
Design, Synthesis, and Antitumor Evaluation of Novel Mono-Carbonyl Curcumin Analogs in Hepatocellular Carcinoma Cell

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Novel Curcumin Analogs Strongly Inhibit Proliferation in HepG2 Cell
2.3. Inhibition Effect on HepG2 Cell Migration
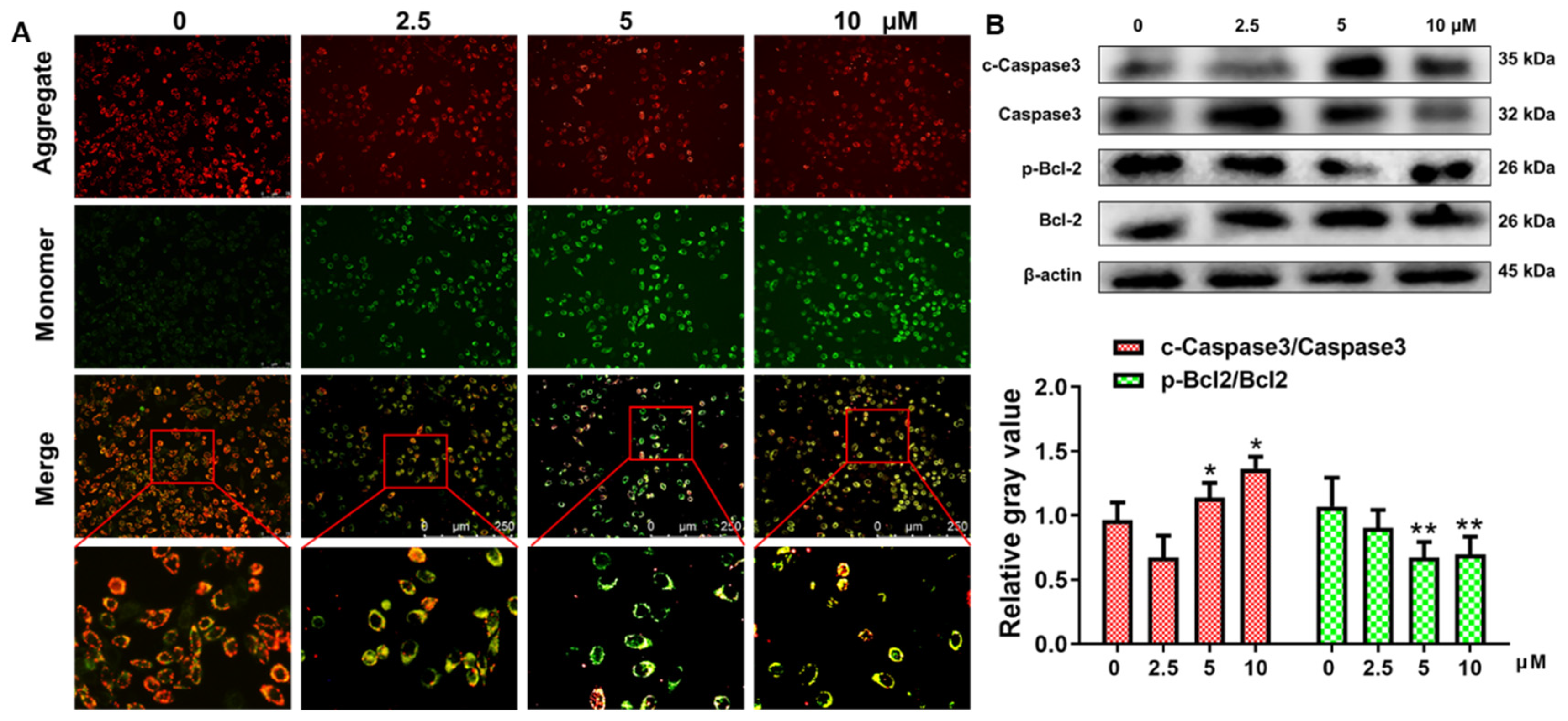
2.4. Apoptosis of HepG2 Cells through Mitochondrial Death Pathway Generation
2.5. Curcumin-Induced Comprehensive Transcriptomes Profile in HepG2 Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Description | Significant | Annotated | Pvalue | Qvalue |
|---|---|---|---|---|---|
| ko05202 | Transcriptional misregulation in cancers | 70/1805 | 169/7736 | 1.00 × 10−7 | 3.00 × 10−5 |
| ko05034 | Alcoholism | 69/1805 | 180/7736 | 3.67 × 10−6 | 0.000551 |
| ko04151 | PI3K-Akt signaling pathway | 109/1805 | 337/7736 | 7.10 × 10−5 | 0.007097 |
| ko05206 | MicroRNAs in cancer | 52/1805 | 138/7736 | 9.58 × 10−5 | 0.007186 |
| ko00532 | Glycosaminoglycan biosynthesis—chondroitin sulfate/dermatan sulfate | 12/1805 | 20/7736 | 0.000469 | 0.027069 |
| ko04068 | FoxO signaling pathway | 46/1805 | 126/7736 | 0.000541 | 0.027069 |
| ko00270 | Cysteine and methionine metabolism | 21/1805 | 46/7736 | 0.000698 | 0.029904 |
| ko04971 | Gastric acid secretion | 29/1805 | 74/7736 | 0.001596 | 0.059848 |
| ko04015 | Rap1 signaling pathway | 67/1805 | 208/7736 | 0.001903 | 0.063443 |
| ko04115 | p53 signaling pathway | 26/1805 | 66/7736 | 0.002502 | 0.075074 |
| ko05219 | Bladder cancer | 18/1805 | 42/7736 | 0.003896 | 0.092958 |
| ko05224 | Breast cancer | 49/1805 | 148/7736 | 0.004009 | 0.092958 |
| ko04975 | Fat digestion and absorption | 17/1805 | 39/7736 | 0.004028 | 0.092958 |
| ko04512 | ECM-receptor interaction | 30/1805 | 84/7736 | 0.006771 | 0.140346 |
| ko04810 | Regulation of actin cytoskeleton | 65/1805 | 211/7736 | 0.007017 | 0.140346 |
| ko04024 | cAMP signaling pathway | 61/1805 | 197/7736 | 0.007824 | 0.144381 |
| ko05215 | Prostate cancer | 30/1805 | 85/7736 | 0.008182 | 0.144381 |
| ko04510 | Focal adhesion | 60/1805 | 195/7736 | 0.009586 | 0.154245 |
| ko04390 | Hippo signaling pathway | 47/1805 | 147/7736 | 0.009769 | 0.154245 |
| ko05200 | Pathways in cancer | 111/1805 | 392/7736 | 0.01089 | 0.161349 |
| ko00052 | Galactose metabolism | 14/1805 | 33/7736 | 0.011553 | 0.161349 |
| ko00900 | Terpenoid backbone biosynthesis | 10/1805 | 21/7736 | 0.012603 | 0.161349 |
| ko04391 | Hippo signaling pathway-fly | 25/1805 | 70/7736 | 0.01275 | 0.161349 |
| ko04066 | HIF-1 signaling pathway | 32/1805 | 95/7736 | 0.013676 | 0.161349 |
| ko04013 | MAPK signaling pathway-fly | 28/1805 | 81/7736 | 0.014076 | 0.161349 |
| ko05143 | African trypanosomiasis | 15/1805 | 37/7736 | 0.014586 | 0.161349 |
| ko04071 | Sphingolipid signaling pathway | 41/1805 | 128/7736 | 0.01462 | 0.161349 |
| ko04725 | Cholinergic synapse | 36/1805 | 110/7736 | 0.015059 | 0.161349 |
| ko04640 | Hematopoietic cell lineage | 28/1805 | 84/7736 | 0.023258 | 0.240598 |
| ko04320 | Dorso-ventral axis formation | 11/1805 | 26/7736 | 0.024647 | 0.245998 |
2.6. The Inhibitory Mechanism of Anti-HCC Activity of Curcumin and B5 toward HepG2 Cells
3. Experiments
3.1. General
3.2. General Procedure for the Preparation of Curcumin Analogs A1–A8
3.2.1. ((1E,6E)-3,5-dioxohepta-1,6-diene-1,7-diyl)bis(2-methoxy-4,1-phenylene) (2E,2′E)-bis(3-(4-fluorophenyl)acrylate) (A1)
3.2.2. ((1E,6E)-3,5-dioxohepta-1,6-diene-1,7-diyl)bis(2-methoxy-4,1-phenylene) (2E,2′E)-bis(3-(2-chlorophenyl)acrylate) (A2)
3.2.3. ((1E,6E)-3,5-dioxohepta-1,6-diene-1,7-diyl)bis(2-methoxy-4,1-phenylene) (2E,2′E)-bis(3-(3-chlorophenyl)acrylate) (A3)
3.2.4. ((1E,6E)-3,5-dioxohepta-1,6-diene-1,7-diyl)bis(2-methoxy-4,1-phenylene) (2E,2′E)-bis(3-(3-(trifluoromethyl)phenyl)acrylate) (A4)
3.2.5. ((1E,6E)-3,5-dioxohepta-1,6-diene-1,7-diyl)bis(2-methoxy-4,1-phenylene) (2E,2′E)-bis(3-(4-(trifluoromethyl)phenyl)acrylate) (A5)
3.2.6. ((1E,6E)-3,5-dioxohepta-1,6-diene-1,7-diyl)bis(2-methoxy-4,1-phenylene) (2E,2′E)-bis(3-(3-nitrophenyl)acrylate) (A6)
3.2.7. ((1E,6E)-3,5-dioxohepta-1,6-diene-1,7-diyl)bis(2-methoxy-4,1-phenylene) (2E,2′E)-bis(3-(2-nitrophenyl)acrylate) (A7)
3.2.8. ((1E,6E)-3,5-dioxohepta-1,6-diene-1,7-diyl)bis(2-methoxy-4,1-phenylene) (2E,2′E)-bis(3-(p-tolyl)acrylate) (A8)
3.3. General Procedure for the Preparation of Curcumin Analogs B1–B7
3.3.1. (1E,4E)-1,5-bis(4-hydroxy-3-methoxyphenyl)penta-1,4-dien-3-one (3)
3.3.2. ((1E,4E)-3-oxopenta-1,4-diene-1,5-diyl)bis(2-methoxy-4,1-phenylene) (2E,2′E)-bis(3-(4-fluorophenyl)acrylate) (B1)
3.3.3. ((E,4E)-3-oxopenta-1,4-diene-1,5-diyl)bis(2-methoxy-4,1-phenylene) (2E,2′E)-bis(3-(2-chlorophenyl)acrylate) (B2)
3.3.4. ((1E,4E)-3-oxopenta-1,4-diene-1,5-diyl)bis(2-methoxy-4,1-phenylene) (2E,2′E)-bis(3-(2-fluorophenyl)acrylate) (B3)
3.3.5. ((1E,4E)-3-oxopenta-1,4-diene-1,5-diyl)bis(2-methoxy-4,1-phenylene) (2E,2′E)-bis(3-(3-(trifluoromethyl)phenyl)acrylate) (B4)
3.3.6. ((1E,4E)-3-oxopenta-1,4-diene-1,5-diyl)bis(2-methoxy-4,1-phenylene) (2E,2′E)-bis(3-(3-nitrophenyl)acrylate) (B5)
3.3.7. ((1E,4E)-3-oxopenta-1,4-diene-1,5-diyl)bis(2-methoxy-4,1-phenylene) (2E,2′E)-bis(3-(p-tolyl)acrylate) (B6)
3.3.8. ((1E,4E)-3-oxopenta-1,4-diene-1,5-diyl)bis(2-methoxy-4,1-phenylene) (2E,2′E)-bis(3-(p-tolyl)acrylate) (B7)
3.4. General Procedure for the Preparation of Curcumin Analogs C1–C6
3.4.1. (E)-1,3-bis(4-hydroxy-3-methoxyphenyl)prop-2-en-1-one (8)
3.4.2. ((E)-3-oxoprop-1-ene-1,3-diyl)bis(2-methoxy-4,1-phenylene) (2E,2′E)-bis(3-(4-fluorophenyl)acrylate) (C1)
3.4.3. ((E)-3-oxoprop-1-ene-1,3-diyl)bis(2-methoxy-4,1-phenylene) (2E,2′E)-bis(3-(2-chlorophenyl)acrylate) (C2)
3.4.4. ((E)-3-oxoprop-1-ene-1,3-diyl)bis(2-methoxy-4,1-phenylene) (2E,2′E)-bis(3-(3-chlorophenyl)acrylate) (C3)
3.4.5. ((E)-3-oxoprop-1-ene-1,3-diyl)bis(2-methoxy-4,1-phenylene) (2E,2′E)-bis(3-(3-(trifluoromethyl)phenyl)acrylate) (C4)
3.4.6. ((E)-3-oxoprop-1-ene-1,3-diyl)bis(2-methoxy-4,1-phenylene) (2E,2′E)-bis(3-(4-(trifluoromethyl)phenyl)acrylate) (C5)
3.4.7. ((E)-3-oxoprop-1-ene-1,3-diyl)bis(2-methoxy-4,1-phenylene) (2E,2′E)-bis(3-(p-tolyl)acrylate) (C6)
3.5. Cell Culture
3.6. MTT Assay
3.7. Clone Formation
3.8. Wound Healing Assay
3.9. Mitochondrial Membrane Potential Monitored by JC-1 Staining
3.10. Western Blot Analysis
3.11. Molecular Docking Studies
3.12. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Zhao, S.; Pi, C.; Ye, Y.; Zhao, L.; Wei, Y. Recent advances of analogues of curcumin for treatment of cancer. Eur. J. Med. Chem. 2019, 180, 524–535. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, D.K.; Mishra, P.K. Curcumin and its analogues: Potential anticancer agents. Med. Res. Rev. 2010, 30, 818–860. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ning, J.; Peng, L.; He, D. Effect of curcumin on Bcl-2 and Bax expression in nude mice prostate cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 9272–9278. [Google Scholar] [PubMed]
- Yang, J.; Cao, Y.; Sun, J.; Zhang, Y. Curcumin reduces the expression of Bcl-2 by upregulating miR-15a and miR-16 in MCF-7 cells. Med. Oncol. 2010, 27, 1114–1118. [Google Scholar] [CrossRef] [PubMed]
- Mou, S.; Zhou, Z.; He, Y.; Liu, F.; Gong, L. Curcumin inhibits cell proliferation and promotes apoptosis of laryngeal cancer cells through Bcl-2 and PI3K/Akt, and by upregulating miR-15a. Oncol. Lett. 2017, 14, 4937–4942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rana, C.; Piplani, H.; Vaish, V.; Nehru, B.; Sanyal, S.N. Downregulation of PI3-K/Akt/PTEN pathway and activation of mitochondrial intrinsic apoptosis by Diclofenac and Curcumin in colon cancer. Mol. Cell Biochem. 2015, 402, 225–241. [Google Scholar] [CrossRef] [PubMed]
- Al-Kahtani, M.; Abdel-Daim, M.M.; Sayed, A.A.; El-Kott, A.; Morsy, K. Curcumin phytosome modulates aluminum-induced hepatotoxicity via regulation of antioxidant, Bcl-2, and caspase-3 in rats. Environ. Sci. Pollut. Res. Int. 2020, 27, 21977–21985. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.; Xing, J.; Yu, X. Curcumin induces osteosarcoma MG63 cells apoptosis via ROS/Cyto-C/Caspase-3 pathway. Tumour. Biol. 2014, 35, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of curcumin: Problems and promises. Mol. Pharm. 2007, 4, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Ireson, C.R.; Jones, D.J.; Orr, S.; Coughtrie, M.W.; Boocock, D.J.; Williams, M.L.; Farmer, P.B.; Steward, W.P.; Gescher, A.J. Metabolism of the cancer chemopreventive agent curcumin in human and rat intestine. Cancer Epidemiol. Biomark. Prev. 2002, 11, 105–111. [Google Scholar]
- Weber, W.M.; Hunsaker, L.A.; Abcouwer, S.F.; Deck, L.M.; Vander Jagt, D.L. Anti-oxidant activities of curcumin and related enones. Bioorg. Med. Chem. 2005, 13, 3811–3820. [Google Scholar] [CrossRef]
- Weber, W.M.; Hunsaker, L.A.; Roybal, C.N.; Bobrovnikova-Marjon, E.V.; Abcouwer, S.F.; Royer, R.E.; Deck, L.M.; Vander Jagt, D.L. Activation of NFkappaB is inhibited by curcumin and related enones. Bioorg. Med. Chem. 2006, 14, 2450–2461. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Zhao, C.G.; He, W.F.; Wang, Z.; Fang, Q.L.; Xiao, B.; Liu, Z.G.; Liang, G.; Yang, S.L. Discovery and evaluation of asymmetrical monocarbonyl analogs of curcumin as anti-inflammatory agents. Drug Des. Dev. Ther. 2014, 8, 373–382. [Google Scholar]
- Zhao, C.; Liu, Z.; Liang, G. Promising curcumin-based drug design: Mono-carbonyl analogues of curcumin (MACs). Curr. Pharm. Des. 2013, 19, 2114–2135. [Google Scholar] [PubMed]
- Quincoces Suarez, J.A.; Rando, D.G.; Santos, R.P.; Goncalves, C.P.; Ferreira, E.; de Carvalho, J.E.; Kohn, L.; Maria, D.A.; Faiao-Flores, F.; Michalik, D.; et al. New antitumoral agents I: In vitro anticancer activity and in vivo acute toxicity of synthetic 1,5-bis(4-hydroxy-3-methoxyphenyl)-1,4-pentadien-3-one and derivatives. Bioorg. Med. Chem. 2010, 18, 6275–6281. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Shih, C.C.; Lee, K.H. Novel anti-prostate cancer curcumin analogues that enhance androgen receptor degradation activity. Anti-Cancer Agents Med. Chem. 2009, 9, 904–912. [Google Scholar] [CrossRef]
- Reddy, A.R.; Dinesh, P.; Prabhakar, A.S.; Umasankar, K.; Shireesha, B.; Raju, M.B. A comprehensive review on SAR of curcumin. Mini Rev. Med. Chem. 2013, 13, 1769–1777. [Google Scholar] [CrossRef]
- Rodrigues, F.C.; Anil Kumar, N.V.; Thakur, G. Developments in the anticancer activity of structurally modified curcumin: An up-to-date review. Eur. J. Med. Chem. 2019, 177, 76–104. [Google Scholar] [CrossRef]
- Feng, L.; Li, Y.; Song, Z.F.; Li, H.J.; Huai, Q.Y. Synthesis and Biological Evaluation of Curcuminoid Derivatives. Chem. Pharm. Bull. 2015, 63, 873–881. [Google Scholar] [CrossRef] [Green Version]
- Yu, P.; Li, D.D.; Ni, J.J.; Xia, C.J.; Wang, Z.Z.; Xiao, W.; Ding, G.; Zhao, L.G. Synthesis and Antitumor Activity of C-3(R) Hydroxy Modified Betulinic Acid Derivatives. Chem. Nat. Compd. 2019, 55, 1080–1084. [Google Scholar] [CrossRef]
- Cao, W.; Gao, J.; Zhang, Y.; Li, A.; Yu, P.; Cao, N.; Liang, J.; Tang, X. Autophagy up-regulated by MEK/ERK promotes the repair of DNA damage caused by aflatoxin B1. Toxicol. Mech. Methods 2021, 32, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Ali, A.; Lv, Z.; Wang, N.; Zhao, X.; Hao, H.; Zhang, Y.; Rahman, F.U. Platinum complexes inhibit HER-2 enriched and triple-negative breast cancer cells metabolism to suppress growth, stemness and migration by targeting PKM/LDHA and CCND1/BCL2/ATG3 signaling pathways. Eur. J. Med. Chem. 2021, 224, 113689. [Google Scholar] [CrossRef] [PubMed]
- Burke, P.J. Mitochondria, Bioenergetics and Apoptosis in Cancer. Trends Cancer 2017, 3, 857–870. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Peng, B.; Xiao, B.B.; Sheng-Li, C.; Yang, C.R.; Wang, W.Z.; Wang, F.C.; Li, H.Y.; Yuan, X.L.; Shi, R.; et al. Synthesis and evaluation of chalcone analogues containing a 4-oxoquinazolin-2-yl group as potential anti-tumor agents. Eur. J. Med. Chem. 2019, 162, 586–601. [Google Scholar] [CrossRef]
- Siddiqui, W.A.; Ahad, A.; Ahsan, H. The mystery of BCL2 family: Bcl-2 proteins and apoptosis: An update. Arch. Toxicol. 2015, 89, 289–317. [Google Scholar] [CrossRef]
- Ola, M.S.; Nawaz, M.; Ahsan, H. Role of Bcl-2 family proteins and caspases in the regulation of apoptosis. Mol. Cell Biochem. 2011, 351, 41–58. [Google Scholar] [CrossRef]
- Xing, Z.; Chu, C.; Chen, L.; Kong, X. The use of Gene Ontology terms and KEGG pathways for analysis and prediction of oncogenes. Biochim. Biophys. Acta 2016, 1860 Pt B, 2725–2734. [Google Scholar] [CrossRef]
- Yu, P.; Xia, C.J.; Li, D.D.; Ni, J.J.; Zhao, L.G.; Ding, G.; Wang, Z.Z.; Xiao, W. Design, synthesis, and anti-inflammatory activity of caffeoyl salicylate analogs as NO production inhibitors. Fitoterapia 2018, 129, 25–33. [Google Scholar] [CrossRef]
- Wu, W.I.; Voegtli, W.C.; Sturgis, H.L.; Dizon, F.P.; Vigers, G.P.; Brandhuber, B.J. Crystal structure of human AKT1 with an allosteric inhibitor reveals a new mode of kinase inhibition. PLoS ONE 2010, 5, e12913. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Zuo, Y.; Huang, J.; Zhou, B.; Wang, S.; Shao, W.; Zhu, C.; Lin, L.; Wen, G.; Wang, H.; Du, J.; et al. Synthesis, cytotoxicity of new 4-arylidene curcumin analogues and their multi-functions in inhibition of both NF-kappaB and Akt signalling. Eur. J. Med. Chem. 2012, 55, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Schuttelkopf, A.W.; van Aalten, D.M. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 2004, 60 Pt 8, 1355–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Q.; Wu, N.; Li, H.L.; Zhang, J.Y.; Li, X.; Deng, M.F.; Zhu, K.; Wang, J.E.; Duan, H.X.; Yang, Q. A Piperine-Based Scaffold as a Novel Starting Point to Develop Inhibitors against the Potent Molecular Target OfChtI. J. Agric. Food Chem. 2021, 69, 7534–7544. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Ong, E.E.S.; Liow, J.L. The temperature-dependent structure, hydrogen bonding and other related dynamic properties of the standard TIP3P and CHARMM-modified TIP3P water models. Fluid Phase Equilibr. 2019, 481, 55–65. [Google Scholar] [CrossRef]
- Yan, S.R.; Toghraie, D.; Hekmatifar, M.; Miansari, M.; Rostami, S. Molecular dynamics simulation of Water-Copper nanofluid flow in a three-dimensional nanochannel with different types of surface roughness geometry for energy economic management. J. Mol. Liq. 2020, 311, 113222. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Gotz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]



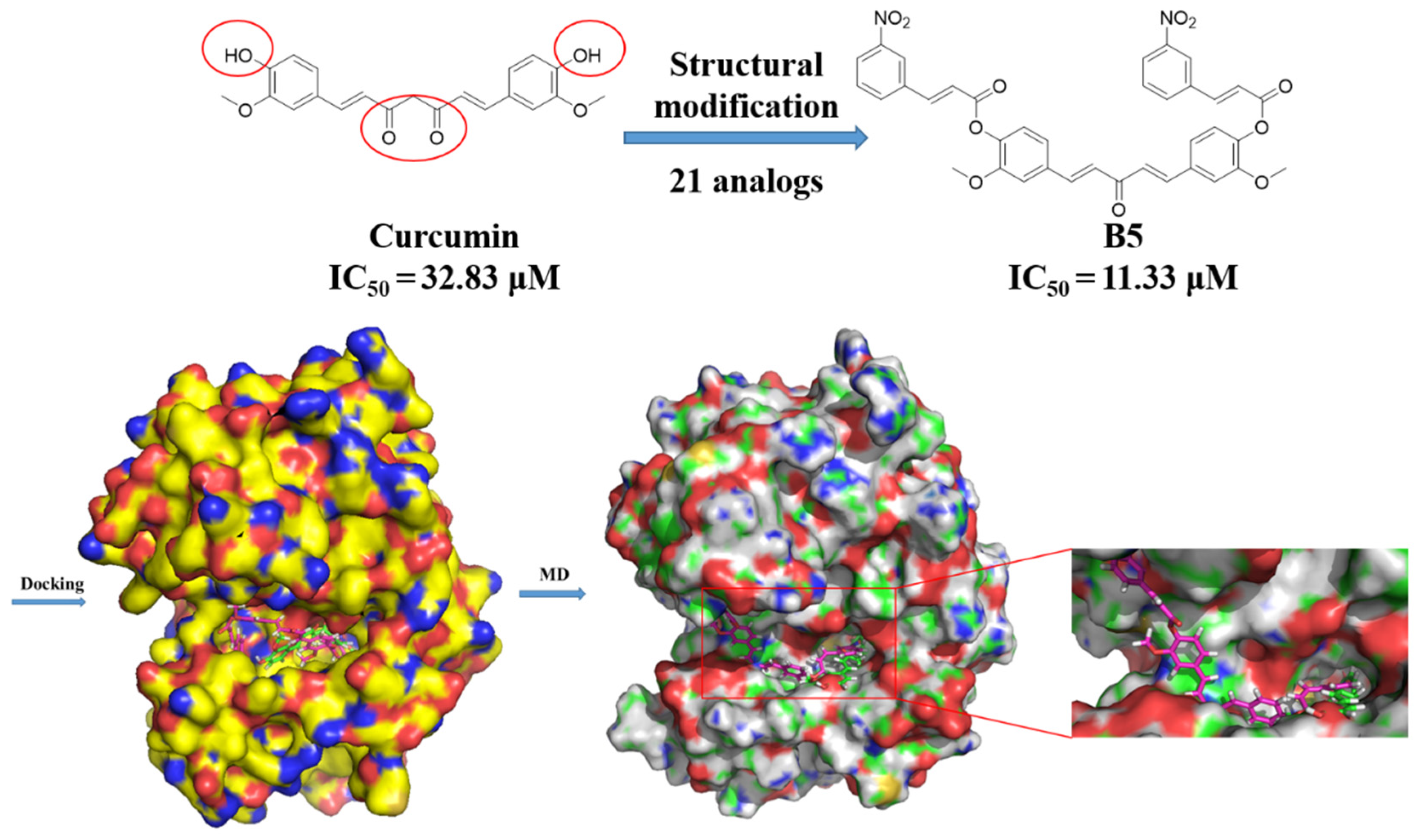

| Predicted ΔG (Kcal/mol) | |||||||
|---|---|---|---|---|---|---|---|
| Compound | Evdw | Eel | Egb | Esurf | ΔGgas | ΔGsolv | ΔGtotal |
| Curcumin | −38.18 ± 3.20 | −25.84 ± 3.43 | 42.16 ± 2.29 | −5.21 ± 0.31 | −64.02 ± 4.12 | 36.96 ± 2.15 | −27.06 ± 3.22 |
| B5 | −73.18 ± 3.97 | −33.57 ± 7.04 | 61.54 ± 8.20 | −9.81 ± 0.52 | −106.76 ± 9.26 | 51.74 ± 7.89 | −55.02 ± 3.22 |
| Compounds | Key Residues (<−1.0 Kcal/mol) |
|---|---|
| Curcumin | LEU156, VAL164, THR211, MET227, GLU228, TYR229, THR291, PHE442 |
| B5 | LEU156, GLY157, PHE161, LYS163, VAL164, HIE194, MET281, LEU295, PHE309, CYS310, PRO313, LEU316 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, P.; Cao, W.; Zhao, L.; Han, Q.; Yang, S.; Yang, K.; Pan, X.; Wang, Q.; Wang, Y. Design, Synthesis, and Antitumor Evaluation of Novel Mono-Carbonyl Curcumin Analogs in Hepatocellular Carcinoma Cell. Pharmaceuticals 2022, 15, 950. https://doi.org/10.3390/ph15080950
Yu P, Cao W, Zhao L, Han Q, Yang S, Yang K, Pan X, Wang Q, Wang Y. Design, Synthesis, and Antitumor Evaluation of Novel Mono-Carbonyl Curcumin Analogs in Hepatocellular Carcinoma Cell. Pharmaceuticals. 2022; 15(8):950. https://doi.org/10.3390/ph15080950
Chicago/Turabian StyleYu, Pan, Weiya Cao, Linguo Zhao, Qing Han, Shilong Yang, Kepeng Yang, Xiaolei Pan, Qianyun Wang, and Yuan Wang. 2022. "Design, Synthesis, and Antitumor Evaluation of Novel Mono-Carbonyl Curcumin Analogs in Hepatocellular Carcinoma Cell" Pharmaceuticals 15, no. 8: 950. https://doi.org/10.3390/ph15080950
APA StyleYu, P., Cao, W., Zhao, L., Han, Q., Yang, S., Yang, K., Pan, X., Wang, Q., & Wang, Y. (2022). Design, Synthesis, and Antitumor Evaluation of Novel Mono-Carbonyl Curcumin Analogs in Hepatocellular Carcinoma Cell. Pharmaceuticals, 15(8), 950. https://doi.org/10.3390/ph15080950