

Leukotrienes vs. Montelukast—Activity, Metabolism, and Toxicity Hints for Repurposing



Abstract
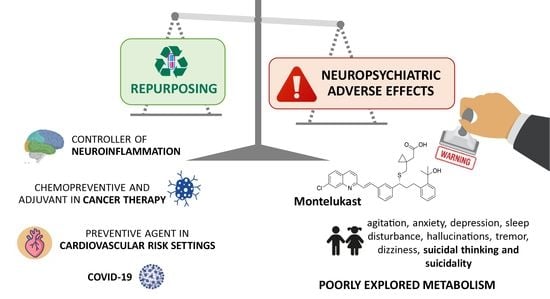
1. Introduction
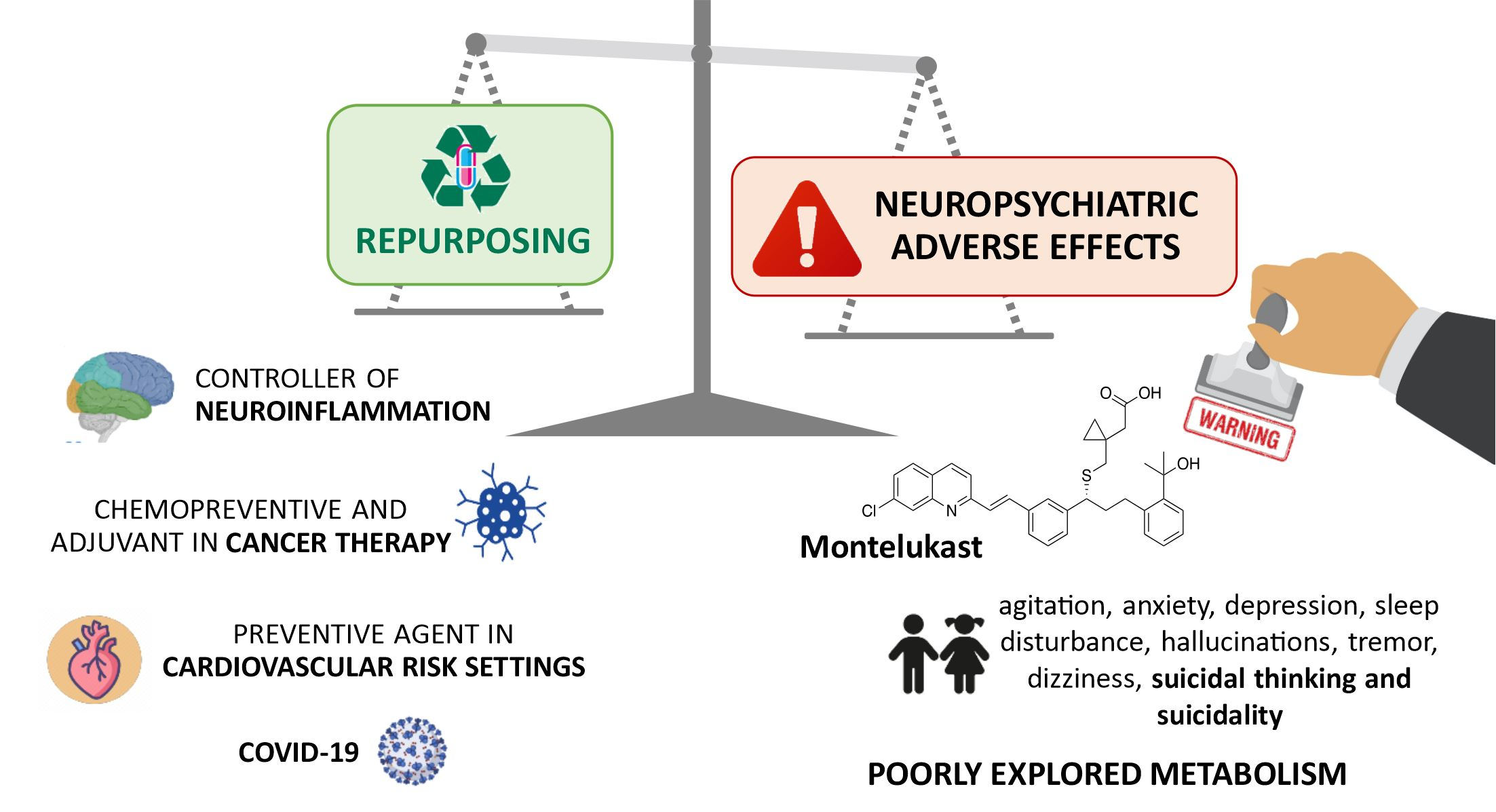
2. Cysteinyl Leukotrienes—Multifunctional Inflammation Mediators
2.1. Cysteinyl Leukotrienes and Their Receptors

2.2. Leukotrienes in the Brain
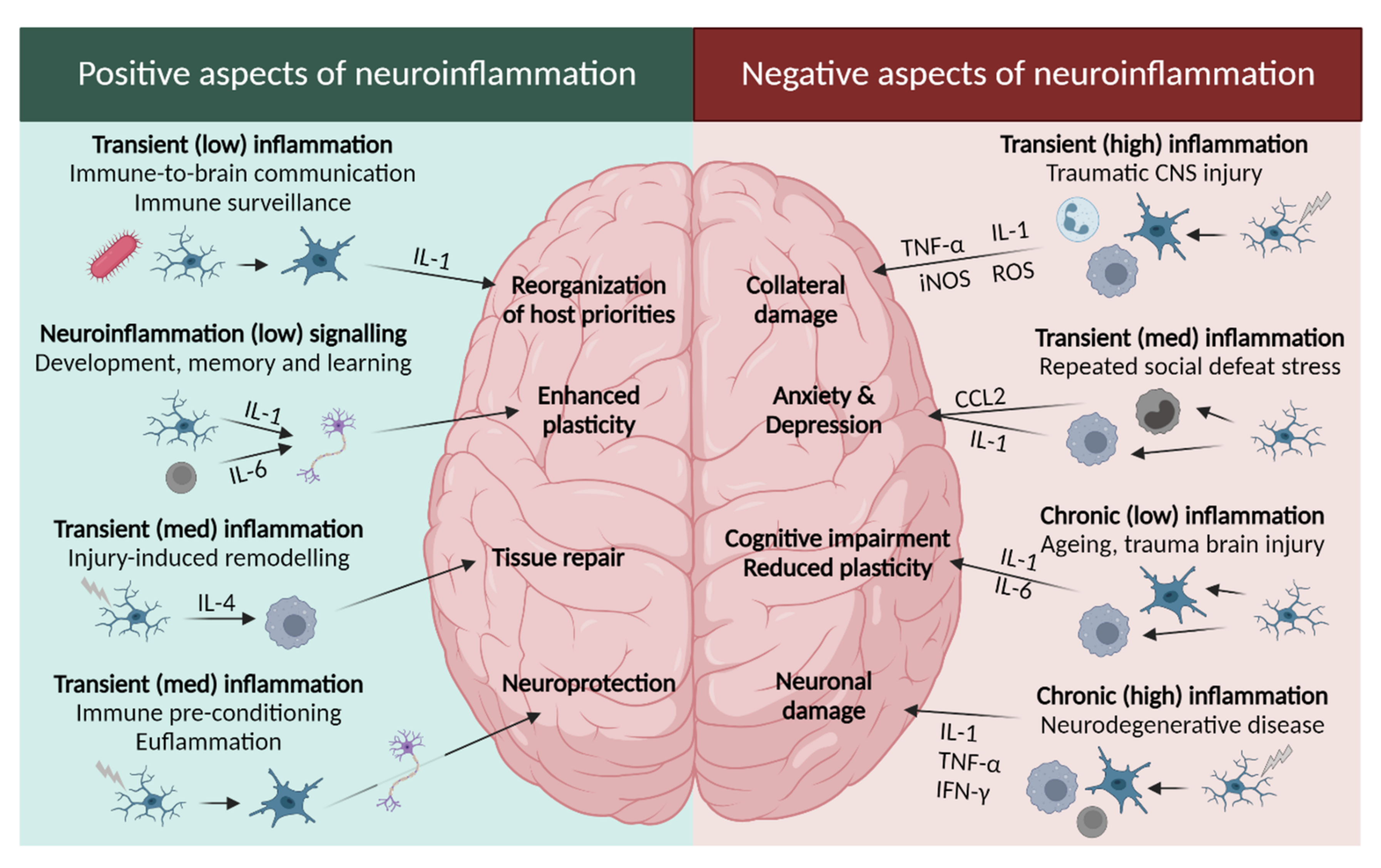
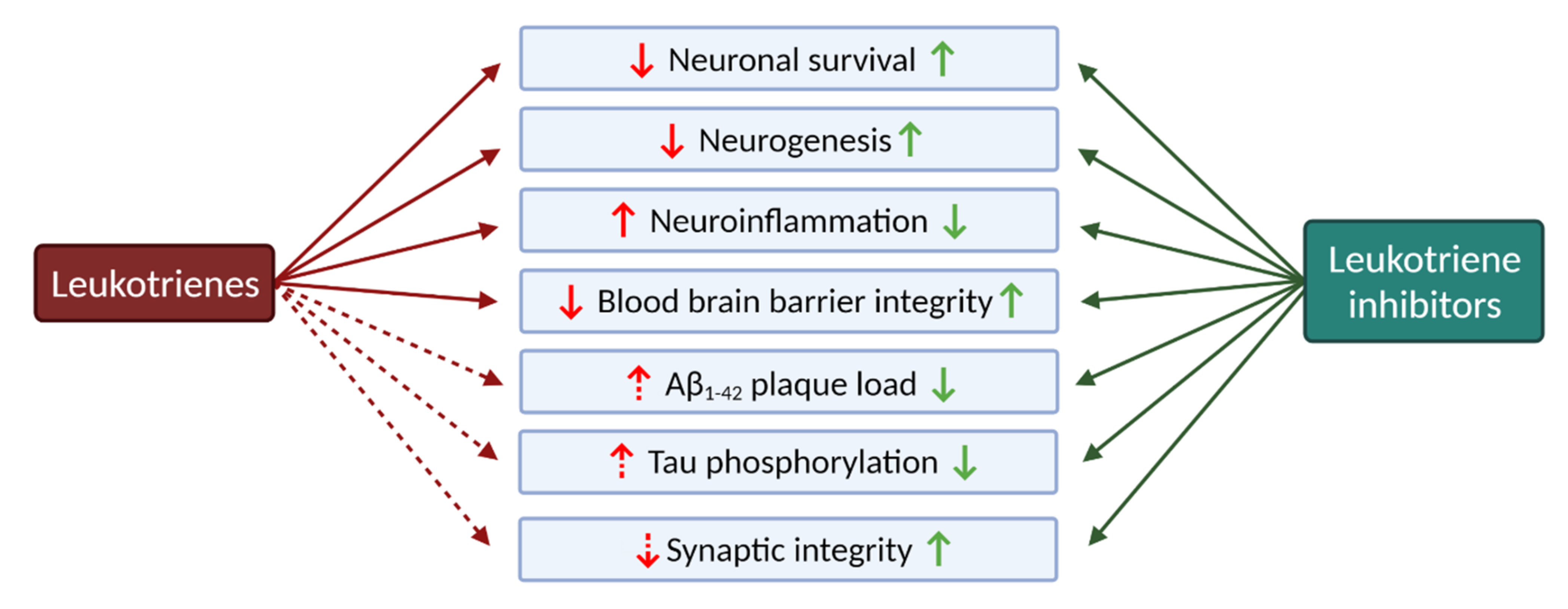
2.2.1. Leukotrienes: Role in Neuroinflammation
2.2.2. Leukotrienes in Neuro-Signalling Pathways
2.2.3. The Leukotriene Link between Stress and Depression
2.2.4. The Role of Leukotrienes in Neurodegenerative Diseases
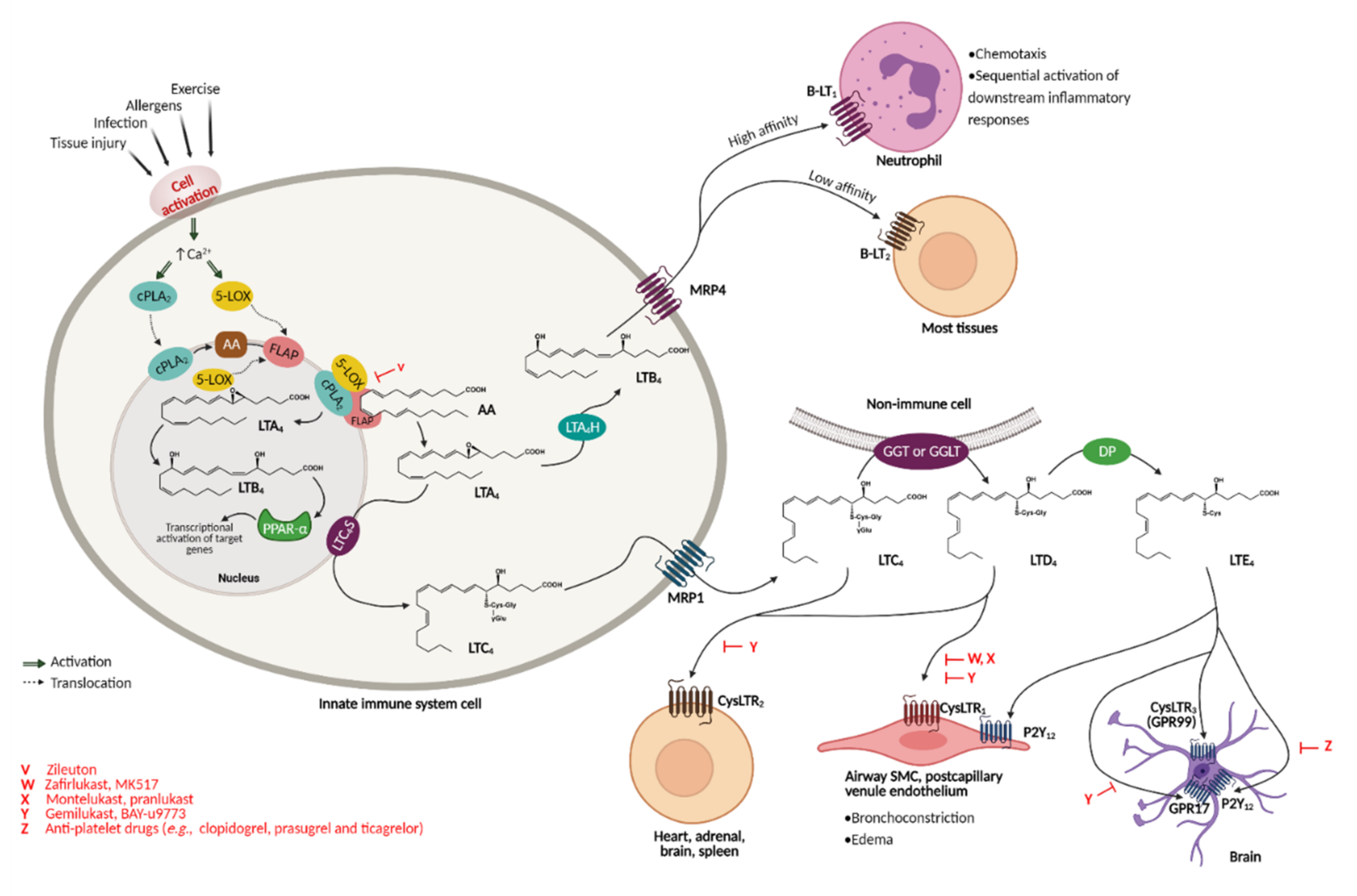
3. A Cysteine Leukotriene Receptor Antagonist Known as Montelukast
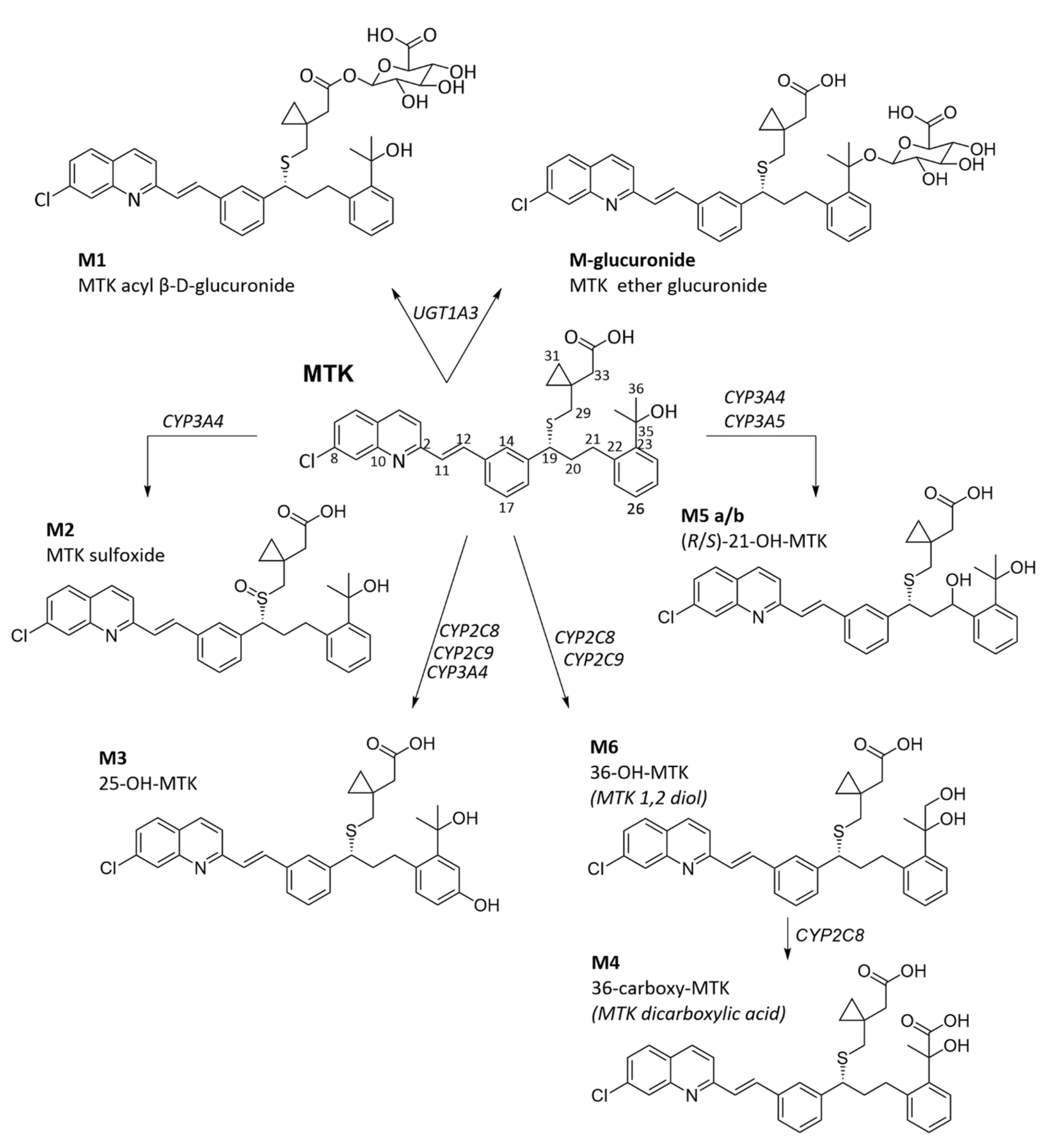
3.1. Montelukast Metabolism and Bioavailability

3.2. Adverse Drug Reactions Related to Montelukast Administration
3.2.1. Neuropsychiatric and Nervous System Disorders
3.2.2. Hepatobiliary, Pancreatic, and Uropoietic Disorders
3.2.3. Skin and Subcutaneous Tissue Disorders
3.2.4. Immune System Disorders
3.2.5. Montelukast Administration during Pregnancy
3.3. Montelukast Repurposing Applications

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Models | Modulation | Outcome | |
|---|---|---|---|
| Bones and joints | |||
| C57B/6 mice with a femoral fracture | Pharmacological treatment with MTK | ↑ chondrocyte proliferation and early bone formation | [201] |
| In vitro osteoarthritis model with chondrocytes (ATDC5) | Pharmacological treatment with MTK | ↓ cartilage degradation; ↓ cell injury, oxidative stress, apoptosis; ↓ CysLTR1 expression; ↑ KLF2 expression | [202] |
| Cancer | |||
| Nationwide population-based study with data from the Taiwan National Health Insurance Research Database | Cancer patients with diagnosed asthma, treated with leukotriene inhibitors | ↓ cancer risk | [192] |
| Human lung cancer cells and Lewis lung-carcinoma-bearing mice | Pharmacological treatment with MTK | Cell proliferation inhibition; ↓ Bcl-2; ↑ Bak; ↑ nuclear translocation of AIF; ↓ phosphorylation of WNK1, Akt, Erk1/2, MEK, and PRAS40 proteins | [191] |
| Prostate cancer cell lines | Pharmacological treatment with MTK | ↓ HIF-1α protein; ↑ phosphorylation of eIF-2α | [193] |
| Phorbol-myristate--acetate-differentiated U937 cells | Pharmacological effect of MTK | ↓ TNF-α-stimulated IL-8 expression; no effect on NF-kB p65 activation; suppressed NF-kB p65-associated HAT activity | [195] |
| Tumour specimens from patients with prostate cancer and prostate cancer cell lines | Pharmacological treatment with MTK | CysLTR1 overexpressed in prostate tissues; ↑ apoptosis of prostate cancer cells | [194] |
| Cardiovascular | |||
| Nationwide population-based study (Swedish population) | Association between MTK use and cardiovascular outcomes | ↓ recurrent cardiovascular events | [203] |
| Nationwide population-based study (Swedish population) | Association between MTK use and cardiovascular outcomes | ↓ risk of aortic stenosis | [204] |
| Asthmatic patients | Pharmacological effect of MTK on cardiovascular risk | ↓ levels of cardiovascular disease-associated inflammatory biomarkers and lipid levels | [205] |
| CNS: Alzheimer’s disease | |||
| Transgenic 5xFAD Mice (AD mouse model) | Pharmacological effect of MTK on neuroinflammation (microglia and CD8+ T cells) | ↑ Tmem119+; ↓ genes related to AD-associated microglia; ↓ infiltration of CD8+ T-cells into the brain parenchyma; ↑ cognitive functions; ↓ 1061 genes (e.g., Gpr17, Entpd1, Mlec); ↑ 744 genes (e.g., Zfp46, Ciart, Dbp); more pronounced effect in females | [189] |
| Transgenic DCX-DsRed2 and wildtype Fisher 344 rats, FoxO1/3/4fl mice, andGPR17_/_GFPmice | Pharmacological treatment with MTK | ↑ learning and memory in old rats; no effect on learning in young rats; ↓ microglia inflammation; ↑ BBB integrity; ↑ hippocampal neurogenesis; ↓ GPR17; ↓ CD68; ↑ claudin-5; ↑ PCNA, DCX, NeuN | [47] |
| Intracerebroventricular infusions of aggregated Aβ1–42 in ICR mice | Rescue effect of MTK on Aβ1–42-induced neurotoxicity | ↓ memory impairment; ↓ inflammation and apoptosis markers; ↓ CysLTR1 mRNA/protein; ↓ IL-1β, TNF-α, NF-κB p65; ↓ caspase-3; ↑ Bcl-2 | [105] |
| Primary mouse neurons (foetal ICR mice) treated with Aβ1–42 | Rescue effect of MTK on Aβ1–42-induced neurotoxicity | ↑ cell viability; ↓ CysLTR1 mRNA/protein; ↓ IL-1β, TNF-α; NF-κB p65; ↓ caspase-3; ↑ Bcl-2 | [183] |
| Intracerebroventricular streptozotocin-induced model of sporadic AD in ICR mice | Pharmacological treatment with MTK | ↓ memory impairment; ↓ neuroinflammation and apoptosis; ↓ CysLTR1 expression; ↓ TNF-α, IL-1β, NF-κB p65; ↓ cleaved caspase-3; ↑ Bcl-2/Bax ratio | [190] |
| CNS: Anti-nociception | |||
| Local antinociception model of pain | Pharmacological treatment with MTK | ↓ local pain behaviour in both phases (neurogenic and inflammatory); Involvement of L-Arg/NO/cGMP/KATP channel pathway and PPARγ receptors | [206] |
| CNS: Brain ischemia | |||
| Middle cerebral artery occlusion model in mice and rats | Pharmacological treatment with MTK | ↓ behavioural dysfunction, brain infarct volume, brain atrophy, and neuron loss | [207] |
| Bilateral carotid artery occlusion model in rats | Pharmacological prophylaxis and treatment with MTK | ↓ oxidative stress, inflammatory and apoptotic markers (myeloperoxidase, NF-κB, TNF-α, and IL-6); ↓ glutamate and lactate dehydrogenase | [83] |
| CNS: Dementia with Lewy bodies | |||
| Human brain specimen and female transgenic mice expressing human wild-type α-synuclein vs. their wild-type litter mates | Pharmacological treatment with MTK | ↑ memory function; ↓ α-synuclein load in the dentate gyrus; ↑ Beclin-1 expression; autophagy as a possible mechanistic pathway | [187] |
| CNS: Epilepsy | |||
| Epilepsy-induced spontaneous recurrent seizures with pentylenetetrazole (PTZ) in mice | Pharmacological treatment with MTK | Prevention of PTZ-induced BBB disruption; ↓ recurrent seizures; ↓ mean amplitude of electroencephalography recording during seizures | [208] |
| ↓ recurrent seizures; ↓ frequency of daily seizures | [209] | ||
| Pilocarpine-induced seizures in mice | |||
| Electrically-induced seizures in mice | |||
| CNS: Huntington’s disease | |||
| Intrastriatal-quinolinic-acid-and malonic-acid-induced Huntington’s-like symptoms in rats | Pharmacological treatment with MTK | ↓ behavioural alterations; ↓ oxidative stress; ↓ mitochondrial dysfunction; ↓ TNF-α level | [186] |
| CNS: Multiple Sclerosis | |||
| MOG35-55-induced experimental autoimmune encephalomyelitis in female mice | Pharmacological treatment with MTK | ↓ CNS infiltration of inflammatory cells; ↓ clinical symptoms; ↓ IL-17; ↓ BBB disruption | [210] |
| CNS: neurological ageing | |||
| Observational study using data from two databases: NorPD and the Tromsø Study | Association between MTK use and neurological health | Improved cognitive and neurologic function | [188] |
| CNS: Parkinson’s disease | |||
| Rotenone-induced model of PD in rats | Pharmacological treatment with MTK | ↑ locomotor activity; ↓ immobility time; ↓brain MDA levels; ↑ GSH levels; ↓ TNF-α levels | [111] |
| ↑ locomotor activity; ↓ p38 MAPK, TNF-α, IL-1β, NF-κB; ↓ CysLTR1 expression; ↓ p53 mRNA, caspase-3; ↑ GSH, SOD; ↓ MDA levels | [82] | ||
| 6-Hydroxydopamine mouse model (C57BL/6 mice) of PD | Therapeutic effects of MTK | ↓ TNF-α levels; ↓ IL-1β | [107] |
| COVID-19 | |||
| Computational methods | Target-based virtual ligand screening and molecular docking | Well-fitted in the active pocket of SARS-CoV-2 3CLpro, Mpro and RdRp | [211,212] |
| Retrospective study of COVID patients | COVID patients treated with or without MTK | ↓ events of clinical deterioration | [213] |
| Glaucoma | |||
| Magnetic microbead injection into the anterior chamber of female Brown Norway rats | Pharmacological treatment with MTK | ↓ intra ocular pressure; ↑ retinal ganglion cell survival in ocular hypertension eyes; ↓ activation of Iba1+ microglial cells in retina; ↓ GPR17+ cells | [214] |
| Lung transplant | |||
| Bronchiolitis obliterans syndrome after lung transplantation in patients | Pharmacological treatment with MTK | ↓ forced expiratory volume in 1 s (FEV1) | [215,216,217] |
| Pulmonary fibrosis | |||
| Bleomycin-induced pulmonary fibrosis in female C57BL/6J mice | Pharmacological prophylaxis and treatment with MTK | ↓ fibrotic area; ↓ IL-6, IL-10, IL-13, and TGF-β1 mRNA levels; ↑ CysLTR2 mRNA expression | [218] |
| Renal failure | |||
| Rhabdomyolysis-induced acute renal failure in Wistar rats | Pharmacological prophylaxis and treatment with MTK | Improved functional and structural renal damage; ↓ tubular damage; ↓ serum creatinine and urea levels; ↓ serum phosphate levels; ↓ GSH and MDA levels; ↑ SOD levels; ↓ serum TNF-α, TGF-β1, Fas, IL-10; ↑ IL-6/ TNF-α ratio | [219] |
| Cisplatin-induced renal dysfunction in male Sprague Dawley rats | Pharmacological prophylaxis and treatment with MTK | Ameliorated renal toxicity; ↓ responsiveness to acetylcholine; ↓ serum creatinine, blood urea nitrogen, LDH; ↑ serum albumin to normal levels; ↑ GSH levels; ↓ SOD levels | [220] |
| Pyelonephritis induced by Escherichia coli in Wistar rats | Pharmacological treatment with MTK | ↓ severity of kidney damage and renal scarring; ↓ serum TNF-α, creatinine, blood urea nitrogen, MDA levels; ↑ GSH levels | [221] |
4. Human Neurodegenerative Diseases
4.1. Alzheimer’s Disease

4.2. Parkinson’s Disease
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 3CLpro | 3-Chymotrypsin-like protease |
| 5-HpETE | 5(S)-Hydroperoxyeicosatetraenoic acid |
| 5-LOX | 5-Lipoxygenase |
| AA | Arachidonic acid |
| AD | Alzheimer’s disease |
| ADRs | Adverse drug reactions |
| AIF | Apoptosis-inducing factor |
| Akt | Protein kinase B |
| APP | Amyloid-beta precursor protein |
| Aβ | β-Amyloid protein |
| Aβ1–42 | β-Amyloid peptide, amino acids 1 to 42 |
| B-LT | Leukotriene B receptors |
| Bak | Bcl-2 homologous antagonist/killer |
| BAX | Bcl-2-associated X protein |
| BBB | Blood–brain barrier |
| Bcl-2 | B-cell lymphoma 2 |
| CCL2 | C-C motif chemokine 2 |
| CD | Cluster of differentiation |
| cGMP/KATP | Cyclic monophosphate/ATP-sensitive potassium |
| Ciart | Circadian associated repressor of transcription |
| CNS | Central nervous system |
| COVID-19 | SARS-CoV-2 disease |
| cPLA2 | Cytosolic phospholipase A2 |
| CSS | Churg–Strauss Syndrome |
| CTF | C terminal fragment |
| CYP | Cytochrome P450 |
| CysLTR | Cysteinyl leukotrienes receptor (isoforms 1, 2, and 3) |
| CysLTs | Cysteinyl leukotrienes |
| Dbp | D site albumin promoter binding protein |
| DCX | Doublecortin |
| DP | Dipeptidase |
| eIF-2α | Eukaryotic initiation factor-2α |
| Entpd1 | Ectonucleoside triphosphate diphosphohydrolase 1 |
| Erk1/2 | Extracellular signal-regulated kinase 1/2 |
| FAD | Familial early-onset Alzheimer’s disease |
| Fas | Tumour necrosis factor receptor superfamily member 6 |
| FLAP | 5-LOX activating protein |
| GGLT | γ-Glutamyl leukotrienase |
| GGT | γ-Glutamyl transpeptidase |
| GPR17 | G Protein-Coupled Receptor 17 |
| GSH | Glutathione |
| GSSG | Glutathione disulphide |
| HAT | Histone acetyltransferase |
| HD | Hungtinton’s disease |
| HIF-1α | Hypoxia-inducible factor-1 |
| Iba1 | Ionized calcium-binding adaptor molecule 1 |
| IFN-γ | Interferon-γ |
| IL | Interleukin |
| iNOS | Inducible nitric oxide synthase |
| KLF2 | Krüppel-like Factor 2 |
| LDH | Lactate dehydrogenase |
| LPS | Lipopolysaccharide |
| LT | Leukotriene |
| LTA4 | Leukotriene A4 |
| LTA4H | Leukotriene A4 hydrolase |
| LTB4 | Leukotriene B4 |
| LTC4 | Leukotriene C4 |
| LTC4S | Leukotriene C4 synthase |
| LTD4 | Leukotriene D4 |
| LTE4 | Leukotriene E4 |
| MAPK | Mitogen-activated protein kinase |
| MDA | Malondialdehyde |
| MEK | Extracellular signal-regulated kinase kinase |
| Mlec | Malectin protein |
| MOG | Myelin oligodendrocyte glycoprotein |
| Mpro | SARS-CoV-2 Main protease |
| mRNA | Messenger RNA |
| MRP | Multidrug resistance proteins (isoforms 1 and 4) |
| MTK | Montelukast |
| NeuN | Neuronal nuclear protein |
| NF-κB | Nuclear factor kappa B |
| P2Y12 | P2Y purinoceptor 12 |
| p38 MAPK | p38 mitogen-activated protein kinase |
| PCNA | Proliferating cell nuclear antigen |
| PD | Parkinson’s disease |
| PG | Prostaglandin |
| PPAR-α | Peroxisomal proliferator-activated receptor α |
| PPARγ | Proliferator-activated receptor γ |
| PRAS40 | Proline-rich Akt substrate of 40 kDa |
| Psen | Presenilin |
| PTZ | Pentylenetetrazole |
| RdRp | RNA dependent RNA polymerase |
| ROS | Reactive oxygen species |
| SAD | Sporadic late-onset Alzheimer’s disease |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| SMC | Smooth muscle cells |
| SOD | Superoxide dismutase |
| TGF-β1 | Transforming growth factor-beta 1 |
| Tmem119 | Transmembrane protein 119 |
| TNF-α | Tumour necrosis factor α |
| UGT | Glucuronosyltransferase |
| WNK1 | WNK lysine deficient protein kinase 1 |
| Zfp46 | Zinc finger protein 46 |
References
- Bäck, M. Leukotriene signaling in atherosclerosis and ischemia. Cardiovasc. Drugs Ther. 2009, 23, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Brocklehurst, W.E. The release of histamine and formation of a slow-reacting substance (SRS-A) during anaphylactic shock. J. Physiol. 1960, 151, 416–435. [Google Scholar] [CrossRef]
- Rius, M.; Hummel-Eisenbeiss, J.; Keppler, D. ATP-dependent transport of leukotrienes B4 and C4 by the multidrug resistance protein ABCC4 (MRP4). J. Pharmacol. Exp. Ther. 2008, 324, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Johnson, Z.L.; Chen, J. Structural Basis of Substrate Recognition by the Multidrug Resistance Protein MRP1. Cell 2017, 168, 1075–1085.e9. [Google Scholar] [CrossRef]
- Samuelsson, B.; Dáhlen, S.E.; Lindgren, J.A.; Rouzer, C.A.; Serhan, C.N. Leukotrienes and Lipoxins—Structures, Biosynthesis, and Biological Effects. Science 1987, 237, 1171–1176. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, K.A.; Corser-Jensen, C.E. Chapter 12-5-Lipoxygenase-Activating Protein Inhibitors: Promising Drugs for Treating Acute and Chronic Neuroinflammation Following Brain Injury. In New Therapeutics for Traumatic Brain Injury; Heidenreich, K.A., Ed.; Academic Press: San Diego, CA, USA, 2017; pp. 199–210. [Google Scholar]
- Dennis, E.A.; Norris, P.C. Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef]
- Massoumi, R.; Sjölander, A. The role of leukotriene receptor signaling in inflammation and cancer. Sci. World J. 2007, 7, 1413–1421. [Google Scholar] [CrossRef]
- Kanaoka, Y.; Boyce, J.A. Cysteinyl leukotrienes and their receptors: Cellular distribution and function in immune and inflammatory responses. J. Immunol. 2004, 173, 1503–1510. [Google Scholar] [CrossRef]
- Capra, V. Molecular and functional aspects of human cysteinyl leukotriene receptors. Pharm. Res. 2004, 50, 1–11. [Google Scholar] [CrossRef]
- Singh, R.K.; Gupta, S.; Dastidar, S.; Ray, A. Cysteinyl leukotrienes and their receptors: Molecular and functional characteristics. Pharmacology 2010, 85, 336–349. [Google Scholar] [CrossRef]
- Singh, R.K.; Tandon, R.; Dastidar, S.G.; Ray, A. A review on leukotrienes and their receptors with reference to asthma. J. Asthma. 2013, 50, 922–931. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Ouyang, J.; Sharma, A.N.; Liu, S.; Yang, B.; Xiong, W.; Xu, R. Leukotriene inhibitors for bronchiolitis in infants and young children. Cochrane Database Syst. Rev. 2015. [Google Scholar] [CrossRef] [PubMed]
- Yokomizo, T.; Nakamura, M.; Shimizu, T. Leukotriene receptors as potential therapeutic targets. J. Clin. Investig. 2018, 128, 2691–2701. [Google Scholar] [CrossRef]
- Smith, W.L.; Murphy, R.C. The Eicosanoids. In Biochemistry of Lipids, Lipoproteins and Membranes, 6th ed.; Ridgway, N.D., McLeod, R.S., Eds.; Academic Press: Cambridge, MA, USA, 2016; pp. 259–296. [Google Scholar]
- Bäck, M. Leukotrienes: Potential therapeutic targets in cardiovascular diseases. Bull. Acad. Natl. Med. 2006, 190, 1511–1521. [Google Scholar]
- Bäck, M. Inhibitors of the 5-lipoxygenase pathway in atherosclerosis. Curr. Pharm. Des. 2009, 15, 3116–3132. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Shimizu, T. Leukotriene receptors. Chem. Rev. 2011, 111, 6231–6298. [Google Scholar] [CrossRef] [PubMed]
- Funk, C.D. Prostaglandins and leukotrienes: Advances in eicosanoid biology. Science 2001, 294, 1871–1875. [Google Scholar] [CrossRef]
- Capra, V.; Accomazzo, M.R.; Gardoni, F.; Barbieri, S.; Rovati, G.E. A role for inflammatory mediators in heterologous desensitization of CysLT1 receptor in human monocytes. J. Lipid. Res. 2010, 51, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yokomizo, T. The role of leukotrienes in allergic diseases. Allergol. Int. 2015, 64, 17–26. [Google Scholar] [CrossRef]
- Wunder, F.; Tinel, H.; Kast, R.; Geerts, A.; Becker, E.M.; Kolkhof, P.; Hutter, J.; Erguden, J.; Härter, M. Pharmacological characterization of the first potent and selective antagonist at the cysteinyl leukotriene 2 (CysLT2) receptor. Br. J. Pharmacol. 2010, 160, 399–409. [Google Scholar] [CrossRef]
- Poff, C.D.; Balazy, M. Drugs that target lipoxygenases and leukotrienes as emerging therapies for asthma and cancer. Curr. Drug Targets Inflamm. Allergy 2004, 3, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Gauvreau, G.M.; Boulet, L.P.; FitzGerald, J.M.; Cockcroft, D.W.; Davis, B.E.; Leigh, R.; Tanaka, M.; Fourre, J.A.; Tanaka, M.; Nabata, T.; et al. A dual CysLT1/2 antagonist attenuates allergen-induced airway responses in subjects with mild allergic asthma. Allergy 2016, 71, 1721–1727. [Google Scholar] [CrossRef] [PubMed]
- Itadani, S.; Yashiro, K.; Aratani, Y.; Sekiguchi, T.; Kinoshita, A.; Moriguchi, H.; Ohta, N.; Takahashi, S.; Ishida, A.; Tajima, Y.; et al. Discovery of Gemilukast (ONO-6950), a Dual CysLT1 and CysLT2 Antagonist As a Therapeutic Agent for Asthma. J. Med. Chem. 2015, 58, 6093–6113. [Google Scholar] [CrossRef] [PubMed]
- Lynch, K.R.; O'Neill, G.P.; Liu, Q.; Im, D.S.; Sawyer, N.; Metters, K.M.; Coulombe, N.; Abramovitz, M.; Figueroa, D.J.; Zeng, Z.; et al. Characterization of the human cysteinyl leukotriene CysLT1 receptor. Nature 1999, 399, 789–793. [Google Scholar] [CrossRef]
- Heise, C.E.; O'Dowd, B.F.; Figueroa, D.J.; Sawyer, N.; Nguyen, T.; Im, D.S.; Stocco, R.; Bellefeuille, J.N.; Abramovitz, M.; Cheng, R.; et al. Characterization of the human cysteinyl leukotriene 2 receptor. J. Biol. Chem. 2000, 275, 30531–30536. [Google Scholar] [CrossRef]
- Sarau, H.M.; Ames, R.S.; Chambers, J.; Ellis, C.; Elshourbagy, N.; Foley, J.J.; Schmidt, D.B.; Muccitelli, R.M.; Jenkins, O.; Murdock, P.R.; et al. Identification, molecular cloning, expression, and characterization of a cysteinyl leukotriene receptor. Mol. Pharmacol. 1999, 56, 657–663. [Google Scholar] [CrossRef]
- J Jones, T.R.; Zamboni, R.; Belley, M.; Champion, E.; Charette, L.; Ford-Hutchinson, A.W.; Frenette, R.; Gauthier, J.Y.; Leger, S.; Masson, P.; et al. Pharmacology of L-660,711 (MK-571): A novel potent and selective leukotriene D4 receptor antagonist. Can. J. Physiol. Pharmacol. 1989, 67, 17–28. [Google Scholar] [CrossRef]
- Ni, N.C.; Yan, D.; Ballantyne, L.L.; Barajas-Espinosa, A.; St Amand, T.; Pratt, D.A.; Funk, C.D. A selective cysteinyl leukotriene receptor 2 antagonist blocks myocardial ischemia/reperfusion injury and vascular permeability in mice. J. Pharmacol. Exp. Ther. 2011, 339, 768–778. [Google Scholar] [CrossRef]
- Abbracchio, M.P.; Burnstock, G.; Boeynaems, J.M.; Barnard, E.A.; Boyer, J.L.; Kennedy, C.; Miras-Portugal, M.T.; King, B.F.; Gachet, C.; Jacobson, K.A.; et al. The recently deorphanized GPR80 (GPR99) proposed to be the P2Y15 receptor is not a genuine P2Y receptor. Trends Pharmacol. Sci. 2005, 26, 8–9. [Google Scholar] [CrossRef]
- Kanaoka, Y.; Maekawa, A.; Austen, K.F. Identification of GPR99 protein as a potential third cysteinyl leukotriene receptor with a preference for leukotriene E4 ligand. J. Biol. Chem. 2013, 288, 10967–10972. [Google Scholar] [CrossRef]
- Cherif, H.; Duhamel, F.; Cécyre, B.; Bouchard, A.; Quintal, A.; Chemtob, S.; Bouchard, J.F. Receptors of intermediates of carbohydrate metabolism, GPR91 and GPR99, mediate axon growth. PLoS Biol. 2018, 16, e2003619. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, P.; Pluznick, J.L. Unsung renal receptors: Orphan G-protein-coupled receptors play essential roles in renal development and homeostasis. Acta Physiol. (Oxf) 2017, 220, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Wittenberger, T.; Hellebrand, S.; Munck, A.; Kreienkamp, H.J.; Schaller, H.C.; Hampe, W. GPR99, a new G protein-coupled receptor with homology to a new subgroup of nucleotide receptors. BMC Genomics. 2002, 3, 17. [Google Scholar] [CrossRef] [PubMed]
- Peebles, R.S. Antileukotriene Therapy in Asthma. In Middleton’s Allergy: Principles and Practice, 9th ed.; Burks, A.W., Holgate, S.T., O'Hehir, R.E., Bacharier, L.B., Broide, D.H., Hershey, G.K.K., Peebles, S., Eds.; Elsevier: Edinburgh, UK, 2020; pp. 1584–1598. [Google Scholar]
- Paruchuri, S.; Tashimo, H.; Feng, C.; Maekawa, A.; Xing, W.; Jiang, Y.; Kanaoka, Y.; Conley, P.; Boyce, J.A. Leukotriene E4-induced pulmonary inflammation is mediated by the P2Y12 receptor. J. Exp. Med. 2009, 206, 2543–2555. [Google Scholar] [CrossRef] [PubMed]
- Haynes, S.E.; Hollopeter, G.; Yang, G.; Kurpius, D.; Dailey, M.E.; Gan, W.B.; Julius, D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 2006, 9, 1512–1519. [Google Scholar] [CrossRef]
- Moore, C.S.; Ase, A.R.; Kinsara, A.; Rao, V.T.; Michell-Robinson, M.; Leong, S.Y.; Butovsky, O.; Ludwin, S.K.; Séguéla, P.; Bar-Or, A.; et al. P2Y12 expression and function in alternatively activated human microglia. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e80. [Google Scholar] [CrossRef]
- Hollopeter, G.; Jantzen, H.M.; Vincent, D.; Li, G.; England, L.; Ramakrishnan, V.; Yang, R.B.; Nurden, P.; Nurden, A.; Julius, D.; et al. Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature 2001, 409, 202–207. [Google Scholar] [CrossRef]
- Gómez Morillas, A.; Besson, V.C.; Lerouet, D. Microglia and Neuroinflammation: What Place for P2RY12? Int. J. Mol. Sci. 2021, 22, 1636. [Google Scholar] [CrossRef]
- Neves, J.S.; Radke, A.L.; Weller, P.F. Cysteinyl leukotrienes acting via granule membrane-expressed receptors elicit secretion from within cell-free human eosinophil granules. J. Allergy Clin. Immunol. 2010, 125, 477–482. [Google Scholar] [CrossRef]
- Suh, D.H.; Trinh, H.K.T.; Liu, J.N.; Pham, L.D.; Park, S.M.; Park, H.S.; Shin, Y.S. P2Y12 antagonist attenuates eosinophilic inflammation and airway hyperresponsiveness in a mouse model of asthma. J. Cell Mol. Med. 2016, 20, 333–341. [Google Scholar] [CrossRef]
- Collet, J.P.; O’Connor, S. Clinical effects and outcomes with new P2Y12 inhibitors in ACS. Fund. Clin. Pharmacol. 2012, 26, 16–18. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, M.; Daniele, S.; Lecca, D.; Lee, P.R.; Parravicini, C.; Fields, R.D.; Rosa, P.; Antonucci, F.; Verderio, C.; Trincavelli, M.L.; et al. Phenotypic changes, signaling pathway, and functional correlates of GPR17-expressing neural precursor cells during oligodendrocyte differentiation. J. Biol. Chem. 2011, 286, 10593–10604. [Google Scholar] [CrossRef] [PubMed]
- Ciana, P.; Fumagalli, M.; Trincavelli, M.L.; Verderio, C.; Rosa, P.; Lecca, D.; Ferrario, S.; Parravicini, C.; Capra, V.; Gelosa, P.; et al. The orphan receptor GPR17 identified as a new dual uracil nucleotides/cysteinyl-leukotrienes receptor. EMBO J. 2006, 25, 4615–4627. [Google Scholar] [CrossRef] [PubMed]
- Marschallinger, J.; Schäffner, I.; Klein, B.; Gelfert, R.; Rivera, F.J.; Illes, S.; Grassner, L.; Janssen, M.; Rotheneichner, P.; Schmuckermair, C.; et al. Structural and functional rejuvenation of the aged brain by an approved anti-asthmatic drug. Nat. Commun. 2015, 6, 8466. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, M.; Lecca, D.; Coppolino, G.T.; Parravicini, C.; Abbracchio, M.P. Pharmacological Properties and Biological Functions of the GPR17 Receptor, a Potential Target for Neuro-Regenerative Medicine. Adv. Exp. Med. Biol. 2017, 1051, 169–192. [Google Scholar] [CrossRef]
- Maekawa, A.; Xing, W.; Austen, K.F.; Kanaoka, Y. GPR17 regulates immune pulmonary inflammation induced by house dust mites. J. Immunol. 2010, 185, 1846–1854. [Google Scholar] [CrossRef]
- Zhao, B.; Zhao, C.Z.; Zhang, X.Y.; Huang, X.Q.; Shi, W.Z.; Fang, S.H.; Lu, Y.B.; Zhang, W.P.; Xia, Q.; Wei, E.Q. The new P2Y-like receptor G protein-coupled receptor 17 mediates acute neuronal injury and late microgliosis after focal cerebral ischemia in rats. Neuroscience 2012, 202, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Ceruti, S.; Villa, G.; Genovese, T.; Mazzon, E.; Longhi, R.; Rosa, P.; Bramanti, P.; Cuzzocrea, S.; Abbracchio, M.P. The P2Y-like receptor GPR17 as a sensor of damage and a new potential target in spinal cord injury. Brain 2009, 132, 2206–2218. [Google Scholar] [CrossRef]
- Burnstock, G. An introduction to the roles of purinergic signalling in neurodegeneration, neuroprotection and neuroregeneration. Neuropharmacology 2016, 104, 4–17. [Google Scholar] [CrossRef]
- Franke, H.; Parravicini, C.; Lecca, D.; Zanier, E.R.; Heine, C.; Bremicker, K.; Fumagalli, M.; Rosa, P.; Longhi, L.; Stocchetti, N.; et al. Changes of the GPR17 receptor, a new target for neurorepair, in neurons and glial cells in patients with traumatic brain injury. Purinergic. Signal. 2013, 9, 451–462. [Google Scholar] [CrossRef]
- Lecca, D.; Trincavelli, M.L.; Gelosa, P.; Sironi, L.; Ciana, P.; Fumagalli, M.; Villa, G.; Verderio, C.; Grumelli, C.; Guerrini, U.; et al. The recently identified P2Y-like receptor GPR17 is a sensor of brain damage and a new target for brain repair. PLoS ONE 2008, 3, e3579. [Google Scholar] [CrossRef] [PubMed]
- Dziedzic, A.; Miller, E.; Saluk-Bijak, J.; Bijak, M. The GPR17 Receptor-A Promising Goal for Therapy and a Potential Marker of the Neurodegenerative Process in Multiple Sclerosis. Int. J. Mol. Sci. 2020, 21, 1852. [Google Scholar] [CrossRef] [PubMed]
- Ou, Z.; Ma, Y.; Sun, Y.; Zheng, G.; Wang, S.; Xing, R.; Chen, X.; Han, Y.; Wang, J.; Lu, Q.R.; et al. A GPR17-cAMP-Lactate Signaling Axis in Oligodendrocytes Regulates Whole-Body Metabolism. Cell Rep. 2019, 26, 2984–2997.e4. [Google Scholar] [CrossRef]
- Maekawa, A.; Balestrieri, B.; Austen, K.F.; Kanaoka, Y. GPR17 is a negative regulator of the cysteinyl leukotriene 1 receptor response to leukotriene D4. Proc. Natl. Acad. Sci. USA 2009, 106, 11685–11690. [Google Scholar] [CrossRef]
- Pugliese, A.M.; Trincavelli, M.L.; Lecca, D.; Coppi, E.; Fumagalli, M.; Ferrario, S.; Failli, P.; Daniele, S.; Martini, C.; Pedata, F.; et al. Functional characterization of two isoforms of the P2Y-like receptor GPR17: [35S]GTPγS binding and electrophysiological studies in 1321N1 cells. Am. J. Physiol. Cell Physiol. 2009, 297, C1028–C1040. [Google Scholar] [CrossRef]
- Hennen, S.; Wang, H.; Peters, L.; Merten, N.; Simon, K.; Spinrath, A.; Blättermann, S.; Akkari, R.; Schrage, R.; Schröder, R.; et al. Decoding signaling and function of the orphan G protein-coupled receptor GPR17 with a small-molecule agonist. Sci. Signal. 2013, 6, ra93. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Chen, F.; Thakur, A.; Hong, H. Cysteinyl Leukotrienes and Their Receptors: Emerging Therapeutic Targets in Central Nervous System Disorders. CNS Neurosci. Ther. 2016, 22, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Gelosa, P.; Colazzo, F.; Tremoli, E.; Sironi, L.; Castiglioni, L. Cysteinyl Leukotrienes as Potential Pharmacological Targets for Cerebral Diseases. Mediat. Inflamm. 2017, 2017, 3454212. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.Z.; Zhao, B.; Zhang, X.Y.; Huang, X.Q.; Shi, W.Z.; Liu, H.L.; Fang, S.H.; Lu, Y.B.; Zhang, W.P.; Tang, F.D.; et al. Cysteinyl leukotriene receptor 2 is spatiotemporally involved in neuron injury, astrocytosis and microgliosis after focal cerebral ischemia in rats. Neuroscience 2011, 189, 1–11. [Google Scholar] [CrossRef]
- Zhang, W.P.; Hu, H.; Zhang, L.; Ding, W.; Yao, H.T.; Chen, K.D.; Sheng, W.W.; Chen, Z.; Wei, E.Q. Expression of cysteinyl leukotriene receptor 1 in human traumatic brain injury and brain tumors. Neurosci. Lett. 2004, 363, 247–251. [Google Scholar] [CrossRef]
- Zhang, Y.-j.; Zhang, L.; Ye, Y.-l.; Fang, S.-h.; Zhou, Y.; Zhang, W.-p.; Lu, Y.-b.; Wei, E.-q. Cysteinyl leukotriene receptors CysLT1 and CysLT2 are upregulated in acute neuronal injury after focal cerebral ischemia in mice. Acta Pharmacol. Sin. 2006, 27, 1553–1560. [Google Scholar] [CrossRef]
- Fang, S.H.; Wei, E.Q.; Zhou, Y.; Wang, M.L.; Zhang, W.P.; Yu, G.L.; Chu, L.S.; Chen, Z. Increased expression of cysteinyl leukotriene receptor-1 in the brain mediates neuronal damage and astrogliosis after focal cerebral ischemia in rats. Neuroscience 2006, 140, 969–979. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Y.; Zhang, S.; Li, C.; Zhang, L. Modulation of neuroinflammation by cysteinyl leukotriene 1 and 2 receptors: Implications for cerebral ischemia and neurodegenerative diseases. Neurobiol. Aging 2020, 87, 1–10. [Google Scholar] [CrossRef]
- Michael, J.; Marschallinger, J.; Aigner, L. The leukotriene signaling pathway: A druggable target in Alzheimer’s disease. Drug Discov. Today 2019, 24, 505–516. [Google Scholar] [CrossRef] [PubMed]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139 (Suppl. S2), 136–153. [Google Scholar] [CrossRef] [PubMed]
- Shabab, T.; Khanabdali, R.; Moghadamtousi, S.Z.; Kadir, H.A.; Mohan, G. Neuroinflammation pathways: A general review. Int. J. Neurosci. 2017, 127, 624–633. [Google Scholar] [CrossRef]
- Skaper, S.D.; Facci, L.; Zusso, M.; Giusti, P. An Inflammation-Centric View of Neurological Disease: Beyond the Neuron. Front. Cell Neurosci. 2018, 12, 72. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, Y.; Zhang, L.; Cheng, J.K.; Ji, R.R. Cytokine mechanisms of central sensitization: Distinct and overlapping role of interleukin-1β, interleukin-6, and tumor necrosis factor-α in regulating synaptic and neuronal activity in the superficial spinal cord. J. Neurosci. 2008, 28, 5189–5194. [Google Scholar] [CrossRef] [PubMed]
- Schonberg, D.L.; Popovich, P.G.; McTigue, D.M. Oligodendrocyte generation is differentially influenced by toll-like receptor (TLR) 2 and TLR4-mediated intraspinal macrophage activation. J. Neuropathol. Exp. Neurol. 2007, 66, 1124–1135. [Google Scholar] [CrossRef]
- Sica, A.; Schioppa, T.; Mantovani, A.; Allavena, P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: Potential targets of anti-cancer therapy. Eur. J. Cancer 2006, 42, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Kotter, M.R.; Setzu, A.; Sim, F.J.; Van Rooijen, N.; Franklin, R.J. Macrophage depletion impairs oligodendrocyte remyelination following lysolecithin-induced demyelination. Glia 2001, 35, 204–212. [Google Scholar] [CrossRef]
- Tarr, A.J.; Liu, X.; Reed, N.S.; Quan, N. Kinetic characteristics of euflammation: The induction of controlled inflammation without overt sickness behavior. Brain Behav. Immun. 2014, 42, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Nemeth, D.P.; Tarr, A.J.; Belevych, N.; Syed, Z.W.; Wang, Y.; Ismail, A.S.; Reed, N.S.; Sheridan, J.F.; Yajnik, A.R.; et al. Euflammation attenuates peripheral inflammation-induced neuroinflammation and mitigates immune-to-brain signaling. Brain Behav. Immun. 2016, 54, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Gyoneva, S.; Traynelis, S.F. Norepinephrine modulates the motility of resting and activated microglia via different adrenergic receptors. J. Biol. Chem. 2013, 288, 15291–15302. [Google Scholar] [CrossRef]
- Iwata, M.; Ota, K.T.; Duman, R.S. The inflammasome: Pathways linking psychological stress, depression, and systemic illnesses. Brain Behav. Immun. 2013, 31, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Goedert, M. Molecular cloning and functional characterization of chicken brain tau: Isoforms with up to five tandem repeats. Biochemistry-Us 2002, 41, 15203–15211. [Google Scholar] [CrossRef]
- Wohleb, E.S.; Powell, N.D.; Godbout, J.P.; Sheridan, J.F. Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. J. Neurosci. 2013, 33, 13820–13833. [Google Scholar] [CrossRef]
- Mansour, R.M.; Ahmed, M.A.E.; El-Sahar, A.E.; El Sayed, N.S. Montelukast attenuates rotenone-induced microglial activation/p38 MAPK expression in rats: Possible role of its antioxidant, anti-inflammatory and antiapoptotic effects. Toxicol. Appl. Pharmacol. 2018, 358, 76–85. [Google Scholar] [CrossRef]
- Saad, M.A.; Abdelsalam, R.M.; Kenawy, S.A.; Attia, A.S. Montelukast, a cysteinyl leukotriene receptor-1 antagonist protects against hippocampal injury induced by transient global cerebral ischemia and reperfusion in rats. Neurochem. Res. 2015, 40, 139–150. [Google Scholar] [CrossRef]
- Tang, S.S.; Hong, H.; Chen, L.; Mei, Z.L.; Ji, M.J.; Xiang, G.Q.; Li, N.; Ji, H. Involvement of cysteinyl leukotriene receptor 1 in Aβ1-42-induced neurotoxicity in vitro and in vivo. Neurobiol. Aging 2014, 35, 590–599. [Google Scholar] [CrossRef]
- Hu, H.; Chen, G.; Zhang, J.M.; Zhang, W.P.; Zhang, L.; Ge, Q.F.; Yao, H.T.; Ding, W.; Chen, Z.; Wei, E.Q. Distribution of cysteinyl leukotriene receptor 2 in human traumatic brain injury and brain tumors. Acta Pharmacol Sin. 2005, 26, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, Y.; Li, C.T.; Zhang, S.R.; Zheng, W.; Wei, E.Q.; Zhang, L.H. CysLT2 receptor mediates lipopolysaccharide-induced microglial inflammation and consequent neurotoxicity in vitro. Brain Res. 2015, 1624, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Ghosh, A.; Wu, F.; Tang, S.; Hu, M.; Sun, H.; Kong, L.; Hong, H. Preventive effect of genetic knockdown and pharmacological blockade of CysLT1R on lipopolysaccharide (LPS)-induced memory deficit and neurotoxicity in vivo. Brain Behav. Immun. 2017, 60, 255–269. [Google Scholar] [CrossRef]
- Shi, Q.J.; Wang, H.; Liu, Z.X.; Fang, S.H.; Song, X.M.; Lu, Y.B.; Zhang, W.P.; Sa, X.Y.; Ying, H.Z.; Wei, E.Q. HAMI 3379, a CysLT2R antagonist, dose- and time-dependently attenuates brain injury and inhibits microglial inflammation after focal cerebral ischemia in rats. Neuroscience 2015, 291, 53–69. [Google Scholar] [CrossRef]
- Goodman & Gilman’s the Pharmacological Basis of Therapeutics, 3rd ed.; Brunton, L.L., Knollmann, B.r.C., Hilal-Dandan, R., Eds.; McGraw Hill Medical: New York, NY, USA, 2018. [Google Scholar]
- Armada-Moreira, A.; Gomes, J.I.; Pina, C.C.; Savchak, O.K.; Gonçalves-Ribeiro, J.; Rei, N.; Pinto, S.; Morais, T.P.; Martins, R.S.; Ribeiro, F.F.; et al. Going the Extra (Synaptic) Mile: Excitotoxicity as the Road Toward Neurodegenerative Diseases. Front. Cell Neurosci. 2020, 14, 90. [Google Scholar] [CrossRef]
- Ding, Q.; Wei, E.Q.; Zhang, Y.J.; Zhang, W.P.; Chen, Z. Cysteinyl leukotriene receptor 1 is involved in N-methyl-D-aspartate-mediated neuronal injury in mice. Acta Pharmacol. Sin. 2006, 27, 1526–1536. [Google Scholar] [CrossRef][Green Version]
- Kang, K.-H.; Liou, H.-H.; Hour, M.-J.; Liou, H.-C.; Fu, W.-M. Protection of dopaminergic neurons by 5-lipoxygenase inhibitor. Neuropharmacology 2013, 73, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Raison, C.L.; Capuron, L.; Miller, A.H. Cytokines sing the blues: Inflammation and the pathogenesis of depression. Trends Immunol. 2006, 27, 24–31. [Google Scholar] [CrossRef]
- Sukoff Rizzo, S.J.; Neal, S.J.; Hughes, Z.A.; Beyna, M.; Rosenzweig-Lipson, S.; Moss, S.J.; Brandon, N.J. Evidence for sustained elevation of IL-6 in the CNS as a key contributor of depressive-like phenotypes. Transl. Psychiatry 2012, 2, e199. [Google Scholar] [CrossRef]
- Dantzer, R.; O’Connor, J.C.; Freund, G.G.; Johnson, R.W.; Kelley, K.W. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat. Rev. Neurosci. 2008, 9, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Bluthé, R.M.; Layé, S.; Michaud, B.; Combe, C.; Dantzer, R.; Parnet, P. Role of interleukin-1β and tumour necrosis factor-α in lipopolysaccharide-induced sickness behaviour: A study with interleukin-1 type I receptor-deficient mice. Eur. J. Neurosci. 2000, 12, 4447–4456. [Google Scholar] [CrossRef] [PubMed]
- Buschbeck, M.; Ghomashchi, F.; Gelb, M.H.; Watson, S.P.; Börsch-Haubold, A.G. Stress stimuli increase calcium-induced arachidonic acid release through phosphorylation of cytosolic phospholipase A2. Biochem. J. 1999, 344 Pt 2, 359–366. [Google Scholar] [CrossRef]
- Yu, X.B.; Dong, R.R.; Wang, H.; Lin, J.R.; An, Y.Q.; Du, Y.; Tang, S.S.; Hu, M.; Long, Y.; Sun, H.B.; et al. Knockdown of hippocampal cysteinyl leukotriene receptor 1 prevents depressive behavior and neuroinflammation induced by chronic mild stress in mice. Psychopharmacology 2016, 233, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.R.; Fang, S.C.; Tang, S.S.; Hu, M.; Long, Y.; Ghosh, A.; Sun, H.B.; Kong, L.Y.; Hong, H. Hippocampal CysLT1R knockdown or blockade represses LPS-induced depressive behaviors and neuroinflammatory response in mice. Acta Pharmacol. Sin. 2017, 38, 477–487. [Google Scholar] [CrossRef]
- Uz, T.; Dimitrijevic, N.; Imbesi, M.; Manev, H.; Manev, R. Effects of MK-886, a 5-lipoxygenase activating protein (FLAP) inhibitor, and 5-lipoxygenase deficiency on the forced swimming behavior of mice. Neurosci. Lett. 2008, 436, 269–272. [Google Scholar] [CrossRef]
- Na, J.Y.; Song, K.; Lee, J.W.; Kim, S.; Kwon, J. 6-Shogaol has anti-amyloidogenic activity and ameliorates Alzheimer’s disease via CysLT1R-mediated inhibition of cathepsin B. Biochem. Biophys. Res. Commun. 2016, 477, 96–102. [Google Scholar] [CrossRef]
- Wang, X.Y.; Tang, S.S.; Hu, M.; Long, Y.; Li, Y.Q.; Liao, M.X.; Ji, H.; Hong, H. Leukotriene D4 induces amyloid-β generation via CysLT1R-mediated NF-κB pathways in primary neurons. Neurochem. Int. 2013, 62, 340–347. [Google Scholar] [CrossRef]
- Tang, S.S.; Wang, X.Y.; Hong, H.; Long, Y.; Li, Y.Q.; Xiang, G.Q.; Jiang, L.Y.; Zhang, H.T.; Liu, L.P.; Miao, M.X.; et al. Leukotriene D4 induces cognitive impairment through enhancement of CysLT1R-mediated amyloid-β generation in mice. Neuropharmacology 2013, 65, 182–192. [Google Scholar] [CrossRef]
- Herbst-Robinson, K.J.; Liu, L.; James, M.; Yao, Y.; Xie, S.X.; Brunden, K.R. Inflammatory Eicosanoids Increase Amyloid Precursor Protein Expression via Activation of Multiple Neuronal Receptors. Sci. Rep. 2015, 5, 18286. [Google Scholar] [CrossRef]
- Lai, J.; Hu, M.; Wang, H.; Hu, M.; Long, Y.; Miao, M.X.; Li, J.C.; Wang, X.B.; Kong, L.Y.; Hong, H. Montelukast targeting the cysteinyl leukotriene receptor 1 ameliorates Aβ1-42-induced memory impairment and neuroinflammatory and apoptotic responses in mice. Neuropharmacology 2014, 79, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Kalra, J.; Kumar, P.; Majeed, A.B.; Prakash, A. Modulation of LOX and COX pathways via inhibition of amyloidogenesis contributes to mitoprotection against β-amyloid oligomer-induced toxicity in an animal model of Alzheimer's disease in rats. Pharmacol. Biochem. Behav. 2016, 146–147, 1–12. [Google Scholar] [CrossRef]
- Jang, H.; Kim, S.; Lee, J.M.; Oh, Y.-S.; Park, S.M.; Kim, S.R. Montelukast treatment protects nigral dopaminergic neurons against microglial activation in the 6-hydroxydopamine mouse model of Parkinson’s disease. Neuroreport 2017, 28, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Bournival, J.; Plouffe, M.; Renaud, J.; Provencher, C.; Martinoli, M.G. Quercetin and sesamin protect dopaminergic cells from MPP+-induced neuroinflammation in a microglial (N9)-neuronal (PC12) coculture system. Oxid. Med. Cell Longev. 2012, 2012, 921941. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.C.; Kim, S.R.; Park, J.Y.; Chung, E.S.; Park, K.W.; Won, S.Y.; Bok, E.; Jin, M.; Park, E.S.; Yoon, S.H.; et al. Fluoxetine prevents MPTP-induced loss of dopaminergic neurons by inhibiting microglial activation. Neuropharmacology 2011, 60, 963–974. [Google Scholar] [CrossRef]
- Sherer, T.B.; Betarbet, R.; Kim, J.H.; Greenamyre, J.T. Selective microglial activation in the rat rotenone model of Parkinson’s disease. Neurosci. Lett. 2003, 341, 87–90. [Google Scholar] [CrossRef]
- Nagarajan, V.B.; Marathe, P.A. Effect of montelukast in experimental model of Parkinson’s disease. Neurosci. Lett. 2018, 682, 100–105. [Google Scholar] [CrossRef]
- GBD 2019 Diseases and Injuries Collaborators. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar] [CrossRef]
- Reiss, T.F.; Altman, L.C.; Chervinsky, P.; Bewtra, A.; Stricker, W.E.; Noonan, G.P.; Kundu, S.; Zhang, J. Effects of montelukast (MK-0476), a new potent cysteinyl leukotriene (LTD4) receptor antagonist, in patients with chronic asthma. J. Allergy Clin. Immunol. 1996, 98, 528–534. [Google Scholar] [CrossRef]
- Knorr, B.; Matz, J.; Bernstein, J.A.; Nguyen, H.; Seidenberg, B.C.; Reiss, T.F.; Becker, A. Montelukast for chronic asthma in 6- to 14-year-old children: A randomized, double-blind trial. Pediatric Montelukast Study Group. JAMA 1998, 279, 1181–1186. [Google Scholar] [CrossRef]
- De Lepeleire, I.; Reiss, T.F.; Rochette, F.; Botto, A.; Zhang, J.; Kundu, S.; Decramer, M. Montelukast causes prolonged, potent leukotriene D4-receptor antagonism in the airways of patients with asthma. Clin. Pharmacol. Ther. 1997, 61, 83–92. [Google Scholar] [CrossRef]
- Araújo, A.C.; Tang, X.; Haeggström, J.Z. Targeting cysteinyl-leukotrienes in abdominal aortic aneurysm. Prostaglandins Other Lipid. Mediat. 2018, 139, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Ramires, R.; Caiaffa, M.F.; Tursi, A.; Haeggström, J.Z.; Macchia, L. Novel inhibitory effect on 5-lipoxygenase activity by the anti-asthma drug montelukast. Biochem. Biophys. Res. Commun. 2004, 324, 815–821. [Google Scholar] [CrossRef]
- Trinh, H.K.T.; Suh, D.H.; Nguyen, T.V.T.; Choi, Y.; Park, H.S.; Shin, Y.S. Characterization of cysteinyl leukotriene-related receptors and their interactions in a mouse model of asthma. Prostaglandins Leukot. Essent. Fatty Acids 2019, 141, 17–23. [Google Scholar] [CrossRef]
- Goshtasbi, K.; Abouzari, M.; Abiri, A.; Ziai, K.; Lehrich, B.M.; Risbud, A.; Bayginejad, S.; Lin, H.W.; Djalilian, H.R. Trends and patterns of neurotology drug prescriptions on a nationwide insurance database. Laryngoscope Investig. Otolaryngol. 2021, 6, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Balani, S.K.; Xu, X.; Pratha, V.; Koss, M.A.; Amin, R.D.; Dufresne, C.; Miller, R.R.; Arison, B.H.; Doss, G.A.; Chiba, M.; et al. Metabolic profiles of montelukast sodium (Singulair), a potent cysteinyl leukotriene1 receptor antagonist, in human plasma and bile. Drug Metab. Dispos. 1997, 25, 1282–1287. [Google Scholar]
- Chiba, M.; Xu, X.; Nishime, J.A.; Balani, S.K.; Lin, J.H. Hepatic microsomal metabolism of montelukast, a potent leukotriene D4 receptor antagonist, in humans. Drug Metab. Dispos. 1997, 25, 1022–1031. [Google Scholar] [PubMed]
- VandenBrink, B.M.; Foti, R.S.; Rock, D.A.; Wienkers, L.C.; Wahlstrom, J.L. Evaluation of CYP2C8 inhibition in vitro: Utility of montelukast as a selective CYP2C8 probe substrate. Drug Metab. Dispos. 2011, 39, 1546–1554. [Google Scholar] [CrossRef]
- Filppula, A.M.; Laitila, J.; Neuvonen, P.J.; Backman, J.T. Reevaluation of the microsomal metabolism of montelukast: Major contribution by CYP2C8 at clinically relevant concentrations. Drug Metab. Dispos. 2011, 39, 904–911. [Google Scholar] [CrossRef]
- Cardoso, J.O.; Oliveira, R.V.; Lu, J.B.; Desta, Z. In Vitro Metabolism of Montelukast by Cytochrome P450s and UDP-Glucuronosyltransferases. Drug Metab. Dispos. 2015, 43, 1905–1916. [Google Scholar] [CrossRef]
- Hirvensalo, P.; Tornio, A.; Neuvonen, M.; Tapaninen, T.; Paile-Hyvärinen, M.; Kärjä, V.; Männistö, V.T.; Pihlajamäki, J.; Backman, J.T.; Niemi, M. Comprehensive Pharmacogenomic Study Reveals an Important Role of UGT1A3 in Montelukast Pharmacokinetics. Clin. Pharmacol. Ther. 2018, 104, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Leff, J.A.; Amin, R.; Gertz, B.J.; De Smet, M.; Noonan, N.; Rogers, J.D.; Malbecq, W.; Meisner, D.; Somers, G. Pharmacokinetics, bioavailability, and safety of montelukast sodium (MK-0476) in healthy males and females. Pharm. Res. 1996, 13, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Merck Sharp & Dohme Corp. Full Prescribing Information: Singulair (Revised 6/2021). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/020829s073,020830s075,021409s051lbl.pdf (accessed on 7 July 2018).
- Mougey, E.B.; Feng, H.; Castro, M.; Irvin, C.G.; Lima, J.J. Absorption of montelukast is transporter mediated: A common variant of OATP2B1 is associated with reduced plasma concentrations and poor response. Pharm. Genom. 2009, 19, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Brännström, M.; Nordell, P.; Bonn, B.; Davis, A.M.; Palmgren, A.P.; Hilgendorf, C.; Rubin, K.; Grime, K. Montelukast Disposition: No Indication of Transporter-Mediated Uptake in OATP2B1 and OATP1B1 Expressing HEK293 Cells. Pharmaceutics 2015, 7, 554–564. [Google Scholar] [CrossRef] [PubMed]
- Bednarczyk, D.; Sanghvi, M.V. Organic anion transporting polypeptide 2B1 (OATP2B1), an expanded substrate profile, does it align with OATP2B1’s hypothesized function? Xenobiotica 2020, 50, 1128–1137. [Google Scholar] [CrossRef] [PubMed]
- Kinzi, J.; Grube, M.; Meyer Zu Schwabedissen, H.E. OATP2B1—The underrated member of the organic anion transporting polypeptide family of drug transporters? Biochem. Pharmacol. 2021, 188, 114534. [Google Scholar] [CrossRef] [PubMed]
- Clarridge, K.; Chin, S.; Eworuke, E.; Seymour, S. A Boxed Warning for Montelukast: The FDA Perspective. J. Allergy Clin. Immunol. Pract. 2021, 9, 2638–2641. [Google Scholar] [CrossRef]
- Calapai, G.; Casciaro, M.; Miroddi, M.; Calapai, F.; Navarra, M.; Gangemi, S. Montelukast-induced adverse drug reactions: A review of case reports in the literature. Pharmacology 2014, 94, 60–70. [Google Scholar] [CrossRef]
- Sansing-Foster, V.; Haug, N.; Mosholder, A.; Cocoros, N.M.; Bradley, M.; Ma, Y.; Pennap, D.; Dee, E.C.; Toh, S.; Pestine, E.; et al. Risk of Psychiatric Adverse Events Among Montelukast Users. J. Allergy Clin. Immunol. Pract. 2021, 9, 385–393.e12. [Google Scholar] [CrossRef]
- Uppsala Monitoring Centre. VigiAccess—WHO Programme for International Drug Monitoring. Available online: https://www.vigiaccess.org/ (accessed on 10 October 2021).
- Glockler-Lauf, S.D.; Finkelstein, Y.; Zhu, J.; Feldman, L.Y.; To, T. Montelukast and Neuropsychiatric Events in Children with Asthma: A Nested Case-Control Study. J. Pediatr. 2019, 209, 176–182.e4. [Google Scholar] [CrossRef]
- Aldea Perona, A.; García-Sáiz, M.; Sanz Álvarez, E. Psychiatric Disorders and Montelukast in Children: A Disproportionality Analysis of the VigiBase®. Drug Saf. 2016, 39, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Kocyigit, A.; Gulcan Oksuz, B.; Yarar, F.; Uzun, F.; Igde, M.; Islek, I. Hallucination development with montelukast in a child with asthma: Case presentation. Iran. J. Allergy Asthma. Immunol. 2013, 12, 397–399. [Google Scholar] [PubMed]
- Byrne, F.; Oluwole, B.; Whyte, V.; Fahy, S.; McGuinness, D. Delayed Onset of Neuropsychiatric Effects Associated with Montelukast. Ir. J. Psychol. Med. 2012, 29, 125–127. [Google Scholar] [CrossRef] [PubMed]
- Bernal Vañó, E.; López Andrés, N. Un caso de síndrome de Alicia en el país de las maravillas en probable relación con el uso de montelukast [A case of Alice-in-Wonderland syndrome probably associated with the use of montelukast]. An. Pediatr. (Barc) 2013, 78, 127–128. [Google Scholar] [CrossRef]
- Carnovale, C.; Gentili, M.; Antoniazzi, S.; Radice, S.; Clementi, E. Montelukast-induced metamorphopsia in a pediatric patient: A case report and a pharmacovigilance database analysis. Ann. Allergy Asthma. Immunol. 2016, 116, 370–371. [Google Scholar] [CrossRef]
- Benard, B.; Bastien, V.; Vinet, B.; Yang, R.; Krajinovic, M.; Ducharme, F.M. Neuropsychiatric adverse drug reactions in children initiated on montelukast in real-life practice. Eur. Respir. J. 2017, 50, 1700148. [Google Scholar] [CrossRef]
- Wallerstedt, S.M.; Brunlöf, G.; Sundström, A.; Eriksson, A.L. Montelukast and psychiatric disorders in children. Pharmacoepidemiol. Drug Saf. 2009, 18, 858–864. [Google Scholar] [CrossRef]
- Caudevilla Lafuente, P.; Garcia Íñiguez, J.P.; Martín de Vicente, C. Reacciones adversas a montelukast: De la teoría a la práctica. Serie de casos [Adverse drug reactions of montelukast: From theory to practice. Case report]. Arch. Argent. Pediatr. 2021, 119, e357–e359. [Google Scholar] [CrossRef]
- Alkhuja, S.; Gazizov, N.; Alexander, M.E. Sleeptalking! Sleepwalking! Side effects of montelukast. Case Rep. Pulmonol. 2013, 2013, 813786. [Google Scholar] [CrossRef]
- Anandan, N.; Ibitoye, F. Montelukast and worsening of hallucinations in paranoid schizophrenia. Psychiatr. Bulletin. 2008, 32, 276. [Google Scholar] [CrossRef][Green Version]
- Ibarra-Barrueta, O.; Palacios-Zabalza, I.; Mora-Atorrasagasti, O.; Mayo-Suarez, J. Effect of concomitant use of montelukast and efavirenz on neuropsychiatric adverse events. Ann. Pharm. 2014, 48, 145–148. [Google Scholar] [CrossRef]
- Philip, G.; Hustad, C.; Noonan, G.; Malice, M.-P.; Ezekowitz, A.; Reiss, T.F.; Knorr, B. Reports of suicidality in clinical trials of montelukast. J. Allergy Clin. Immunol. 2009, 124, 691–696.e6. [Google Scholar] [CrossRef]
- Jick, H.; Hagberg, K.W.; Egger, P. Rate of suicide in patients taking montelukast. Pharmacotherapy 2009, 29, 165–166. [Google Scholar] [CrossRef] [PubMed]
- Manalai, P.; Woo, J.-M.; Postolache, T.T. Suicidality and montelukast. Expert Opin. Drug Saf. 2009, 8, 273–282. [Google Scholar] [CrossRef]
- Schumock, G.T.; Stayner, L.T.; Valuck, R.J.; Joo, M.J.; Gibbons, R.D.; Lee, T.A. Risk of suicide attempt in asthmatic children and young adults prescribed leukotriene-modifying agents: A nested case-control study. J. Allergy Clin. Immunol. 2012, 130, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Mondal, S.; Dey, J.K.; Bandyopadhyay, S.; Saha, I.; Tripathi, S.K. A case of montelukast induced hypercholesterolemia, severe hypertriglyceridemia and pancreatitis. J. Young Pharm. 2013, 5, 64–66. [Google Scholar] [CrossRef]
- Xie, J.-X.; Wei, J.-F.; Meng, L. Montelukast sodium-induced hematuria: A case report and literature review of 19 cases in mainland China. Int J. Clin. Pharm. Ther. 2013, 51, 958–962. [Google Scholar] [CrossRef]
- Harugeri, A.; Parthasarathi, G.; Sharma, J.; D’Souza, G.A.; Ramesh, M. Montelukast induced acute hepatocellular liver injury. J. Postgrad. Med. 2009, 55, 141–142. [Google Scholar] [CrossRef]
- Russmann, S.; Iselin, H.U.; Meier, D.; Zimmermann, A.; Simon, H.-U.; Caduff, P.; Reichen, J. Acute hepatitis associated with montelukast. J. Hepatol. 2003, 38, 694–695. [Google Scholar] [CrossRef]
- Incecik, F.; Onlen, Y.; Sangun, O.; Akoglu, S. Probable montelukast-induced hepatotoxicity in a pediatric patient: Case report. Ann. Saudi. Med. 2007, 27, 462–463. [Google Scholar] [CrossRef] [PubMed]
- Lebensztejn, D.M.; Bobrus-Chociej, A.; Kłusek, M.; Uscinowicz, M.; Lotowska, J.; Sobaniec-Lotowska, M.; Kaczmarski, M. Hepatotoxicity caused by montelukast in a paediatric patient. Prz. Gastroenterol. 2014, 9, 121–123. [Google Scholar] [CrossRef]
- Sabbagh, R.; Sheikh-Taha, M. Possible montelukast-induced angioedema. Am. J. Health Syst. Pharm. 2009, 66, 1705–1706. [Google Scholar] [CrossRef]
- Minciullo, P.L.; Saija, A.; Bonanno, D.; Ferlazzo, E.; Gangemi, S. Montelukast-induced generalized urticaria. Ann. Pharm. 2004, 38, 999–1001. [Google Scholar] [CrossRef] [PubMed]
- Cetkovská, P.; Pizinger, K. Childhood pemphigus associated with montelukast administration. Clin. Exp. Dermatol. 2003, 28, 328–329. [Google Scholar] [CrossRef] [PubMed]
- Price, D. Tolerability of montelukast. Drugs 2000, 59 (Suppl. S1), 35–42; discussion 43–45. [Google Scholar] [CrossRef] [PubMed]
- Haarman, M.G.; van Hunsel, F.; de Vries, T.W. Adverse drug reactions of montelukast in children and adults. Pharm. Res. Perspect. 2017, 5, e00341. [Google Scholar] [CrossRef]
- Hauser, T.; Mahr, A.; Metzler, C.; Coste, J.; Sommerstein, R.; Gross, W.L.; Guillevin, L.; Hellmich, B. The leucotriene receptor antagonist montelukast and the risk of Churg-Strauss syndrome: A case-crossover study. Thorax 2008, 63, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Anar, C.; Ünsal, I.; Ozanturk, M.E.; Halilçolar, H.; Yucel, N. A case of Churg-Strauss syndrome treated with montelukast. Med. Princ. Pract. 2012, 21, 186–189. [Google Scholar] [CrossRef]
- Mateo, M.L.; Cortés, C.M.; Berisa, F. Síndrome de Churg-Strauss asociado a la administración de montelukast en un paciente asmático sin tratamiento esteroide de base. Arch. Bronconeumol. 2002, 38, 56. [Google Scholar] [CrossRef]
- Jennings, L.; Ho, W.L.; Gulmann, C.; Murphy, G.M. Churg-Strauss syndrome secondary to antileucotriene therapy in a patient not receiving oral corticosteroids. Clin. Exp. Dermatol. 2009, 34, e430–e431. [Google Scholar] [CrossRef]
- Churg, J.; Strauss, L. Allergic granulomatosis, allergic angiitis, and periarteritis nodosa. Am. J. Pathol 1951, 27, 277–301. [Google Scholar]
- Franco, J.; Artés, M.J. Pulmonary eosinophilia associated with montelukast. Thorax 1999, 54, 558–560. [Google Scholar] [CrossRef][Green Version]
- Wechsler, M.E.; Finn, D.; Gunawardena, D.; Westlake, R.; Barker, A.; Haranath, S.P.; Pauwels, R.A.; Kips, J.C.; Drazen, J.M. Churg-Strauss syndrome in patients receiving montelukast as treatment for asthma. Chest 2000, 117, 708–713. [Google Scholar] [CrossRef]
- Villena, V.; Hidalgo, R.; Sotelo, M.T.; Martin-Escribano, P. Montelukast and Churg-Strauss syndrome. Eur. Respir. J. 2000, 15, 626. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Uyar, M.; Elbek, O.; Bakır, K.; Kibar, Y.; Bayram, N.; Dikensoy, Ö. Churg-Strauss syndrome related to montelukast. Tuberk. Toraks 2012, 60, 56–58. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.B.; Yosipovitch, G. Acute Churg-Strauss syndrome in an asthmatic patient receiving montelukast therapy. Arch. Dermatol. 2003, 139, 715–718. [Google Scholar] [CrossRef]
- Cuchacovich, R.; Justiniano, M.; Espinoza, L.R. Churg-Strauss syndrome associated with leukotriene receptor antagonists (LTRA). Clin. Rheumatol. 2007, 26, 1769–1771. [Google Scholar] [CrossRef]
- Kaliterna, D.M.; Perković, D.; Radić, M. Churg-Strauss syndrome associated with montelukast therapy. J. Asthma. 2009, 46, 604–605. [Google Scholar] [CrossRef] [PubMed]
- Black, J.G.; Bonner, J.R.; Boulware, D.; Andea, A.A. Montelukast-associated Churg-Strauss vasculitis: Another associated report. Ann. Allergy Asthma. Immunol. 2009, 102, 351–352. [Google Scholar] [CrossRef]
- Kanda, T.; Tanio, H.; Wu, C.; Nishihara, H.; Nogaki, F.; Ono, T. Churg-Strauss syndrome with severe granulomatous angiitis and crescentic glomerulonephritis, which developed during therapy with a leukotriene receptor antagonist. Clin. Exp. Nephrol. 2010, 14, 602–607. [Google Scholar] [CrossRef]
- Camozzi, P.; Milani, G.P.; Bianchetti, M.G. Leukotriene receptor antagonists in Henoch-Schonlein syndrome: Friends or foes? Pediatr. Int. 2014, 56, 802. [Google Scholar] [CrossRef] [PubMed]
- Rejnö, G.; Lundholm, C.; Gong, T.; Larsson, K.; Saltvedt, S.; Almqvist, C. Asthma during pregnancy in a population-based study--pregnancy complications and adverse perinatal outcomes. PLoS ONE 2014, 9, e104755. [Google Scholar] [CrossRef]
- Nelsen, L.M.; Shields, K.E.; Cunningham, M.L.; Stoler, J.M.; Bamshad, M.J.; Eng, P.M.; Smugar, S.S.; Gould, A.L.; Philip, G. Congenital malformations among infants born to women receiving montelukast, inhaled corticosteroids, and other asthma medications. J. Allergy Clin. Immunol. 2012, 129, 251–254.e6. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, M.; Koren, G.; Kalra, S.; Ying, A.; Smorlesi, C.; De Santis, M.; Diav-Citrin, O.; Avgil, M.; Lavigne, S.V.; Berkovich, M.; et al. Montelukast use during pregnancy: A multicentre, prospective, comparative study of infant outcomes. Eur. J. Clin. Pharmacol. 2009, 65, 1259–1264. [Google Scholar] [CrossRef]
- Bakhireva, L.N.; Jones, K.L.; Schatz, M.; Klonoff-Cohen, H.S.; Johnson, D.; Slymen, D.J.; Chambers, C.D.; Organization of Teratology Information Specialists Collaborative Research Group. Safety of leukotriene receptor antagonists in pregnancy. J. Allergy Clin. Immunol. 2007, 119, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Cavero-Carbonell, C.; Vinkel-Hansen, A.; Rabanque-Hernández, M.J.; Martos, C.; Garne, E. Fetal Exposure to Montelukast and Congenital Anomalies: A Population Based Study in Denmark. Birth Defects Res. 2017, 109, 452–459. [Google Scholar] [CrossRef]
- Lai, J.; Mei, Z.L.; Wang, H.; Hu, M.; Long, Y.; Miao, M.X.; Li, N.; Hong, H. Montelukast rescues primary neurons against Aβ1–42-induced toxicity through inhibiting CysLT1R-mediated NF-κB signaling. Neurochem. Int. 2014, 75, 26–31. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Chen, L.; Yang, Y.; Xu, D.M.; Zhang, S.R.; Li, C.T.; Zheng, W.; Yu, S.Y.; Wei, E.Q.; Zhang, L.H. Regulation of rotenone-induced microglial activation by 5-lipoxygenase and cysteinyl leukotriene receptor 1. Brain Res. 2014, 1572, 59–71. [Google Scholar] [CrossRef]
- Wallin, J.; Svenningsson, P. Potential Effects of Leukotriene Receptor Antagonist Montelukast in Treatment of Neuroinflammation in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 5606. [Google Scholar] [CrossRef]
- Kalonia, H.; Kumar, P.; Kumar, A.; Nehru, B. Protective effect of montelukast against quinolinic acid/malonic acid induced neurotoxicity: Possible behavioral, biochemical, mitochondrial and tumor necrosis factor-α level alterations in rats. Neuroscience 2010, 171, 284–299. [Google Scholar] [CrossRef]
- Marschallinger, J.; Altendorfer, B.; Rockenstein, E.; Holztrattner, M.; Garnweidner-Raith, J.; Pillichshammer, N.; Leister, I.; Hutter-Paier, B.; Strempfl, K.; Unger, M.S.; et al. The Leukotriene Receptor Antagonist Montelukast Reduces Alpha-Synuclein Load and Restores Memory in an Animal Model of Dementia with Lewy Bodies. Neurotherapeutics 2020, 17, 1061–1074. [Google Scholar] [CrossRef]
- Grinde, B.; Schirmer, H.; Eggen, A.E.; Aigner, L.; Engdahl, B. A possible effect of montelukast on neurological aging examined by the use of register data. Int. J. Clin. Pharm. 2021, 43, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Michael, J.; Zirknitzer, J.; Unger, M.S.; Poupardin, R.; Rieß, T.; Paiement, N.; Zerbe, H.; Hutter-Paier, B.; Reitsamer, H.; Aigner, L. The Leukotriene Receptor Antagonist Montelukast Attenuates Neuroinflammation and Affects Cognition in Transgenic 5xFAD Mice. Int. J. Mol. Sci. 2021, 22, 2782. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.T.; Lin, J.R.; Wu, F.; Ghosh, A.; Tang, S.S.; Hu, M.; Long, Y.; Sun, H.B.; Hong, H. Montelukast ameliorates streptozotocin-induced cognitive impairment and neurotoxicity in mice. Neurotoxicology 2016, 57, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.J.; Chang, W.A.; Tsai, P.H.; Wu, C.Y.; Ho, Y.W.; Yen, M.C.; Lin, Y.S.; Kuo, P.L.; Hsu, Y.L. Montelukast Induces Apoptosis-Inducing Factor-Mediated Cell Death of Lung Cancer Cells. Int. J. Mol. Sci. 2017, 18, 1353. [Google Scholar] [CrossRef]
- Tsai, M.J.; Wu, P.H.; Sheu, C.C.; Hsu, Y.L.; Chang, W.A.; Hung, J.Y.; Yang, C.J.; Yang, Y.H.; Kuo, P.L.; Huang, M.S. Cysteinyl Leukotriene Receptor Antagonists Decrease Cancer Risk in Asthma Patients. Sci. Rep. 2016, 6, 23979. [Google Scholar] [CrossRef]
- Tang, C.; Lei, H.; Zhang, J.; Liu, M.; Jin, J.; Luo, H.; Xu, H.; Wu, Y. Montelukast inhibits hypoxia inducible factor-1α translation in prostate cancer cells. Cancer Biol. Ther. 2018, 19, 715–721. [Google Scholar] [CrossRef]
- Matsuyama, M.; Hayama, T.; Funao, K.; Kawahito, Y.; Sano, H.; Takemoto, Y.; Nakatani, T.; Yoshimura, R. Overexpression of cysteinyl LT1 receptor in prostate cancer and CysLT1R antagonist inhibits prostate cancer cell growth through apoptosis. Oncol. Rep. 2007, 18, 99–104. [Google Scholar] [CrossRef]
- Tahan, F.; Jazrawi, E.; Moodley, T.; Rovati, G.E.; Adcock, I.M. Montelukast inhibits tumour necrosis factor-alpha-mediated interleukin-8 expression through inhibition of nuclear factor-κB p65-associated histone acetyltransferase activity. Clin. Exp. Allergy 2008, 38, 805–811. [Google Scholar] [CrossRef]
- Sanghai, N.; Tranmer, G.K. Taming the cytokine storm: Repurposing montelukast for the attenuation and prophylaxis of severe COVID-19 symptoms. Drug Discov. Today 2020, 25, 2076–2079. [Google Scholar] [CrossRef]
- Funk, C.D.; Ardakani, A. A Novel Strategy to Mitigate the Hyperinflammatory Response to COVID-19 by Targeting Leukotrienes. Front. Pharmacol. 2020, 11, 1214. [Google Scholar] [CrossRef]
- Kow, C.S.; Hasan, S.S. Montelukast in children with allergic rhinitis amid COVID-19 pandemic. Acta Paediatr. 2020, 109, 2151. [Google Scholar] [CrossRef]
- Dey, M.; Singh, R.K. Possible Therapeutic Potential of Cysteinyl Leukotriene Receptor Antagonist Montelukast in Treatment of SARS-CoV-2-Induced COVID-19. Pharmacology 2021, 106, 469–476. [Google Scholar] [CrossRef]
- Aigner, L.; Pietrantonio, F.; Bessa de Sousa, D.M.; Michael, J.; Schuster, D.; Reitsamer, H.A.; Zerbe, H.; Studnicka, M. The Leukotriene Receptor Antagonist Montelukast as a Potential COVID-19 Therapeutic. Front. Mol. Biosci. 2020, 7, 610132. [Google Scholar] [CrossRef]
- Wixted, J.J.; Fanning, P.J.; Gaur, T.; O'Connell, S.L.; Silva, J.; Mason-Savas, A.; Ayers, D.C.; Stein, G.S.; Lian, J.B. Enhanced fracture repair by leukotriene antagonism is characterized by increased chondrocyte proliferation and early bone formation: A novel role of the cysteinyl LT-1 receptor. J. Cell Physiol. 2009, 221, 31–39. [Google Scholar] [CrossRef]
- Li, Z.; Wang, J.; Ma, Y. Montelukast attenuates interleukin IL-1β-induced oxidative stress and apoptosis in chondrocytes by inhibiting CYSLTR1 (Cysteinyl Leukotriene Receptor 1) and activating KLF2 (Kruppel Like Factor 2). Bioengineered 2021, 12, 8476–8484. [Google Scholar] [CrossRef] [PubMed]
- Ingelsson, E.; Yin, L.; Bäck, M. Nationwide cohort study of the leukotriene receptor antagonist montelukast and incident or recurrent cardiovascular disease. J. Allergy Clin. Immunol. 2012, 129, 702–707.e2. [Google Scholar] [CrossRef]
- Bäck, M.; Yin, L.; Nagy, E.; Ingelsson, E. The leukotriene receptor antagonist montelukast and aortic stenosis. Br. J. Clin. Pharmacol. 2013, 75, 280–281. [Google Scholar] [CrossRef]
- Allayee, H.; Hartiala, J.; Lee, W.; Mehrabian, M.; Irvin, C.G.; Conti, D.V.; Lima, J.J. The effect of montelukast and low-dose theophylline on cardiovascular disease risk factors in asthmatics. Chest 2007, 132, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Alizamani, E.; Ghorbanzadeh, B.; Naserzadeh, R.; Mansouri, M.T. Montelukast, a cysteinyl leukotriene receptor antagonist, exerts local antinociception in animal model of pain through the L-arginine/nitric oxide/cyclic GMP/KATP channel pathway and PPARgamma receptors. Int. J. Neurosci. 2021, 131, 1004–1011. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Shi, W.-Z.; Zhang, Y.-M.; Fang, S.-H.; Wei, E.-Q. Montelukast, a cysteinyl leukotriene receptor-1 antagonist, attenuates chronic brain injury after focal cerebral ischaemia in mice and rats. J. Pharm. Pharmacol. 2011, 63, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Lenz, Q.F.; Arroyo, D.S.; Temp, F.R.; Poersch, A.B.; Masson, C.J.; Jesse, A.C.; Marafiga, J.R.; Reschke, C.R.; Iribarren, P.; Mello, C.F. Cysteinyl leukotriene receptor (CysLT) antagonists decrease pentylenetetrazol-induced seizures and blood-brain barrier dysfunction. Neuroscience 2014, 277, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Rehni, A.K.; Singh, T.G. Modulation of leukotriene D4 attenuates the development of seizures in mice. Prostaglandins Leukot. Essent. Fatty Acids 2011, 85, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Du, C.; Lv, J.; Wei, W.; Cui, Y.; Xie, X. Antiasthmatic drugs targeting the cysteinyl leukotriene receptor 1 alleviate central nervous system inflammatory cell infiltration and pathogenesis of experimental autoimmune encephalomyelitis. J. Immunol. 2011, 187, 2336–2345. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef] [PubMed]
- Copertino, D.C.; Duarte, R.R.R.; Powell, T.R.; de Mulder Rougvie, M.; Nixon, D.F. Montelukast drug activity and potential against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). J. Med. Virol. 2021, 93, 187–189. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.R.; Misdary, C.; Yegya-Raman, N.; Kim, S.; Narayanan, N.; Siddiqui, S.; Salgame, P.; Radbel, J.; Groote, F.; Michel, C.; et al. Montelukast in hospitalized patients diagnosed with COVID-19. J. Asthma. 2021, 59, 1–7. [Google Scholar] [CrossRef]
- Tr Trost, A.; Motloch, K.; Koller, A.; Bruckner, D.; Runge, C.; Schroedl, F.; Bogner, B.; Kaser-Eichberger, A.; Strohmaier, C.; Ladek, A.M.; et al. Inhibition of the cysteinyl leukotriene pathways increases survival of RGCs and reduces microglial activation in ocular hypertension. Exp. Eye Res. 2021, 213, 108806. [Google Scholar] [CrossRef]
- Verleden, G.M.; Verleden, S.E.; Vos, R.; De Vleeschauwer, S.I.; Dupont, L.J.; Van Raemdonck, D.E.; Vanaudenaerde, B.M. Montelukast for bronchiolitis obliterans syndrome after lung transplantation: A pilot study. Transpl. Int. 2011, 24, 651–656. [Google Scholar] [CrossRef]
- Ruttens, D.; Verleden, S.E.; Demeyer, H.; Van Raemdonck, D.E.; Yserbyt, J.; Dupont, L.J.; Vanaudenaerde, B.M.; Vos, R.; Verleden, G.M. Montelukast for bronchiolitis obliterans syndrome after lung transplantation: A randomized controlled trial. PLoS ONE 2018, 13, e0193564. [Google Scholar] [CrossRef]
- Vos, R.; Eynde, R.V.; Ruttens, D.; Verleden, S.E.; Vanaudenaerde, B.M.; Dupont, L.J.; Yserbyt, J.; Verbeken, E.K.; Neyrinck, A.P.; Van Raemdonck, D.E.; et al. Montelukast in chronic lung allograft dysfunction after lung transplantation. J. Heart Lung Transplant. 2019, 38, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Shimbori, C.; Shiota, N.; Okunishi, H. Effects of montelukast, a cysteinyl-leukotriene type 1 receptor antagonist, on the pathogenesis of bleomycin-induced pulmonary fibrosis in mice. Eur. J. Pharmacol. 2011, 650, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Helmy, M.M.; El-Gowelli, H.M. Montelukast abrogates rhabdomyolysis-induced acute renal failure via rectifying detrimental changes in antioxidant profile and systemic cytokines and apoptotic factors production. Eur. J. Pharmacol. 2012, 683, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Suddek, G.M. Montelukast ameliorates kidney function and urinary bladder sensitivity in experimentally induced renal dysfunction in rats. Fundam. Clin. Pharmacol. 2013, 27, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Tuğtepe, H.; Şener, G.; Çetinel, S.; Velioğlu-Öğünç, A.; Yeğen, B.C. Oxidative renal damage in pyelonephritic rats is ameliorated by montelukast, a selective leukotriene CysLT1 receptor antagonist. Eur. J. Pharmacol. 2007, 557, 69–75. [Google Scholar] [CrossRef]
- Kort, E.; Jovinge, S. Drug Repurposing: Claiming the Full Benefit from Drug Development. Curr. Cardiol. Rep. 2021, 23, 62. [Google Scholar] [CrossRef]
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef]
- Wyss-Coray, T. Ageing, neurodegeneration and brain rejuvenation. Nature 2016, 539, 180–186. [Google Scholar] [CrossRef]
- Elobeid, A.; Libard, S.; Leino, M.; Popova, S.N.; Alafuzoff, I. Altered Proteins in the Aging Brain. J. Neuropathol. Exp. Neurol. 2016, 75, 316–325. [Google Scholar] [CrossRef]
- Müller, U.C.; Deller, T.; Korte, M. Not just amyloid: Physiological functions of the amyloid precursor protein family. Nat. Rev. Neurosci. 2017, 18, 281–298. [Google Scholar] [CrossRef]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2020 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2020, 16, 391–460. [Google Scholar] [CrossRef]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef]
- L Lambert, J.C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef]
- Bellenguez, C.; Charbonnier, C.; Grenier-Boley, B.; Quenez, O.; Le Guennec, K.; Nicolas, G.; Chauhan, G.; Wallon, D.; Rousseau, S.; Richard, A.C.; et al. Contribution to Alzheimer’s disease risk of rare variants in TREM2, SORL1, and ABCA7 in 1779 cases and 1273 controls. Neurobiol. Aging 2017, 59, 220.e1–220.e9. [Google Scholar] [CrossRef] [PubMed]
- Mandelkow, E.M.; Mandelkow, E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2012, 2, a006247. [Google Scholar] [CrossRef]
- Viswanathan, A.; Greenberg, S.M. Cerebral amyloid angiopathy in the elderly. Ann. Neurol. 2011, 70, 871–880. [Google Scholar] [CrossRef]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef]
- Robakis, N.K.; Wisniewski, H.M.; Jenkins, E.C.; Devine-Gage, E.A.; Houck, G.E.; Yao, X.L.; Ramakrishna, N.; Wolfe, G.; Silverman, W.P.; Brown, W.T. Chromosome 21q21 sublocalisation of gene encoding beta-amyloid peptide in cerebral vessels and neuritic (senile) plaques of people with Alzheimer disease and Down syndrome. Lancet 1987, 1, 384–385. [Google Scholar] [CrossRef]
- Carrodeguas, J.A.; Rodolosse, A.; Garza, M.V.; Sanz-Clemente, A.; Peréz-Pé, R.; Lacosta, A.M.; Domínguez, L.; Monleon, I.; Sánchez-Díaz, R.; Sorribas, V.; et al. The chick embryo appears as a natural model for research in beta-amyloid precursor protein processing. Neuroscience 2005, 134, 1285–1300. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Vassar, R.; Golde, T. The secretases: Enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Sun, Y.; Guo, Y.; Chen, Y.; Yu, B.; Zhang, H.; Wu, J.; Yu, X.; Kong, W.; Wu, H. Comparison of neurotoxicity of different aggregated forms of Aβ40, Aβ42 and Aβ43 in cell cultures. J. Pept. Sci. 2017, 23, 245–251. [Google Scholar] [CrossRef]
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s disease. Lancet 2006, 368, 387–403. [Google Scholar] [CrossRef]
- Nalivaeva, N.N.; Turner, A.J. Targeting amyloid clearance in Alzheimer’s disease as a therapeutic strategy. Br. J. Pharmacol. 2019, 176, 3447–3463. [Google Scholar] [CrossRef]
- Xin, S.H.; Tan, L.; Cao, X.; Yu, J.T.; Tan, L. Clearance of Amyloid Beta and Tau in Alzheimer’s Disease: From Mechanisms to Therapy. Neurotox. Res. 2018, 34, 733–748. [Google Scholar] [CrossRef]
- Tsigelny, I.F.; Crews, L.; Desplats, P.; Shaked, G.M.; Sharikov, Y.; Mizuno, H.; Spencer, B.; Rockenstein, E.; Trejo, M.; Platoshyn, O.; et al. Mechanisms of hybrid oligomer formation in the pathogenesis of combined Alzheimer’s and Parkinson’s diseases. PLoS ONE 2008, 3, e3135. [Google Scholar] [CrossRef]
- Twohig, D.; Nielsen, H.M. α-synuclein in the pathophysiology of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 23. [Google Scholar] [CrossRef]
- Danysz, W.; Parsons, C.G.; Möbius, H.J.; Stöffler, A.; Quack, G. Neuroprotective and symptomatological action of memantine relevant for Alzheimer’s disease—a unified glutamatergic hypothesis on the mechanism of action. Neurotox. Res. 2000, 2, 85–97. [Google Scholar] [CrossRef]
- Mintun, M.A.; Lo, A.C.; Duggan Evans, C.; Wessels, A.M.; Ardayfio, P.A.; Andersen, S.W.; Shcherbinin, S.; Sparks, J.; Sims, J.R.; Brys, M.; et al. Donanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2021, 384, 1691–1704. [Google Scholar] [CrossRef]
- Fields, C.R.; Bengoa-Vergniory, N.; Wade-Martins, R. Targeting Alpha-Synuclein as a Therapy for Parkinson’s Disease. Front. Mol. Neurosci. 2019, 12, 299. [Google Scholar] [CrossRef]
- Maiti, P.; Manna, J.; Dunbar, G.L. Current understanding of the molecular mechanisms in Parkinson’s disease: Targets for potential treatments. Transl. Neurodegener. 2017, 6, 28. [Google Scholar] [CrossRef]
- Politis, M.; Niccolini, F. Serotonin in Parkinson’s disease. Behav. Brain Res. 2015, 277, 136–145. [Google Scholar] [CrossRef]
- Grosch, J.; Winkler, J.; Kohl, Z. Early Degeneration of Both Dopaminergic and Serotonergic Axons—A Common Mechanism in Parkinson’s Disease. Front. Cell Neurosci. 2016, 10, 293. [Google Scholar] [CrossRef] [PubMed]
- Mallajosyula, J.K.; Kaur, D.; Chinta, S.J.; Rajagopalan, S.; Rane, A.; Nicholls, D.G.; Di Monte, D.A.; Macarthur, H.; Andersen, J.K. MAO-B elevation in mouse brain astrocytes results in Parkinson’s pathology. PLoS ONE 2008, 3, e1616. [Google Scholar] [CrossRef] [PubMed]
- Datta, P.; Rewers-Felkins, K.; Baker, T.; Hale, T.W. Transfer of Montelukast into Human Milk During Lactation. Breastfeed. Med. 2017, 12, 54–57. [Google Scholar] [CrossRef] [PubMed]



| Recombinant CYP Isoform | ||||||||||||||
| 1A2 | 2A6 | 2B6 | 2C8 | 2C9 | 2C19 | 2D6 | 2E1 | 3A4 | 3A5 | |||||
| M2a | [121] | |||||||||||||
| [123] | ||||||||||||||
| [124] * | ||||||||||||||
| [122] * | ||||||||||||||
| M2b | [121] | |||||||||||||
| [123] | ||||||||||||||
| M3 | [121] | |||||||||||||
| [123] | ||||||||||||||
| [124] | ||||||||||||||
| M4 | [123] | |||||||||||||
| M5a | [121] | |||||||||||||
| [123] | ||||||||||||||
| [124] | ||||||||||||||
| [122] δ | ||||||||||||||
| M5b | [121] | |||||||||||||
| [123] | ||||||||||||||
| [124] | ||||||||||||||
| M6 | [121] | |||||||||||||
| [123] | ||||||||||||||
| [124] | ||||||||||||||
| [122] | ||||||||||||||
| Metabolite formation rate (no shade—lower; darker shade—higher) | ||||||||||||||
| Not included in the study | ||||||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marques, C.F.; Marques, M.M.; Justino, G.C. Leukotrienes vs. Montelukast—Activity, Metabolism, and Toxicity Hints for Repurposing. Pharmaceuticals 2022, 15, 1039. https://doi.org/10.3390/ph15091039
Marques CF, Marques MM, Justino GC. Leukotrienes vs. Montelukast—Activity, Metabolism, and Toxicity Hints for Repurposing. Pharmaceuticals. 2022; 15(9):1039. https://doi.org/10.3390/ph15091039
Chicago/Turabian StyleMarques, Cátia F., Maria Matilde Marques, and Gonçalo C. Justino. 2022. "Leukotrienes vs. Montelukast—Activity, Metabolism, and Toxicity Hints for Repurposing" Pharmaceuticals 15, no. 9: 1039. https://doi.org/10.3390/ph15091039
APA StyleMarques, C. F., Marques, M. M., & Justino, G. C. (2022). Leukotrienes vs. Montelukast—Activity, Metabolism, and Toxicity Hints for Repurposing. Pharmaceuticals, 15(9), 1039. https://doi.org/10.3390/ph15091039