

Oxy210, a Semi-Synthetic Oxysterol, Inhibits Profibrotic Signaling in Cellular Models of Lung and Kidney Fibrosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
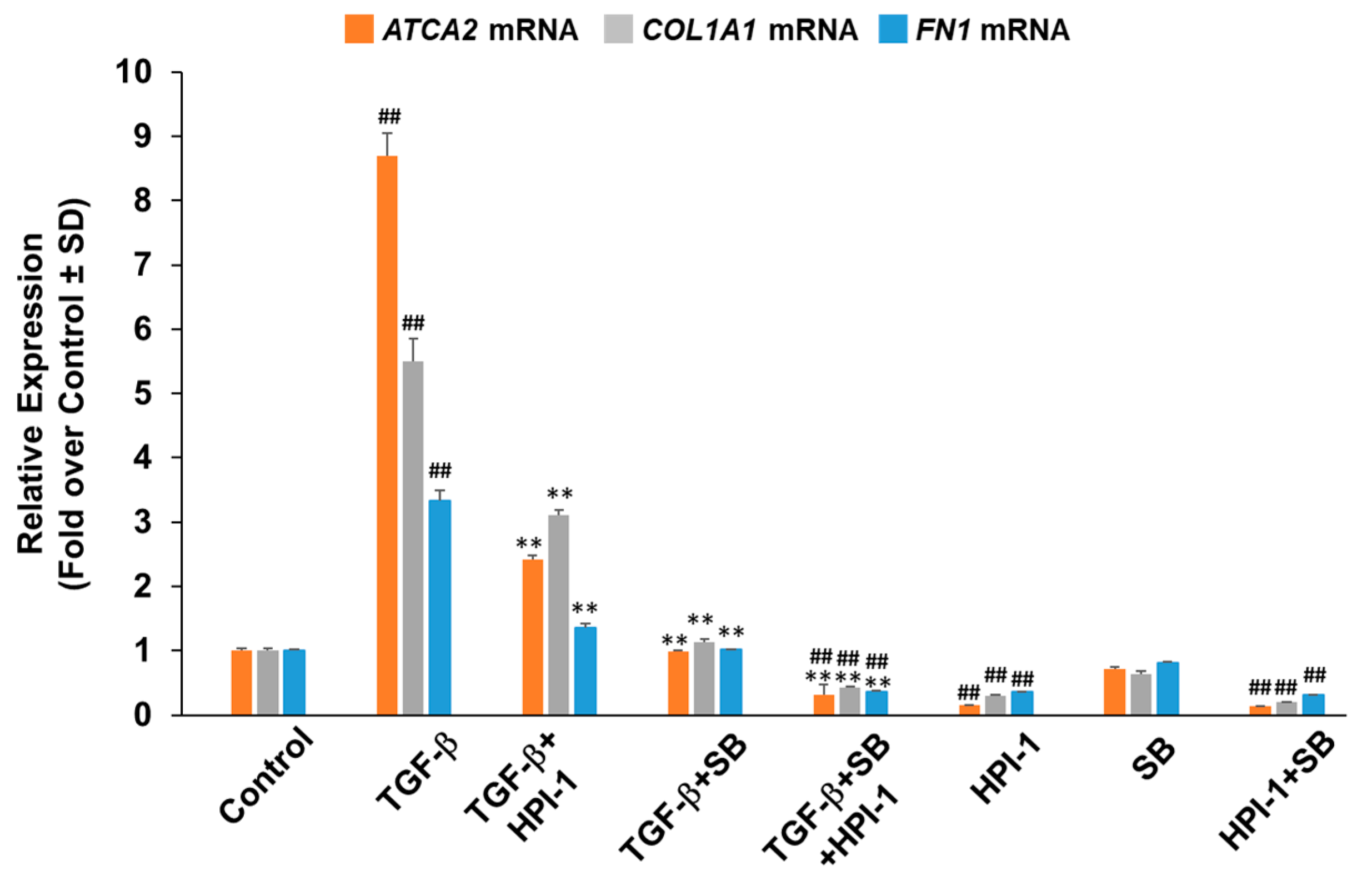
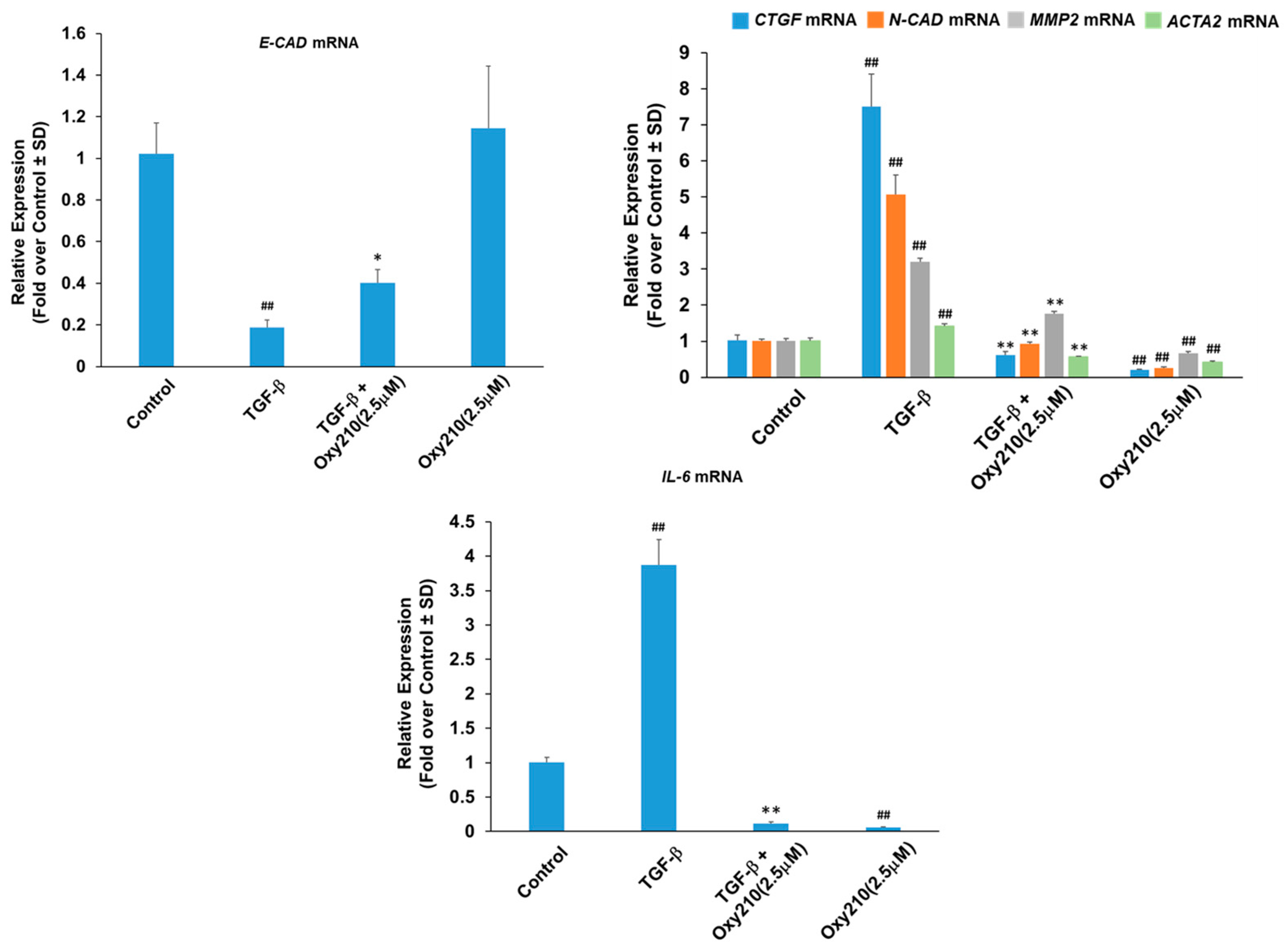
2.1. Oxy210 Inhibits Mediators of Fibrosis in Cultures of Lung Fibroblasts
2.2. Profibrotic Gene Expression in Lung Fibroblasts Is Regulated by Hh and TGF-β Signaling
2.3. Oxy210 Inhibits Proliferation of Pulmonary Fibroblasts
2.4. Oxy210 Inhibits Expression of Epithelial–Mesenchymal Transition (EMT) Genes and Interleukin-6 in Human Primary Small Airway Epithelial (HSAE) Cells
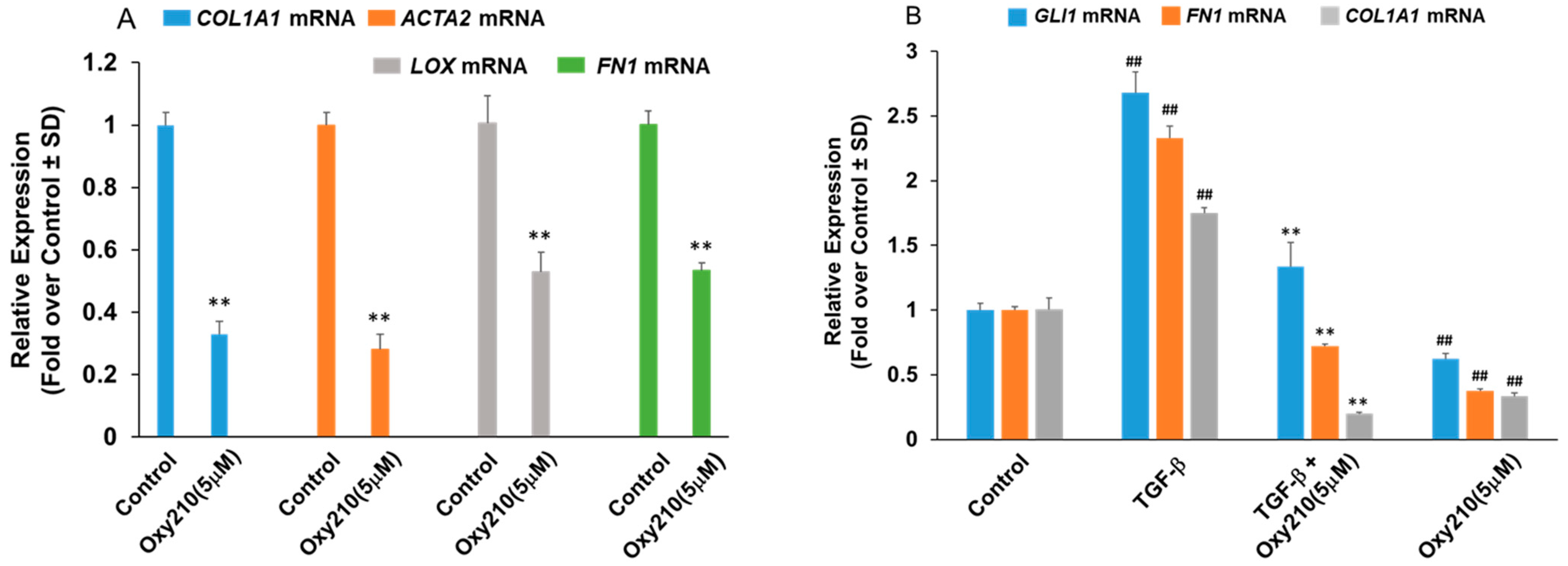
2.5. Oxy210 Inhibits Mediators of Fibrosis in Cultures of Kidney Cells
2.6. Oxy210 Inhibits Proliferation of Primary Human Pericytes, Renal Fibroblasts and Renal Mesangial Cells
2.7. Oxy210 Inhibits TGF-β-Induced EMT Gene Expression of Human Primary Renal Tubular Epithelial Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Quantitative RT-PCR
4.3. ELISA Assay
4.4. Cell Counting Assay
4.5. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Pakshir, P.; Noskovicova, N.; Lodyga, M.; Son, D.O.; Schuster, R.; Goodwin, A.; Karvonen, H.; Hinz, B. The myofibroblast at a glance. J. Cell Sci. 2020, 133, jcs227900. [Google Scholar] [CrossRef] [PubMed]
- Plikus, M.V.; Wang, X.; Sinha, S.; Forte, E.; Thompson, S.M.; Herzog, E.L.; Driskell, R.R.; Rosenthal, N.; Biernaskie, J.; Horsley, V. Fibroblasts: Origins, definitions, and functions in health and disease. Cell 2021, 184, 3852–3872. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Fibroblast-Extracellular Matrix Interactions in Tissue Fibrosis. Curr. Pathobiol. Rep. 2016, 4, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, T. Extracellular Interactions between Fibulins and Transforming Growth Factor (TGF)-β in Physiological and Pathological Conditions. Int. J. Mol. Sci. 2018, 19, 2787. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Transforming growth factor-β in tissue fibrosis. J. Exp. Med. 2020, 217, e20190103. [Google Scholar] [CrossRef]
- Edeling, M.; Ragi, G.; Huang, S.; Pavenstädt, H.; Susztak, K. Developmental signalling pathways in renal fibrosis: The roles of Notch, Wnt and Hedgehog. Nat. Rev. Nephrol. 2016, 12, 426–439. [Google Scholar] [CrossRef]
- Xie, G.; Karaca, G.; Swiderska-Syn, M.; Michelotti, G.A.; Krüger, L.; Chen, Y.; Premont, R.T.; Choi, S.S.; Diehl, A.M. Cross-talk between Notch and Hedgehog regulates hepatic stellate cell fate in mice. Hepatology 2013, 58, 1801–1813. [Google Scholar] [CrossRef]
- Chen, X.; Shi, C.; Cao, H.; Chen, L.; Hou, J.; Xiang, Z.; Hu, K.; Han, X. The hedgehog and Wnt/β-catenin system machinery mediate myofibroblast differentiation of LR-MSCs in pulmonary fibrogenesis. Cell Death Dis. 2018, 9, 639. [Google Scholar] [CrossRef]
- Hu, B.; Phan, S.H. Notch in fibrosis and as a target of anti-fibrotic therapy. Pharmacol. Res. 2016, 108, 57–64. [Google Scholar] [CrossRef]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-Y.; Qin, L.; Li, G.; Wang, Z.; Dahlman, J.E.; Malagon-Lopez, J.; Gujja, S.; Cilfone, N.A.; Kauffman, K.J.; Sun, L.; et al. Endothelial TGF-β signalling drives vascular inflammation and atherosclerosis. Nat. Metab. 2019, 1, 912–926. [Google Scholar] [CrossRef] [PubMed]
- Marek, A.; Brodzicki, J.; Liberek, A.; Korzon, M. TGF-beta (transforming growth factor-beta) in chronic inflammatory conditions—A new diagnostic and prognostic marker? Med. Sci. Monit. 2002, 8, RA145–RA151. [Google Scholar] [PubMed]
- Shi, N.; Wang, Z.; Zhu, H.; Liu, W.; Zhao, M.; Jiang, X.; Zhao, J.; Ren, C.; Zhang, Y.; Luo, L. Research progress on drugs targeting the TGF-β signaling pathway in fibrotic diseases. Immunol. Res. 2022, 70, 276–288. [Google Scholar] [CrossRef]
- Mallat, Z.; Gojova, A.; Marchiol-Fournigault, C.; Esposito, B.; Kamaté, C.; Merval, R.; Fradelizi, D.; Tedgui, A. Inhibition of transforming growth factor-beta signaling accelerates atherosclerosis and induces an unstable plaque phenotype in mice. Circ. Res. 2001, 89, 930–934. [Google Scholar] [CrossRef]
- Dardare, J.; Witz, A.; Merlin, J.L.; Bochnakian, A.; Toussaint, P.; Gilson, P.; Harlé, A. Epithelial to Mesenchymal Transition in Patients with Pancreatic Ductal Adenocarcinoma: State-of-the-Art and Therapeutic Opportunities. Pharmaceuticals 2021, 14, 740. [Google Scholar] [CrossRef]
- Tie, Y.; Tang, F.; Peng, D.; Zhang, Y.; Shi, H. TGF-beta signal transduction: Biology, function and therapy for diseases. Mol. Biomed. 2022, 3, 45. [Google Scholar]
- Park, C.H.; Yoo, T.H. TGF-β Inhibitors for Therapeutic Management of Kidney Fibrosis. Pharmaceuticals 2022, 15, 1485. [Google Scholar] [CrossRef]
- Kramer, E.L.; Clancy, J.P. TGFβ as a therapeutic target in cystic fibrosis. Expert Opin. Ther. Targets 2018, 22, 177–189. [Google Scholar] [CrossRef]
- Righetti, G.; Casale, M.; Tonelli, M.; Liessi, N.; Fossa, P.; Pedemonte, N.; Millo, E.; Cichero, E. New Insights into the Binding Features of F508del CFTR Potentiators: A Molecular Docking, Pharmacophore Mapping and QSAR Analysis Approach. Pharmaceuticals 2020, 13, 445. [Google Scholar] [CrossRef]
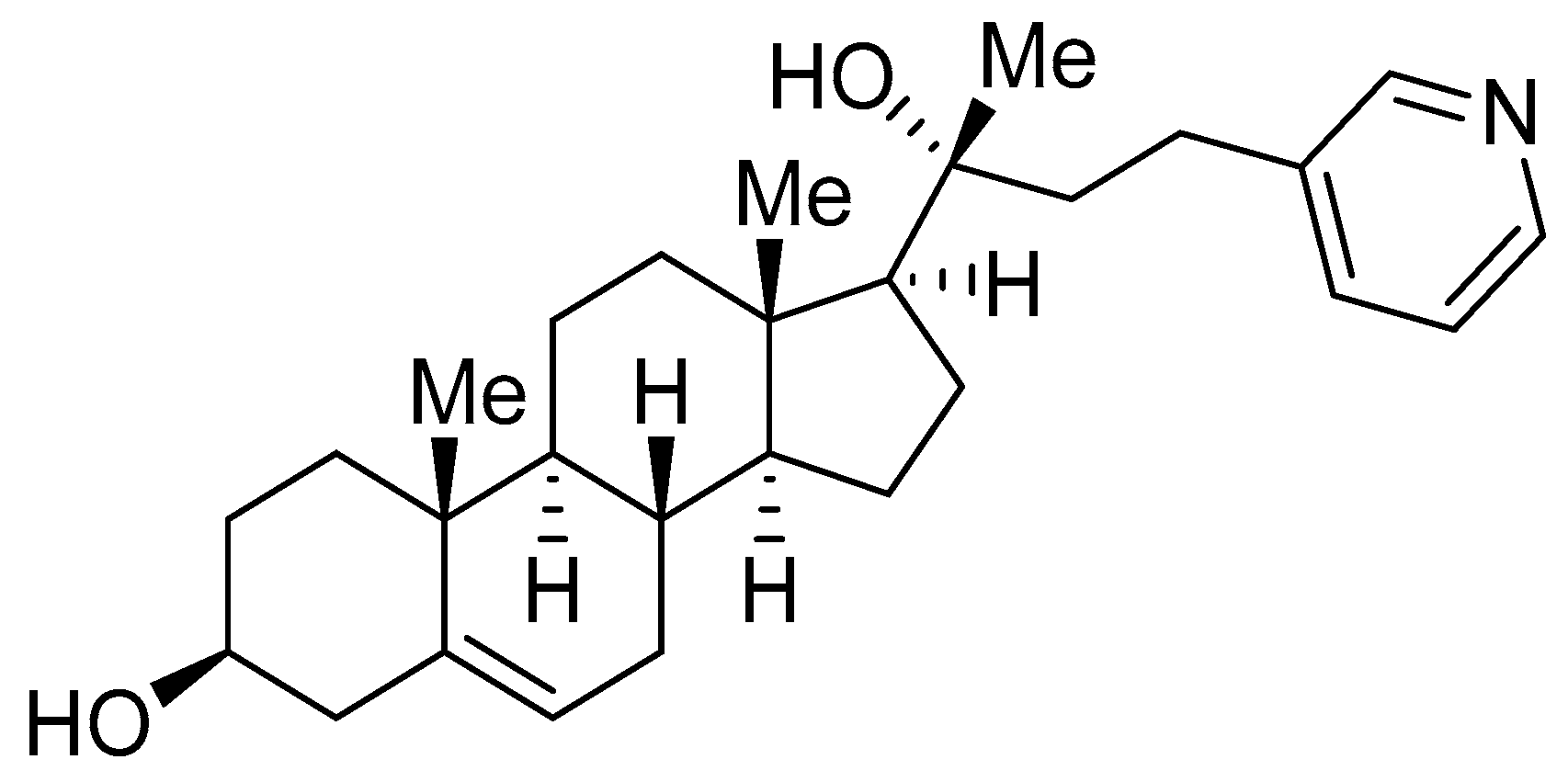
- Stappenbeck, F.; Wang, F.; Tang, L.Y.; Zhang, Y.E.; Parhami, F. Inhibition of Non-Small Cell Lung Cancer Cells by Oxy210, an Oxysterol-Derivative that Antagonizes TGFβ and Hedgehog Signaling. Cells 2019, 8, 1297. [Google Scholar] [CrossRef] [PubMed]
- Hui, S.T.; Wang, F.; Stappenbeck, F.; French, S.W.; Magyar, C.E.; Parhami, F.; Lusis, A.J. Oxy210, a novel inhibitor of hedgehog and TGF-β signalling, ameliorates hepatic fibrosis and hypercholesterolemia in mice. Endocrinol. Diabetes Metab. 2021, 4, e00296. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Stappenbeck, F.; Tang, L.-Y.; Zhang, Y.E.; Hui, S.T.; Lusis, A.J.; Parhami, F. Oxy210, a Semi-Synthetic Oxysterol, Exerts Anti-Inflammatory Effects in Macrophages via Inhibition of Toll-like Receptor (TLR) 4 and TLR2 Signaling and Modulation of Macrophage Polarization. Int. J. Mol. Sci. 2022, 23, 5478. [Google Scholar] [CrossRef] [PubMed]
- Terashima, H.; Aonuma, M.; Tsuchida, H.; Sugimoto, K.; Yokoyama, M.; Kato, M. Attenuation of pulmonary fibrosis in type I collagen-targeted reporter mice with ALK-5 inhibitors. Pulm. Pharmacol. Ther. 2019, 54, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Bülow, R.D.; Boor, P. Extracellular Matrix in Kidney Fibrosis: More Than Just a Scaffold. J. Histochem. Cytochem. 2019, 67, 643–661. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Zapater, E.; Signes-Costa, J.; Montero, P.; Roger, I. Lung Fibrosis and Fibrosis in the Lungs: Is It All about Myofibroblasts? Biomedicines 2022, 10, 1423. [Google Scholar] [CrossRef] [PubMed]
- Horn, A.; Palumbo, K.; Cordazzo, C.; Dees, C.; Akhmetshina, A.; Tomcik, M.; Zerr, P.; Avouac, J.; Gusinde, J.; Zwerina, J.; et al. Hedgehog signaling controls fibroblast activation and tissue fibrosis in systemic sclerosis. Arthritis Rheum. 2012, 64, 2724–2733. [Google Scholar] [CrossRef]
- Effendi, W.I.; Nagano, T. The Hedgehog Signaling Pathway in Idiopathic Pulmonary Fibrosis: Resurrection Time. Int. J. Mol. Sci. 2022, 23, 171. [Google Scholar] [CrossRef]
- Rockey, D.C.; Weymouth, N.; Shi, Z. Smooth muscle α actin (Acta2) and myofibroblast function during hepatic wound healing. PLoS ONE 2013, 8, e77166. [Google Scholar] [CrossRef]
- Hyman, J.M.; Firestone, A.J.; Heine, V.M.; Zhao, Y.; Ocasio, C.A.; Han, K.; Sun, M.; Rack, P.G.; Sinha, S.; Wu, J.J.; et al. Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proc. Natl. Acad. Sci. USA 2009, 106, 14132–14137. [Google Scholar] [CrossRef]
- Halder, S.K.; Beauchamp, R.D.; Datta, P.K. A specific inhibitor of TGF-beta receptor kinase, SB-431542, as a potent antitumor agent for human cancers. Neoplasia 2005, 7, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Kendall, R.T.; Feghali-Bostwick, C.A. Fibroblasts in fibrosis: Novel roles and mediators. Front. Pharmacol. 2014, 5, 123. [Google Scholar] [CrossRef] [PubMed]
- Correll, K.A.; Edeen, K.E.; Redente, E.F.; Zemans, R.L.; Edelman, B.L.; Danhorn, T.; Curran-Everett, U.; Mikels-Vigdal, A.; Mason, R.J. TGF beta inhibits HGF, FGF7, and FGF10 expression in normal and IPF lung fibroblasts. Physiol. Rep. 2018, 6, e13794. [Google Scholar] [CrossRef] [PubMed]
- Shang, L.; Hosseini, M.; Liu, X.; Kisseleva, T.; Brenner, D.A. Human hepatic stellate cell isolation and characterization. J. Gastroenterol. 2018, 53, 6–17. [Google Scholar] [CrossRef]
- El Taghdouini, A.; Najimi, M.; Sancho-Bru, P.; Sokal, E.; van Grunsven, L.A. In vitro reversion of activated primary human hepatic stellate cells. Fibrogenesis Tissue Repair. 2015, 8, 14. [Google Scholar] [CrossRef]
- Lokmic, Z.; Musyoka, J.; Hewitson, T.D.; Darby, I.A. Hypoxia and hypoxia signaling in tissue repair and fibrosis. Int. Rev. Cell Mol. Biol. 2012, 296, 139–185. [Google Scholar] [PubMed]
- Chakraborty, A.; Mastalerz, M.; Ansari, M.; Schiller, H.B.; Staab-Weijnitz, C.A. Emerging Roles of Airway Epithelial Cells in Idiopathic Pulmonary Fibrosis. Cells 2022, 11, 1050. [Google Scholar] [CrossRef]
- Camelo, A.; Dunmore, R.; Sleeman, M.A.; Clarke, D.L. The epithelium in idiopathic pulmonary fibrosis: Breaking the barrier. Front. Pharmacol. 2014, 4, 173. [Google Scholar] [CrossRef]
- Lovisa, S. Epithelial-to-Mesenchymal Transition in Fibrosis: Concepts and Targeting Strategies. Front. Pharmacol. 2021, 12, 737570. [Google Scholar] [CrossRef]
- Liu, L.; Sun, Q.; Davis, F.; Mao, J.; Zhao, H.; Ma, D. Epithelial-mesenchymal transition in organ fibrosis development: Current understanding and treatment strategies. Burn. Trauma 2022, 10, tkac011. [Google Scholar] [CrossRef]
- Jonckheere, S.; Adams, J.; De Groote, D.; Campbell, K.; Berx, G.; Goossens, S. Epithelial-Mesenchymal Transition (EMT) as a Therapeutic Target. Cells Tissues Organs 2022, 211, 157–182. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.; Jones, M.G.; Davies, D.E.; Wang, Y. Epithelial-mesenchymal transition contributes to pulmonary fibrosis via aberrant epithelial/fibroblastic cross-talk. J. Lung Health Dis. 2019, 3, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Tanaka, K.; Fujita, T.; Umezawa, H.; Amano, H.; Yoshioka, K.; Naito, Y.; Hatano, M.; Kimura, S.; Tatsumi, K.; et al. Bidirectional role of IL-6 signal in pathogenesis of lung fibrosis. Respir. Res. 2015, 16, 99. [Google Scholar] [CrossRef] [PubMed]
- Kramann, R.; Humphreys, B.D. Kidney pericytes: Roles in regeneration and fibrosis. Semin. Nephrol. 2014, 34, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Strutz, F.; Zeisberg, M. Renal fibroblasts and myofibroblasts in chronic kidney disease. J. Am. Soc. Nephrol. 2006, 17, 2992–2998. [Google Scholar] [CrossRef] [PubMed]
- Cove-Smith, A.; Hendry, B.M. The regulation of mesangial cell proliferation. Nephron Exp. Nephrol. 2008, 108, e74–e79. [Google Scholar] [CrossRef]
- Lu, H.; Chen, B.; Hong, W.; Liang, Y.; Bai, Y. Transforming growth factor-β1 stimulates hedgehog signaling to promote epithelial-mesenchymal transition after kidney injury. FEBS J. 2016, 283, 3771–3790. [Google Scholar] [CrossRef] [PubMed]
- Sheng, L.; Zhuang, S. New Insights into the Role and Mechanism of Partial Epithelial-Mesenchymal Transition in Kidney Fibrosis. Front. Physiol. 2020, 11, 569322. [Google Scholar] [CrossRef]
- Loh, C.-Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef]
- Deyell, M.; Garris, C.S.; Laughney, A.M. Cancer metastasis as a non-healing wound. Br. J. Cancer 2021, 124, 1491–1502. [Google Scholar] [CrossRef]
- Fabregat, I.; Moreno-Càceres, J.; Sánchez, A.; Dooley, S.; Dewidar, B.; Giannelli, G.; Ten Dijke, P.; IT-LIVER Consortium. TGF-β signalling and liver disease. FEBS J. 2016, 283, 2219–2232. [Google Scholar] [CrossRef] [PubMed]
- Tatler, A.L.; Jenkins, G. TGF-β activation and lung fibrosis. Proc. Am. Thorac. Soc. 2012, 9, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Isaka, Y. Targeting TGF-β Signaling in Kidney Fibrosis. Int. J. Mol. Sci. 2018, 19, 2532. [Google Scholar] [CrossRef] [PubMed]
- Giroux-Leprieur, E.; Costantini, A.; Ding, V.W.; He, B. Hedgehog Signaling in Lung Cancer: From Oncogenesis to Cancer Treatment Resistance. Int. J. Mol. Sci. 2018, 19, 2835. [Google Scholar] [CrossRef]
- Saito, A.; Horie, M.; Nagase, T. TGF-β Signaling in Lung Health and Disease. Int. J. Mol. Sci. 2018, 19, 2460. [Google Scholar] [CrossRef] [PubMed]
- Dormoy, V.; Danilin, S.; Lindner, V.; Thomas, L.; Rothhut, S.; Coquard, C.; Helwig, J.-J.; Jacqmin, D.; Lang, H.; Massfelder, T. The sonic hedgehog signaling pathway is reactivated in human renal cell carcinoma and plays orchestral role in tumor growth. Mol. Cancer 2009, 8, 123. [Google Scholar] [CrossRef] [PubMed]
- Sitaram, R.T.; Mallikarjuna, P.; Landström, M.; Ljungberg, B. Transforming growth factor-β promotes aggressiveness and invasion of clear cell renal cell carcinoma. Oncotarget 2016, 7, 35917–35931. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Li, H.-Y.; Zhang, L.; Zhou, Y.; Wu, J. Hedgehog Signaling, a Critical Pathway Governing the Development and Progression of Hepatocellular Carcinoma. Cells 2021, 10, 123. [Google Scholar] [CrossRef]
- Tu, S.; Huang, W.; Huang, C.; Luo, Z.; Yan, X. Contextual Regulation of TGF-β Signaling in Liver Cancer. Cells 2019, 8, 1235. [Google Scholar] [CrossRef]
- Raghu, G.; Amatto, V.C.; Behr, J.; Stowasser, S. Comorbidities in idiopathic pulmonary fibrosis patients: A systematic literature review. Eur. Respir. J. 2015, 46, 1113–1130. [Google Scholar] [CrossRef]
- Ruaro, B.; Pozzan, R.; Confalonieri, P.; Tavano, S.; Hughes, M.; Matucci Cerinic, M.; Baratella, E.; Zanatta, E.; Lerda, S.; Geri, P.; et al. Gastroesophageal Reflux Disease in Idiopathic Pulmonary Fibrosis: Viewer or Actor? To Treat or Not to Treat? Pharmaceuticals 2022, 15, 1033. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Nie, P.; Lou, Y.; Zhu, Y.; Jiang, S.; Li, B.; Luo, P. Pirfenidone is a renal protective drug: Mechanisms, signalling pathways, and preclinical evidence. Eur. J. Pharmacol. 2021, 911, 174503. [Google Scholar] [CrossRef]
- Cho, M.E.; Smith, D.C.; Branton, M.H.; Penzak, S.R.; Kopp, J.B. Pirfenidone slows renal function decline in patients with focal segmental glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2007, 2, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Philips, C.A.; Padsalgi, G.; Ahamed, R.; Paramaguru, R.; Rajesh, S.; George, T.; Mahadevan, P.; Augustine, P. Repurposing Pirfenidone for Nonalcoholic Steatohepatitis-related Cirrhosis: A Case Series. J. Clin. Transl. Hepatol. 2020, 8, 100–105. [Google Scholar] [CrossRef]
- Didiasova, M.; Singh, R.; Wilhelm, J.; Kwapiszewska, G.; Wujak, L.; Zakrzewicz, D.; Schaefer, L.; Markart, P.; Seeger, W.; Lauth, M.; et al. Pirfenidone exerts antifibrotic effects through inhibition of GLI transcription factors. FASEB J. 2017, 31, 1916–1928. [Google Scholar] [CrossRef]
- Rangarajan, S.; Kurundkar, A.; Kurundkar, D.; Bernard, K.; Sanders, Y.Y.; Ding, Q.; Antony, V.B.; Zhang, J.; Zmijewski, J.; Thannickal, V.J. Novel Mechanisms for the Antifibrotic Action of Nintedanib. Am. J. Respir. Cell Mol. Biol. 2016, 54, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Zisser, A.; Ipsen, D.H.; Tveden-Nyborg, P. Hepatic Stellate Cell Activation and Inactivation in NASH-Fibrosis—Roles as Putative Treatment Targets? Biomedicines 2021, 9, 365. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primers 2017, 3, 17074. [Google Scholar] [CrossRef]
- Baratella, E.; Ruaro, B.; Giudici, F.; Wade, B.; Santagiuliana, M.; Salton, F.; Confalonieri, P.; Simbolo, M.; Scarpa, A.; Tollot, S.; et al. Evaluation of Correlations between Genetic Variants and High-Resolution Computed Tomography Patterns in Idiopathic Pulmonary Fibrosis. Diagnostics 2021, 11, 762. [Google Scholar] [CrossRef]
- Zhao, J.H. Mesangial Cells and Renal Fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 165–194. [Google Scholar]
- Lovisa, S.; LeBleu, V.S.; Tampe, B.; Sugimoto, H.; Vadnagara, K.; Carstens, J.; Wu, C.-C.; Hagos, Y.; Burckhardt, B.; Pentcheva-Hoang, T.; et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 2015, 21, 998–1009. [Google Scholar] [CrossRef]
- Zhao, Y.L.; Zhu, R.T.; Sun, Y.L. Epithelial-mesenchymal transition in liver fibrosis. Biomed. Rep. 2016, 4, 269–274. [Google Scholar] [CrossRef]
- Tsoukalas, N.; Aravantinou-Fatorou, E.; Tolia, M.; Giaginis, C.; Galanopoulos, M.; Kiakou, M.; Kostakis, I.D.; Dana, E.; Vamvakaris, I.; Korogiannos, A.; et al. Epithelial-Mesenchymal Transition in Non Small-cell Lung Cancer. Anticancer Res. 2017, 37, 1773–1778. [Google Scholar] [PubMed]
- Ohashi, H.; Wang, F.; Stappenbeck, F.; Tsuchimoto, K.; Kobayashi, C.; Saso, W.; Kataoka, M.; Yamasaki, M.; Kuramochi, K.; Muramatsu, M.; et al. Identification of Anti-Severe Acute Respiratory Syndrome-Related Coronavirus 2 (SARS-CoV-2) Oxysterol Derivatives In Vitro. Int. J. Mol. Sci. 2021, 22, 3163. [Google Scholar] [CrossRef]
- Yue, X.; Shan, B.; Lasky, J.A. TGF-β: Titan of Lung Fibrogenesis. Curr. Enzym. Inhib. 2010, 6, 67–77. [Google Scholar] [CrossRef]
- Arribillaga, L.; Dotor, J.; Basagoiti, M.; Riezu-Boj, J.I.; Borrás-Cuesta, F.; Lasarte, J.J.; Sarobe, P.; Cornet, M.E.; Feijoó, E. Therapeutic effect of a peptide inhibitor of TGF-β on pulmonary fibrosis. Cytokine 2011, 53, 327–333. [Google Scholar] [CrossRef]
- McGaraughty, S.; Davis-Taber, R.A.; Zhu, C.Z.; Cole, T.B.; Nikkel, A.L.; Chhaya, M.; Doyle, K.J.; Olson, L.M.; Preston, G.M.; Grinnell, C.M.; et al. Targeting Anti-TGF-β Therapy to Fibrotic Kidneys with a Dual Specificity Antibody Approach. J. Am. Soc. Nephrol. 2017, 28, 3616–3626. [Google Scholar] [CrossRef]
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From mechanisms to medicines. Nature 2020, 587, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Diebold, R.J.; Eis, M.J.; Yin, M.; Ormsby, I.; Boivin, G.; Darrow, B.; Saffitz, J.; Doetschman, T. Early-onset multifocal inflammation in the transforming growth factor beta 1-null mouse is lymphocyte mediated. Proc. Natl. Acad. Sci. USA 1995, 92, 12215–12219. [Google Scholar] [CrossRef]
- Wei, Y.; Kim, T.J.; Peng, D.H.; Duan, D.; Gibbons, D.L.; Yamauchi, M.; Jackson, J.R.; Le Saux, C.J.; Calhoun, C.; Peters, J.; et al. Fibroblast-specific inhibition of TGF-β1 signaling attenuates lung and tumor fibrosis. J. Clin. Investig. 2017, 127, 3675–3688. [Google Scholar] [CrossRef]
- Drexler, S.K.; Foxwell, B.M. The role of toll-like receptors in chronic inflammation. Int. J. Biochem. Cell Biol. 2010, 42, 506–518. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Friedman, S.L. Toll-like receptor 4 signaling in liver injury and hepatic fibrogenesis. Fibrogenes. Tissue Repair. 2010, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Yang, L.; van Rooijen, N.; Brenner, D.A.; Ohnishi, H.; Seki, E. Toll-like receptor 2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis through inflammasome activation in mice. Hepatology 2013, 57, 577–589. [Google Scholar] [CrossRef]
- Karampitsakos, T.; Woolard, T.; Bouros, D.; Tzouvelekis, A. Toll-like receptors in the pathogenesis of pulmonary fibrosis. Eur. J. Pharmacol. 2017, 808, 35–43. [Google Scholar] [CrossRef]
- You, W.H.; Lin, M.; Tang, S.C. Toll-like receptor activation: From renal inflammation to fibrosis. Kidney Int. Suppl. 2014, 4, 20–25. [Google Scholar] [CrossRef]
- Xie, H.; Paradise, B.D.; Ma, W.W.; Fernandez-Zapico, M.E. Recent Advances in the Clinical Targeting of Hedgehog/GLI Signaling in Cancer. Cells 2019, 8, 394. [Google Scholar] [CrossRef] [PubMed]
- Prasse, A.; Ramaswamy, M.; Mohan, S.; Pan, L.; Kenwright, A.; Neighbors, M.; Belloni, P.; LaCamera, P.P. A Phase 1b Study of Vismodegib with Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis. Pulm. Ther. 2019, 5, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Arensdorf, A.M.; Marada, S.; Ogden, S.K. Smoothened Regulation: A Tale of Two Signals. Trends Pharmacol. Sci. 2016, 37, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Morinello, E.; Pignatello, M.; Villabruna, L.; Goelzer, P.; Bürgin, H. Embryofetal development study of vismodegib, a hedgehog pathway inhibitor, in rats. Birth Defects Res. B Dev. Reprod. Toxicol. 2014, 101, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.M.; Jacobs, D.T.; Allard, B.A.; Fields, T.A.; Sharma, M.; Wallace, D.P.; Tran, P.V. Inhibition of Hedgehog signaling suppresses proliferation and microcyst formation of human Autosomal Dominant Polycystic Kidney Disease cells. Sci. Rep. 2018, 8, 4985. [Google Scholar] [CrossRef]
- Zhou, D.; Tan, R.J.; Liu, Y. Sonic hedgehog signaling in kidney fibrosis: A master communicator. Sci. China Life Sci. 2016, 59, 920–929. [Google Scholar] [CrossRef] [PubMed]
- Kramann, R. Hedgehog Gli signalling in kidney fibrosis. Nephrol. Dial. Transplant. 2016, 31, 1989–1995. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sun, N.; Mo, N.; Lu, S.; Song, E.; Ren, C.; Li, Z. Quercetin inhibits kidney fibrosis and the epithelial to mesenchymal transition of the renal tubular system involving suppression of the Sonic Hedgehog signaling pathway. Food Funct. 2019, 10, 3782–3797. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, F.; Stappenbeck, F.; Parhami, F. Oxy210, a Semi-Synthetic Oxysterol, Inhibits Profibrotic Signaling in Cellular Models of Lung and Kidney Fibrosis. Pharmaceuticals 2023, 16, 114. https://doi.org/10.3390/ph16010114
Wang F, Stappenbeck F, Parhami F. Oxy210, a Semi-Synthetic Oxysterol, Inhibits Profibrotic Signaling in Cellular Models of Lung and Kidney Fibrosis. Pharmaceuticals. 2023; 16(1):114. https://doi.org/10.3390/ph16010114
Chicago/Turabian StyleWang, Feng, Frank Stappenbeck, and Farhad Parhami. 2023. "Oxy210, a Semi-Synthetic Oxysterol, Inhibits Profibrotic Signaling in Cellular Models of Lung and Kidney Fibrosis" Pharmaceuticals 16, no. 1: 114. https://doi.org/10.3390/ph16010114
APA StyleWang, F., Stappenbeck, F., & Parhami, F. (2023). Oxy210, a Semi-Synthetic Oxysterol, Inhibits Profibrotic Signaling in Cellular Models of Lung and Kidney Fibrosis. Pharmaceuticals, 16(1), 114. https://doi.org/10.3390/ph16010114