
Immunogenicity of Current and New Therapies for Hemophilia A


Abstract
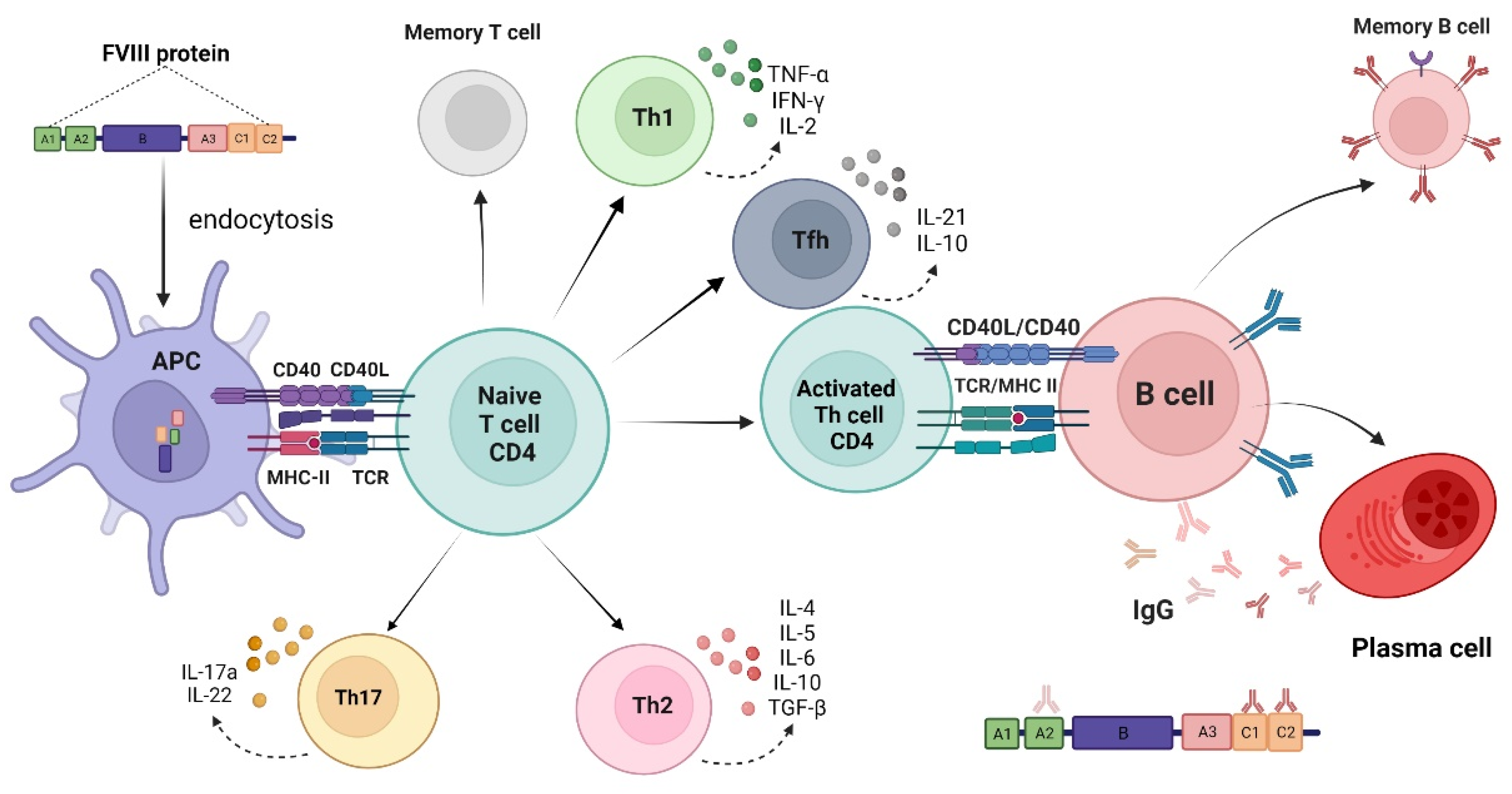
:1. Introduction
2. The Development of Anti-Factor VIII Neutralizing Antibodies (Inhibitors)
3. Immunogenicity Aspects of Factor VIII Products
4. Current Therapies for Hemophilia A
4.1. Replacement Therapy: Plasma-Derived Factor VIII Concentrates
4.2. Replacement Therapy: Standard Half-Life (SHL) Recombinant FVIII Products

{kind=link}
| Product | Company | Year of First Licensing | Half-Life (Hours) | VWF:RCo/FVIII:C Ratio | Immunogenicity PTPs (%) | Immunogenicity PUPs (%) | Ref. |
|---|---|---|---|---|---|---|---|
| Hemofil M | Takeda | 1966 | 15 | NA | 0 | 2.77 | [37] |
| Alphanate | Grifols | 1978 | 18 | 1.21 | NA | NA | [38] |
| Humate-P | CSL Behring | 1981 | 12.6 | 2.4 | NA | NA | [39] |
| Koate-DVI | Kedrion | 1992 | 16 | 1.1 | NA | NA | [38] |
| Octanate | Octapharma | 1998 | 11–14 | 0.4 | 0 | 9.8 All inhibitors 7.8 HT inhibitors | [40,41] |
| Wilate | Octapharma | 2009 | 13.1 h (OSA) 11.2 h (CSA) | 1.1 | 0 | 0 * | [42,43] |
4.3. Replacement Therapy: Extended Half-Life (EHL) Recombinant FVIII Products
4.4. Non-Replacement Therapy for Hemophilia A: Emicizumab
| Product (Brand) | Company | Year of First Licensing | Technology | Cell Line | FVIII | Half-Life (Hours) | Immunogenicity PTPs (%) | Immunogenicity PUPs (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Efmoroctocog alfa (Elocta, Eloctate) | Sanofi | 2014 | IgG1-Fc-fusion | HEK | B-domain deleted | 19 (OSA) 20.9 (CSA) | No inhibitor No anaphylaxis | 31.1 All inhibitors 15.6 HT inhibitors No anaphylaxis | [66,67,77,78] |
| Rurioctocog alfa pegol (Adynovi, Adynovate) | Takeda | 2015 | Random PEGylation | CHO | full-length | 14.3–16 (OSA) | No inhibitor No anaphylaxis | 19.2 All inhibitors | [63,73,79] |
| Damoctocog alfa pegol (JIVI) | Bayer | 2018 | Site-specific PEGylation | BHK | B-domain deleted | 19 (OSA) (>12 yo) 15–16 (OSA) (<12 yo) | No inhibitor 1.5 hypersensibility 3.7 anti-PEG Ab | NA | [64,72] |
| Turoctocog alfa pegol (N8-GP, Esperoct) | Novo Nordisk | 2019 | Site-specific glycoPEGylation | CHO | B-domain truncated | 15.8–19.9 (CSA) (>12 yo) 13.2–14.2 (CSA) (<12 yo) | 0.6 All inhibitors 12.3 anti-PEG Ab (>12 yo) 29.4 anti-PEG Ab (<12 yo) | 29.9 All inhibitors 14.9 HT inhibitors No anaphylaxis | [65,71,80] |
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berntorp, E.; Fischer, K.; Hart, D.P.; Mancuso, M.E.; Stephensen, D.; Shapiro, A.D.; Blanchette, V. Haemophilia. Nat. Rev. Dis. Primers 2021, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Santagostino, E.; Dougall, A.; Kitchen, S.; Sutherland, M.; Pipe, S.W.; Carcao, M.; Mahlangu, J.; Ragni, M.V.; Windyga, J.; et al. WFH Guidelines for the Management of Hemophilia, 3rd edition. Haemophilia 2020, 26 (Suppl. 6), 1–158. [Google Scholar] [CrossRef]
- Mahlangu, J.; Young, G.; Hermans, C.; Blanchette, V.; Berntorp, E.; Santagostino, E. Defining extended half-life rFVIII-A critical review of the evidence. Haemophilia 2018, 24, 348–358. [Google Scholar] [CrossRef]
- Mancuso, M.E.; Mahlangu, J.N.; Pipe, S.W. The changing treatment landscape in haemophilia: From standard half-life clotting factor concentrates to gene editing. Lancet 2021, 397, 630–640. [Google Scholar] [CrossRef]
- Peyvandi, F.; Mannucci, P.M.; Garagiola, I.; El-Beshlawy, A.; Elalfy, M.; Ramanan, V.; Eshghi, P.; Hanagavadi, S.; Varadarajan, R.; Karimi, M.; et al. A Randomized Trial of Factor VIII and Neutralizing Antibodies in Hemophilia A. New Engl. J. Med. 2016, 374, 2054–2064. [Google Scholar] [CrossRef]
- Berg, H.M.V.D.; Fischer, K.; Carcao, M.; Chambost, H.; Kenet, G.; Kurnik, K.; Königs, C.; Male, C.; Santagostino, E.; Ljung, R.; et al. Timing of inhibitor development in more than 1000 previously untreated patients with severe hemophilia A. Blood 2019, 134, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Gouw, S.C.; van der Bom, J.G.; Ljung, R.; Escuriola, C.; Cid, A.R.; Claeyssens-Donadel, S.; van Geet, C.; Kenet, G.; Mäkipernaa, A.; Molinari, A.C.; et al. Factor VIII Products and Inhibitor Development in Severe Hemophilia A. N. Engl. J. Med. 2013, 368, 231–239. [Google Scholar] [CrossRef]
- Montalvão, S.A.L.; Tucunduva, A.C.; Siqueira, L.H.; Sambo, A.L.A.; Medina, S.S.; Ozelo, M.C. A longitudinal evaluation of anti-FVIII antibodies demonstrated IgG4 subclass is mainly correlated with high-titre inhibitor in haemophilia A patients. Haemophilia 2015, 21, 686–692. [Google Scholar] [CrossRef]
- Abshire, T.C.; Brackmann, H.H.; Scharrer, I.; Hoots, K.; Gazengel, C.; Powell, J.S.; Gorina, E.; Kellermann, E.; Vosburgh, E. Sucrose formulated recombinant human antihemophilic factor VIII is safe and efficacious for treatment of hemophilia A in home therapy—International Kogenate-FS Study Group. Thromb. Haemost. 2000, 83, 811–816. [Google Scholar]
- Montalvão, S.A.D.L.; Tucunduva, A.C.; Sambo, A.L.D.A.; De Paula, E.V.; Medina, S.D.S.; Ozelo, M.C. Heat treatment of samples improve the performance of the Nijmegen–Bethesda assay in hemophilia A patients undergoing immune tolerance induction. Thromb. Res. 2015, 136, 1280–1284. [Google Scholar] [CrossRef]
- Whelan, S.F.J.; Hofbauer, C.J.; Horling, F.M.; Allacher, P.; Wolfsegger, M.; Oldenburg, J.; Male, C.; Windyga, J.; Tiede, A.; Schwarz, H.P.; et al. Distinct characteristics of antibody responses against factor VIII in healthy individuals and in different cohorts of hemophilia A patients. Blood 2013, 121, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Schweiger, H.; Rejtő, J.; Hofbauer, C.J.; Berg, V.; Allacher, P.; Zwiauer, K.; Feistritzer, C.; Schuster, G.; Ay, C.; Reipert, B.M.; et al. Nonneutralizing FVIII-specific antibody signatures in patients with hemophilia A and in healthy donors. Blood Adv. 2022, 6, 946–958. [Google Scholar] [CrossRef] [PubMed]
- Hofbauer, C.J.; Kepa, S.; Schemper, M.; Quehenberger, P.; Reitter-Pfoertner, S.; Mannhalter, C.; Reipert, B.M.; Pabinger, I. FVIII-binding IgG modulates FVIII half-life in patients with severe and moderate hemophilia A without inhibitors. Blood 2016, 128, 293–296. [Google Scholar] [CrossRef]
- Jing, W.; Chen, J.; Cai, Y.; Chen, Y.; Schroeder, J.A.; Johnson, B.D.; Cui, W.; Shi, Q. Induction of activated T follicular helper cells is critical for anti-FVIII inhibitor development in hemophilia A mice. Blood Adv. 2019, 3, 3099–3110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacroix-Desmazes, S.; Voorberg, J.; Lillicrap, D.; Scott, D.W.; Pratt, K.P. Tolerating Factor VIII: Recent Progress. Front. Immunol. 2020, 10, 2991. [Google Scholar] [CrossRef] [PubMed]
- Gouw, S.C.; Berg, H.M.V.D.; Oldenburg, J.; Astermark, J.; de Groot, P.G.; Margaglione, M.; Thompson, A.R.; van Heerde, W.; Boekhorst, J.; Miller, C.H.; et al. F8 gene mutation type and inhibitor development in patients with severe hemophilia A: Systematic review and meta-analysis. Blood 2012, 119, 2922–2934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astermark, J.; Donfield, S.M.; Gomperts, E.D.; Schwarz, J.; Menius, E.D.; Pavlova, A.; Oldenburg, J.; Kessing, B.; DiMichele, D.M.; Shapiro, A.D.; et al. The polygenic nature of inhibitors in hemophilia A: Results from the Hemophilia Inhibitor Genetics Study (HIGS) Combined Cohort. Blood 2013, 121, 1446–1454. [Google Scholar] [CrossRef] [Green Version]
- Carcao, M.; Re, W.; Ewenstein, B. The role of previously untreated patient studies in understanding the development of FVIII inhibitors. Haemophilia 2015, 22, 22–31. [Google Scholar] [CrossRef]
- Gouw, S.C.; Berg, H.M.V.D.; Fischer, K.; Auerswald, G.; Carcao, M.; Chalmers, E.; Chambost, H.; Kurnik, K.; Liesner, R.; Petrini, P.; et al. Intensity of factor VIII treatment and inhibitor development in children with severe hemophilia A: The RODIN study. Blood 2013, 121, 4046–4055. [Google Scholar] [CrossRef]
- Lai, J.; Hough, C.; Tarrant, J.; Lillicrap, D. Biological considerations of plasma-derived and recombinant factor VIII immunogenicity. Blood 2017, 129, 3147–3154. [Google Scholar] [CrossRef] [Green Version]
- Aledort, L.M.; Navickis, R.J.; Wilkes, M.M. Can B-domain deletion alter the immunogenicity of recombinant factor VIII? A meta-analysis of prospective clinical studies. J. Thromb. Haemost. 2011, 9, 2180–2192. [Google Scholar] [CrossRef]
- Hay, C.R.; Palmer, B.; Chalmers, E.; Liesner, R.; Maclean, R.; Rangarajan, S.; Williams, M.; Collins, P.W.; United Kingdom Haemophilia Centre Doctors’ Organisation (UKHCDO). Incidence of factor VIII inhibitors throughout life in severe hemophilia A in the United Kingdom. Blood 2011, 117, 6367–6370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvez, T.; Chambost, H.; D’Oiron, R.; Dalibard, V.; Demiguel, V.; Doncarli, A.; Gruel, Y.; Huguenin, Y.; Lutz, P.; Rothschild, C.; et al. Analyses of the FranceCoag cohort support differences in immunogenicity among one plasma-derived and two recombinant factor VIII brands in boys with severe hemophilia A. Haematologica 2017, 103, 179–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvez, T.; Chambost, H.; Claeyssens-Donadel, S.; D’Oiron, R.; Goulet, V.; Guillet, B.; Héritier, V.; Milien, V.; Rothschild, C.; Roussel-Robert, V.; et al. Recombinant factor VIII products and inhibitor development in previously untreated boys with severe hemophilia A. Blood 2014, 124, 3398–3408. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.W.; Palmer, B.P.; Chalmers, E.A.; Hart, D.P.; Liesner, R.; Rangarajan, S.; Talks, K.; Williams, M.; Hay, C.R.M. Factor VIII brand and the incidence of factor VIII inhibitors in previously untreated UK children with severe hemophilia A, 2000–2011. Blood 2014, 124, 3389–3397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, K.; Lassila, R.; Peyvandi, F.; Calizzani, G.; Gatt, A.; Lambert, T.; Windyga, J.; Iorio, A.; Gilman, E.; Makris, M.; et al. Inhibitor development in haemophilia according to concentrate. Four-year results from the European Haemophilia Safety Surveillance (EUHASS) project. Thromb. Haemost. 2015, 113, 968–975. [Google Scholar] [CrossRef] [Green Version]
- Iorio, A.; Puccetti, P.; Makris, M. Clotting factor concentrate switching and inhibitor development in hemophilia A. Blood 2012, 120, 720–727. [Google Scholar] [CrossRef] [Green Version]
- Kempton, C.L.; Soucie, J.M.; Abshire, T.C. Incidence of inhibitors in a cohort of 838 males with hemophilia A previously treated with factor VIII concentrates. J. Thromb. Haemost. 2006, 4, 2576–2581. [Google Scholar] [CrossRef] [Green Version]
- Xi, M.; Makris, M.; Marcucci, M.; Santagostino, E.; Mannucci, P.M.; Iorio, A. Inhibitor development in previously treated hemophilia A patients: A systematic review, meta-analysis, and meta-regression. J. Thromb. Haemost. 2013, 11, 1655–1662. [Google Scholar] [CrossRef]
- Peyvandi, F.; Miri, S.; Garagiola, I. Immune Responses to Plasma-Derived Versus Recombinant FVIII Products. Front. Immunol. 2021, 11, 591878. [Google Scholar] [CrossRef]
- Mannucci, P.M. Clinical evaluation of viral safety of coagulation factor VIII and IX concentrates. Vox Sang. 1993, 64, 197–203. [Google Scholar] [CrossRef] [PubMed]
- D’Amici, G.M.; Blasi, B.; D’Alessandro, A.; Vaglio, S.; Zolla, L. Plasma-derived clotting factor VIII: Heterogeneity evaluation in the quest for potential inhibitory-antibody stimulating factors. Electrophoresis 2011, 32, 2941–2950. [Google Scholar] [CrossRef] [PubMed]
- Goudemand, J.; Rothschild, C.; DeMiguel, V.; Vinciguerra, C.; Lambert, T.; Chambost, H.; Borel-Derlon, A.; Claeyssens, S.; Laurian, Y.; Calvez, T.; et al. Influence of the type of factor VIII concentrate on the incidence of factor VIII inhibitors in previously untreated patients with severe hemophilia A. Blood 2006, 107, 46–51. [Google Scholar] [CrossRef]
- Pipe, S. Recombinant clotting factors. Thromb. Haemost. 2008, 99, 840–850. [Google Scholar] [CrossRef] [PubMed]
- Schiavoni, M.; Napolitano, M.; Giuffrida, G.; Coluccia, A.; Siragusa, S.; Calafiore, V.; Lassandro, G.; Giordano, P. Status of Recombinant Factor VIII Concentrate Treatment for Hemophilia A in Italy: Characteristics and Clinical Benefits. Front. Med. 2019, 6, 261. [Google Scholar] [CrossRef]
- Lissitchkov, T.; Klukowska, A.; Pasi, J.; Kessler, C.M.; Klamroth, R.; Liesner, R.J.; Belyanskaya, L.; Walter, O.; Knaub, S.; Bichler, J.; et al. Efficacy and safety of simoctocog alfa (Nuwiq®) in patients with severe hemophilia A: A review of clinical trial data from the GENA program. Ther. Adv. Hematol. 2019, 10, 2040620719858471. [Google Scholar] [CrossRef]
- Lusher, J.M. Viral safety and inhibitor development associated with monoclonal antibody-purified F VIII C. Ann. Hematol. 1991, 63, 138–141. [Google Scholar] [CrossRef]
- Batlle, J.; Fernandez, M.F.L.; Fraga, E.L.; Tillo, A.R.; Rodríguez, A.P. Von Willebrand factor/factor VIII concentrates in the treatment of von Willebrand disease. Blood Coagul. Fibrinolysis 2009, 20, 89–100. [Google Scholar] [CrossRef]
- Berntorp, E.; Archey, W.; Auerswald, G.; Federici, A.B.; Franchini, M.; Knaub, S.; Kreuz, W.; Lethagen, S.; Mannucci, P.M.; Pollmann, H.; et al. A systematic overview of the first pasteurised VWF/FVIII medicinal product, Haemate®P/ Humate®-P: History and clinical performance. Eur. J. Haematol. 2008, 80, 3–35. [Google Scholar] [CrossRef]
- Klukowska, A.; Komrska, V.; Vdovin, V.; Pavlova, A.; Jansen, M.; Lowndes, S.; Belyanskaya, L.; Walter, O.; Laguna, P. Low incidence of factor VIII inhibitors in previously untreated patients with severe haemophilia A treated with octanate®: Final report from a prospective study. Haemophilia 2018, 24, 221–228. [Google Scholar] [CrossRef] [Green Version]
- Klukowska, A.; Komrska, V.; Vdovin, V.; Zozulya, N.; Lissitchkov, T.; Oldenburg, J.; Ettingshausen, C.E. octanate®: Over 20 years of clinical experience in overcoming challenges in haemophilia A treatment. Ther. Adv. Hematol. 2020, 11, 2040620720914692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vézina, C.; Carcao, M.; Infante-Rivard, C.; Lillicrap, D.; Stain, A.M.; Paradis, E.; Teitel, J.; Rivard, G.E. The Association of Hemophilia Clinic Directors of Canada and of the Canadian Association of Nurses in Hemophilia Care Incidence and risk factors for inhibitor development in previously untreated severe haemophilia A patients born between 2005 and 2010. Haemophilia 2014, 20, 771–776. [Google Scholar] [CrossRef]
- Klukowska, A.; Windyga, J.; Batorova, A. Clinical efficacy of a novel VWF-containing FVIII concentrate, Wilate®, in the prophylaxis and treatment of bleeding episodes in previously treated haemophilia A patients. Thromb. Res. 2011, 127, 247–253. [Google Scholar] [CrossRef]
- Bray, G.L.; Gomperts, E.D.; Courter, S.; Gruppo, R.; Gordon, E.M.; Manco-Johnson, M.; Shapiro, A.; Scheibel, E.; White, G.; Lee, M. A multicenter study of recombinant factor VIII (recombinate): Safety, efficacy, and inhibitor risk in previously untreated patients with hemophilia A. The Recombinate Study Group. Blood 1994, 83, 2428–2435. [Google Scholar] [PubMed]
- Laurian, Y.; Satre, E.P.; Derlon, A.B.; Chambost, H.; Moreau, P.; Goudemand, J.; Parquet, A.; Peynet, J.; Vicariot, M.; Beurrier, P.; et al. French Previously Untreated Patients with Severe Hemophilia A after Exposure to Recombinant Factor VIII: Incidence of Inhibitor and Evaluation of Immune Tolerance. Thromb. Haemost. 1998, 80, 779–783. [Google Scholar] [CrossRef]
- Ewenstein, B.M.; Gomperts, E.D.; Pearson, S.; O’Banion, M.E. Inhibitor development in patients receiving recombinant factor VIII (Recombinate rAHF/Bioclate): A prospective pharmacovigilance study. Haemophilia 2004, 10, 491–498. [Google Scholar] [CrossRef]
- Kreuz, W.; Gill, J.C.; Rothschild, C.; Manco-Johnson, M.J.; Lusher, J.M.; Kellermann, E.; Gorina, E.; Larson, P.J.; International Kogenate-FS Study Group. Full-length sucrose-formulated recombinant factor VIII for treatment of previously untreated or minimally treated young children with severe haemophilia A: Results of an international clinical investigation. Thromb. Haemost. 2005, 93, 457–467. [Google Scholar] [CrossRef]
- Tarantino, M.D.; Collins, P.W.; Hay, C.R.M.; Shapiro, A.; Gruppo, R.A.; Berntorp, E.; Bray, G.L.; Tonetta, S.A.; Schroth, P.C.; Retzios, A.D.; et al. Clinical evaluation of an advanced category antihaemophilic factor prepared using a plasma/albumin-free method: Pharmacokinetics, efficacy, and safety in previously treated patients with haemophilia A1. Haemophilia 2004, 10, 428–437. [Google Scholar] [CrossRef]
- Auerswald, G.; Thompson, A.A.; Recht, M.; Brown, D.; Liesner, R.; Guzmán-Becerra, N.; Dyck-Jones, J.; Ewenstein, B.; Abbuehl, B. Experience of Advate rAHF-PFM in previously untreated patients and minimally treated patients with haemophilia A. Thromb. Haemost. 2012, 107, 1072–1082. [Google Scholar] [CrossRef] [Green Version]
- Jardim, L.L.; Santana, M.P.; Chaves, D.G.; van der Bom, J.; Rezende, S.M. Risk factors for antibody formation in children with hemophilia: Methodological aspects and clinical characteristics of the HEMFIL cohort study. Blood Coagul. Fibrinolysis 2021, 32, 443–450. [Google Scholar] [CrossRef]
- Recht, M.; Nemes, L.; Matysiak, M.; Manco-Johnson, M.; Lusher, J.; Smith, M.; Mannucci, P.; Hay, C.; Abshire, T.; O’Brien, A.; et al. Clinical evaluation of moroctocog alfa (AF-CC), a new generation of B-domain deleted recombinant factor VIII (BDDrFVIII) for treatment of haemophilia A: Demonstration of safety, efficacy, and pharmacokinetic equivalence to full-length recombinant factor VIII. Haemophilia 2009, 15, 869–880. [Google Scholar] [CrossRef]
- Mathias, M.C.; Collins, P.W.; Palmer, B.P.; Chalmers, E.; Alamelu, J.; Richards, M.; Will, A.; Hay, C.R.M.; Party, T.U.K.H.C.D.O.I.W. The immunogenicity of ReFacto AF (moroctocog alfa AF-CC) in previously untreated patients with haemophilia A in the United Kingdom. Haemophilia 2018, 24, 896–901. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, R.; Karim, F.A.; Glamocanin, S.; Janic, D.; Vdovin, V.; Ozelo, M.; Rageliene, L.; Carboni, E.; Laguna, P.; Dobaczewski, G.; et al. Results from a large multinational clinical trial (guardianTM3) using prophylactic treatment with turoctocog alfa in paediatric patients with severe haemophilia A: Safety, efficacy and pharmacokinetics. Haemophilia 2013, 19, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Yaish, H.; Matsushita, T.; Belhani, M.; Jiménez-Yuste, V.; Kavakli, K.; Korsholm, L.; Matytsina, I.; Philipp, C.; Reichwald, K.; Wu, R. Safety and efficacy of turoctocog alfa in the prevention and treatment of bleeds in previously untreated paediatric patients with severe haemophilia A: Results from the guardian 4 multinational clinical trial. Haemophilia 2019, 26, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Liesner, R.J.; Abraham, A.; Altisent, C.; Belletrutti, M.J.; Carcao, M.; Carvalho, M.; Chambost, H.; Chan, A.K.C.; Dubey, L.; Ducore, J.; et al. Simoctocog Alfa (Nuwiq) in Previously Untreated Patients with Severe Haemophilia A: Final Results of the NuProtect Study. Thromb. Haemost. 2021, 121, 1400–1408. [Google Scholar] [CrossRef]
- Saxena, K.; Lalezari, S.; Oldenburg, J.; Tseneklidou-Stoeter, D.; Beckmann, H.; Yoon, M.; Maas Enriquez, M. Efficacy and safety of BAY 81-8973, a full-length recombinant factor VIII: Results from the LEOPOLD I trial. Haemophilia 2016, 22, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Kenet, G.; Ljung, R.; Rusen, L.; Kerlin, B.A.; Blanchette, V.; Trakymienė, S.S.; Uscatescu, V.; Beckmann, H.; Tseneklidou-Stoeter, D.; Church, N. Continued benefit demonstrated with BAY 81-8973 prophylaxis in previously treated children with severe haemophilia A: Interim analysis from the LEOPOLD Kids extension study. Thromb. Res. 2020, 189, 96–101. [Google Scholar] [CrossRef] [Green Version]
- Mahlangu, J.; Kuliczkowski, K.; Karim, F.A.; Stasyshyn, O.; Kosinova, M.V.; Lepatan, L.M.; Skotnicki, A.; Boggio, L.N.; Klamroth, R.; Oldenburg, J.; et al. Efficacy and safety of rVIII-Single Chain: Results of a phase 1/3 multicenter clinical trial in severe hemophilia A. Blood 2016, 128, 630–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannucci, P.M. Hemophilia therapy: The future has begun. Haematologica 2020, 105, 545–553. [Google Scholar] [CrossRef]
- Ozelo, M.C.; Yamaguti-Hayakawa, G.G. Impact of novel hemophilia therapies around the world. Res. Pract. Thromb. Haemost. 2022, 6, e12695. [Google Scholar] [CrossRef]
- Björkman, S.; Oh, M.; Spotts, G.; Schroth, P.; Fritsch, S.; Ewenstein, B.M.; Casey, K.; Fischer, K.; Blanchette, V.S.; Collins, P.W. Population pharmacokinetics of recombinant factor VIII: The relationships of pharmacokinetics to age and body weight. Blood 2012, 119, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Kepa, S.; Horvath, B.; Reitter-Pfoertner, S.; Schemper, M.; Quehenberger, P.; Grundbichler, M.; Heistinger, M.; Neumeister, P.; Mannhalter, C.; Pabinger, I. Parameters influencing FVIII pharmacokinetics in patients with severe and moderate haemophilia A. Haemophilia 2015, 21, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Konkle, B.A.; Stasyshyn, O.; Chowdary, P.; Bevan, D.H.; Mant, T.; Shima, M.; Engl, W.; Dyck-Jones, J.; Fuerlinger, M.; Patrone, L.; et al. Pegylated, full-length, recombinant factor VIII for prophylactic and on-demand treatment of severe hemophilia A. Blood 2015, 126, 1078–1085. [Google Scholar] [CrossRef] [Green Version]
- Reding, M.T.; Ng, H.J.; Poulsen, L.H.; Eyster, M.E.; Pabinger, I.; Shin, H.-J.; Walsch, R.; Lederman, M.; Wang, M.; Hardtke, M.; et al. Safety and efficacy of BAY 94-9027, a prolonged-half-life factor VIII. J. Thromb. Haemost. 2017, 15, 411–419. [Google Scholar] [CrossRef]
- Giangrande, P.; Andreeva, T.; Chowdary, P.; Ehrenforth, S.; Hanabusa, H.; Leebeek, F.W.G.; Lentz, S.; Nemes, L.; Poulsen, L.H.; Santagostino, E.; et al. Clinical evaluation of glycoPEGylated recombinant FVIII: Efficacy and safety in severe haemophilia A. Thromb. Haemost. 2017, 117, 252–261. [Google Scholar] [CrossRef]
- Mahlangu, J.; Powell, J.S.; Ragni, M.V.; Chowdary, P.; Josephson, N.C.; Pabinger, I.; Hanabusa, H.; Gupta, N.; Kulkarni, R.; Fogarty, P.; et al. Phase 3 study of recombinant factor VIII Fc fusion protein in severe hemophilia A. Blood 2014, 123, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Nolan, B.; Mahlangu, J.; Pabinger, I.; Young, G.; Konkle, B.A.; Barnes, C.; Nogami, K.; Santagostino, E.; Pasi, K.J.; Khoo, L.; et al. Recombinant factor VIII Fc fusion protein for the treatment of severe haemophilia A: Final results from the ASPIRE extension study. Haemophilia 2020, 26, 494–502. [Google Scholar] [CrossRef]
- Lissitchkov, T.J.; Willemze, A.; Katragadda, S.; Rice, K.; Poloskey, S.; Benson, C. Efanesoctocog alfa for hemophilia A: Results from a phase 1 repeat-dose study. Blood Adv. 2022, 6, 1089–1094. [Google Scholar] [CrossRef]
- Kozma, G.T.; Shimizu, T.; Ishida, T.; Szebeni, J. Anti-PEG antibodies: Properties, formation, testing and role in adverse immune reactions to PEGylated nano-biopharmaceuticals. Adv. Drug Deliv. Rev. 2020, 154-155, 163–175. [Google Scholar] [CrossRef]
- Yang, Q.; Lai, S.K. Anti-PEG immunity: Emergence, characteristics, and unaddressed questions. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2015, 7, 655–677. [Google Scholar] [CrossRef] [Green Version]
- Meunier, S.; Alamelu, J.; Ehrenforth, S.; Hanabusa, H.; Karim, F.A.; Kavakli, K.; Khodaie, M.; Staber, J.; Stasyshyn, O.; Yee, D.L.; et al. Safety and efficacy of a glycoPEGylated rFVIII (turoctocog alpha pegol, N8-GP) in paediatric patients with severe haemophilia A. Thromb. Haemost. 2017, 117, 1705–1713. [Google Scholar] [CrossRef] [PubMed]
- Santagostino, E.; Kenet, G.; Fischer, K.; Biss, T.; Ahuja, S.; Steele, M.; Martínez, M.; Male, C.; Van Geet, C.; Mondelaers, V.; et al. PROTECT VIII Kids: BAY 94-9027 (PEGylated Recombinant Factor VIII) safety and efficacy in previously treated children with severe haemophilia A. Haemophilia 2020, 26, e55–e65. [Google Scholar] [CrossRef] [PubMed]
- Klamroth, R.; Windyga, J.; Radulescu, V.; Collins, P.W.; Stasyshyn, O.; Ibrahim, H.M.; Engl, W.; Tangada, S.D.; Savage, W.; Ewenstein, B.M. Rurioctocog alfa pegol PK-guided prophylaxis in hemophilia A: Results from the phase 3 PROPEL study. Blood 2021, 137, 1818–1827. [Google Scholar] [CrossRef] [PubMed]
- Oldenburg, J.; Mahlangu, J.N.; Kim, B.; Schmitt, C.; Callaghan, M.U.; Young, G.; Santagostino, E.; Kruse-Jarres, R.; Negrier, C.; Kessler, C.; et al. Emicizumab Prophylaxis in Hemophilia A with Inhibitors. New Engl. J. Med. 2017, 377, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, C.; Emrich, T.; Chebon, S.; Fernandez, E.; Petry, C.; Yoneyama, K.; Kiialainen, A.; Howard, M.; Niggli, M.; Paz-Priel, I.; et al. Low immunogenicity of emicizumab in persons with haemophilia A. Haemophilia 2021, 27, 984–992. [Google Scholar] [CrossRef]
- Mahlangu, J.; Iorio, A.; Kenet, G. Emicizumab state-of-the-art update. Haemophilia 2022, 28 (Suppl. 4), 103–110. [Google Scholar] [CrossRef]
- Young, G.; Mahlangu, J.; Kulkarni, R.; Nolan, B.; Liesner, R.; Pasi, J.; Barnes, C.; Neelakantan, S.; Gambino, G.; Cristiano, L.M.; et al. Recombinant factor VIII Fc fusion protein for the prevention and treatment of bleeding in children with severe hemophilia A. J. Thromb. Haemost. 2015, 13, 967–977. [Google Scholar] [CrossRef]
- Königs, C.; Ozelo, M.C.; Dunn, A.; Kulkarni, R.; Nolan, B.; Brown, S.A.; Schiavulli, M.; Gunawardena, S.; Mukhopadhyay, S.; Jayawardene, D.; et al. First study of extended half-life rFVIIIFc in previously untreated patients with hemophilia A: PUPs A-LONG final results. Blood 2022, 139, 3699–3707. [Google Scholar] [CrossRef]
- Sidonio, R.F.; Knoll, C.; Peyvandi, F.; Stasyshyn, O.; Antmen, A.B.; Yeoh, S.L.; Sosothikul, D.; Maggiore, C.; Engl, W.; Allen, G.; et al. Immunogenicity, Efficacy and Safety of Rurioctocog Alfa Pegol in Previously Untreated Patients with Severe Hemophilia a: Interim Results from an Open-Label Multicenter Clinical Trial. Blood 2021, 138, 3184. [Google Scholar] [CrossRef]
- Chowdary, P.; Carcao, M.; Holme, P.A.; Jiménez-Yuste, V.; Lentz, S.R.; Møss, J.; Poulsen, L.H.; Shen, C.; Tosetto, A.; Wheeler, A.; et al. Fixed doses of N8-GP prophylaxis maintain moderate-to-mild factor VIII levels in the majority of patients with severe hemophilia A. Res. Pract. Thromb. Haemost. 2019, 3, 542–554. [Google Scholar] [CrossRef] [Green Version]
| Product (Brand) | Company | Year of First Licensing | rFVIII Generation | Cell Line | Stabilizer | FVIII | Half-Life (Hours) | Immunogenicity PTPs (%) | Immunogenicity PUPs (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| Octocog alfa (Recombinate) | Takeda | 1992 | First | CHO | Human albumin | full-length | 15 | 0.12 All inhibitors 0.06 HT inhibitors | 23.9 All inhibitors 11.3 HT Inhibitors | [44,45,46] |
| Octocog alfa (Kogenate FS) | Bayer | 1993 | Second | BHK | Sucrose | full-length | 11 | No inhibitors | 15–50.1 All inhibitors 9.8–31.6 HT inhibitor | [9,23,47] |
| Octocog alfa (Advate) | Takeda | 2003 | Third | CHO | Trehalose | full-length | 9–12 | 0.92 All inhibitors | 29.1–38 All inhibitors 12.7–26 HT inhibitors | [48,49,50] |
| Moroctocog alfa (Xyntha/ReFacto AF) | Pfizer | 2008 | Third | CHO | Sucrose | B-domain deleted | 8–11 | 1.47 All inhibitors | 33 All inhibitors 14.5 HT inhibitors | [51,52] |
| Turoctocog alfa (Novoeight) | Novo Nordisk | 2013 | Third | CHO | Sucrose | B-domain truncated | 11 | No inhibitors | 43.1 All inhibitors 27.6 HT inhibitors | [53,54] |
| Simoctocog alfa (Nuwiq) | Octapharma | 2015 | Fourth | HEK | Sucrose/ arginine | full-length | 12–17 | No inhibitors | 26.7 All inhibitors 16.2 HT inhibitors | [36,55] |
| Octogog alfa (Kovaltry) | Bayer | 2016 | Third | BHK | Sucrose | full-length | 12.2–14.2 | 0.93 All inhibitors | 54.8 All inhibitors 40.5 HT inhibitors * | [56,57] |
| Lonoctocog alfa (Afstyla) | CSL Behring | 2016 | Third | CHO | Sucrose/ L-histidine, | B-domain truncated single chain | 14.5 | No inhibitors | 52 All inhibitors 26 HT inhibitors ** | [58] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prezotti, A.N.L.; Frade-Guanaes, J.O.; Yamaguti-Hayakawa, G.G.; Ozelo, M.C. Immunogenicity of Current and New Therapies for Hemophilia A. Pharmaceuticals 2022, 15, 911. https://doi.org/10.3390/ph15080911
Prezotti ANL, Frade-Guanaes JO, Yamaguti-Hayakawa GG, Ozelo MC. Immunogenicity of Current and New Therapies for Hemophilia A. Pharmaceuticals. 2022; 15(8):911. https://doi.org/10.3390/ph15080911
Chicago/Turabian StylePrezotti, Alessandra N. L., Jéssica O. Frade-Guanaes, Gabriela G. Yamaguti-Hayakawa, and Margareth C. Ozelo. 2022. "Immunogenicity of Current and New Therapies for Hemophilia A" Pharmaceuticals 15, no. 8: 911. https://doi.org/10.3390/ph15080911
APA StylePrezotti, A. N. L., Frade-Guanaes, J. O., Yamaguti-Hayakawa, G. G., & Ozelo, M. C. (2022). Immunogenicity of Current and New Therapies for Hemophilia A. Pharmaceuticals, 15(8), 911. https://doi.org/10.3390/ph15080911