
Interaction between Mesenchymal Stem Cells and the Immune System in Rheumatoid Arthritis

 , , , and
, , , and
Abstract
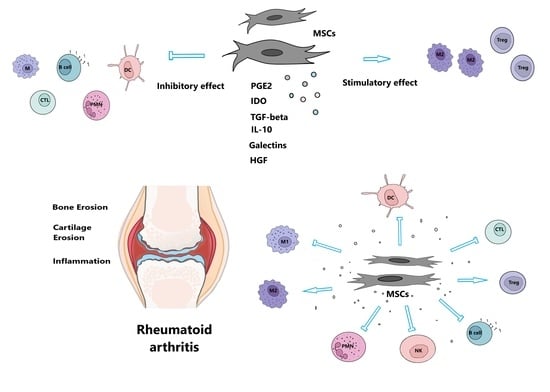
:
1. Introduction
1.1. The Immunology of Rheumatoid Arthritis
1.2. Autoantibodies in Rheumatoid Arthritis
1.3. Post-Translational Modifications of Proteins ECM
2. Mesenchymal Stem Cells
2.1. Mesenchymal Stem Cells in Synovium, in Their Native Environment
2.2. MSCs and Immune Cells in the Inflammatory Environment of Damaged Tissue
2.2.1. Homing and Migration of MSCs
2.2.2. The Modulation of T Cells by MSCs
2.2.3. Interactions between MSCs and Dendritic Cells
2.2.4. Macrophages and MSCs
2.2.5. B Cells and MSCs
2.3. Fibroblast-like Synoviocytes’ Interactions with Immune Cells
2.4. Immunomodulatory Potential of Mesenchymal Stem Cells
2.5. Mesenchymal Stem Cells for RA Treatment
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O., III. 2010 rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010, 62, 2569–2581. [Google Scholar] [CrossRef] [PubMed]
- Trentham, D.E.; Dynesius-Trentham, R.A.; Orav, E.J.; Combitchi, D. Effects of oral administration of type II collagen on rheumatoid arthritis. Science 1993, 261, 1727–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firestein, G.S. Evolving concepts of rheumatoid arthritis. Nature 2003, 423, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Sarsenova, M.; Issabekova, A.; Abisheva, S.; Rutskaya-Moroshan, K.; Ogay, V. Mesenchymal Stem Cell-Based Therapy for Rheumatoid Arthritis. Int. J. Mol. Sci. 2021, 22, 11592. [Google Scholar] [CrossRef]
- De Bari, C. Are mesenchymal stem cells in rheumatoid arthritis the good or bad guys? Arthritis Res. Ther. 2015, 17, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Delft, M.A.; Huizinga, T.W. An overview of autoantibodies in rheumatoid arthritis. J. Autoimmun. 2020, 110, 102392. [Google Scholar] [CrossRef] [PubMed]
- Holoshitz, J. The rheumatoid arthritis HLA-DRB1 shared epitope. Curr. Opin. Rheumatol. 2010, 22, 293. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, K.; Sugiyama, D.; Kogata, Y.; Tsuji, G.; Nakazawa, T.; Kawano, S. Meta-analysis: Diagnostic accuracy of anti–cyclic citrullinated peptide antibody and rheumatoid factor for rheumatoid arthritis. Ann. Intern. Med. 2007, 146, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Van den Broek, M.; Dirven, L.; Klarenbeek, N.B.; Han, K.H.; Kerstens, P.J.S.M.; Huizinga, T.W.J. The association of treatment response and joint damage with ACPA-status in recent-onset RA: A subanalysis of the 8-year follow-up of the BeSt study. Ann. Rheum. Dis. 2012, 71, 245–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Boekel, M.A.; Vossenaar, E.R.; Van den Hoogen, F.H.; van Venrooij, W.J. Autoantibody systems in rheumatoid arthritis: Specificity, sensitivity and diagnostic value. Arthritis Res. Ther. 2001, 4, 7. [Google Scholar]
- Van Steendam, K.; Tilleman, K.; Deforce, D. The relevance of citrullinated vimentin in the production of antibodies against citrullinated proteins and the pathogenesis of rheumatoid arthritis. Rheumatology 2011, 50, 830–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musaelyan, A.; Lapin, S.; Nazarov, V.; Tkachenko, O.; Gilburd, B.; Mazing, A.; Mikhailova, L.; Shoenfeld, Y. Vimentin as antigenic target in autoimmunity. A comprehensive review. Autoimmun. Rev. 2018, 17, 926–934. [Google Scholar] [CrossRef] [PubMed]
- Brentville, V.A.; Vankemmelbeke, M.; Metheringham, R.L.; Durrant, L.G. Post-translational modifications such as citrullination are excellent targets for cancer therapy. In Seminars in Immunology; Academic Press: Cambridge, MA, USA, 2020; Volume 47, p. 101393. [Google Scholar] [CrossRef]
- Uysal, H.; Bockermann, R.; Nandakumar, K.S.; Sehnert, B.; Bajtner, E. Structure and pathogenicity of antibodies specific for citrullinated collagen type II in experimental arthritis. J. Exp. Med. 2009, 206, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Utz, P.J.; Genovese, M.C.; Robinson, W.H. Unlocking the “PAD” lock on rheumatoid arthritis. Ann. Rheum. Dis. 2004, 63, 330–332. [Google Scholar] [CrossRef] [Green Version]
- Zeltz, C.; Gullberg, D. Post-translational modifications of integrin ligands as pathogenic mechanisms in disease. Matrix Biol. 2014, 40, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Vossenaar, E.R.; Radstake, T.R.; van der Heijden, A.; van Mansum, M.A.; Dieteren, C. Expression and activity of citrullinating peptidylarginine deiminase enzymes in monocytes and macrophages. Ann. Rheum. Dis. 2004, 63, 373–381. [Google Scholar] [CrossRef] [Green Version]
- Ostrowska-Podhorodecka, Z.; Ding, I.; Norouzi, M. Impact of Vimentin on Regulation of Cell Signaling and Matrix Remodeling. Front. Cell Dev. Biol. 2022, 10, 869069. [Google Scholar] [CrossRef] [PubMed]
- Klareskog, L.; Rönnelid, J.; Lundberg, K.; Padyukov, L.; Alfredsson, L. Immunity to citrullinated proteins in rheumatoid arthritis. Annu. Rev. Immunol. 2008, 26, 651–675. [Google Scholar] [CrossRef] [Green Version]
- Demoruelle, M.K.; Deane, K. Antibodies to citrullinated protein antigens (ACPAs): Clinical and pathophysiologic significance. Curr. Rheumatol. Rep. 2011, 13, 421–430. [Google Scholar] [CrossRef]
- Hill, J.A.; Bell, D.A.; Brintnell, W.; Yue, D. Arthritis induced by posttranslationally modified (citrullinated) fibrinogen in DR4-IE transgenic mice. J. Exp. Med. 2008, 205, 967–979. [Google Scholar] [CrossRef]
- Luban, S.; Li, Z.G. Citrullinated peptide and its relevance to rheumatoid arthritis: An update. Int. J. Rheum. Dis. 2010, 13, 284–287. [Google Scholar] [CrossRef]
- Machado, C.D.V.; Telles, P.D.D.S. Immunological characteristics of mesenchymal stem cells. Rev. Bras. De Hematol. E Hemoter. 2013, 35, 62–67. [Google Scholar] [CrossRef]
- Friedenstein, A.J.; Latzinik, N.V.; Gorskaya, Y.F.; Luria, E.A.; Moskvina, I.L. Bone marrow stromal colony formation requires stimulation by haemopoietic cells. Bone Miner. 1992, 18, 199–213. [Google Scholar] [CrossRef]
- Owen, M. Marrow stromal stem cells. J. Cell Sci. 1988, 10, 63–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caplan, A. Mesenchymal stem cells. J. Orthop. Res. 1991, 9, 641–650. [Google Scholar] [CrossRef]
- Procop, D.J. Marrow stromal cells as stem cells for nonhematopoietic tissues. Science 1997, 276, 71–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes, M.; Lund, T.; Lenvik, T.; Aguiar, D.; Koodie, L.; Verfaillie, C.M. Purification and ex vivo expansion of postnatal human marrow mesodermal progenitor cells. Blood J. Am. Soc. Hematol. 2001, 98, 2615–2625. [Google Scholar]
- Si, Y.L.; Zhao, Y.L.; Hao, H.J.; Fu, X.B. MSCs: Biological characteristics, clinical applications and their outstanding concerns. Ageing Res. Rev. 2011, 10, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Azizi, S.A.; Stokes, D.; Augelli, B.J.; DiGirolamo, C. Engraftment and migration of human bone marrow stromal cells implanted in the brains of albino rats—similarities to astrocyte grafts. Proc. Natl. Acad. Sci. USA 1998, 95, 3908–3913. [Google Scholar] [CrossRef] [Green Version]
- Scherzed, A.; Hackenberg, S. The differentiation of hMSCs counteracts their migration capability and pro-angiogenic effects in vitro. Oncol. Rep. 2016, 35, 219–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominici, M.L.B.K.; Le Blanc, K.; Mueller, I. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, S.A.; Mankani, M.H.; Bianco, P. Enumeration of the colony-forming units–fibroblast from mouse and human bone marrow in normal and pathological conditions. Stem Cell Res. 2009, 2, 83–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosna, F.; Sensebe, L.; Krampera, M. Human bone marrow and adipose tissue mesenchymal stem cells: A user’s guide. Stem Cells Dev. 2010, 19, 1449–1470. [Google Scholar] [CrossRef] [PubMed]
- Parolini, O.; Alviano, F.; Bagnara, G.P. Concise review: Isolation and characterization of cells from human term placenta: Outcome of the first international Workshop on Placenta Derived Stem Cells. Stem Cells 2008, 26, 300–311. [Google Scholar] [CrossRef] [Green Version]
- Soncini, M.; Vertua, E.; Gibelli, L.; Zorzi, F.; Denegri, M. Isolation and characterization of mesenchymal cells from human fetal membranes. J. Tissue Eng. Regen. Med. 2007, 1, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Bailo, M.; Soncini, M.; Vertua, E.; Signoroni, P.B. Engraftment potential of human amnion and chorion cells derived from term placenta. Transplantation 2004, 78, 1439–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alviano, F.; Fossati, V.; Marchionni, C.; Arpinati, M. Term amniotic membrane is a high throughput source for multipotent mesenchymal stem cells with the ability to differentiate into endothelial cells in vitro. BMC Dev. Biol. 2007, 7, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bačenková, D.; Rosocha, J.; Tóthová, T. Isolation and basic characterization of human term amnion and chorion mesenchymal stromal cells. Cytotherapy 2011, 13, 1047–1056. [Google Scholar] [CrossRef]
- Shariatzadeh, S.; Shafiee, S.; Zafari, A.; Tayebi, T.; Yazdanpanah, G. Developing a pro-angiogenic placenta derived amniochorionic scaffold with two exposed basement membranes as substrates for cultivating endothelial cells. Sci. Rep. 2021, 11, 1–14. [Google Scholar] [CrossRef]
- Jaramillo-Ferrada, P.A.; Wolvetang, E.J. Differential mesengenic potential and expression of stem cell-fate modulators in mesenchymal stromal cells from human-term placenta and bone marrow. J. Cell. Physiol. 2012, 227, 3234–3242. [Google Scholar] [CrossRef]
- Koo, B.K.; Park, I.Y.; Kim, J.; Kim, J.H. Isolation and characterization of chorionic mesenchymal stromal cells from human full term placenta. J. Korean Med. Sci. 2012, 27, 857–863. [Google Scholar] [CrossRef] [Green Version]
- Kurth, T.B.; Dell’Accio, F.; Crouch, V.; Augello, A.; Sharpe, P.T. Functional mesenchymal stem cell niches in adult mouse knee joint synovium in vivo. Arthritis Rheum. 2011, 63, 1289–1300. [Google Scholar] [CrossRef]
- Lee, D.M.; Kiener, H.P.; Agarwal, S.K.; Noss, E.H.; Watts, G.F. Cadherin-11 in synovial lining formation and pathology in arthritis. Science 2007, 315, 1006–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefèvre, S.; Knedla, A.; Tennie, C.; Kampmann, A.; Wunrau, C. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat. Med. 2009, 15, 1414–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gumbiner, B.M. Cell adhesion: The molecular basis of tissue architecture and morphogenesis. Cell 1996, 84, 345–357. [Google Scholar] [CrossRef] [Green Version]
- Müller-Ladner, U.; Kriegsmann, J.; Franklin, B.N.; Matsumoto, S.; Geiler, T. Synovial fibroblasts of patients with rheumatoid arthritis attach to and invade normal human cartilage when engrafted into SCID mice. Am. J. Pathol. 1996, 149, 1607. [Google Scholar] [PubMed]
- Yoshitomi, H. Regulation of immune responses and chronic inflammation by fibroblast-like synoviocytes. Front. Immunol. 2019, 10, 1395. [Google Scholar] [CrossRef] [PubMed]
- Asif Amin, M.; Fox, D.A.; Ruth, J.H. Synovial cellular and molecular markers in rheumatoid arthritis. In Seminars in Immunopathology; Springer: Berlin/Heidelberg, Germany, 2017; pp. 385–393. [Google Scholar]
- Nepom, G.T.; Erlich, H. MHC class-II molecules and autoimmunity. Annu. Rev. Immunol. 1991, 9, 493–525. [Google Scholar] [CrossRef]
- Filer, A. The fibroblast as a therapeutic target in rheumatoid arthritis. Curr. Opin. Pharmacol. 2013, 13, 413–419. [Google Scholar] [CrossRef] [PubMed]
- López-García, L.; Castro-Manrreza, M.E. TNF-α and IFN-γ Participate in Improving the Immunoregulatory Capacity of Mesenchymal Stem/Stromal Cells: Importance of Cell–Cell Contact and Extracellular Vesicles. Int. J. Mol. Sci. 2021, 22, 9531. [Google Scholar] [CrossRef] [PubMed]
- Szydlak, R. Biological, chemical and mechanical factors regulating migration and homing of mesenchymal stem cells. World J. Stem Cells 2021, 13, 619. [Google Scholar] [CrossRef] [PubMed]
- Zachar, L.; Bačenková, D. Activation, homing, and role of the mesenchymal stem cells in the inflammatory environment. J. Inflamm. Res. 2016, 9, 231. [Google Scholar] [CrossRef] [Green Version]
- Le Blanc, K.; Mougiakakos, D. Multipotent mesenchymal stromal cells and the innate immune system. Nat. Rev. Immunol. 2012, 12, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Xu, J. Immune modulation by mesenchymal stem cells. Cell Prolif. 2020, 53, e12712. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, T. Human bone marrow mesenchymal stromal cells attenuate tissue injury and reduce inflammation in experimental acute pancreatitis. Adv. Pharm. Bull. 2022, 12, 375. [Google Scholar] [CrossRef] [PubMed]
- Clanchy, F.I.; Borghese, F.; Bystrom, J.; Balog, A.; Penn, H. TLR expression profiles are a function of disease state in rheumatoid arthritis and experimental arthritis. J. Autoimmun. 2021, 118, 102597. [Google Scholar] [CrossRef] [PubMed]
- Andreeva, E.R.; Buravkova, L.B. The role of interplay of mesenchymal stromal cells and macrophages in physiological and reparative tissue remodel-ing. Hum. Physiol. 2018, 44, 102–114. [Google Scholar] [CrossRef]
- Ruhl, T.; Corsten, C.; Beier, J.P.; Kim, B.S. The immunosuppressive effect of the endocannabinoid system on the inflammatory phenotypes of macrophages and mesenchymal stromal cells: A comparative study. Pharmacol. Rep. 2021, 73, 143–153. [Google Scholar] [CrossRef] [PubMed]
- De Miguel, M.; Fuentes-Julian, S.; Blazquez-Martinez, A.; Pascual, C.Y.; Aller, M.A. Immunosuppressive properties of mesenchymal stem cells: Advances and applications. Curr. Mol. Med. 2012, 12, 574–591. [Google Scholar] [CrossRef]
- Bačenková, D.; Trebuňová, M.; Zachar, L.; Hudák, R.; Ižaríková, G. Analysis of same selected immunomodulatory properties of chorionic mesenchymal stem cells. Appl. Sci. 2020, 10, 9040. [Google Scholar] [CrossRef]
- Su, J.; Chen, X.; Huang, Y.; Li, W.; Li, J. Phylogenetic distinction of iNOS and IDO function in mesenchymal stem cell-mediated immunosuppression in mammalian species. Cell Death Differ. 2014, 21, 388–396. [Google Scholar] [CrossRef] [Green Version]
- Tatara, R.; Ozaki, K.; Kikuchi, Y.; Hatanaka, K.; Oh, I. Mesenchymal stromal cells inhibit Th17 but not regulatory T-cell differentiation. Cytotherapy 2011, 13, 686–694. [Google Scholar] [CrossRef]
- Galland, S.; Stamenkovic, I. Mesenchymal stromal cells in can-cer: A review of their immunomodulatory functions and dual effects on tumor progression. J. Pathol. 2020, 250, 555–572. [Google Scholar] [CrossRef] [Green Version]
- Ren, G.; Zhao, X.; Zhang, L.; Zhang, J.; L’Huillier, A.; Ling, W. Inflammatory cytokine-induced intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 in mesenchymal stem cells are critical for immunosuppression. J. Immunol. 2010, 184, 2321–2328. [Google Scholar] [CrossRef] [PubMed]
- Lefevre, S.; Meier, F.M.P.; Neumann, E.; Muller-Ladner, U. Role of synovial fibroblasts in rheumatoid arthritis. Curr. Pharm. Des. 2015, 21, 130–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najar, M.; Raicevic, G.; Fayyad-Kazan, H.; De Bruyn, C.; Bron, D. Impact of different mesenchymal stromal cell types on T-cell activation, proliferation and migration. Int. Immunopharmacol. 2013, 15, 693–702. [Google Scholar] [CrossRef]
- Luque-Campos, N.; Contreras-López, R.A.; Jose Paredes-Martínez, M.; Torres, M.J.; Bahraoui, S. Mesenchymal stem cells improve rheumatoid arthritis progression by controlling memory T cell response. Front. Immunol. 2019, 10, 798. [Google Scholar] [CrossRef]
- Berthelot, J.M.; Le Goff, B.; Maugars, Y. Bone marrow mesenchymal stem cells in rheumatoid arthritis, spondyloarthritis, and ankylosing spondylitis: Problems rather than solutions? Arthritis Res. Ther. 2019, 21, 9. [Google Scholar] [CrossRef] [Green Version]
- Pardali, E.; Goumans, M.J.; ten Dijke, P. Signaling by members of the TGF-β family in vascular morphogenesis and disease. Trends Cell Biol. 2010, 20, 556–567. [Google Scholar] [CrossRef]
- Bačenková, D.; Trebuňová, M.; Čížková, D.; Hudák, R.; Dosedla, E. In Vitro Model of Human Trophoblast in Early Placentation. Biomedicines 2022, 10, 904. [Google Scholar] [CrossRef]
- de Araújo Farias, V.; Carrillo-Gálvez, A.B.; Martin, F.; Anderson, P. TGF-β and mesenchymal stromal cells in regenerative medicine, autoimmunity and cancer. Cytokine Growth Factor Rev. 2018, 43, 25–37. [Google Scholar] [CrossRef]
- Yang, Y.H.; Hsieh, T.L.; Ji, A.T.Q.; Hsu, W.T.; Liu, C.Y. Stromal tissue rigidity promotes mesenchymal stem cell-mediated corneal wound healing through the transforming growth factor β signaling pathway. Stem Cells 2016, 34, 2525–2535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Santalla, M.; Fernandez-Perez, R. Mesenchymal stem/stromal cells for rheumatoid arthritis treatment: An update on clinical applications. Cells 2020, 9, 1852. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Mu, R.; Wang, S.; Long, L.; Liu, X. Therapeutic potential of human umbilical cord mesenchymal stem cells in the treatment of rheumatoid arthritis. Arthritis Res. Ther. 2010, 12, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shadmanfar, S.; Labibzadeh, N.; Emadedin, M. Intra-articular knee implantation of autologous bone marrow–derived mesenchymal stromal cells in rheumatoid arthritis patients with knee involvement: Results of a randomized, triple-blind, placebo-controlled phase 1/2 clinical trial. Cytotherapy 2018, 20, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Djouad, F.; Bouffi, C.; Ghannam, S.; Noël, D.; Jorgensen, C. Mesenchymal stem cells: Innovative therapeutic tools for rheumatic diseases. Nat. Rev. Rheumatol. 2009, 5, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Shalini, P.U.; Vidyasagar, J.V.S.; Kona, L.K.; Ponnana, M.; Chelluri, L.K. In vitro allogeneic immune cell response to mesenchymal stromal cells derived from human adipose in patients with rheumatoid arthritis. Cell. Immunol. 2017, 314, 18–25. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, X.; Wang, H.; Liu, X.; Zhang, T. The challenges and promises of allogeneic mesenchymal stem cells for use as a cell-based therapy. Stem Cell Res. Ther. 2015, 6, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulin, D.; Lilienbaum, A.; Kardjian, S. Vimentin: Regulation and pathogenesis. Biochimie 2022, 197, 96–112. [Google Scholar] [CrossRef] [PubMed]
- Brentville, V.A.; Metheringham, R.L.; Gunn, B.; Symonds, P.; Daniels, I. Citrullinated vimentin presented on MHC-II in tumor cells is a target for CD4+ T-cell–mediated antitumor immunity. Cancer Res. 2016, 76, 548–560. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.J.; Rand, E.; Webster, A.J.; Genever, P.G. Characterisation of mesenchymal stromal cells in clinical trial reports: Analysis of published descriptors. Stem Cell Res. Ther. 2021, 12, 15. [Google Scholar] [CrossRef]
- Orozco, L.; Munar, A.; Soler, R.; Alberca, M.; Soler, F. Treatment of knee osteoarthritis with autologous mesenchymal stem cells: Two-year follow-up results. Transplantation 2014, 97, 66–68. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Li, X.; Zhang, H.; Wang, D.; Feng, X. Allogeneic mesenchymal stem cells transplantation in patients with refractory RA. Clin. Rheumatol. 2012, 31, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Janockova, J.; Matejova, J.; Moravek, M.; Homolova, L.; Slovinska, L. Small Extracellular Vesicles Derived from Human Chorionic MSCs as Modern Perspective towards Cell-Free Therapy. Int. J. Mol. Sci. 2021, 22, 13581. [Google Scholar] [CrossRef]
- Moran-Moguel, M.C.; Petarra-del Rio, S. Rheumatoid arthritis and miRNAs: A critical review through a functional view. J. Immunol. Res. 2018, 2018, 2474529. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, H.; Xia, Y.; Yan, F.; Lu, Y. Therapeutic potential of mesenchymal cell–derived miR-NA-150-5p–expressing exosomes in rheumatoid arthritis mediated by the modulation of MMP14 and VEGF. J. Immunol. 2018, 201, 2472–2482. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.L.; Ang, D.C.; Almeida-Porada, G. Targeting mesenchymal stromal cells/pericytes (MSCs) with pulsed electromagnetic field (PEMF) has the potential to treat rheumatoid arthritis. Front. Immunol. 2019, 10, 12. [Google Scholar] [CrossRef] [PubMed]
- Yamada, R.; Suzuki, A.; Chang, X. Citrullinated proteins in rheumatoid arthritis. Front. Biosci. 2005, 10, 54–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Deng, W.; Yao, G.; Chen, W.; Tang, X. Citrullinated fibrinogen impairs immunomodulatory function of bone marrow mesenchymal stem cells by triggering toll-like receptor. Clin. Immunol. 2018, 193, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Hagmann, S.; Gotterbarm, T.; Müller, T.; Bäsig, A.M.; Gantz, S. The influence of bone marrow-and synovium-derived mesenchymal stromal cells from osteoarthritis patients on regulatory T cells in co-culture. Clin. Exp. Immunol. 2013, 173, 454–462. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviations | Acronym |
|---|---|
| Anti-citrullinated protein antibodies | ACPAs |
| C-X-C chemokine receptor | CXCR |
| Dendritic cells | DC |
| Extracellular matrix | ECM |
| Extracellular vesicles | EVs |
| Fibroblast-like synoviocytes | FLSs |
| Immunoglobulins | Ig |
| Indolamine 2,3-dioxygenase | IDO |
| Interferon gamma | IFN-γ |
| Interleukin | IL |
| Intracellular adhesion molecule 1 | ICAM1 |
| Macrophage inflammatory proteins | MIP |
| Matrix metalloproteinases | MMP |
| Mesenchymal stem cells | MSCs |
| Natural killer cells | NK cells |
| Peptidyl arginine deiminase | PAD |
| Prostaglandin E2 | PGE2 |
| Rheumatoid arthritis | RA |
| Synovial fluid | SF |
| T regulatory cells | Treg |
| Toll-like receptors | TLR |
| Transforming growth factor beta | TGF-β |
| Tumor necrosis factor alpha | TNF-α |
| Vascular adhesion molecule 1 | VCAM1 |
| Positive ≥ 95% | Negative ≤ 2% |
|---|---|
| CD105 | CD34 |
| CD73 | CD14, CD11b |
| CD90 | CD79 alpha, CD19 |
| HLA-DR |
| Markers | Human MCSs | Properties/Functions |
|---|---|---|
| CD105/Endoglin | Bone marrow MSCs (BM MSCs), adipose tissue MSCs (ADSC), umbilical blood cord MSCs (UCB MSCs) | A type I transmembrane protein reported to induce activation and proliferation of endothelial cells and co-receptor for (transforming growth factor beta) TGF-β [34] |
| CD90/Thy-1 | BM MSCs, ADSCs, UCB MSCs | Surface marker hypothesized to function in cell-cell and cell-matrix interactions, nerve regeneration, apoptosis, inflammation [32] |
| CD73/Ecto-5′-nucleotidase | BM MSCs, ADSC, UCB MSCs | Catalyzes the conversion at neutral pH of purine 5-prime mononucleotides to nucleosides, the preferred substrate being adenosine 5′-monophosphate (AMP) [32] |
| Stro-1 | BM MSCs, Amnion MSCs (AMSCs), Synovial membrane derived MSCs | Cell surface antigen in human bone marrow cells capable of differentiating stromal cells with a vascular smooth muscle-like phenotype, adipocytes, osteoblasts and chondrocytes [35] |
| CD271/LNGFR/Low-affinity nerve growth factor receptor | BM MSCs, ADSC, Placenta MSCs, Wharton Jely derived MSCs (WJ MSCs), AMSCs, Chorion MSCs (CMSCs) | The specific markers for the purification of human BM-MSCs [36] |
| Oct-4/Octamer-binding protein 4 | AMSCs, CMSCs | Transcription factors for pluripotency and self-renewal [35] |
| SSEA-4/Stage-specific embryonic antigen-4 | BM MSCs, Synovial membrane derived MSCs | Stage-specific embryonic antigen and MSCs from whole human bone marrow [35,41] |
| CD146/MCAM/Melanoma cell adhesion molecule | BM MSCs, Synovial membrane derived MSCs, Pericytes | Receptor for laminin alpha 4, a matrix molecule that is broadly expressed within the vascular wall [37] |
| Sox11/SRY-Box Transcription Factor 11 | BM MSCs | Marker downregulated during culture. Knockdown affect proliferation and osteogenesis potential [41] |
| CD349/Frizzled-9 | AMSCs, CMSCs | A novel marker for isolation of MSC from placenta. Members of the ‘frizzled’ gene family encode 7-transmembrane domain proteins that are receptors for Wnt signaling proteins [35] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bačenková, D.; Trebuňová, M.; Morochovič, R.; Dosedla, E.; Findrik Balogová, A.; Gašparová, P.; Živčák, J. Interaction between Mesenchymal Stem Cells and the Immune System in Rheumatoid Arthritis. Pharmaceuticals 2022, 15, 941. https://doi.org/10.3390/ph15080941
Bačenková D, Trebuňová M, Morochovič R, Dosedla E, Findrik Balogová A, Gašparová P, Živčák J. Interaction between Mesenchymal Stem Cells and the Immune System in Rheumatoid Arthritis. Pharmaceuticals. 2022; 15(8):941. https://doi.org/10.3390/ph15080941
Chicago/Turabian StyleBačenková, Darina, Marianna Trebuňová, Radoslav Morochovič, Erik Dosedla, Alena Findrik Balogová, Petra Gašparová, and Jozef Živčák. 2022. "Interaction between Mesenchymal Stem Cells and the Immune System in Rheumatoid Arthritis" Pharmaceuticals 15, no. 8: 941. https://doi.org/10.3390/ph15080941
APA StyleBačenková, D., Trebuňová, M., Morochovič, R., Dosedla, E., Findrik Balogová, A., Gašparová, P., & Živčák, J. (2022). Interaction between Mesenchymal Stem Cells and the Immune System in Rheumatoid Arthritis. Pharmaceuticals, 15(8), 941. https://doi.org/10.3390/ph15080941