
Anti-AMPA Receptor Autoantibodies Reduce Excitatory Currents in Rat Hippocampal Neurons



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
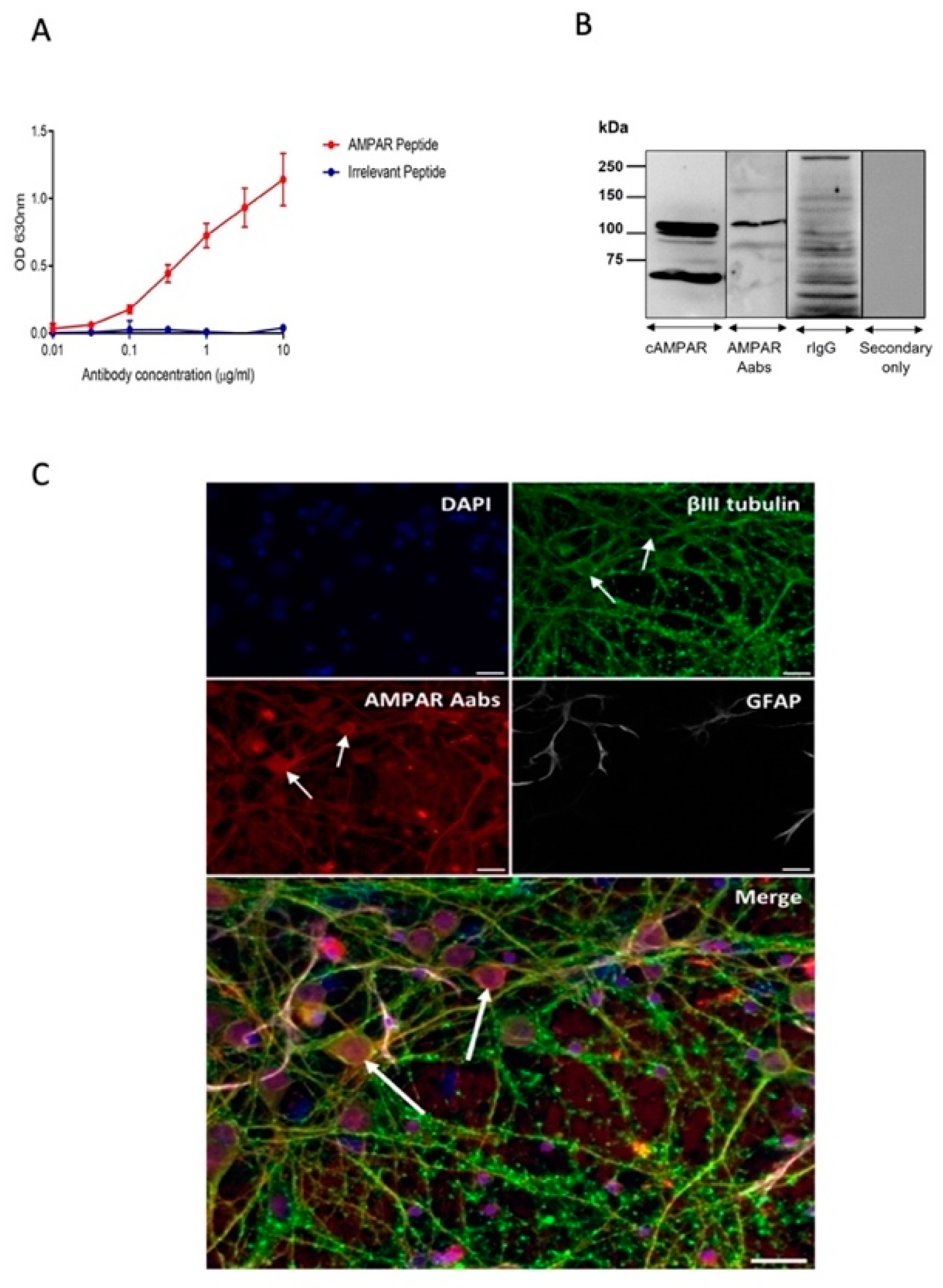
2.1. Generation and Characterisation of AMPAR Aabs
2.2. Assessing the Functionality of Anti-AMPAR Aabs on Spontaneous Excitatory Postsynaptic Currents (sEPSCs)
2.2.1. Effects of Acute Anti-AMPAR Aab Incubation
2.2.2. Effects of 24 h Anti-AMPAR Aab Incubation
2.3. Effects of Anti-AMPAR Aab Incubation on Miniature Excitatory Postsynaptic Currents (mEPSCs)
2.3.1. Effects of Acute Anti-AMPAR Aab Incubation
2.3.2. Effects of 24 h Anti-AMPAR Aab Incubation
3. Discussion
3.1. Anti-AMPAR Aabs Bind to Native AMPARs
3.2. Anti-AMPAR Aabs Exhibit a Functional Inhibitory Effect
3.3. Clinical Relevance
4. Materials and Methods
4.1. Rabbit Immunisation and Antibody Production and Purification
4.1.1. Serum Screening and Antibody Titre
4.1.2. Protein A Purification of Polyclonal IgG Antibody from Rabbit Serum
4.1.3. SDS-PAGE Analysis of Aab Purity
4.2. Immunocytochemistry
SDS PAGE and Western Blotting
4.3. Animals
E18 Primary Neuronal Cell Culture
4.4. In Vitro Electrophysiology
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parkin, J.; Cohen, B. An overview of the immune system. Lancet 2001, 357, 1777–1789. [Google Scholar] [CrossRef] [PubMed]
- Siloşi, I.; Siloşi, C.A.; Boldeanu, M.V.; Cojocaru, M.; Biciuşcă, V.; Avrămescu, C.S.; Cojocaru, I.M.; Bogdan, M.; FolcuŢi, R.M. The role of autoantibodies in health and disease. Rom. J. Morphol. Embryol. 2016, 57, 633–638. [Google Scholar]
- Ludwig, R.J.; Vanhoorelbeke, K.; Leypoldt, F.; Kaya, Z.; Bieber, K.; McLachlan, S.M.; Komorowski, L.; Luo, J.; Cabral-Marques, O.; Hammers, C.M.; et al. Mechanisms of autoantibody-induced pathology. Front. Immunol. 2017, 8, 603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gold, M.; Pul, R.; Bach, J.P.; Stangel, M.; Dodel, R. Pathogenic and physiological autoantibodies in the central nervous system. Immunol. Rev. 2012, 248, 68–86. [Google Scholar] [CrossRef]
- Dalmau, J.; Geis, C.; Graus, F. Autoantibodies to synaptic receptors and neuronal cell surface proteins in autoimmune diseases of the Central Nervous System. Physiol. Rev. 2017, 97, 839–887. [Google Scholar] [CrossRef] [Green Version]
- Gardoni, F.; Stanic, J.; Scheggia, D.; Benussi, A.; Borroni, B.; Di Luca, M. NMDA and AMPA receptor autoantibodies in brain disorders: From molecular mechanisms to clinical features. Cells 2021, 10, 77. [Google Scholar] [CrossRef] [PubMed]
- Gouaux, E. Structure and function of AMPA receptors. J. Physiol. 2004, 554, 249–253. [Google Scholar] [CrossRef]
- Chater, T.E.; Goda, Y. The role of AMPA receptors in postsynaptic mechanisms of synaptic plasticity. Front. Cell. Neurosci. 2014, 8, 401. [Google Scholar] [CrossRef]
- Rogers, S.W.; Andrews, P.I.; Gahring, L.C.; Whisenand, T.; Cauley, K.; Crain, B.; Hughes, T.E.; Heinemann, S.F.; McNamara, J.O. Autoantibodies to glutamate receptor GluR3 in Rasmussen’s encephalitis. Science 1994, 265, 648–651. [Google Scholar] [CrossRef]
- Levite, M. Glutamate receptor antibodies in neurological diseases: Anti-AMPA-GluR3 antibodies, anti-NMDA-NR1 antibodies, anti-NMDA-NR2A/B antibodies, anti-mGluR1 antibodies or anti-mGluR5 antibodies are present in subpopulations of patients with either: Epilepsy, encephalitis, cerebellar ataxia, systemic lupus erythematosus (SLE) and neuropsychiatric SLE, Sjogren’s syndrome, schizophrenia, mania or stroke. These autoimmune anti-glutamate receptor antibodies can bind neurons in few brain regions, activate glutamate receptors, decrease glutamate receptor’s expression, impair glutamate-induced signaling and function, activate blood brain barrier endothelial cells, kill neurons, damage the brain, induce behavioral/psychiatric/cognitive abnormalities and ataxia in animal models, and can be removed or silenced in some patients by immunotherapy. J. Neural. Transm. 2014, 121, 1029–1075. [Google Scholar]
- Borroni, B.; Stanic, J.; Verpelli, C.; Mellone, M.; Bonomi, E.; Alberici, A.; Bernasconi, P.; Culotta, L.; Zianni, E.; Archetti, S.; et al. Anti-AMPA GluR3 antibodies in frontotemporal dementia: A new molecular target. Sci. Rep. 2017, 7, 6723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollak, T.A.; Beck, K.; Irani, S.R.; Howes, O.D.; David, A.S.; McGuire, P.K. Autoantibodies to central nervous system neuronal surface antigens: Psychiatric symptoms and psychopharmacological implications. Psychopharmacology 2016, 233, 1605–1621. [Google Scholar] [CrossRef] [PubMed]
- Ganor, Y.; Goldberg-Stern, H.; Cohen, R.; Teichberg, V.; Levite, M. Glutamate receptor antibodies directed against AMPA receptors subunit 3 peptide B (GluR3B) can be produced in DBA/2J mice, lower seizure threshold and induce abnormal behavior. Psychoneuroendocrinology 2014, 42, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Levite, M.; Hermelin, A. Autoimmunity to the glutamate receptor in mice—A model for Rasmussen’s encephalitis? J. Autoimmun. 1999, 13, 73–82. [Google Scholar] [CrossRef]
- Levite, M.; Fleidervish, I.A.; Schwarz, A.; Pelled, D.; Futerman, A.H. Autoantibodies to the glutamate receptor kill neurons via activation of the receptor ion channel. J. Autoimmun. 1999, 13, 61–72. [Google Scholar] [CrossRef]
- Ganor, Y.; Gottlieb, M.; Eilam, R.; Otmy, H.; Teichberg, V.I.; Levite, M. Immunization with the glutamate receptor-derived peptide GluR3B induces neuronal death and reactive gliosis, but confers partial protection from pentylenetetrazole-induced seizures. Exp. Neurol. 2005, 195, 92–102. [Google Scholar] [CrossRef]
- Twyman, R.E.; Gahring, L.C.; Spiess, J.; Rogers, S.W. Glutamate receptor antibodies activate a subset of receptors and reveal an agonist binding site. Neuron 1995, 14, 755–762. [Google Scholar] [CrossRef] [Green Version]
- Palese, F.; Bonomi, E.; Nuzzo, T.; Benussi, A.; Mellone, M.; Zianni, E.; Cisani, F.; Casamassa, A.; Alberici, A.; Scheggia, D.; et al. Anti-GluR3 antibodies in frontotemporal dementia: Effects on glutamatergic neurotransmission and synaptic failure. Neurobiol. Aging 2020, 86, 143–155. [Google Scholar] [CrossRef] [Green Version]
- Scheggia, D.; Stanic, J.; Italia, M.; La Greca, F.; Zianni, E.; Benussi, A.; Borroni, B.; Di Luca, M.; Gardoni, F. GluR3 autoantibodies induce alterations in dendritic spine and behavior in mice. Brain Behav. Immun. 2021, 97, 89–101. [Google Scholar] [CrossRef]
- Frassoni, C.; Spreafico, R.; Franceschetti, S.; Aurisano, N.; Bernasconi, P.; Garbelli, R.; Antozzi, C.; Taverna, S.; Granata, T.; Mantegazza, R. Labeling of rat neurons by anti-GluR3 IgG from patients with Rasmussen encephalitis. Neurology 2001, 57, 324–327. [Google Scholar] [CrossRef]
- Stern-Bach, Y.; Bettler, B.; Hartley, M.; Sheppard, P.O.; O’Hara, P.J.; Heinemann, S.F. Agonist selectivity of glutamate receptors is specified by two domains structurally related to bacterial amino acid-binding proteins. Neuron 1994, 13, 1345–1357. [Google Scholar] [CrossRef] [PubMed]
- Hnasko, R.M.; McGarvey, J.A. Affinity purification of antibodies. Methods Mol. Biol. 2015, 1318, 29–41. [Google Scholar] [PubMed]
- Meyer, E.L.; Strutz, N.; Gahring, L.C.; Rogers, S.W. Glutamate receptor subunit 3 is modified by site-specific limited proteolysis including cleavage by gamma-secretase. J. Biol. Chem. 2003, 278, 23786–23796. [Google Scholar] [CrossRef] [Green Version]
- Renner, M.C.; Albers, E.H.; Gutierrez-Castellanos, N.; Reinders, N.R.; van Huijstee, A.N.; Xiong, H.; Lodder, T.R.; Kessels, H.W. Synaptic plasticity through activation of GluR3-containing AMPA-receptors. Elife 2017, 6, e25462. [Google Scholar] [CrossRef] [PubMed]
- Zucker, R.S.; Regehr, W.G. Short-term synaptic plasticity. Annu. Rev. Physiol. 2002, 64, 355–405. [Google Scholar] [CrossRef] [Green Version]
- Haselmann, H.; Mannara, F.; Werner, C.; Planaguma, J.; Miguez-Cabello, F.; Schmidl, L.; Grunewald, B.; Petit-Pedrol, M.; Kirmse, K.; Classen, J.; et al. Human autoantibodies against the AMPA receptor subunit GluA2 induce receptor reorganization and memory dysfunction. Neuron 2018, 100, 91–105.e9. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Hughes, E.G.; Moscato, E.H.; Parsons, T.D.; Dalmau, J.; Balice-Gordon, R.J. Cellular plasticity induced by anti-alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor encephalitis antibodies. Ann. Neurol. 2015, 77, 381–398. [Google Scholar] [CrossRef] [Green Version]
- Hughes, E.G.; Peng, X.; Gleichman, A.J.; Lai, M.; Zhou, L.; Tsou, R.; Parsons, T.D.; Lynch, D.R.; Dalmau, J.; Balice-Gordon, R.J. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J. Neurosci. 2010, 30, 5866–5875. [Google Scholar] [CrossRef] [Green Version]
- Cohen-Kashi Malina, K.; Ganor, Y.; Levite, M.; Teichberg, V.I. Autoantibodies against an extracellular peptide of the GluR3 subtype of AMPA receptors activate both homomeric and heteromeric AMPA receptor channels. Neurochem. Res. 2006, 31, 1181–1190. [Google Scholar] [CrossRef]
- Partin, K.M. AMPA receptor potentiators: From drug design to cognitive enhancement. Curr. Opin. Pharmacol. 2015, 20, 46–53. [Google Scholar] [CrossRef] [Green Version]
- Zanetti, L.; Regoni, M.; Ratti, E.; Valtorta, F.; Sassone, J. Presynaptic AMPA receptors in health and disease. Cells 2021, 10, 2260. [Google Scholar] [CrossRef] [PubMed]
- Wenthold, R.J.; Petralia, R.S.; Niedzielski, A.S. Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J. Neurosci. 1996, 16, 1982–1989. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.-H.; Hayashi, Y.; Esteban, J.A.; Malinow, R. Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell 2001, 105, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Goldberg-Stern, H.; Ganor, Y.; Cohen, R.; Pollak, L.; Teichberg, V.; Levite, M. Glutamate receptor antibodies directed against AMPA receptors subunit 3 peptide B (GluR3B) associate with some cognitive/psychiatric/behavioral abnormalities in epilepsy patients. Psychoneuroendocrinology 2014, 40, 221–231. [Google Scholar] [CrossRef]
- Percie du Sert, N.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biol. 2020, 40, e3000410. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Day, C.; Silva, J.-P.; Munro, R.; Baker, T.S.; Wolff, C.; Bithell, A.; Stephens, G.J. Anti-AMPA Receptor Autoantibodies Reduce Excitatory Currents in Rat Hippocampal Neurons. Pharmaceuticals 2023, 16, 77. https://doi.org/10.3390/ph16010077
Day C, Silva J-P, Munro R, Baker TS, Wolff C, Bithell A, Stephens GJ. Anti-AMPA Receptor Autoantibodies Reduce Excitatory Currents in Rat Hippocampal Neurons. Pharmaceuticals. 2023; 16(1):77. https://doi.org/10.3390/ph16010077
Chicago/Turabian StyleDay, Charlotte, John-Paul Silva, Rebecca Munro, Terry S. Baker, Christian Wolff, Angela Bithell, and Gary J. Stephens. 2023. "Anti-AMPA Receptor Autoantibodies Reduce Excitatory Currents in Rat Hippocampal Neurons" Pharmaceuticals 16, no. 1: 77. https://doi.org/10.3390/ph16010077