
Redrawing Urokinase Receptor (uPAR) Signaling with Cancer Driver Genes for Exploring Possible Anti-Cancer Targets and Drugs

 ,
,
Abstract
:1. Introduction
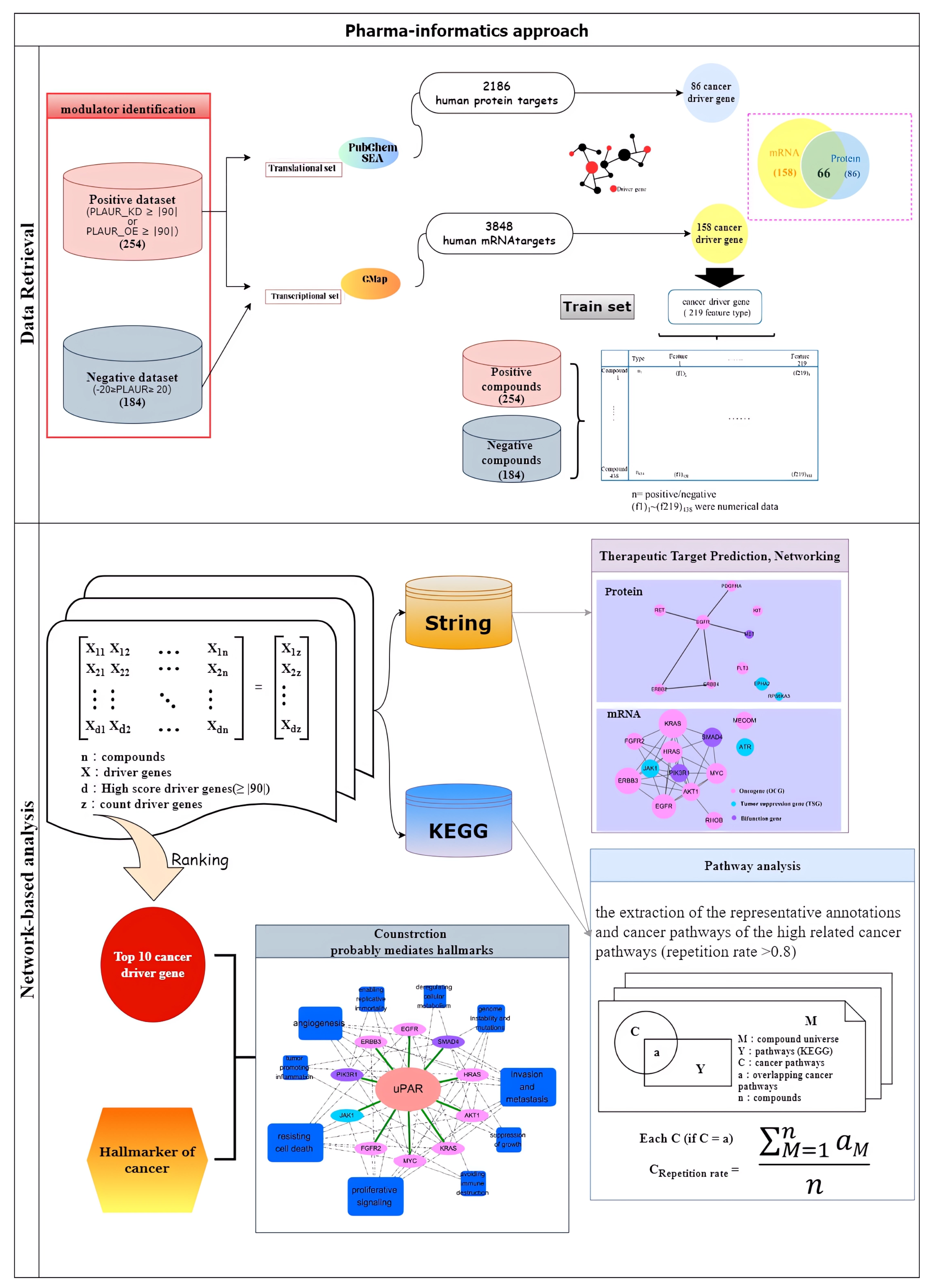
2. Results
2.1. uPAR Modulators and Their Cancerous Signaling Networks
2.2. Essences of uPAR-Mediated Signaling (Table 1) in Cancer Pathways

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway ID | KEGG Pathway Description | Modulators (254) | Counts of Top 10 Driver Genes |
|---|---|---|---|
| Repetition Rate (Number/254) | |||
| hsa04010 | MAPK signaling pathway | 0.97 (246/254) | 7 |
| hsa04630 | JAK-STAT signaling pathway | 0.86 (219/254) | 9 |
| hsa04151 | PI3K-Akt signaling pathway | 0.96 (243/254) | 6 |
| hsa04150 | mTOR signaling pathway | 0.89 (227/254) | 4 |
| hsa04510 | Focal adhesion | 0.82 (208/254) | 4 |
| hsa04066 | HIF-1 signaling pathway | 0.85 (216/254) | 3 |
| hsa04210 | Apoptosis | 0.89 (226/254) | 4 |
| hsa04370 | VEGF signaling pathway | 0.86 (219/254) | 4 |
| hsa04110 | Cell cycle | 0.86 (219/254) | 2 |
| hsa04915 | Estrogen signaling pathway | 0.85 (215/254) | 5 |
2.3. Virtual Screening of uPAR Modulators by Machine Learning
2.4. Implication of uPAR Modulators in Anti-Cancer Therapy
3. Discussion
4. Materials and Methods
4.1. Modulator Identification
4.2. Target/Cancer Driver Gene Ontology Networking
4.3. Therapeutic Target Prediction, Networking and Pathway Analysis
4.4. Data Mining and Predictive Model Construction
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blasi, F. Proteolysis, Cell Adhesion, Chemotaxis, and Invasiveness Are Regulated by the u-PA-u-PAR-PAI-1 System. Thromb. Haemost. 1999, 82, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Rasch, M.G.; Lund, I.K.; Almasi, C.E.; Hoyer-Hansen, G. Intact and cleaved uPAR forms: Diagnostic and prognostic value in cancer. Front. Biosci. Landmark 2008, 13, 6752–6762. [Google Scholar] [CrossRef] [PubMed]
- Bifulco, K.; Votta, G.; Ingangi, V.; Di Carluccio, G.; Rea, D.; Losito, S.; Montuori, N.; Ragno, P.; Stoppelli, M.P.; Arra, C.; et al. Urokinase receptor promotes ovarian cancer cell dissemination through its 84–95 sequence. Oncotarget 2014, 5, 4154–4169. [Google Scholar] [CrossRef]
- Eden, G.; Archinti, M.; Furlan, F.; Murphy, R.; Degryse, B. The Urokinase Receptor Interactome. Curr. Pharm. Des. 2011, 17, 1874–1889. [Google Scholar] [CrossRef] [PubMed]
- Blasi, F.; Carmeliet, P. uPAR: A versatile signalling orchestrator. Nat. Rev. Mol. Cell Biol. 2002, 3, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.W.; Marshall, C.J. Regulation of cell signalling by uPAR. Nat. Rev. Mol. Cell Biol. 2010, 11, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Leurer, C.; Rabbani, S.A. Plasminogen Activator System—Diagnostic, Prognostic and Therapeutic Implications in Breast Cancer; IntechOpen: London, UK, 2015. [Google Scholar] [CrossRef]
- Montuori, N.; Pesapane, A.; Rossi, F.W.; Giudice, V.; De Paulis, A.; Selleri, C.; Ragno, P. Urokinase type plasminogen activator receptor (uPAR) as a new therapeutic target in cancer. Transl. Med. UniSa 2016, 15, 15–21. [Google Scholar] [PubMed]
- Yuan, C.; Guo, Z.; Yu, S.; Jiang, L.; Huang, M. Development of inhibitors for uPAR: Blocking the interaction of uPAR with its partners. Drug Discov. Today 2021, 26, 1076–1085. [Google Scholar] [CrossRef]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385.e18. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Wei, L.; Chen, J.-S.; Lin, H.-T.; Wu, C.-Z.; Zheng, C.-M.; Chang, Y.-C.; Chang, L.-C.; Lin, Y.-F. Atorvastatin from target screening attenuates endothelial cell tube formation and migration by regulating urokinase receptor-related signaling pathway and F/G actin. J. Chin. Med. Assoc. 2017, 80, 86–95. [Google Scholar] [CrossRef]
- Crescencio, M.E.; Rodriguez, E.; Paez, A.; Masso, F.A.; Montano, L.F.; Lopez-Marure, R. Statins inhibit the proliferation and induce cell death of human papilloma virus positive and negative cervical cancer cells. Int. J. Biomed. Sci. 2009, 5, 411–420. [Google Scholar] [PubMed]
- Miller, C.M.; O’Sullivan, E.C.; McCarthy, F.O. Novel 11-Substituted Ellipticines as Potent Anticancer Agents with Divergent Activity against Cancer Cells. Pharmaceuticals 2019, 12, 90. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.J.; Han, J.M.; Choi, Y.S.; Jung, H.J. Pterostilbene Suppresses both Cancer Cells and Cancer Stem-Like Cells in Cervical Cancer with Superior Bioavailability to Resveratrol. Molecules 2020, 25, 228. [Google Scholar] [CrossRef] [PubMed]
- Obrador, E.; Salvador-Palmer, R.; Jihad-Jebbar, A.; Lopez-Blanch, R.; Dellinger, T.H.; Dellinger, R.W.; Estrela, J.M. Pterostilbene in Cancer Therapy. Antioxidants 2021, 10, 492. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.; Lei, F.J.; Chang, H.J.; Wei, S.T.; Wang, C.C.; Huang, Y.C.; Wang, H.L.; Chuang, C.F.; Hu, S.Y.; Hsieh, C.H. Haloperidol Instigates Endometrial Carcinogenesis and Cancer Progression by the NF-kappaB/CSF-1 Signaling Cascade. Cancers 2022, 14, 3089. [Google Scholar] [CrossRef]
- National Toxicology Program. Phenazopyridine Hydrochloride: CAS No. 136-40-3. In 15th Report on Carcinogens; National Toxicology Program: Research Triangle Park, NC, USA, 2021. [Google Scholar]
- Binder, B.R.; Mihaly, J.; Prager, G.W. uPAR–uPA–PAI-1 interactions and signaling: A vascular biologist’s view. Thromb. Haemost. 2007, 97, 336–342. [Google Scholar]
- Resnati, M.; Pallavicini, I.; Wang, J.M.; Oppenheim, J.; Serhan, C.N.; Romano, M.; Blasi, F. The fibrinolytic receptor for urokinase activates the G protein-coupled chemotactic receptor FPRL1/LXA4R. Proc. Natl. Acad. Sci. USA 2002, 99, 1359–1364. [Google Scholar] [CrossRef]
- Montuori, N.; Bifulco, K.; Carriero, M.V.; La Penna, C.; Visconte, V.; Alfano, D.; Pesapane, A.; Rossi, F.W.; Salzano, S.; Rossi, G.; et al. The cross-talk between the urokinase receptor and fMLP receptors regulates the activity of the CXCR4 chemokine receptor. Cell. Mol. Life Sci. 2011, 68, 2453–2467. [Google Scholar] [CrossRef]
- Huang, C.; Xie, D.; Cui, J.; Li, Q.; Gao, Y.; Xie, K. FOXM1c promotes pancreatic cancer epithelial-to-mesenchymal transition and metastasis via upregulation of expression of the urokinase plasminogen activator system. Clin. Cancer Res. 2014, 20, 1477–1488. [Google Scholar] [CrossRef]
- Gupta, R.; Chetty, C.; Bhoopathi, P.; Lakka, S.; Mohanam, S.; Rao, J.S.; Dinh, D.H. Downregulation of uPA/uPAR inhibits intermittent hypoxia-induced epithelial-mesenchymal transition (EMT) in DAOY and D283 medulloblastoma cells. Int. J. Oncol. 2011, 38, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Laurenzana, A.; Biagioni, A.; Bianchini, F.; Peppicelli, S.; Chillà, A.; Margheri, F.; Luciani, C.; Pimpinelli, N.; Del Rosso, M.; Calorini, L.; et al. Inhibition of uPAR-TGFβ crosstalk blocks MSC-dependent EMT in melanoma cells. J. Mol. Med. 2015, 93, 783–794. [Google Scholar] [CrossRef]
- Mahmood, N.; Mihalcioiu, C.; Rabbani, S.A. Multifaceted Role of the Urokinase-Type Plasminogen Activator (uPA) and Its Receptor (uPAR): Diagnostic, Prognostic, and Therapeutic Applications. Front. Oncol. 2018, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Chounta, A.; Ellinas, C.; Tzanetakou, V.; Pliarhopoulou, F.; Mplani, V.; Oikonomou, A.; Leventogiannis, K.; Giamarellos-Bourboulis, E.J. Serum soluble urokinase plasminogen activator receptor as a screening test for the early diagnosis of hepatocellular carcinoma. Liver Int. 2015, 35, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.K.; Mukhopadhyay, A.K. Tyrosine kinase—Role and significance in Cancer. Int. J. Med. Sci. 2004, 1, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Arteaga, C.L.; Engelman, J.A. ERBB Receptors: From Oncogene Discovery to Basic Science to Mechanism-Based Cancer Therapeutics. Cancer Cell 2014, 25, 282–303. [Google Scholar] [CrossRef] [PubMed]
- Hirsh, V. Next-Generation Covalent Irreversible Kinase Inhibitors in NSCLC: Focus on Afatinib. BioDrugs 2015, 29, 167–183. [Google Scholar] [CrossRef]
- Zhou, J.; Kwak, K.J.; Wu, Z.; Yang, D.; Li, J.; Chang, M.; Song, Y.; Zeng, H.; Lee, L.J.; Hu, J.; et al. PLAUR Confers Resistance to Gefitinib Through EGFR/P-AKT/Survivin Signaling Pathway. Cell. Physiol. Biochem. 2018, 47, 1909–1924. [Google Scholar] [CrossRef]
- Laurenzana, A.; Margheri, F.; Biagioni, A.; Chillà, A.; Pimpinelli, N.; Ruzzolini, J.; Peppicelli, S.; Andreucci, E.; Calorini, L.; Serratì, S.; et al. EGFR/uPAR interaction as druggable target to overcome vemurafenib acquired resistance in melanoma cells. EBioMedicine 2019, 39, 194–206. [Google Scholar] [CrossRef]
- Cortés, I.; Sánchez-Ruíz, J.; Zuluaga, S.; Calvanese, V.; Marqués, M.; Hernández, C.; Rivera, T.; Kremer, L.; González-García, A.; Carrera Ana, C. p85β phosphoinositide 3-kinase subunit regulates tumor progression. Proc. Natl. Acad. Sci. USA 2012, 109, 11318–11323. [Google Scholar] [CrossRef]
- Taniguchi, C.M.; Winnay, J.; Kondo, T.; Bronson, R.T.; Guimaraes, A.R.; Alemán, J.O.; Luo, J.; Stephanopoulos, G.; Weissleder, R.; Cantley, L.C.; et al. The Phosphoinositide 3-Kinase Regulatory Subunit p85α Can Exert Tumor Suppressor Properties through Negative Regulation of Growth Factor Signaling. Cancer Res. 2010, 70, 5305–5315. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Snapper, S.B.; Yballe, C.M.; Davidson, L.; Yu, J.Y.; Alt, F.W.; Cantley, L.C. Impaired B Cell Development and Proliferation in Absence of Phosphoinositide 3-Kinase p85α. Science 1999, 283, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Sabbineni, H.; Alwhaibi, A.; Goc, A.; Gao, F.; Pruitt, A.; Somanath, P.R. Genetic deletion and pharmacological inhibition of Akt1 isoform attenuates bladder cancer cell proliferation, motility and invasion. Eur. J. Pharmacol. 2015, 764, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Somanath, P.R.; Razorenova, O.V.; Chen, J.; Byzova, T.V. Akt1 in endothelial cell and angiogenesis. Cell Cycle 2006, 5, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Testa, J.R.; Tsichlis, P.N. AKT signaling in normal and malignant cells. Oncogene 2005, 24, 7391–7393. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Pan, Y.F.; Xue, Y.-Q.; Fang, L.-K.; Guo, X.-H.; Guo, X.; Liu, M.; Mo, B.-Y.; Yang, M.-R.; Liu, F.; et al. uPAR promotes tumor-like biologic behaviors of fibroblast-like synoviocytes through PI3K/Akt signaling pathway in patients with rheumatoid arthritis. Cell. Mol. Immunol. 2018, 15, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Gondi, C.S.; Kandhukuri, N.; Dinh, D.H.; Gujrati, M.; Rao, J.S. Down-regulation of uPAR and uPA activates caspase-mediated apoptosis and inhibits the PI3K/AKT pathway. Int. J. Oncol. 2007, 31, 19–27. [Google Scholar] [CrossRef]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef]
- Mauro, C.D.; Pesapane, A.; Formisano, L.; Rosa, R.; D’Amato, V.; Ciciola, P.; Servetto, A.; Marciano, R.; Orsini, R.C.; Monteleone, F.; et al. Urokinase-type plasminogen activator receptor (uPAR) expression enhances invasion and metastasis in RAS mutated tumors. Sci. Rep. 2017, 7, 9388. [Google Scholar] [CrossRef] [PubMed]
- Alfano, D.; Votta, G.; Schulze, A.; Downward, J.; Caputi, M.; Stoppelli, M.P.; Iaccarino, I. Modulation of cellular migration and survival by c-Myc through the downregulation of urokinase (uPA) and uPA receptor. Mol. Cell. Biol. 2010, 30, 1838–1851. [Google Scholar] [CrossRef] [PubMed]
- Laurenzana, A.; Chillà, A.; Luciani, C.; Peppicelli, S.; Biagioni, A.; Bianchini, F.; Tenedini, E.; Torre, E.; Mocali, A.; Calorini, L.; et al. uPA/uPAR system activation drives a glycolytic phenotype in melanoma cells. Int. J. Cancer 2017, 141, 1190–1200. [Google Scholar] [CrossRef] [PubMed]
- Biagioni, A.; Chillà, A.; Del Rosso, M.; Fibbi, G.; Scavone, F.; Andreucci, E.; Peppicelli, S.; Bianchini, F.; Calorini, L.; Li Santi, A.; et al. CRISPR/Cas9 uPAR Gene Knockout Results in Tumor Growth Inhibition, EGFR Downregulation and Induction of Stemness Markers in Melanoma and Colon Carcinoma Cell Lines. Front. Oncol. 2021, 11, 663225. [Google Scholar] [CrossRef] [PubMed]
- Qureshy, Z.; Johnson, D.E.; Grandis, J.R. Targeting the JAK/STAT pathway in solid tumors. J. Cancer Metastasis Treat. 2020, 6, 27. [Google Scholar] [CrossRef] [PubMed]
- Wehde, B.L.; Rädler, P.D.; Shrestha, H.; Johnson, S.J.; Triplett, A.A.; Wagner, K.-U. Janus Kinase 1 Plays a Critical Role in Mammary Cancer Progression. Cell Rep. 2018, 25, 2192–2207.e5. [Google Scholar] [CrossRef] [PubMed]
- Koshelnick, Y.; Ehart, M.; Hufnagl, P.; Heinrich, P.C.; Binder, B.R. Urokinase Receptor Is Associated with the Components of the JAK1/STAT1 Signaling Pathway and Leads to Activation of This Pathway upon Receptor Clustering in the Human Kidney Epithelial Tumor Cell Line TCL-598. J. Biol. Chem. 1997, 272, 28563–28567. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.; Verstovsek, S. JAK2 inhibitors for myeloproliferative neoplasms: What is next? Blood 2017, 130, 115–125. [Google Scholar] [CrossRef]
- Huynh, J.; Etemadi, N.; Hollande, F.; Ernst, M.; Buchert, M. The JAK/STAT3 axis: A comprehensive drug target for solid malignancies. Semin. Cancer Biol. 2017, 45, 13–22. [Google Scholar] [CrossRef]
- Connolly, E.C.; Freimuth, J.; Akhurst, R.J. Complexities of TGF-β targeted cancer therapy. Int. J. Biol. Sci. 2012, 8, 964–978. [Google Scholar] [CrossRef]
- Zhao, M.; Mishra, L.; Deng, C.-X. The role of TGF-β/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Dennler, S.; Itoh, S.; Vivien, D.; ten Dijke, P.; Huet, S.; Gauthier, J.M. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998, 17, 3091–3100. [Google Scholar] [CrossRef] [PubMed]
- Sanfelice, R.A.; da Silva, S.S.; Bosqui, L.R.; Miranda-Sapla, M.M.; Barbosa, B.F.; Silva, R.J.; Ferro, E.A.V.; Panagio, L.A.; Navarro, I.T.; Bordignon, J.; et al. Pravastatin and simvastatin inhibit the adhesion, replication and proliferation of Toxoplasma gondii (RH strain) in HeLa cells. Acta Trop. 2017, 167, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Weis, M.; Heeschen, C.; Glassford, A.J.; Cooke, J.P. Statins have biphasic effects on angiogenesis. Circulation 2002, 105, 739–745. [Google Scholar] [CrossRef]
- Rao, P.S.; Rao, U.S. Statins decrease the expression of c-Myc protein in cancer cell lines. Mol. Cell Biochem. 2021, 476, 743–755. [Google Scholar] [CrossRef] [PubMed]
- Kodach, L.L.; Bleuming, S.A.; Peppelenbosch, M.P.; Hommes, D.W.; van den Brink, G.R.; Hardwick, J.C. The effect of statins in colorectal cancer is mediated through the bone morphogenetic protein pathway. Gastroenterology 2007, 133, 1272–1281. [Google Scholar] [CrossRef] [PubMed]
- Gawecka, J.E.; Young-Robbins, S.S.; Sulzmaier, F.J.; Caliva, M.J.; Heikkilä, M.M.; Matter, M.L.; Ramos, J.W. RSK2 protein suppresses integrin activation and fibronectin matrix assembly and promotes cell migration. J. Biol. Chem. 2012, 287, 43424–43437. [Google Scholar] [CrossRef]
- Houles, T.; Roux, P.P. Defining the role of the RSK isoforms in cancer. Semin. Cancer Biol. 2018, 48, 53–61. [Google Scholar] [CrossRef]
- Doehn, U.; Hauge, C.; Frank, S.R.; Jensen, C.J.; Duda, K.; Nielsen, J.V.; Cohen, M.S.; Johansen, J.V.; Winther, B.R.; Lund, L.R.; et al. RSK is a principal effector of the RAS-ERK pathway for eliciting a coordinate promotile/invasive gene program and phenotype in epithelial cells. Mol. Cell 2009, 35, 511–522. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [PubMed]
- Keiser, M.J.; Roth, B.L.; Armbruster, B.N.; Ernsberger, P.; Irwin, J.J.; Shoichet, B.K. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 2007, 25, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452.e17. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef] [PubMed]
- Demšar, J.; Curk, T.; Erjavec, A.; Gorup, C.; Hocevar, T.; Milutinovic, M.; Možina, M.; Polajnar, M.; Toplak, M.; Staric, A.; et al. Orange: Data Mining Toolbox in Python. J. Mach. Learn. Res. 2013, 14, 2349–2353. [Google Scholar]




| Active Modulators | Cancer Driver Genes |
|---|---|
| Suppression | |
| Homatropine | AKT1, ARHGAP35, ATR, AXIN2, BTG2, CCND1, CDKN1B, CHEK2, DICER1, DNMT3A, EGFR, EPAS1, ERBB2, ERBB3, FAT1, FBXW7, FGFR2, HRAS, JAK1, KRAS, MECOM, MSH6, MYD88, PDS5B, PMS1, RAC1, RASA1, RET, RHOA, RHOB, RXRA, SF3B1, SMAD4, TCF7L2, ZBTB20, ZFP36L1 |
| Hydralazine | ARHGAP35, B2M, BTG2, CCND1, CDK4, CDKN2C, CHEK2, EGFR, ERBB3, FLT3, KRAS, MET, NIPBL, PIK3CA, PIK3R1, PLCB4, PLCG1, RAC1, RASA1, RET, RPS6KA3, SMAD4, SMARCB1, WT1, ZBTB20 |
| Salbutamol | ARHGAP35, ATR, BRAF, CCND1, CTNNB1, EGFR, ERBB3, ERBB4, FGFR2, HRAS, IDH2, KRAS, MAP2K4, MECOM, MEN1, MTOR, MYC, PRKAR1A, RAC1, RHOA, SF3B1, SMAD4, SMARCB1, TRAF3 |
| Spironolactone | ARHGAP35, AXIN2, CDKN2C, EGFR, ERBB2, ERBB3, FOXQ1, GNA13, HRAS, NRAS, RHOB, SMAD4, TSC2, ZFP36L2 |
| Stimulation | |
| Aspirin | APOB, ARHGAP35, ATXN3, AXIN2, B2M, CHEK2, EGFR, ERBB4, FLT3, FOXQ1, GNA13, GNAS, HRAS, IDH1, JAK1, MAP2K4, MECOM, MET, MYC, PIK3CA, PIK3R1, POLRMT, PPP2R1A, PSIP1, RASA1, RB1, RET, RHOB, RPL22, SMARCB1, TCF7L2, TSC1, TSC2, WT1 |
| Atorvastatin | ARID5B, ATM, ATR, BRCA1, CDKN1A, CDKN2C, CTNND1, ERBB3, FLT3, IL6ST, IRF2, KRAS, MECOM, MEN1, MTOR, MYC, NRAS, PIK3R1, PIK3R1, PIM1, RHOB, RPS6KA3, RXRA, SF1, SMAD2, TSC2, WT1, ZBTB20, ZFP36L1, ZMYM2, ZNF133 |
| Methotrexate | AKT1, ARHGAP35, ATR, AXIN2, BRCA1, CHEK2, EGFR, ERBB3, KRAS, MAX, MECOM, MEN1, MTOR, MYC, PIK3CA, PIK3CG, RHOA, RXRA, TCF12, TCF7L2 |
| Quinine | ATM, BRCA1, CDKN2C, CHEK2, ERBB3, ERBB4, FGFR2, GNAS, HRAS, IRF6, MAP2K4, MAX, MECOM, MET, MTOR, NRAS, PIK3CG, PIK3R1, PRKAR1A, RAC1, RB1, SMARCB1, TNFAIP3, TSC2, TXNIP, USP9X, VHL, ZFP36L2 |
| Simvastatin | CDK4, CDKN1A, CDKN2C, ERBB3, IL6ST, KRAS, MAP2K4, MYC, MYD88, RAC1, RHOB, RXRA, TXNIP, ZFP36L1, ZMYM2 |
| Classifier | Performance | |||
|---|---|---|---|---|
| AUC | Accuracy | Sensitivity | Precision | |
| Total Features (218) | ||||
| kNN | 0.850 | 0.727 | 0.727 | 0.717 |
| SVM | 0.921 | 0.775 | 0.775 | 0.770 |
| Random Forest | 0.853 | 0.702 | 0.702 | 0.692 |
| Neural Network | 0.920 | 0.788 | 0.788 | 0.784 |
| Naive Bayes | 0.868 | 0.698 | 0.698 | 0.708 |
| Logistic Regression | 0.912 | 0.778 | 0.778 | 0.776 |
| Gradient Boosting | 0.891 | 0.730 | 0.730 | 0.722 |
| Strictly Associated Features (31) | ||||
| kNN | 0.872 | 0.738 | 0.738 | 0.731 |
| SVM | 0.917 | 0.774 | 0.774 | 0.770 |
| Random Forest | 0.877 | 0.728 | 0.728 | 0.720 |
| Neural Network | 0.913 | 0.778 | 0.778 | 0.779 |
| Naive Bayes | 0.905 | 0.747 | 0.747 | 0.748 |
| Logistic Regression | 0.896 | 0.749 | 0.749 | 0.749 |
| Gradient Boosting | 0.905 | 0.755 | 0.755 | 0.750 |
| Total Features (218) | Strictly Associated Features (31) | ||||
|---|---|---|---|---|---|
| SVM | Neural Network | SVM | Neural Network | ||
| uPAR activity | 9 | 9 | 9 | 9 | |
| No activity | 0 | 0 | 0 | 0 | |
| Hallmarks | Driver Gene | Preferred Marker |
|---|---|---|
| proliferative signaling | AKT1, EGFR, ERBB3, FGFR2, HRAS, JAK1, KRAS, MYC | MYC |
| resisting cell death | AKT1, EGFR, ERBB3, FGFR2, HRAS, JAK1, KRAS, MYC | MYC |
| angiogenesis | AKT1, EGFR, HRAS, KRAS, SMAD4 | SMAD4 |
| invasion and metastasis | AKT1, EGFR, ERBB3, HRAS, KRAS, MYC, PIK3R1, SMAD4 | PIK3R1, SMAD4 |
| Predicted Drug | CMap Expression Score * | Drug Effects | Referred Validation | ||
|---|---|---|---|---|---|
| uPAR | MYC | SMAD4 | |||
| Statins (atorvastatin) | 88.31(KD) | 99.15(KD) | Anti-cancer | In-house data, [12,13] | |
| Ellipticine | 91.97(KD) | 90.64(KD) | Anti-cancer | [14] | |
| Pterostilbene | 95.19(KD) | 94.29(KD) | Anti-cancer | [15,16] | |
| haloperidol | 94.07(OE) | 98.59(KD) | Carcinogenic | [17] | |
| phenazopyridine | 92.97(OE) | 94.41(KD) | Carcinogenic | [18] | |
| Driver Gene | Gene Description | Hallmarks |
|---|---|---|
| AKT1 | AKT serine/threonine kinase 1 | proliferative signaling, evading growth suppressors, invasion and metastasis, angiogenesis, resisting cell death, deregulating cellular metabolism, genome instability and mutations |
| EGFR | ErbB (epidermal growth factor) receptor family, epidermal growth factor receptor | proliferative signaling, avoiding immune destruction, invasion and metastasis, angiogenesis, resisting cell death, deregulating cellular metabolism |
| ERBB3 | ErbB (epidermal growth factor) receptor family, v-erb-b2 erythroblastic leukemia viral oncogene homolog 3 | proliferative signaling, invasion and metastasis, resisting cell death |
| FGFR2 | Type V RTKs: FGF (fibroblast growth factor) receptor family, fibroblast growth factor receptor 2 | proliferative signaling, resisting cell death |
| HRAS | RAS subfamily, v-Ha-ras Harvey rat sarcoma viral oncogene homolog | proliferative signaling, tumor-promoting inflammation, invasion and metastasis, angiogenesis, genome instability and mutations, resisting cell death, avoiding immune destruction |
| JAK1 | Janus kinase (JakA) family, Janus kinase 1 | proliferative signaling, avoiding immune destruction, resisting cell death |
| KRAS | RAS subfamily, v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog | proliferative signaling, enabling replicative immortality, tumor-promoting inflammation, invasion and metastasis, angiogenesis, resisting cell death, deregulating cellular metabolism |
| MYC | Basic helix-loop-helix proteins, v-myc myelocytomatosis viral oncogene homolog | proliferative signaling, angiogenesis, avoiding immune destruction, genome instability and mutations, deregulating cellular metabolism, resisting cell death, invasion and metastasis, enabling replicative immortality |
| PIK3R1 | Phosphatidylinositol kinases, phosphoinositide-3-kinase, regulatory subunit 1 (alpha) | evading growth suppressors, enabling replicative immortality, invasion and metastasis |
| SMAD4 | SMADs, SMAD family member 4 | evading growth suppressors, tumor-promoting inflammation, invasion and metastasis, angiogenesis, genome instability and mutations |
| Resource | Description | Website | Ref. |
|---|---|---|---|
| Translational (protein) | |||
| PubChem | A web-based informatics environment for data from small molecules and their biological activities. | https://pubchem.ncbi.nlm.nih.gov/ (accessed on 1 July 2023) | [63] |
| Similarity ensemble approach (SEA) | An open resource related to proteins based on the set-wise chemical similarity among their ligands. | http://sea.bkslab.org/ (accessed on 1 July 2023) | [64] |
| Transcriptional (gene) | |||
| Connectivity Map (CMap) | A public catalog of gene expression data collected from human cells treated with chemical compounds and genetic reagents | https://clue.io/cmap (accessed on 1 July 2023) | [65] |
| KEGG | A curated database collecting comprehensive data including genes, reactions, pathways, drugs and diseases, for studying functions and utilities of the biological systems | http://www.kegg.jp/kegg/ (accessed on 1 July 2023) | [11] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, Y.-C.; Wu, C.-Z.; Cheng, C.-W.; Chen, J.-S.; Chang, L.-C. Redrawing Urokinase Receptor (uPAR) Signaling with Cancer Driver Genes for Exploring Possible Anti-Cancer Targets and Drugs. Pharmaceuticals 2023, 16, 1435. https://doi.org/10.3390/ph16101435
Chang Y-C, Wu C-Z, Cheng C-W, Chen J-S, Chang L-C. Redrawing Urokinase Receptor (uPAR) Signaling with Cancer Driver Genes for Exploring Possible Anti-Cancer Targets and Drugs. Pharmaceuticals. 2023; 16(10):1435. https://doi.org/10.3390/ph16101435
Chicago/Turabian StyleChang, Yu-Ching, Chung-Ze Wu, Chao-Wen Cheng, Jin-Shuen Chen, and Li-Chien Chang. 2023. "Redrawing Urokinase Receptor (uPAR) Signaling with Cancer Driver Genes for Exploring Possible Anti-Cancer Targets and Drugs" Pharmaceuticals 16, no. 10: 1435. https://doi.org/10.3390/ph16101435