2.1. Siccanin Strongly Inhibits P. falciparum SQR Activity
| Assay | Siccanin IC50 [μM] | Selectivity a | |
|---|---|---|---|
| P. falciparum | Mammal | ||
| Complex II | 0.016 and 8.93 | 861 b | 57,400 and 96 |
| Complex III | 8.39 | >500 | >60 |
| Growth | 8.4 | 34.2 c 16.1 d | 4.1 1.9 |
2.2. Inhibition of P. falciparum Growth by Siccanin
2.3. Effect of Succinate or Fumarate, and Pfsdha Disruption, on Growth Inhibition by Siccanin
2.4. Siccanin Inhibits P. falciparum Complex III
2.5. Interaction of Atovaquone with Siccanin against P. falciparum In Vitro
2.6. Effect of Siccanin on P. falciparum yDHODH-3D7 and Dd2 Drug-Resistant-Panel Strains
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Malaria Parasite Strains and Cultivation
4.3. Assessment of Parasite Survival
4.4. Drug sensitivity Assay
4.5. Mammalian Cell Cytotoxicity Assay
4.6. Preparation of Crude P. falciparum Mitochondrial Fraction
4.7. Measurement of P. falciparum ETC Enzyme Activities
4.8. Measurement of NADH-Cytochrome c Reductase Activity in Porcine Mitochondria
4.9. Isobologram Analysis with Atovaquone and Siccanin
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. World Malaria Report: 2020; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Dondorp, A.M.; Nosten, F.; Yi, P.; Das, D.; Phyo, A.P.; Tarning, J.; Lwin, K.M.; Ariey, F.; Hanpithakpong, W.; Lee, S.J.; et al. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2009, 361, 455–467. [Google Scholar] [CrossRef]
- Balikagala, B.; Fukuda, N.; Ikeda, M.; Katuro, O.T.; Tachibana, S.I.; Yamauchi, M.; Opio, W.; Emoto, S.; Anywar, D.A.; Kimura, E.; et al. Evidence of Artemisinin-Resistant Malaria in Africa. N. Engl. J. Med. 2021, 385, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Kaneko, M.; Tachibana, S.I.; Balikagala, B.; Sakurai-Yatsushiro, M.; Yatsushiro, S.; Takahashi, N.; Yamauchi, M.; Sekihara, M.; Hashimoto, M.; et al. Artemisinin-Resistant Plasmodium falciparum with High Survival Rates, Uganda, 2014–2016. Emerg. Infect. Dis. 2018, 24, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Uwimana, A.; Umulisa, N.; Venkatesan, M.; Svigel, S.S.; Zhou, Z.; Munyaneza, T.; Habimana, R.M.; Rucogoza, A.; Moriarty, L.F.; Sandford, R.; et al. Association of Plasmodium falciparum kelch13 R561H genotypes with delayed parasite clearance in Rwanda: An open-label, single-arm, multicentre, therapeutic efficacy study. Lancet Infect. Dis. 2021, 21, 1120–1128. [Google Scholar] [CrossRef]
- Blagborough, A.M.; Churcher, T.S.; Upton, L.M.; Ghani, A.C.; Gething, P.W.; Sinden, R.E. Transmission-blocking interventions eliminate malaria from laboratory populations. Nat. Commun. 2013, 4, 1812. [Google Scholar] [CrossRef]
- Biagini, G.A.; Viriyavejakul, P.; O’Neill, P.M.; Bray, P.G.; Ward, S.A. Functional characterization and target validation of alternative complex I of Plasmodium falciparum mitochondria. Antimicrob. Agents Chemother. 2006, 50, 1841–1851. [Google Scholar] [CrossRef]
- Hino, A.; Hirai, M.; Tanaka, T.Q.; Watanabe, Y.; Matsuoka, H.; Kita, K. Critical roles of the mitochondrial complex II in oocyst formation of rodent malaria parasite Plasmodium berghei. J. Biochem. 2012, 152, 259–268. [Google Scholar] [CrossRef]
- Tanaka, T.Q.; Hirai, M.; Watanabe, Y.; Kita, K. Toward understanding the role of mitochondrial complex II in the intraerythrocytic stages of Plasmodium falciparum: Gene targeting of the Fp subunit. Parasitol. Int. 2012, 61, 726–728. [Google Scholar] [CrossRef]
- Bulusu, V.; Jayaraman, V.; Balaram, H. Metabolic fate of fumarate, a side product of the purine salvage pathway in the intraerythrocytic stages of Plasmodium falciparum. J. Biol. Chem. 2011, 286, 9236–9245. [Google Scholar] [CrossRef]
- Painter, H.J.; Morrisey, J.M.; Mather, M.W.; Vaidya, A.B. Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature 2007, 446, 88–91. [Google Scholar] [CrossRef]
- Shears, M.J.; Botte, C.Y.; McFadden, G.I. Fatty acid metabolism in the Plasmodium apicoplast: Drugs, doubts and knockouts. Mol. Biochem. Parasitol. 2015, 199, 34–50. [Google Scholar] [CrossRef]
- Stocks, P.A.; Barton, V.; Antoine, T.; Biagini, G.A.; Ward, S.A.; O’Neill, P.M. Novel inhibitors of the Plasmodium falciparum electron transport chain. Parasitology 2014, 141, 50–65. [Google Scholar] [CrossRef]
- Kawahara, K.; Mogi, T.; Tanaka, T.Q.; Hata, M.; Miyoshi, H.; Kita, K. Mitochondrial dehydrogenases in the aerobic respiratory chain of the rodent malaria parasite Plasmodium yoelii yoelii. J. Biochem. 2009, 145, 229–237. [Google Scholar] [CrossRef]
- Belard, S.; Ramharter, M. DSM265: A novel drug for single-dose cure of Plasmodium falciparum malaria. Lancet Infect. Dis. 2018, 18, 819–820. [Google Scholar] [CrossRef]
- Collins, K.A.; Ruckle, T.; Elliott, S.; Marquart, L.; Ballard, E.; Chalon, S.; Griffin, P.; Mohrle, J.J.; McCarthy, J.S. DSM265 at 400 Milligrams Clears Asexual Stage Parasites but Not Mature Gametocytes from the Blood of Healthy Subjects Experimentally Infected with Plasmodium falciparum. Antimicrob. Agents Chemother. 2019, 63, e01837-18. [Google Scholar] [CrossRef]
- Llanos-Cuentas, A.; Casapia, M.; Chuquiyauri, R.; Hinojosa, J.C.; Kerr, N.; Rosario, M.; Toovey, S.; Arch, R.H.; Phillips, M.A.; Rozenberg, F.D.; et al. Antimalarial activity of single-dose DSM265, a novel plasmodium dihydroorotate dehydrogenase inhibitor, in patients with uncomplicated Plasmodium falciparum or Plasmodium vivax malaria infection: A proof-of-concept, open-label, phase 2a study. Lancet Infect. Dis. 2018, 18, 874–883. [Google Scholar] [CrossRef]
- Murphy, S.C.; Duke, E.R.; Shipman, K.J.; Jensen, R.L.; Fong, Y.; Ferguson, S.; Janes, H.E.; Gillespie, K.; Seilie, A.M.; Hanron, A.E.; et al. A Randomized Trial Evaluating the Prophylactic Activity of DSM265 against Preerythrocytic Plasmodium falciparum Infection during Controlled Human Malarial Infection by Mosquito Bites and Direct Venous Inoculation. J. Infect. Dis. 2018, 217, 693–702. [Google Scholar] [CrossRef]
- Phillips, M.A.; Lotharius, J.; Marsh, K.; White, J.; Dayan, A.; White, K.L.; Njoroge, J.W.; El Mazouni, F.; Lao, Y.; Kokkonda, S.; et al. A long-duration dihydroorotate dehydrogenase inhibitor (DSM265) for prevention and treatment of malaria. Sci. Transl. Med. 2015, 7, 296ra111. [Google Scholar] [CrossRef]
- Sulyok, M.; Ruckle, T.; Roth, A.; Murbeth, R.E.; Chalon, S.; Kerr, N.; Samec, S.S.; Gobeau, N.; Calle, C.L.; Ibanez, J.; et al. DSM265 for Plasmodium falciparum chemoprophylaxis: A randomised, double blinded, phase 1 trial with controlled human malaria infection. Lancet Infect. Dis. 2017, 17, 636–644. [Google Scholar] [CrossRef]
- Goodman, C.D.; Siregar, J.E.; Mollard, V.; Vega-Rodríguez, J.; Syafruddin, D.; Matsuoka, H.; Matsuzaki, M.; Toyama, T.; Sturm, A.; Cozijnsen, A.; et al. Parasites resistant to the antimalarial atovaquone fail to transmit by mosquitoes. Science 2016, 352, 349–353. [Google Scholar] [CrossRef]
- Takashima, E.; Takamiya, S.; Takeo, S.; Mi-ichi, F.; Amino, H.; Kita, K. Isolation of mitochondria from Plasmodium falciparum showing dihydroorotate dependent respiration. Parasitol. Int. 2001, 50, 273–278. [Google Scholar] [CrossRef]
- Shapiro, T.A.; Ranasinha, C.D.; Kumar, N.; Barditch-Crovo, P. Prophylactic activity of atovaquone against Plasmodium falciparum in humans. Am. J. Trop. Med. Hyg. 1999, 60, 831–836. [Google Scholar] [CrossRef] [PubMed]
- Cecchini, G. Function and structure of complex II of the respiratory chain. Annu. Rev. Biochem. 2003, 72, 77–109. [Google Scholar] [CrossRef] [PubMed]
- Harada, S.; Inaoka, D.K.; Ohmori, J.; Kita, K. Diversity of parasite complex II. Biochim. Biophys. Acta 2013, 1827, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Maclean, A.E.; Bridges, H.R.; Silva, M.F.; Ding, S.; Ovciarikova, J.; Hirst, J.; Sheiner, L. Complexome profile of Toxoplasma gondii mitochondria identifies divergent subunits of respiratory chain complexes including new subunits of cytochrome bc1 complex. PLoS Pathog. 2021, 17, e1009301. [Google Scholar] [CrossRef]
- Matsubayashi, M.; Inaoka, D.K.; Komatsuya, K.; Hatta, T.; Kawahara, F.; Sakamoto, K.; Hikosaka, K.; Yamagishi, J.; Sasai, K.; Shiba, T.; et al. Novel Characteristics of Mitochondrial Electron Transport Chain from Eimeria tenella. Genes 2019, 10, 29. [Google Scholar] [CrossRef]
- Millar, A.H.; Eubel, H.; Jänsch, L.; Kruft, V.; Heazlewood, J.L.; Braun, H.P. Mitochondrial cytochrome c oxidase and succinate dehydrogenase complexes contain plant specific subunits. Plant Mol. Biol. 2004, 56, 77–90. [Google Scholar] [CrossRef]
- Miyadera, H.; Shiomi, K.; Ui, H.; Yamaguchi, Y.; Masuma, R.; Tomoda, H.; Miyoshi, H.; Osanai, A.; Kita, K.; Omura, S. Atpenins, potent and specific inhibitors of mitochondrial complex II (succinate-ubiquinone oxidoreductase). Proc. Natl. Acad. Sci. USA 2003, 100, 473–477. [Google Scholar] [CrossRef]
- Suraveratum, N.; Krungkrai, S.R.; Leangaramgul, P.; Prapunwattana, P.; Krungkrai, J. Purification and characterization of Plasmodium falciparum succinate dehydrogenase. Mol. Biochem. Parasitol. 2000, 105, 215–222. [Google Scholar] [CrossRef]
- Nose, K.; Endo, A. Mode of action of the antibiotic siccanin on intact cells and mitochondria of Trichophyton mentagrophytes. J. Bacteriol. 1971, 105, 176–184. [Google Scholar] [CrossRef]
- Hartman, T.; Weinrick, B.; Vilchèze, C.; Berney, M.; Tufariello, J.; Cook, G.M.; Jacobs, W.R. Succinate dehydrogenase is the regulator of respiration in Mycobacterium tuberculosis. PLoS Pathog. 2014, 10, e1004510. [Google Scholar] [CrossRef] [PubMed]
- Mendz, G.L.; Hazell, S.L.; Srinivasan, S. Fumarate reductase: A target for therapeutic intervention against Helicobacter pylori. Arch. Biochem. Biophys. 1995, 321, 153–159. [Google Scholar] [CrossRef]
- Mogi, T.; Kawakami, T.; Arai, H.; Igarashi, Y.; Matsushita, K.; Mori, M.; Shiomi, K.; Omura, S.; Harada, S.; Kita, K. Siccanin rediscovered as a species-selective succinate dehydrogenase inhibitor. J. Biochem. 2009, 146, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Hirai, K.; Nozoe, S.; Tsuda, K.; Iitaka, Y.; Ishibashi, K.; Shirasaka, M. The structure of siccainin. Tetrahedron. Lett. 1967, 23, 2177–2179. [Google Scholar] [CrossRef]
- Ishibashi, K. Studies on Antibiotics from Helminthosporium sp. Fungi. VII Siccanin, a New Antifungal Antibiotic Produced by Helminthosporium Siccans. J. Antibiot. Ser. A 1962, 15, 161–167. [Google Scholar] [CrossRef]
- Arai, M.; Ishibashi, K.; Okazaki, H. Siccanin, a new antifungal antibiotic. I. In vitro studies. Antimicrob. Agents Chemother. (Bethesda) 1969, 9, 247–252. [Google Scholar]
- Nihashi, N.; Inaoka, D.K.; Tsuge, C.; Balogun, E.O.; Osada, Y.; Goto, Y.; Matsumoto, Y.; Nara, T.; Mogi, T.; Harada, S.; et al. Siccanin is a novel selective inhibitor of trypanosomatid complex II (succinate-ubiquinone reductase) and a potent broad-spectrum anti-trypanosomatid drug candidate. In Kala Azar in South Asia: Current Status and Sustainable Challenges; Noiri, E., Jha, T.K., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 101–122. [Google Scholar] [CrossRef]
- Sakuda, S.; Prabowo, D.F.; Takagi, K.; Shiomi, K.; Mori, M.; Omura, S.; Nagasawa, H. Inhibitory effects of respiration inhibitors on aflatoxin production. Toxins 2014, 6, 1193–1200. [Google Scholar] [CrossRef]
- Yamamuro, D.; Uchida, R.; Takahashi, Y.; Masuma, R.; Tomoda, H. Screening for microbial metabolites affecting phenotype of Caenorhabditis elegans. Biol. Pharm. Bull. 2011, 34, 1619–1623. [Google Scholar] [CrossRef][Green Version]
- Kröger, A.; Geisler, V.; Lemma, E.; Theis, F.; Lenger, R. Bacterial fumarate respiration. Arch. Microbiol. 1992, 158, 311–314. [Google Scholar] [CrossRef]
- Cook, G.M.; Greening, C.; Hards, K.; Berney, M. Energetics of pathogenic bacteria and opportunities for drug development. Adv. Microb. Physiol. 2014, 65, 1–62. [Google Scholar] [CrossRef]
- Eoh, H.; Rhee, K.Y. Multifunctional essentiality of succinate metabolism in adaptation to hypoxia in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2013, 110, 6554–6559. [Google Scholar] [CrossRef]
- Watanabe, S.; Zimmermann, M.; Goodwin, M.B.; Sauer, U.; Barry, C.E., 3rd; Boshoff, H.I. Fumarate reductase activity maintains an energized membrane in anaerobic Mycobacterium tuberculosis. PLoS Pathog. 2011, 7, e1002287. [Google Scholar] [CrossRef] [PubMed]
- Enkai, S.; Inaoka, D.K.; Kouguchi, H.; Irie, T.; Yagi, K.; Kita, K. Mitochondrial complex III in larval stage of Echinococcus multilocularis as a potential chemotherapeutic target and in vivo efficacy of atovaquone against primary hydatid cysts. Parasitol. Int. 2020, 75, 102004. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Landry, A.P.; Guha, A.; Vitvitsky, V.; Lee, H.J.; Seike, K.; Reddy, P.; Lyssiotis, C.A.; Banerjee, R. A redox cycle with complex II prioritizes sulfide quinone oxidoreductase-dependent H2S oxidation. J. Biol. Chem. 2022, 298, 101435. [Google Scholar] [CrossRef] [PubMed]
- Ke, H.; Lewis, I.A.; Morrisey, J.M.; McLean, K.J.; Ganesan, S.M.; Painter, H.J.; Mather, M.W.; Jacobs-Lorena, M.; Llinás, M.; Vaidya, A.B. Genetic investigation of tricarboxylic acid metabolism during the Plasmodium falciparum life cycle. Cell Rep. 2015, 11, 164–174. [Google Scholar] [CrossRef]
- McConnell, E.V.; Bruzual, I.; Pou, S.; Winter, R.; Dodean, R.A.; Smilkstein, M.J.; Krollenbrock, A.; Nilsen, A.; Zakharov, L.N.; Riscoe, M.K.; et al. Targeted Structure-Activity Analysis of Endochin-like Quinolones Reveals Potent Qi and Qo Site Inhibitors of Toxoplasma gondii and Plasmodium falciparum Cytochrome bc1 and Identifies ELQ-400 as a Remarkably Effective Compound against Acute Experimental Toxoplasmosis. ACS Infect. Dis. 2018, 4, 1574–1584. [Google Scholar] [CrossRef]
- Song, Z.; Iorga, B.I.; Mounkoro, P.; Fisher, N.; Meunier, B. The antimalarial compound ELQ-400 is an unusual inhibitor of the bc1 complex, targeting both Qo and Qi sites. FEBS Lett. 2018, 592, 1346–1356. [Google Scholar] [CrossRef]
- Stickles Allison, M.; Smilkstein Martin, J.; Morrisey Joanne, M.; Li, Y.; Forquer Isaac, P.; Kelly Jane, X.; Pou, S.; Winter Rolf, W.; Nilsen, A.; Vaidya Akhil, B.; et al. Atovaquone and ELQ-300 Combination Therapy as a Novel Dual-Site Cytochrome bc1 Inhibition Strategy for Malaria. Antimicrob. Agents Chemother. 2016, 60, 4853–4859. [Google Scholar] [CrossRef]
- Fivelman, Q.L.; Adagu, I.S.; Warhurst, D.C. Modified fixed-ratio isobologram method for studying in vitro interactions between atovaquone and proguanil or dihydroartemisinin against drug-resistant strains of Plasmodium falciparum. Antimicrob. Agents Chemother. 2004, 48, 4097–4102. [Google Scholar] [CrossRef]
- Hartuti, E.D.; Inaoka, D.K.; Komatsuya, K.; Miyazaki, Y.; Miller, R.J.; Xinying, W.; Sadikin, M.; Prabandari, E.E.; Waluyo, D.; Kuroda, M.; et al. Biochemical studies of membrane bound Plasmodium falciparum mitochondrial L-malate:quinone oxidoreductase, a potential drug target. Biochim. Biophys. Acta. Bioenerg. 2018, 1859, 191–200. [Google Scholar] [CrossRef]
- Ameyaw, E.O.; Asmah, K.B.; Biney, R.P.; Henneh, I.T.; Owusu-Agyei, P.; Prah, J.; Forkuo, A.D. Isobolographic analysis of co-administration of two plant-derived antiplasmodial drug candidates, cryptolepine and xylopic acid, in Plasmodium berghei. Malar. J. 2018, 17, 153. [Google Scholar] [CrossRef] [PubMed]
- Co, E.M.; Dennull, R.A.; Reinbold, D.D.; Waters, N.C.; Johnson, J.D. Assessment of malaria in vitro drug combination screening and mixed-strain infections using the malaria Sybr green I-based fluorescence assay. Antimicrob. Agents Chemother. 2009, 53, 2557–2563. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gorka, A.P.; Jacobs, L.M.; Roepe, P.D. Cytostatic versus cytocidal profiling of quinoline drug combinations via modified fixed-ratio isobologram analysis. Malar. J. 2013, 12, 332. [Google Scholar] [CrossRef] [PubMed]
- Kemirembe, K.; Cabrera, M.; Cui, L. Interactions between tafenoquine and artemisinin-combination therapy partner drug in asexual and sexual stage Plasmodium falciparum. Int. J. Parasitol. Drugs Drug Resist. 2017, 7, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Paquet, T.; Le Manach, C.; Cabrera Diego, G.; Younis, Y.; Henrich Philipp, P.; Abraham Tara, S.; Lee Marcus, C.S.; Basak, R.; Ghidelli-Disse, S.; Lafuente-Monasterio María, J.; et al. Antimalarial efficacy of MMV390048, an inhibitor of Plasmodium phosphatidylinositol 4-kinase. Sci. Transl. Med. 2017, 9, eaad9735. [Google Scholar] [CrossRef]
- Baragaña, B.; Hallyburton, I.; Lee, M.C.S.; Norcross, N.R.; Grimaldi, R.; Otto, T.D.; Proto, W.R.; Blagborough, A.M.; Meister, S.; Wirjanata, G.; et al. A novel multiple-stage antimalarial agent that inhibits protein synthesis. Nature 2015, 522, 315–320. [Google Scholar] [CrossRef]
- Jiménez-Díaz María, B.; Ebert, D.; Salinas, Y.; Pradhan, A.; Lehane Adele, M.; Myrand-Lapierre, M.-E.; O’Loughlin Kathleen, G.; Shackleford David, M.; Justino de Almeida, M.; Carrillo Angela, K.; et al. (+)-SJ733, a clinical candidate for malaria that acts through ATP4 to induce rapid host-mediated clearance of Plasmodium. Proc. Natl. Acad. Sci. USA 2014, 111, E5455–E5462. [Google Scholar] [CrossRef]
- Meister, S.; Plouffe, D.M.; Kuhen, K.L.; Bonamy, G.M.; Wu, T.; Barnes, S.W.; Bopp, S.E.; Borboa, R.; Bright, A.T.; Che, J.; et al. Imaging of Plasmodium liver stages to drive next-generation antimalarial drug discovery. Science 2011, 334, 1372–1377. [Google Scholar] [CrossRef]
- Stickles Allison, M.; de Almeida Mariana, J.; Morrisey Joanne, M.; Sheridan Kayla, A.; Forquer Isaac, P.; Nilsen, A.; Winter Rolf, W.; Burrows Jeremy, N.; Fidock David, A.; Vaidya Akhil, B.; et al. Subtle Changes in Endochin-Like Quinolone Structure Alter the Site of Inhibition within the Cytochrome bc1 Complex of Plasmodium falciparum. Antimicrob. Agents Chemother. 2015, 59, 1977–1982. [Google Scholar] [CrossRef]
- Hatefi, Y. The mitochondrial electron transport and oxidative phosphorylation system. Annu. Rev. Biochem. 1985, 54, 1015–1069. [Google Scholar] [CrossRef]
- Hines, V.; Keys, L.D.; Johnston, M. Purification and properties of the bovine liver mitochondrial dihydroorotate dehydrogenase. J. Biol. Chem. 1986, 261, 11386–11392. [Google Scholar] [CrossRef]
- Bryant, C.; Voller, A.; Smith, M.J. The incorporation of radioactivity from (14c)glucose into the soluble metabolic intermediates of malaria parasites. Am. J. Trop. Med. Hyg. 1964, 13, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Scheibel, L.W.; Pflaum, W.K. Cytochrome oxidase activity in platelet-free preparations of Plasmodium falciparum. J. Parasitol. 1970, 56, 1054. [Google Scholar] [CrossRef]
- Sakata-Kato, T.; Wirth, D.F. A Novel Methodology for Bioenergetic Analysis of Plasmodium falciparum Reveals a Glucose-Regulated Metabolic Shift and Enables Mode of Action Analyses of Mitochondrial Inhibitors. ACS Infect. Dis. 2016, 2, 903–916. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, C.; Otto, T.D.; Oberstaller, J.; Liao, X.; Adapa, S.R.; Udenze, K.; Bronner, I.F.; Casandra, D.; Mayho, M.; et al. Uncovering the essential genes of the human malaria parasite Plasmodium falciparum by saturation mutagenesis. Science 2018, 360, eaap7847. [Google Scholar] [CrossRef]
- Boysen, K.E.; Matuschewski, K. Arrested oocyst maturation in Plasmodium parasites lacking type II NADH:ubiquinone dehydrogenase. J. Biol. Chem. 2011, 286, 32661–32671. [Google Scholar] [CrossRef] [PubMed]
- Oyedotun, K.S.; Lemire, B.D. The Quinone-binding sites of the Saccharomyces cerevisiae succinate-ubiquinone oxidoreductase. J. Biol. Chem. 2001, 276, 16936–16943. [Google Scholar] [CrossRef]
- Lancaster, C.R.; Kröger, A.; Auer, M.; Michel, H. Structure of fumarate reductase from Wolinella succinogenes at 2.2 A resolution. Nature 1999, 402, 377–385. [Google Scholar] [CrossRef]
- Salerno, J.C.; Harmon, H.J.; Blum, H.; Leigh, J.S.; Ohnishi, T. A transmembrane quinone pair in the succinate dehydrogenase--Cytochrome b region. FEBS Lett. 1977, 82, 179–182. [Google Scholar] [CrossRef]
- Hägerhäll, C.; Magnitsky, S.; Sled, V.D.; Schröder, I.; Gunsalus, R.P.; Cecchini, G.; Ohnishi, T. An Escherichia coli mutant quinol:fumarate reductase contains an EPR-detectable semiquinone stabilized at the proximal quinone-binding site. J. Biol. Chem. 1999, 274, 26157–26164. [Google Scholar] [CrossRef]
- Westenberg, D.J.; Gunsalus, R.P.; Ackrell, B.A.; Cecchini, G. Electron transfer from menaquinol to fumarate. Fumarate reductase anchor polypeptide mutants of Escherichia coli. J. Biol. Chem. 1990, 265, 19560–19567. [Google Scholar] [CrossRef]
- Iverson, T.M.; Luna-Chavez, C.; Croal, L.R.; Cecchini, G.; Rees, D.C. Crystallographic studies of the Escherichia coli quinol-fumarate reductase with inhibitors bound to the quinol-binding site. J. Biol. Chem. 2002, 277, 16124–16130. [Google Scholar] [CrossRef] [PubMed]
- Fry, M.; Beesley, J.E. Mitochondria of mammalian Plasmodium spp. Parasitology 1991, 102 Pt 1, 17–26. [Google Scholar] [CrossRef]
- Trager, W.; Jensen, J.B. Human malaria parasites in continuous culture. Science 1976, 193, 673–675. [Google Scholar] [CrossRef] [PubMed]
- Lambros, C.; Vanderberg, J.P. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 1979, 65, 418–420. [Google Scholar] [CrossRef]
- Ganesan, S.M.; Morrisey, J.M.; Ke, H.; Painter, H.J.; Laroiya, K.; Phillips, M.A.; Rathod, P.K.; Mather, M.W.; Vaidya, A.B. Yeast dihydroorotate dehydrogenase as a new selectable marker for Plasmodium falciparum transfection. Mol. Biochem. Parasitol. 2011, 177, 29–34. [Google Scholar] [CrossRef]
- Hartuti, E.D.; Sakura, T.; Tagod, M.S.O.; Yoshida, E.; Wang, X.; Mochizuki, K.; Acharjee, R.; Matsuo, Y.; Tokumasu, F.; Mori, M.; et al. Identification of 3,4-Dihydro-2H,6H-pyrimido[1,2-c][1,3]benzothiazin-6-imine Derivatives as Novel Selective Inhibitors of Plasmodium falciparum Dihydroorotate Dehydrogenase. Int. J. Mol. Sci. 2021, 22, 7236. [Google Scholar] [CrossRef]
- Duffey, M.; Blasco, B.; Burrows, J.N.; Wells, T.N.C.; Fidock, D.A.; Leroy, D. Assessing risks of Plasmodium falciparum resistance to select next-generation antimalarials. Trends Parasitol. 2021, 37, 709–721. [Google Scholar] [CrossRef]
- Gamo, F.J.; Sanz, L.M.; Vidal, J.; de Cozar, C.; Alvarez, E.; Lavandera, J.L.; Vanderwall, D.E.; Green, D.V.; Kumar, V.; Hasan, S.; et al. Thousands of chemical starting points for antimalarial lead identification. Nature 2010, 465, 305–310. [Google Scholar] [CrossRef]
- Smith, A.L.; Hansen, M. Evidence for P/O ratios approaching 6 in mitochondrial oxidative phosphorylation. Biochem. Biophys. Res. Commun. 1964, 15, 431–435. [Google Scholar] [CrossRef]
- Canfield, C.J.; Pudney, M.; Gutteridge, W.E. Interactions of atovaquone with other antimalarial drugs against Plasmodium falciparum in vitro. Exp. Parasitol. 1995, 80, 373–381. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komatsuya, K.; Sakura, T.; Shiomi, K.; Ōmura, S.; Hikosaka, K.; Nozaki, T.; Kita, K.; Inaoka, D.K. Siccanin Is a Dual-Target Inhibitor of Plasmodium falciparum Mitochondrial Complex II and Complex III. Pharmaceuticals 2022, 15, 903. https://doi.org/10.3390/ph15070903
Komatsuya K, Sakura T, Shiomi K, Ōmura S, Hikosaka K, Nozaki T, Kita K, Inaoka DK. Siccanin Is a Dual-Target Inhibitor of Plasmodium falciparum Mitochondrial Complex II and Complex III. Pharmaceuticals. 2022; 15(7):903. https://doi.org/10.3390/ph15070903
Chicago/Turabian StyleKomatsuya, Keisuke, Takaya Sakura, Kazuro Shiomi, Satoshi Ōmura, Kenji Hikosaka, Tomoyoshi Nozaki, Kiyoshi Kita, and Daniel Ken Inaoka. 2022. "Siccanin Is a Dual-Target Inhibitor of Plasmodium falciparum Mitochondrial Complex II and Complex III" Pharmaceuticals 15, no. 7: 903. https://doi.org/10.3390/ph15070903
APA StyleKomatsuya, K., Sakura, T., Shiomi, K., Ōmura, S., Hikosaka, K., Nozaki, T., Kita, K., & Inaoka, D. K. (2022). Siccanin Is a Dual-Target Inhibitor of Plasmodium falciparum Mitochondrial Complex II and Complex III. Pharmaceuticals, 15(7), 903. https://doi.org/10.3390/ph15070903