
Huntington’s Disease Drug Development: A Phase 3 Pipeline Analysis



Abstract
:1. Introduction
2. Results
2.1. Metformin
2.1.1. Overview of Evidence Supporting Use of Metformin in HD
2.1.2. Overview of Current Phase III Trial
2.2. Dextromethorphan/Quinidine
2.2.1. Overview of Evidence Supporting Use of Dextromethorphan/Quinidine in HD
2.2.2. Overview of Current Phase III Trial
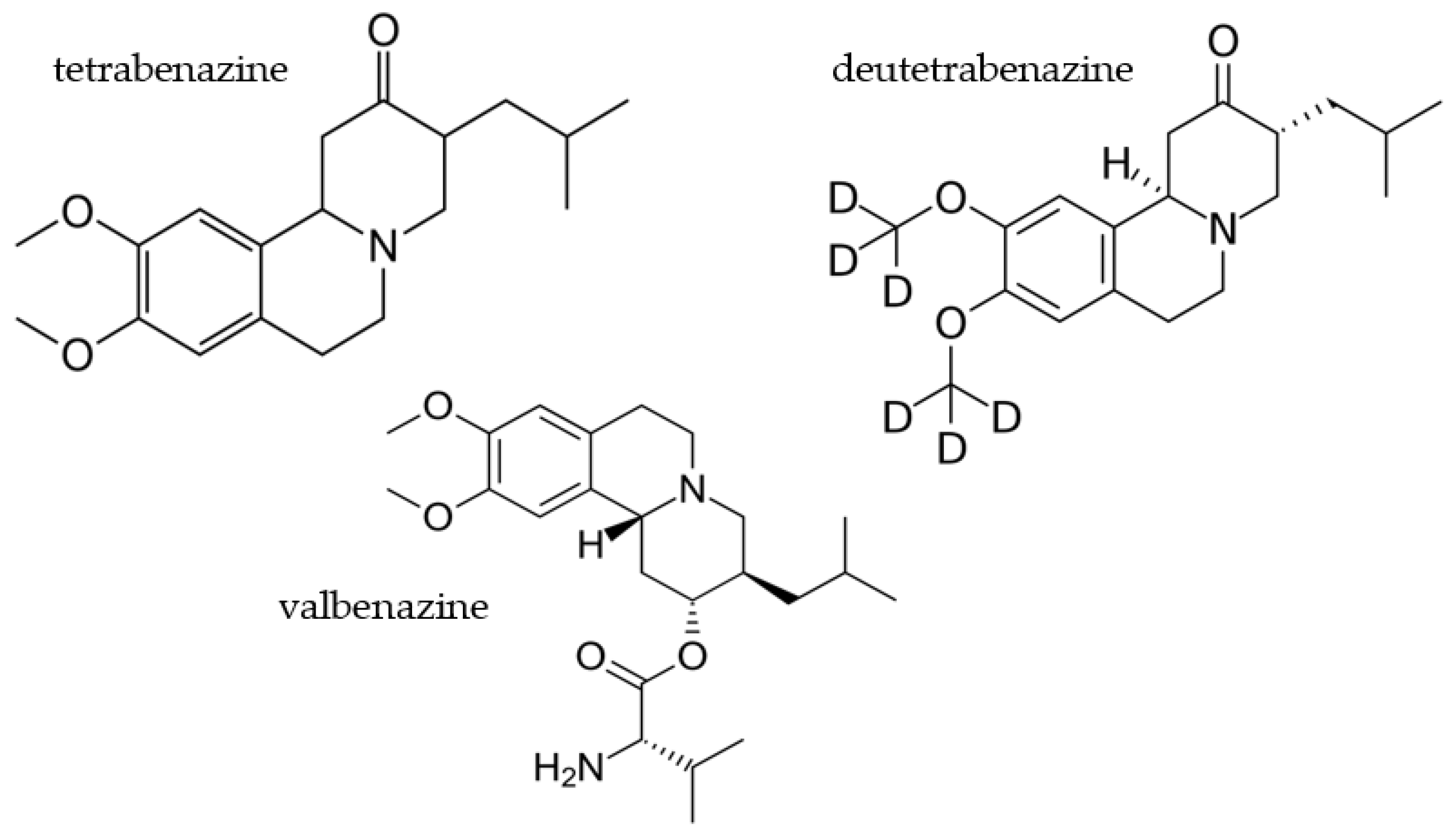
2.3. Deutetrabenazine
2.3.1. Overview of Evidence Supporting Use of Deutetrabenazine in HD
2.3.2. Overview of Current Phase III Trial
2.4. Valbenazine
2.4.1. Overview of Evidence Supporting Use of Valbenazine in HD
2.4.2. Overview of Phase III Clinical Trials
2.5. Cellavita HD
2.5.1. Overview of Evidence Supporting Use of Cellavita HD in HD
2.5.2. Overview of Phase III Clinical Trial
2.6. Pridopidine
2.6.1. Overview of Evidence Supporting Use of Pridopidine in HD
2.6.2. Overview of Phase III Clinical Trial
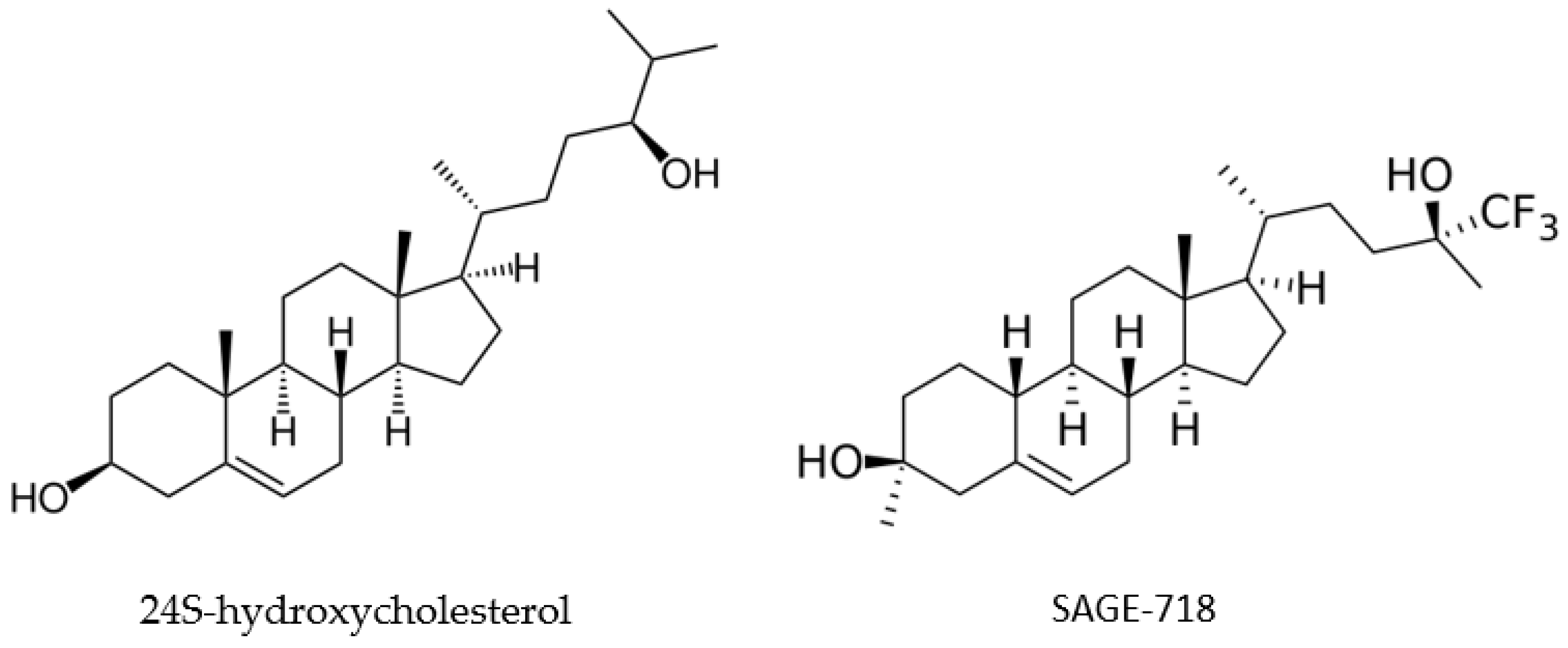
2.7. SAGE-718
2.7.1. Overview of Evidence Supporting Use of SAGE-718 in HD
2.7.2. Overview of Phase III Clinical Trial
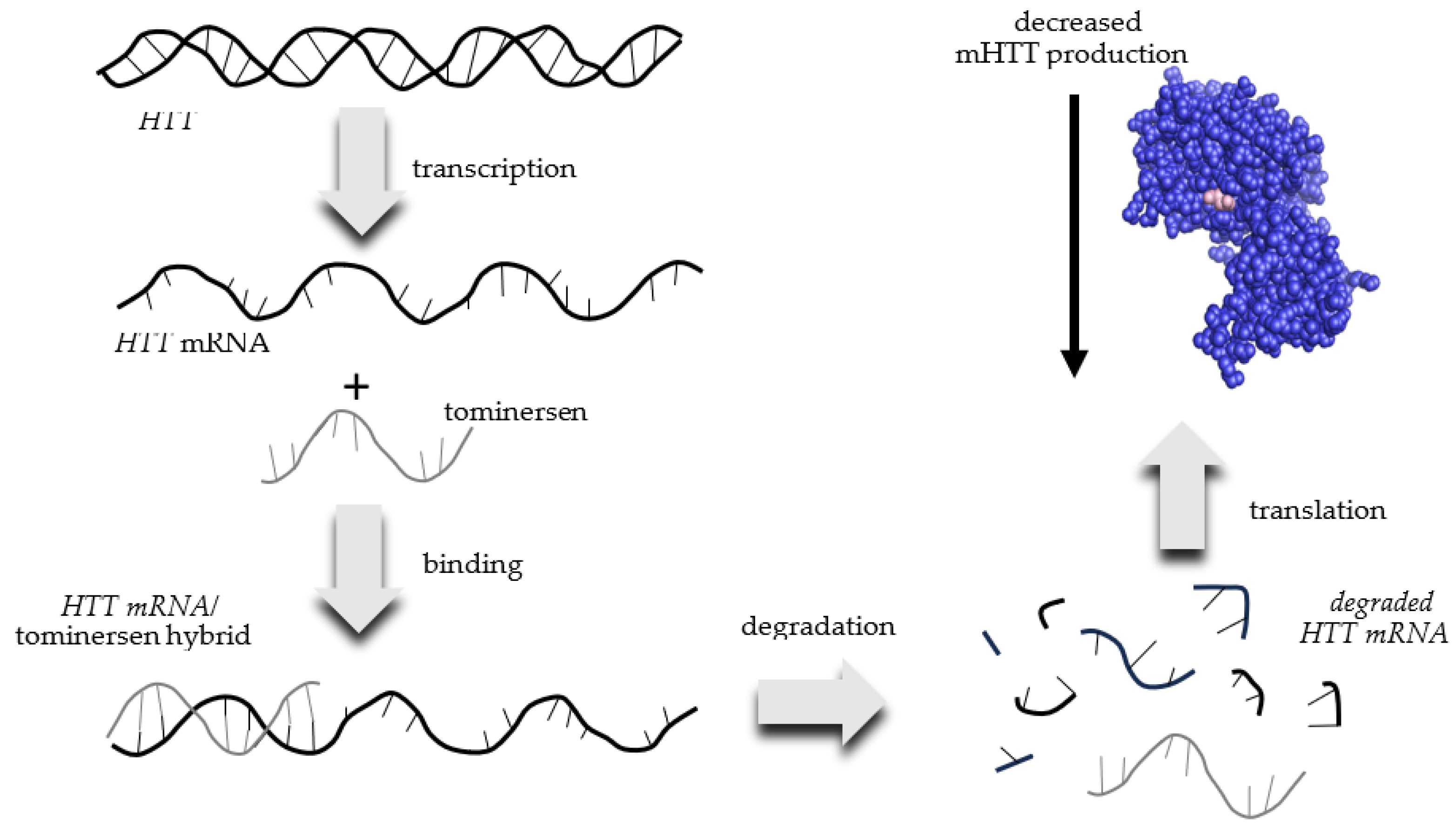
2.8. Tominersen (RO7234292 or RG6042)
2.8.1. Overview of Evidence Supporting Use of Tominersen for HD
2.8.2. Overview of Phase III Clinical Trial
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anil, M.; Mason, S.L.; Barker, R.A. The clinical features and progression of late-onset versus younger-onset in an adult cohort of Huntington’s Disease patients. J. Huntington’s Dis. 2020, 9, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Aylward, E.H.; Wild, E.J.; Langbehn, D.R.; Long, J.D.; Warner, J.H.; Scahill, R.I.; Leavitt, B.R.; Stout, J.C.; Paulsen, J.S.; et al. Huntington disease: Natural history, biomarkers and prospects for therapeutics. Nat. Rev. Neurol. 2014, 10, 204–216. [Google Scholar] [CrossRef] [PubMed]
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Rawlins, M.D.; Wexler, N.S.; Wexler, A.R.; Tabrizi, S.J.; Douglas, I.; Evans, S.J.W.; Smeeth, L. The prevalence of Huntington’s disease. Neuroepidemiology 2016, 46, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Santana, I.; Mestre, T.; Squitieri, F.; Willock, R.; Arnesen, A.; Clarke, A.; D’Alessio, B.; Fisher, A.; Fuller, R.; Hamilton, J.L.; et al. Economic burden of Huntington disease in Europe and the USA: Results from the Huntington’s Disease Burden of Illness study. Eur. J. Neurol. 2023, 30, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Shaw, E.; Mayer, M.; Ekwaru, P.; McMullen, S.; Graves, E.; Wu, J.W.; Budd, N.; Maturi, B.; Cowling, T.; Mestre, T.A. Epidemiology and economic burden of Huntington’s disease: A Canadian provincial public health system perspective. J. Med. Econ. 2022, 1, 212–219. [Google Scholar] [CrossRef]
- Myers, R.H. Huntington’s disease genetics. NeuroRx 2004, 1, 255–262. [Google Scholar] [CrossRef]
- van der Zwaan, K.F.; Mentink, M.D.C.; Jacobs, M.; Roos, R.A.C.; de Bot, S.T. Huntington’s disease influences employment before and during clinical manifestation: A systematic review. Park. Relat. Disord. 2022, 96, 100–108. [Google Scholar] [CrossRef]
- Wright, G.E.B.; Black, H.F.; Collins, J.A.; Gall-Duncan, T.; Caron, N.S.; Pearson, C.E.; Hayden, M.R. Interrupting sequence variants and age of onset in Huntington’s disease: Clinical implications and emerging therapies. Lancet Neurol. 2020, 19, 930–939. [Google Scholar] [CrossRef]
- Li, S.H.; Schilling, G.; Young, W.S., 3rd; Li, X.J.; Margolis, R.L.; Stine, O.C.; Wagster, M.V.; Abbott, M.H.; Franz, M.L.; Ranen, N.G.; et al. Huntington’s disease gene (IT15) is widely expressed in human and rat tissues. Neuron 1993, 11, 985–993. [Google Scholar] [CrossRef]
- Schulte, J.; Littleton, J.T. The biological function of the Huntingtin protein and its relevance to Huntington’s Disease pathology. Curr. Trends Neurol. 2011, 5, 65–78. [Google Scholar] [PubMed]
- Saudou, F.; Humbert, S. The biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef] [PubMed]
- Hackam, A.S.; Hodgson, J.G.; Singaraja, R.; Zhang, T.; Gan, L.; Gutekunst, C.A.; Hersch, S.M.; Hayden, M.R. Evidence for both the nucleus and cytoplasm as subcellular sites of pathogenesis in Huntington’s disease in cell culture and in transgenic mice expressing mutant huntingtin. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1999, 354, 1047–1055. [Google Scholar] [CrossRef]
- Davies, S.W.; Turmaine, M.; Cozens, B.A.; DiFiglia, M.; Sharp, A.H.; Ross, C.A.; Scherzinger, E.; Wanker, E.E.; Mangiarini, L.; Bates, G.P. Formation of Neuronal Intranuclear Inclusions Underlies the Neurological Dysfunction in Mice Transgenic for the HD Mutation. Cell 1997, 90, 537–548. [Google Scholar] [CrossRef]
- Reiner, A.; Dragatsis, I.; Dietrich, P. Genetics and Neuropathology of Huntington’s Disease. Int. Rev. Neurobiol. 2011, 98, 325–372. [Google Scholar]
- Almqvist, E.W.; Bloch, M.; Brinkman, R.; Craufurd, D.; Hayden, M.R. A worldwide assessment of the frequency of suicide, suicide attempts, or psychiatric hospitalization after predictive testing for Huntington disease. Am. J. Hum. Genet. 1999, 64, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Petrella, L.I.; Castelhano, J.M.; Ribeiro, M.; Sereno, J.V.; Gonçalves, S.I.; Laço, M.N.; Hayden, M.R.; Rego, A.C.; Castelo-Branco, M. A whole brain longitudinal study in the YAC128 mouse model of Huntington’s disease shows distinct trajectories of neurochemical, structural connectivity and volumetric changes. Hum. Mol. Genet. 2018, 27, 2125–2137. [Google Scholar] [CrossRef]
- Blumenstock, S.; Dudanova, I. Cortical and striatal circuits in Huntington’s disease. Front. Neurosci. 2020, 14, 82–99. [Google Scholar] [CrossRef]
- Rocha, G.S.; Freire, M.A.M.; Britto, A.M.; Paiva, K.M.; Oliveira, R.F.; Fonseca, I.A.T.; Araújo, D.P.; Oliveira, L.C.; Guzen, F.P.; Morais, P.L.A.G.; et al. Basal ganglia for beginners: The basic concepts you need to know and their role in movement control. Front. Syst. Neurosci. 2023, 17, 1242929–1242936. [Google Scholar] [CrossRef]
- Zielonka, D.; Piotrowska, I.; Marcinkowski, J.T.; Mielcarek, M. Skeletal muscle pathology in Huntington’s disease. Front. Physiol. 2014, 5, 380–384. [Google Scholar] [CrossRef]
- Bonomo, R.; Elia, A.E.; Bonomo, G.; Romito, L.M.; Mariotti, C.; Devigili, G.; Cilia, R.; Giossi, R.; Eleopra, R. Deep brain stimulation in Huntington’s disease: A literature review. Neurol. Sci. 2021, 42, 4447–4457. [Google Scholar] [CrossRef] [PubMed]
- Bonelli, R.M.; Cummings, J.L. Frontal-subcortical dementias. Neurologist 2008, 14, 100–107. [Google Scholar] [CrossRef] [PubMed]
- van Lonkhuizen, P.J.C.; Frank, W.; Heemskerk, A.W.; van Duijn, E.; de Bot, S.T.; Mühlbäck, A.; Landwehrmeyer, G.B.; Chavannes, N.H.; Meijer, E.; HEALTHE-RND Consortium. Quality of life, health-related quality of life, and associated factors in Huntington’s disease: A systematic review. J. Neurol. 2023, 270, 2416–2437. [Google Scholar] [CrossRef] [PubMed]
- Galyan, S.M.; Ewald, C.Y.; Jalencas, X.; Masrani, S.; Meral, S.; Mestres, J. Fragment-based virtual screening identifies a first-in class preclinical drug for Huntington’s disease. Sci. Rep. 2022, 12, 19642–19655. [Google Scholar] [CrossRef]
- Huntington’s Disease. Available online: https://www.mayoclinic.org/diseases-conditions/huntingtons-disease/diagnosis-treatment/drc-20356122 (accessed on 12 September 2023).
- Estevez-Fraga, C.; Tabrizi, S.J.; Wild, E.J. Huntington’s Disease clinical trials corner: November 2022. J. Huntington’s Dis. 2022, 11, 351–367. [Google Scholar] [CrossRef]
- Blonde, L. Management of type 2 diabetes: Update on new pharmacological options. Manag. Care 2000, 9, 11–17. [Google Scholar]
- Kajbaf, F.; De Broe, M.E.; Lalau, J.D. Therapeutic concentrations of metformin: A systematic review. Clin. Pharmacokinet. 2016, 55, 439–459. [Google Scholar] [CrossRef]
- Song, Y.; Wu, Z.; Zhao, P. The function of metformin in aging-related musculoskeletal disorders. Front. Pharmacol. 2022, 13, 865524–865537. [Google Scholar] [CrossRef]
- Trujillo-Del Río, C.; Tortajada-Pérez, J.; Gómez-Escribano, A.P.; Casterá, F.; Peiró, C.; Millán, J.M.; Herrero, M.J.; Vázquez-Manrique, R.P. Metformin to treat Huntington disease: A pleiotropic drug against a multi-system disorder. Mech. Ageing Dev. 2022, 204, 111670–111682. [Google Scholar] [CrossRef]
- Faria, J.; Negalha, G.; Azevedo, A.; Martel, F. Metformin and breast cancer: Molecular targets. J. Mammary Gland Biol. Neoplasia 2019, 24, 111–123. [Google Scholar] [CrossRef]
- Ronnett, G.V.; Ramamurthy, S.; Kleman, A.M.; Landree, L.E.; Aja, S. AMPK in the brain: Its roles in energy balance and neuroprotection. J. Neurochem. 2009, 109 (Suppl. S1), 17–23. [Google Scholar] [CrossRef]
- Sanchis, A.; García-Gimeno, M.A.; Cañada-Martínez, A.J.; Sequedo, M.D.; Millán, J.M.; Sanz, P.; Vázquez-Manrique, R.P. Metformin treatment reduces motor and neuropsychiatric phenotypes in the zQ175 mouse model of Huntington disease. Exp. Mol. Med. 2019, 51, 1–16. [Google Scholar] [CrossRef]
- Hervás, D.; Fornés-Ferrer, V.; Gómez-Escribano, A.P.; Sequedo, M.D.; Peiró, C.; Millán, J.M.; Vázquez-Manrique, R.P. Metformin intake associates with better cognitive function in patients with Huntington’s disease. PLoS ONE 2017, 12, e0179283. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Cong, W.N.; Ji, S.; Rothman, S.; Maudsley, S.; Martin, B. Metabolic dysfunction in Alzheimer’s disease and related neurodegenerative disorders. Curr. Alzheimer Res. 2012, 9, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Lange, J.; Gillham, O.; Flower, M.; Ging, H.; Eaton, S.; Kapadia, S.; Neueder, A.; Duchen, M.R.; Ferretti, P.; Tabrizi, S.J. PolyQ length-dependent metabolic alterations and DNA damage drive human astrocyte dysfunction in Huntington’s disease. Prog. Neurobiol. 2023, 225, 102448–102466. [Google Scholar] [CrossRef] [PubMed]
- Almikhlafi, M.A.; Karami, M.M.; Jana, A.; Alqurashi, T.M.; Majrashi, M.; Alghamdi, B.S.; Ashraf, G.M. Mitochondrial medicine: A promising therapeutic option against various neurodegenerative disorders. Curr. Neuropharmacol. 2023, 21, 1165–1183. [Google Scholar] [CrossRef]
- Solís-Maldonado, M.; Miró, M.P.; Acuña, A.I.; Covarrubias-Pinto, A.; Loaiza, A.; Mayorga, G.; Beltrán, F.A.; Cepeda, C.; Levine, M.S.; Concha, I.I.; et al. Altered lactate metabolism in Huntington’s disease is dependent on GLUT3 expression. CNS Neurosci. Ther. 2018, 24, 343–352. [Google Scholar] [CrossRef]
- Szablewski, L. Glucose transporters in brain: In health and in Alzheimer’s disease. J. Alzheimer’s Dis. 2017, 55, 1307–1320. [Google Scholar] [CrossRef]
- Chaves, G.; Stanley, J.; Pourmand, N. Mutant huntingtin affects diabetes and Alzheimer’s markers in human and cell models of Huntington’s Disease. Cells 2019, 8, 962. [Google Scholar] [CrossRef]
- Andreassen, O.A.; Dedeoglu, A.; Stanojevic, V.; Hughes, D.B.; Browne, S.E.; Leech, C.A.; Ferrante, R.J.; Habener, J.F.; Beal, M.F.; Thomas, M.K. Huntington’s disease of the endocrine pancreas: Insulin deficiency and diabetes mellitus due to impaired insulin gene expression. Neurobiol. Dis. 2002, 11, 410–424. [Google Scholar] [CrossRef]
- Siesling, S.; van Vugt, J.P.; Zwinderman, K.A.; Kieburtz, K.; Roos, R.A. Unified Huntington’s disease rating scale: A follow up. Mov. Disord. 1998, 13, 915–919. [Google Scholar] [CrossRef]
- Gómez-Ansón, B.; Alegret, M.; Muñoz, E.; Sainz, A.; Monte, G.C.; Tolosa, E. Decreased frontal choline and neuropsychological performance in preclinical Huntington disease. Neurology 2007, 68, 906–910. [Google Scholar] [CrossRef] [PubMed]
- Zilliox, L.A.; Chadrasekaran, K.; Kwan, J.Y.; Russell, J.W. Diabetes and cognitive impairment. Curr. Diabetes Rep. 2016, 16, 87–97. [Google Scholar] [CrossRef]
- FDA Center for Drug Evaluation and Research Medical Review Application Number: 021879Orig1s000. 30 April 2010. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2010/021879Orig1s000MedR.pdf (accessed on 5 September 2023).
- Price, D.D.; Mao, J.; Frenk, H.; Mayer, D.J. The N-methyl-D-asparate receptor antagonist dextromethorphan selectively reduces temporal summation of second pain in man. Pain 1994, 59, 165–174. [Google Scholar] [CrossRef]
- Patatanian, E.; Casselman, J. Dextromethorphan/quinidine for the treatment of pseudobulbar affect. Consult. Pharm. 2014, 29, 264–269. [Google Scholar] [CrossRef]
- Nabizadeh, F.; Nikfarjam, M.; Azami, M.; Sharifkazemi, H.; Sodeifian, F. Pseudobulbar affect in neurodegenerative diseases: A systematic review and meta-analysis. J. Clin. Neurosci. 2022, 100, 100–107. [Google Scholar] [CrossRef]
- Persaud, A.; Fares, M.A.; Giles, S.; Gaitour, E.; Shneyder, N. Laughing and dancing: A case report of Pseudobulbar Affect in late onset Huntington’s Disease. Neurology 2018, 90 (Suppl. S15). Available online: https://n.neurology.org/content/90/15_Supplement/P4.047.abstract (accessed on 14 September 2023).
- Scorr, L.M.; Factor, S.A. VMAT2 inhibitors for the treatment of tardive dyskinesia. J. Neurol. Sci. 2018, 389, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Yaffe, D.; Forrest, L.R.; Schuldiner, S. The ins and outs of vesicular monoamine transporters. J. Gen. Physiol. 2018, 150, 671–682. [Google Scholar] [CrossRef]
- Harriott, N.D.; Williams, J.P.; Smith, E.B.; Bozigian, H.P.; Grigoriadis, D.E. VMAT2 inhibitors and the path to Ingrezza (valbenazine). Prog. Med. Chem. 2018, 57, 87–111. [Google Scholar]
- Dean, M.; Sung, V.W. Review of deutetrabenazine: A novel treatment for chorea associated with Huntington’s disease. Drug Des. Dev. Ther. 2018, 12, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Sung, V.W.; Iyer, R.G.; Gandhi, S.K.; Abler, V.; Davis, B.; Irwin, D.E.; Anderson, K.E. Retrospective analysis of healthcare resource use, treatment patterns, and treatment-related events in patients with Huntington’s Disease-associated chorea initiated on tetrabenazine. J. Health Econ. Outcomes Res. 2018, 6, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Claassen, D.O.; Carroll, B.; De Boer, L.M.; Wu, E.; Ayyagari, R.; Gandhi, S.; Stamler, D. Indirect tolerability comparison of deutetrabenazine and tetrabenazine for Huntington disease. J. Clin. Mov. Disord. 2017, 4, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.drugs.com/history/austedo.html (accessed on 20 September 2023).
- Impact of Deutetrabenazine on Functional Speech and Gait Dynamics in Huntington Disease. Available online: https://classic.clinicaltrials.gov/ct2/results?cond=&term=Impact+of+Deutetrabenazine+on+Functional+Speech+and+Gait+Dynamics+in+Huntington+Disease&cntry=&state=&city=&dist= (accessed on 12 September 2023).
- Stipancic, K.L.; Tjaden, K.; Wilding, G. Comparison of intelligibility measures for adults with Parkinson’s Disease, adults with Multiple Sclerosis and healthy control. J. Speech Lang. Hear. Res. 2016, 59, 230–238. [Google Scholar] [CrossRef]
- Furr Stimming, E.; Claassen, D.O.; Kayson, E.; Goldstein, J.; Mehanna, R.; Zhang, H.; Liang, G.S.; Haubenberger, D.; Huntington Study Group KINECT-HD Collaborators. Safety and efficacy of valbenazine for the treatment of chorea associated with Huntington’s disease (KINECT-HD): A phase 3, randomized, double-blind, placebo-controlled trial. Lancet Neurol. 2023, 22, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Szczakowska, A.; Gabryelska, A.; Gawlik-Kotelnicka, O.; Strzelecki, D. Deep brain stimulation in the treatment of tardive dyskinesia. J. Clin. Med. 2023, 12, 1868. [Google Scholar] [CrossRef]
- Available online: https://www.drugs.com/history/ingrezza.html (accessed on 13 September 2023).
- Koch, J.; Shi, W.X.; Dashtipour, K. VMAT2 inhibitors for the treatment of hyperkinetic movement disorders. Pharmacol. Ther. 2020, 212, 107580–107590. [Google Scholar] [CrossRef]
- Frank, S.; Testa, C.; Edmondson, M.C.; Goldstein, J.; Kayson, E.; Leavitt, B.R.; Oakes, D.; O’Neill, C.; Vaughan, C.; Whaley, J.; et al. The safety of deutetrabenazine for chorea in Huntington Disease: An open-label extension study. CNS Drugs 2022, 36, 1207–1216. [Google Scholar] [CrossRef]
- Available online: https://classic.clinicaltrials.gov/ct2/show/NCT04400331 (accessed on 10 September 2023).
- Ferguson, M.W.; Kennedy, C.J.; Palpagama, T.H.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. Current and possible future therapeutic options for Huntington’s Disease. J. Cent. Nerv. Syst. Dis. 2022, 14, 11795735221092517–11795735221092553. [Google Scholar] [CrossRef]
- Song, W.P.; Jin, L.Y.; Zhu, M.D.; Wang, H.; Xia, D.S. Clinical trials using dental stem cells: 2022 update. World J. Stem Cells 2023, 15, 31–51. [Google Scholar] [CrossRef]
- Ueda, T.; Inden, M.; Ito, T.; Kurita, H.; Hozumi, I. Characteristics and therapeutic potential of dental pulp stem cells on neurodegenerative diseases. Front. Neurosci. 2020, 14, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ouchi, T.; Cao, Y.; Zhao, Z.; Men, Y. Dental-derived mesenchymal stem cells: State of the art. Front. Cell Dev. Biol. 2021, 9, 654559–654577. [Google Scholar] [CrossRef]
- Wenceslau, C.V.; de Souza, D.M.; Mambelli-Lisboa, N.C.; Ynoue, L.H.; Araldi, R.P.; da Silva, J.M.; Pagani, E.; Haddad, M.S.; Kerkis, I. Restoration of BDNF, DARPP32, and D2R expression following intravenous infusion of human immature dental pulp stem cells in Huntington’s Disease 3-NP rat model. Cells 2022, 11, 1664. [Google Scholar] [CrossRef]
- Sramkó, B.; Földes, A.; Kádár, K.; Varga, G.; Zsembery, Á.; Pircs, K. The wisdom in teeth: Neuronal differentiation of dental pulp cells. Cell Reprogram. 2023, 25, 32–44. [Google Scholar] [CrossRef] [PubMed]
- Clinical Extension Study for Safety and Efficacy Evaluation of Cellavita-HD Administration in Huntington’s Patients. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT04219241?term=Clinical+Extension+Study+for+Safety+and+Efficacy+Evaluation+of+Cellavita-HD+Administration+in+Huntington%E2%80%99s+Patients&draw=2&rank=1 (accessed on 17 September 2023).
- Waters, S.; Tedroff, J.; Ponten, H.; Klamer, D.; Sonesson, C.; Waters, N. Pridopidine: Overview of pharmacology and rationale for its use in Huntington’s Disease. J. Huntington’s Dis. 2018, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Grachev, I.D.; Meyer, P.M.; Becker, G.A.; Bronzel, M.; Marsteller, D.; Pastino, G.; Voges, O.; Rabinovich, L.; Knebel, H.; Zientek, F.; et al. Sigma-1 and dopamine D2/D3 receptor occupancy of pridopidine in healthy volunteers and patients with Huntington disease: A [18F] fluspidine and [18F] fallypride PET study. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 1103–1115. [Google Scholar] [CrossRef]
- Squitieri, F.; Di Pardo, A.; Favellato, M.; Amico, E.; Maglione, V.; Frati, L. Pridopidine, a dopamine stabilizer, improves motor performance and shows neuroprotective effects in Huntington disease R6/2 mouse model. J. Cell. Mol. Med. 2015, 19, 2540–2548. [Google Scholar] [CrossRef]
- What is Pridopidine? Available online: https://www.Prilenia.com (accessed on 17 September 2023).
- Eddings, C.R.; Arbez, N.; Akimov, S.; Geva, M.; Hayden, M.R.; Ross, C.A. Pridopidine protects neurons from mutant-huntingtin toxicity via the sigma-1 receptor. Neurobiol. Dis. 2019, 129, 118–129. [Google Scholar] [CrossRef]
- Reilmann, R.; McGarry, A.; Grachev, I.D.; Savola, J.M.; Borowsky, B.; Eyal, E.; Gross, N.; Langbehn, D.; Schubert, R.; Wickenberg, A.T.; et al. Safety and efficacy of pridopidine in patients with Huntington’s disease (PRIDE-HD): A phase 2, randomized, placebo-controlled, multicentre, dose-ranging study. Lancet Neurol. 2019, 18, 165–176. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Estevez-Fraga, C.; van Roon-Mom, W.M.C.; Flower, M.D.; Scahill, R.I.; Wild, E.J.; Muñoz-Sanjuan, I.; Sampaio, C.; Rosser, A.E.; Leavitt, B.R. Potential disease-modifying therapies for Huntington’s disease: Lessons learned and future opportunities. Lancet Neurol. 2022, 21, 645–658. [Google Scholar] [CrossRef]
- Available online: https://clinicaltrials.gov/ct2/show/NCT04556656 (accessed on 15 September 2023).
- Hill, M.D.; Blanco, M.J.; Salituro, F.G.; Bai, Z.; Beckley, J.T.; Ackley, M.A.; Dai, J.; Doherty, J.J.; Harrison, B.L.; Hoffmann, E.C.; et al. SAGE-718: A first-in-class N-methyl-d-aspartate receptor positive allosteric modulator for the potential treatment of cognitive impairment. J. Med. Chem. 2022, 65, 9063–9075. [Google Scholar] [CrossRef]
- Leoni, V.; Mariotti, C.; Tabrizi, S.J.; Valenza, M.; Wild, E.J.; Henley, S.M.; Hobbs, N.Z.; Mandelli, M.L.; Grisoli, M.; Björkhem, I.; et al. Plasma 24S-hydroxycholesterol and caudate MRI in pre-manifest and early Huntington’s disease. Brain 2008, 131 Pt 11, 2851–2859. [Google Scholar] [CrossRef] [PubMed]
- Leoni, V.; Long, J.D.; Mills, J.A.; Di Donato, S.; Paulsen, J.S.; PREDICT-HD Study Group. Plasma 24S-hydroxycholeteral correlation with markers of Huntington disease progression. Neurobiol. Dis. 2013, 55, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Byun, S.; Lee, M.; Kim, M. Gene therapy for Huntington’s disease: The final strategy for a cure? J. Mov. Disord. 2022, 15, 15–20. [Google Scholar] [CrossRef]
- Rasul, M.F.; Hussen, B.M.; Salihi, A.; Ismael, B.S.; Jalal, P.J.; Zanichelli, A.; Jamali, E.; Baniahmad, A.; Ghafouri-Fard, S.; Basiri, A.; et al. Strategies to overcome the main challenges of the use of CRISPR/Cas9 as a replacement for cancer therapy. Mol. Cancer 2022, 21, 64–93. [Google Scholar] [CrossRef] [PubMed]
- Scoles, D.R.; Pulst, S.M. Antisense therapies for movement disorders. Mov. Disord. 2019, 34, 1112–1119. [Google Scholar] [CrossRef]
- Available online: https://www.clinicaltrialsarena.com/comment/roche-tominersen-huntingtons-asset/?cf-view (accessed on 18 September 2023).
- Tabrizi, S.J.; Leavitt, B.R.; Landwehrmeyer, G.B.; Wild, E.J.; Saft, C.; Barker, R.A.; Blair, N.F.; Craufurd, D.; Priller, J.; Rickards, H.; et al. Targeting huntingtin expression in patients with Huntington’s Disease. N. Engl. J. Med. 2019, 380, 2307–2316. [Google Scholar] [CrossRef]
- Tang, B.L. Could metformin be therapeutically useful in Huntington’s disease? Rev. Neurosci. 2020, 31, 297–317. [Google Scholar] [CrossRef]
- Russell, R.D.; Black, L.J.; Begley, A. Nutrition education programs for adults with neurological diseases are lacking: A scoping review. Nutrients 2022, 14, 1577. [Google Scholar] [CrossRef]
- Marder, K.; Zhao, H.; Eberly, S.; Tanner, C.M.; Oakes, D.; Shoulson, I.; Huntington Study Group. Dietary intake in adults at risk for Huntington disease: Analysis of PHAROS research participants. Neurology 2009, 73, 385–392. [Google Scholar] [CrossRef]
- Available online: https://hdsa.org/find-help/living-well-with-hd/nutrition/ (accessed on 11 October 2023).
- Kumar, R.R.; Singh, L.; Thakur, A.; Singh, S.; Kumar, B. Role of vitamins in neurodegenerative diseases: A review. CNS Neurol. Disord. Drug Targets 2022, 21, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://hdsa.org/wp-content/uploads/2015/02/11695.pdf (accessed on 11 October 2023).
- Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell 2015, 162, 516–526. [Google Scholar] [CrossRef]
- Moss, D.J.H.; Pardiñas, A.F.; Langbehn, D.; Lo, K.; Leavitt, B.R.; Roos, R.; Durr, A.; Mead, S.; TRACK-HD Investigators; REGISTRY Investigators; et al. Identification of genetic variants associated with Huntington’s disease progression: A genome-wide association study. Lancet Neurol. 2017, 16, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, T.; Suart, C.E.; Hung, C.L.K.; Graham, K.J.; Barba Bazan, C.A.; Truant, R. DNA damage repair in Huntington’s Disease and other neurodegenerative diseases. Neurotherapeutics 2019, 16, 948–956. [Google Scholar] [CrossRef]
- Essa, M.M.; Moghadas, M.; Ba-Omar, T.; Walid Qoronfleh, M.; Guillemin, G.J.; Manivasagam, T.; Justin-Thenmozhi, A.; Ray, B.; Bhat, A.; Chidambaram, S.B.; et al. Protective effects of antioxidants in Huntington’s Disease: An extensive review. Neurotox. Res. 2019, 35, 739–774. [Google Scholar] [CrossRef] [PubMed]
- Gioia, U.; Francia, S.; Cabrini, M.; Brambillasca, S.; Michelini, F.; Jones-Weinert, C.W.; d’Adda di Fagagna, F. Pharmacological boost of DNA damage response and repair by enhanced biogenesis of DNA damage response RNAs. Sci. Rep. 2019, 9, 6460–6474. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Drug | Clinical Trial Name | Identifier | Primary Outcome |
|---|---|---|---|
| Metformin | Testing METformin against cognitive decline in HD | NCT04826692 | Evaluate drug’s effect on scores obtained in different cognitive subtests that make up the Unified Huntington’s Disease Rating Scale (UHDRS) |
| Dextromethorphan/ quinidine | Evaluating the Efficacy of Dextromethorphan/Quinidine in Treating Irritability in HD | NCT03854019 | Measure patient’s irritability using The Irritability Scale |
| Deutetrabenazine | Impact of Deutetrabenazine on Functional Speech and Gait Dynamics in Huntington Disease | NCT04713982 | Improvement on Sentence Intelligibility Test and Motor Speech Evaluation |
| Valbenazine | Efficacy, Safety, and Tolerability of Valbenazine for the Treatment of Chorea Associated with Huntington Disease (KINECT-HD) | NCT04102579 | Improvement in chorea symptoms and evaluation of treatment-emergent adverse events |
| An Open-Label Rollover Study for Continuing Valbenazine Administration for the Treatment of Chorea Associated With Huntington Disease | NCT04400331 | Evaluation of treatment-emergent adverse events (TEAEs) | |
| Cellavita HD | Clinical Extension Study for Safety and Efficacy Evaluation of the Cellavita HD Product in Huntington’s Patients (ADORE-EXT) | NCT04219241 | Evaluate treatment’s effectiveness as verified by comparing the total UHDRS |
| Pridopidine | Pridopidine’s Outcome on Function in Huntington Disease, PROOF-HD | NCT04556656 | Measure change from baseline in UHDRS-Total Functional Capacity score |
| SAGE-718 | A Study to Evaluate the Safety and Tolerability of SAGE-718 in Participants With Huntington’s Disease | NCT05655520 | Measure number and severity of TEAEs Measure number of participants that withdraw due to adverse events Measure change from baseline in clinical laboratory and electrocardiogram parameters Measure change in baseline in C-SSRS responses |
| RO7234292 (RG6042) | An Open-Label Extension Study to Evaluate Long-Term Safety and Tolerability of RO7234292 (RG6042) in Huntington’s Disease | NCT03842969 | Measure number and severity of TEAEs Measure number of participants with suicidal ideation/behavior and self-injurious behavior without suicidal intent based on C-SSRS scale Measure change from baseline in MoCA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van de Roovaart, H.J.; Nguyen, N.; Veenstra, T.D. Huntington’s Disease Drug Development: A Phase 3 Pipeline Analysis. Pharmaceuticals 2023, 16, 1513. https://doi.org/10.3390/ph16111513
Van de Roovaart HJ, Nguyen N, Veenstra TD. Huntington’s Disease Drug Development: A Phase 3 Pipeline Analysis. Pharmaceuticals. 2023; 16(11):1513. https://doi.org/10.3390/ph16111513
Chicago/Turabian StyleVan de Roovaart, Hannah J., Nguyen Nguyen, and Timothy D. Veenstra. 2023. "Huntington’s Disease Drug Development: A Phase 3 Pipeline Analysis" Pharmaceuticals 16, no. 11: 1513. https://doi.org/10.3390/ph16111513
APA StyleVan de Roovaart, H. J., Nguyen, N., & Veenstra, T. D. (2023). Huntington’s Disease Drug Development: A Phase 3 Pipeline Analysis. Pharmaceuticals, 16(11), 1513. https://doi.org/10.3390/ph16111513