
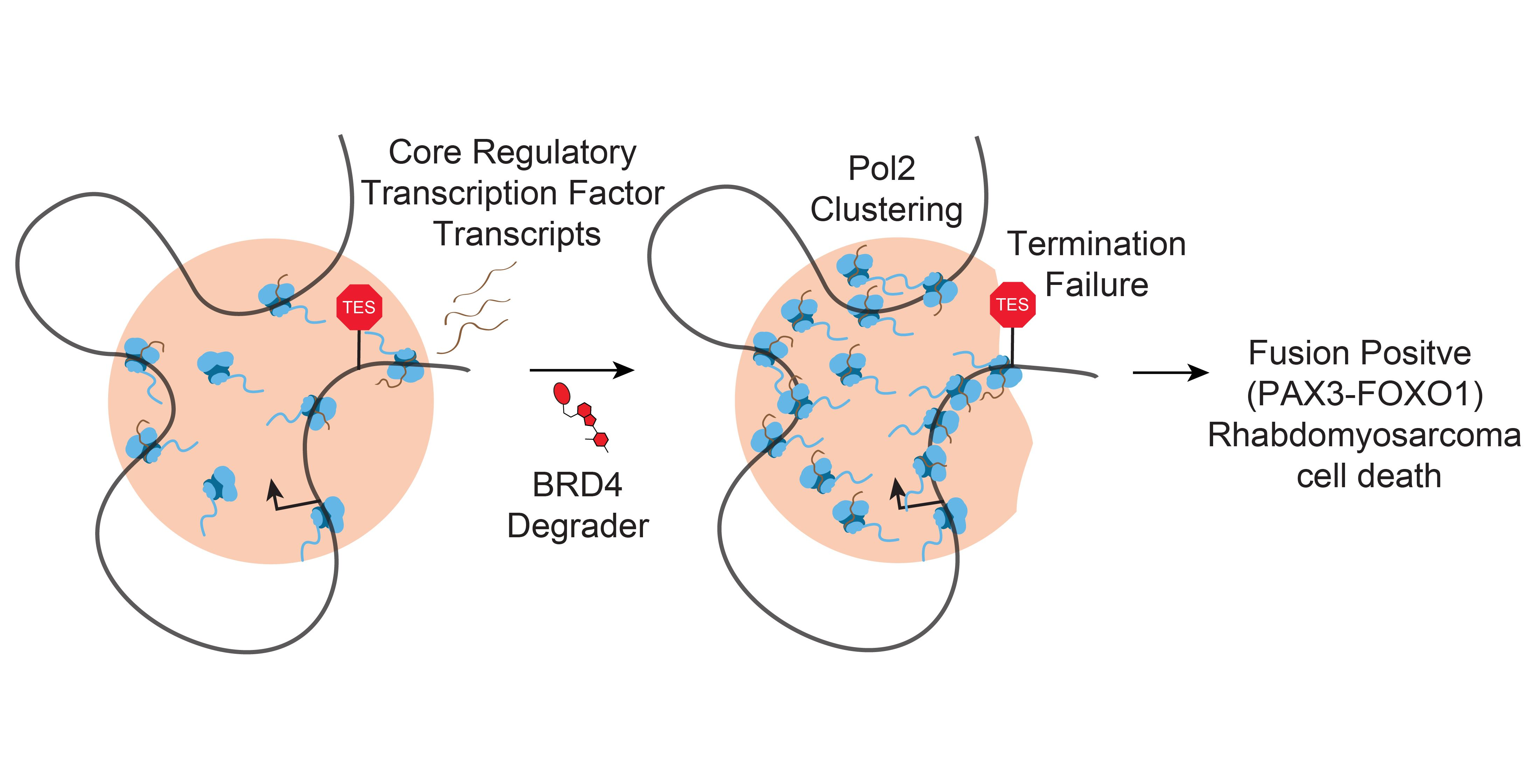
BET Bromodomain Degradation Disrupts Function but Not 3D Formation of RNA Pol2 Clusters
 , , ,
, , ,
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
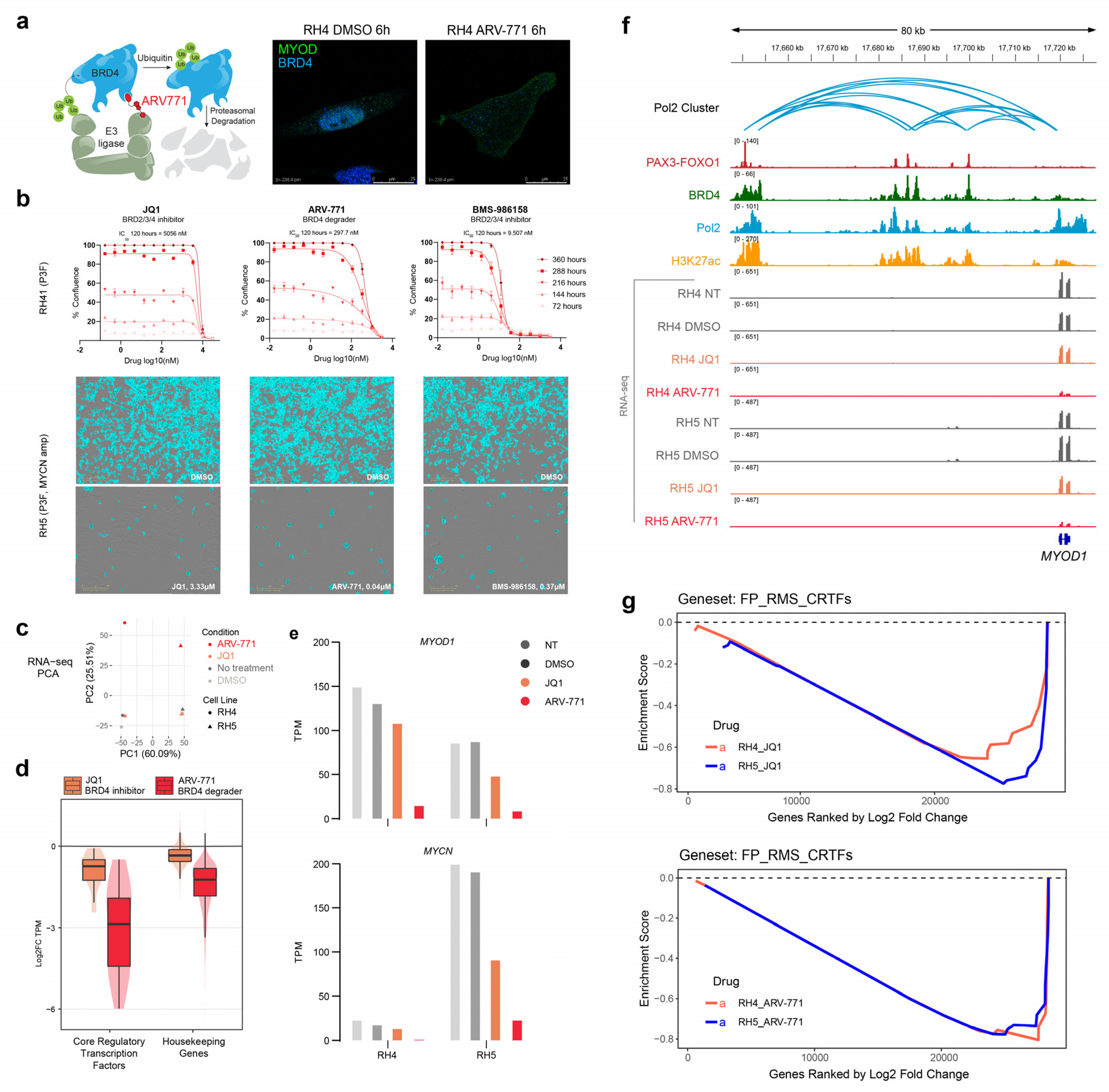
2.1. BET Bromodomain Inhibition and Degradation Halt Cell Growth and Core Regulatory Circuitry in Rhabdomyosarcoma
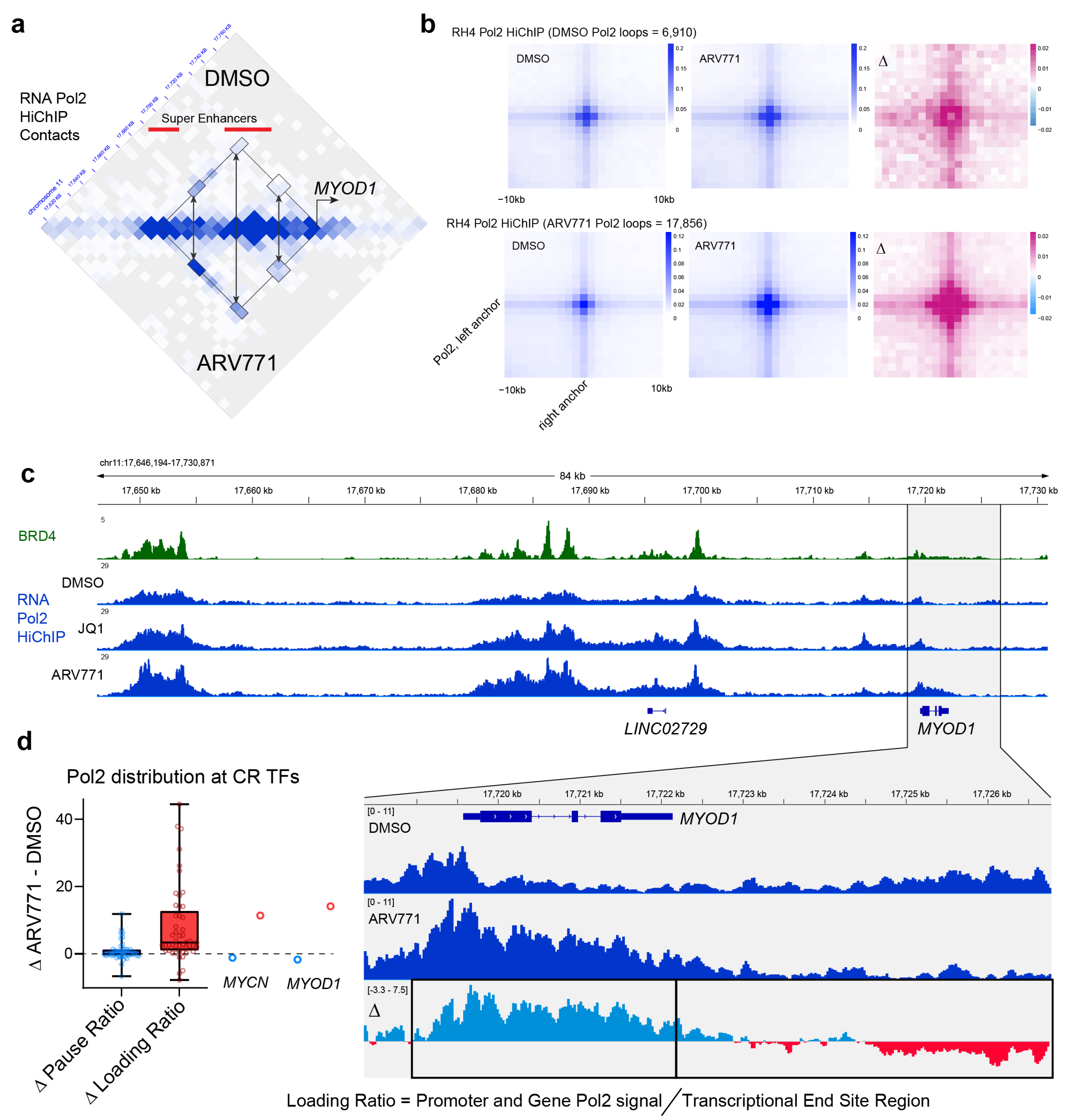
2.2. RNA Polymerase II Loops Increase after Degradation of BET Bromodomains
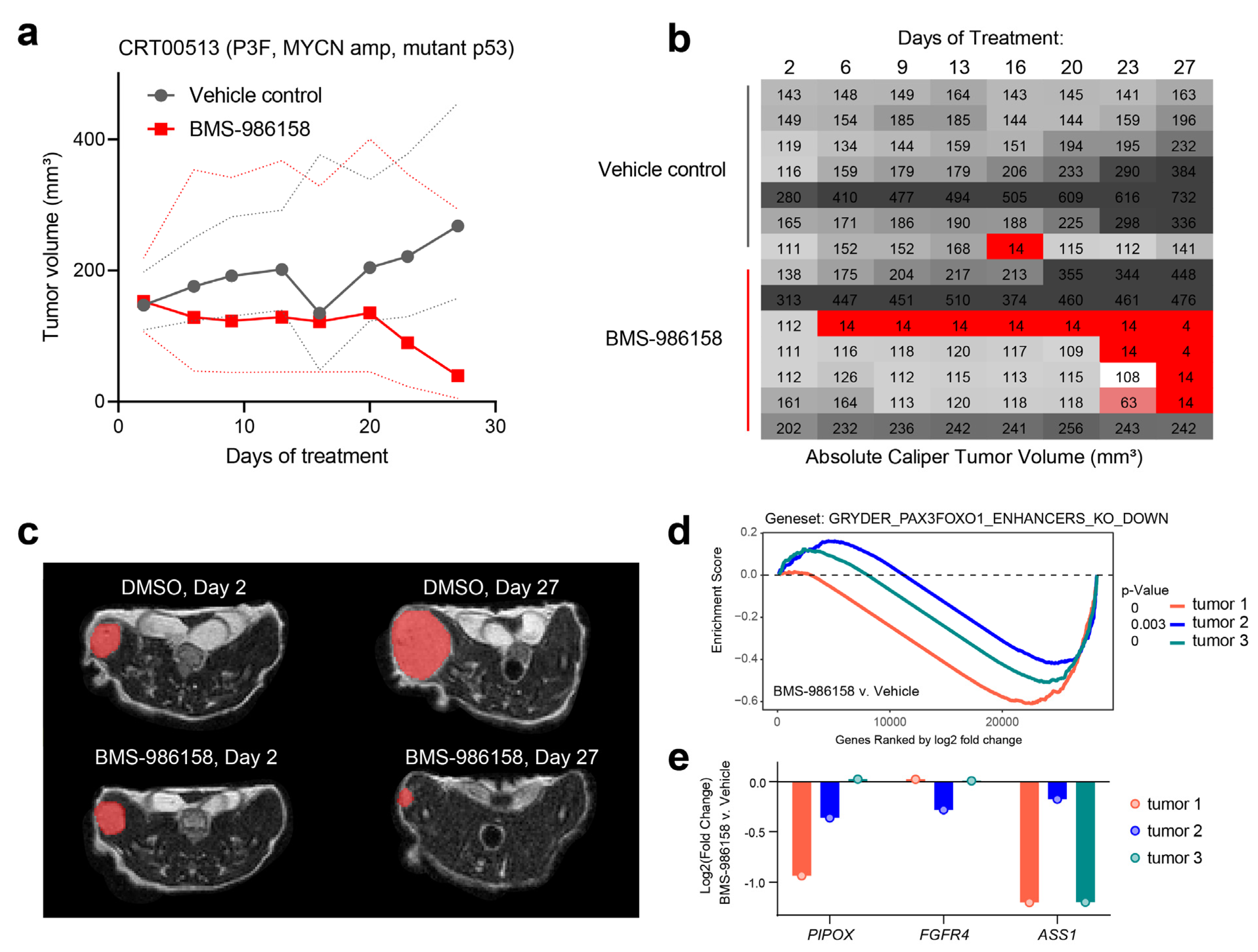
2.3. BET Bromodomain Inhibitor BMS-986158 in an FP-RMS In Vivo Model
3. Discussion
4. Methods
4.1. Chemicals
4.2. Tissue Culture
4.3. Cell Proliferation
4.4. Immunocytochemistry
4.5. RNA-Seq Sample and Library Preparation
4.6. RNA-Seq Analysis
4.7. ChIP-Seq
4.8. AQuA-HiChIP Sample Preparation
4.9. HiChIP Analysis
4.10. In Vivo PDX Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gryder, B.; Scacheri, P.C.; Ried, T.; Khan, J. Chromatin Mechanisms Driving Cancer. Cold Spring Harb. Perspect. Biol. 2021, 14, a040956. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, A.; Chisholm, J.C.; Jenney, M.; Minard-Colin, V.; Orbach, D.; Casanova, M.; Guillen, G.; Glosli, H.; van Rijn, R.R.; A Schoot, R.; et al. Adolescents and young adults with rhabdomyosarcoma treated in the European paediatric Soft tissue sarcoma Study Group (EpSSG) protocols: A cohort study. Lancet Child Adolesc. Health 2022, 6, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Yohe, M.E.; Heske, C.M.; Stewart, E.; Adamson, P.C.; Ahmed, N.; Antonescu, C.R.; Chen, E.; Collins, N.; Ehrlich, A.; Galindo, R.L.; et al. Insights into pediatric rhabdomyosarcoma research: Challenges and goals. Pediatr. Blood Cancer 2019, 66, e27869. [Google Scholar] [CrossRef]
- Shern, J.F.; Selfe, J.; Izquierdo, E.; Patidar, R.; Chou, H.-C.; Song, Y.K.; Yohe, M.E.; Sindiri, S.; Wei, J.; Wen, X.; et al. Genomic Classification and Clinical Outcome in Rhabdomyosarcoma: A Report From an International Consortium. J. Clin. Oncol. 2021, 39, 2859–2871. [Google Scholar] [CrossRef] [PubMed]
- Galili, N.; Davis, R.J.; Fredericks, W.J.; Mukhopadhyay, S.; Rauscher, F.J.; Emanuel, B.S.; Rovera, G.; Barr, F.G. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat. Genet. 1993, 5, 230–235. [Google Scholar] [CrossRef]
- Skapek, S.X.; Anderson, J.; Barr, F.G.; Bridge, J.A.; Gastier-Foster, J.M.; Parham, D.M.; Rudzinski, E.R.; Triche, T.; Hawkins, D.S. PAX-FOXO1 fusion status drives unfavorable outcome for children with rhabdomyosarcoma: A children’s oncology group report. Pediatr. Blood Cancer 2013, 60, 1411–1417. [Google Scholar] [CrossRef]
- Gryder, B.E.; Yohe, M.E.; Chou, H.-C.; Zhang, X.; Marques, J.; Wachtel, M.; Schaefer, B.; Sen, N.; Song, Y.; Gualtieri, A.; et al. PAX3-FOXO1 Establishes Myogenic Super Enhancers and Confers BET Bromodomain Vulnerability. Cancer Discov. 2017, 7, 884–899. [Google Scholar] [CrossRef]
- Milewski, D.; Shukla, S.; Gryder, B.E.; Pradhan, A.; Donovan, J.; Sudha, P.; Vallabh, S.; Pyros, A.; Xu, Y.; Barski, A.; et al. FOXF1 is required for the oncogenic properties of PAX3-FOXO1 in rhabdomyosarcoma. Oncogene 2021, 40, 2182–2199. [Google Scholar] [CrossRef]
- Gryder, B.E.; Pomella, S.; Sayers, C.; Wu, X.S.; Song, Y.; Chiarella, A.M.; Bagchi, S.; Chou, H.-C.; Sinniah, R.S.; Walton, A.; et al. Histone hyperacetylation disrupts core gene regulatory architecture in rhabdomyosarcoma. Nat. Genet. 2019, 51, 1714–1722. [Google Scholar] [CrossRef]
- Gryder, B.E.; Wachtel, M.; Chang, K.; El Demerdash, O.; Aboreden, N.G.; Mohammed, W.; Ewert, W.; Pomella, S.; Rota, R.; Wei, J.S.; et al. Miswired Enhancer Logic Drives a Cancer of the Muscle Lineage. iScience 2020, 23, 101103. [Google Scholar] [CrossRef]
- Ramadan, F.; Saab, R.; Hussein, N.; Clézardin, P.; Cohen, P.A.; Ghayad, S.E. Non-coding RNA in rhabdomyosarcoma progression and metastasis. Front. Oncol. 2022, 12, 971174. [Google Scholar] [CrossRef] [PubMed]
- Hanna, J.A.; Garcia, M.R.; Lardennois, A.; Leavey, P.J.; Maglic, D.; Fagnan, A.; Go, J.C.; Roach, J.; Wang, Y.-D.; Finkelstein, D.; et al. PAX3-FOXO1 drives miR-486-5p and represses miR-221 contributing to pathogenesis of alveolar rhabdomyosarcoma. Oncogene 2018, 37, 1991–2007. [Google Scholar] [CrossRef] [PubMed]
- Phelps, M.P.; Bailey, J.N.; Vleeshouwer-Neumann, T.; Chen, E.Y. CRISPR screen identifies the NCOR/HDAC3 complex as a major suppressor of differentiation in rhabdomyosarcoma. Proc. Natl. Acad. Sci. USA 2016, 113, 15090–15095. [Google Scholar] [CrossRef] [PubMed]
- Marques, J.G.; Gryder, B.E.; Pavlovic, B.; Chung, Y.; A Ngo, Q.; Frommelt, F.; Gstaiger, M.; Song, Y.; Benischke, K.; Laubscher, D.; et al. NuRD subunit CHD4 regulates super-enhancer accessibility in rhabdomyosarcoma and represents a general tumor dependency. Elife 2020, 9, e54993. [Google Scholar] [CrossRef]
- Laubscher, D.; Gryder, B.E.; Sunkel, B.D.; Andresson, T.; Wachtel, M.; Das, S.; Roschitzki, B.; Wolski, W.; Wu, X.S.; Chou, H.-C.; et al. BAF complexes drive proliferation and block myogenic differentiation in fusion-positive rhabdomyosarcoma. Nat. Commun. 2021, 12, 6924. [Google Scholar] [CrossRef]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective Inhibition of Tumor Oncogenes by Disruption of Super-Enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef]
- Bid, H.K.; Phelps, D.A.; Xaio, L.; Guttridge, D.C.; Lin, J.; London, C.; Baker, L.H.; Mo, X.; Houghton, P.J. The Bromodomain BET Inhibitor JQ1 Suppresses Tumor Angiogenesis in Models of Childhood Sarcoma. Mol. Cancer Ther. 2016, 15, 1018–1028. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef]
- Odore, E.; Lokiec, F.; Cvitkovic, E.; Bekradda, M.; Herait, P.; Bourdel, F.; Kahatt, C.; Raffoux, E.; Stathis, A.; Thieblemont, C.; et al. Phase I Population Pharmacokinetic Assessment of the Oral Bromodomain Inhibitor OTX015 in Patients with Haematologic Malignancies. Clin. Pharmacokinet. 2015, 55, 397–405. [Google Scholar] [CrossRef]
- Berthon, C.; Raffoux, E.; Thomas, X.; Vey, N.; Gomez-Roca, C.; Yee, K.; Taussig, D.C.; Rezai, K.; Roumier, C.; Herait, P.; et al. Bromodomain inhibitor OTX015 in patients with acute leukaemia: A dose-escalation, phase 1 study. Lancet Haematol. 2016, 3, e186–e195. [Google Scholar] [CrossRef]
- Postel-Vinay, S.; Herbschleb, K.; Massard, C.; Woodcock, V.; Soria, J.-C.; Walter, A.O.; Ewerton, F.; Poelman, M.; Benson, N.; Ocker, M.; et al. First-in-human phase I study of the bromodomain and extraterminal motif inhibitor BAY 1238097: Emerging pharmacokinetic/pharmacodynamic relationship and early termination due to unexpected toxicity. Eur. J. Cancer 2019, 109, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Piha-Paul, S.A.; Hann, C.L.; French, C.A.; Cousin, S.; Braña, I.; Cassier, P.A.; Moreno, V.; De Bono, J.S.; Harward, S.D.; Ferron-Brady, G.; et al. Phase 1 Study of Molibresib (GSK525762), a Bromodomain and Extra-Terminal Domain Protein Inhibitor, in NUT Carcinoma and Other Solid Tumors. JNCI Cancer Spectr. 2019, 4, pkz093. [Google Scholar] [CrossRef] [PubMed]
- Raina, K.; Lu, J.; Qian, Y.; Altieri, M.; Gordon, D.; Rossi, A.M.K.; Wang, J.; Chen, X.; Dong, H.; Siu, K.; et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 7124–7129. [Google Scholar] [CrossRef] [PubMed]
- Gavai, A.V.; Norris, D.; Delucca, G.; Tortolani, D.; Tokarski, J.S.; Dodd, D.; O’Malley, D.; Zhao, Y.; Quesnelle, C.; Gill, P.; et al. Discovery and Preclinical Pharmacology of an Oral Bromodomain and Extra-Terminal (BET) Inhibitor Using Scaffold-Hopping and Structure-Guided Drug Design. J. Med. Chem. 2021, 64, 14247–14265. [Google Scholar] [CrossRef]
- Williamson, D.; Lu, Y.-J.; Gordon, T.; Sciot, R.; Kelsey, A.; Fisher, C.; Poremba, C.; Anderson, J.; Pritchard-Jones, K.; Shipley, J. Relationship Between MYCN Copy Number and Expression in Rhabdomyosarcomas and Correlation with Adverse Prognosis in the Alveolar Subtype. J. Clin. Oncol. 2005, 23, 880–888. [Google Scholar] [CrossRef]
- Gryder, B.E.; Wu, L.; Woldemichael, G.M.; Pomella, S.; Quinn, T.R.; Park, P.M.C.; Cleveland, A.; Stanton, B.Z.; Song, Y.; Rota, R.; et al. Chemical genomics reveals histone deacetylases are required for core regulatory transcription. Nat. Commun. 2019, 10, 3004. [Google Scholar] [CrossRef]
- Gryder, B.; Khan, J.; Stanton, B. Absolute Quantification of Architecture (AQuA-HiChIP) Enables Measurement of Differential Chromatin Interactions. Nat. Protoc. 2019. [Google Scholar] [CrossRef]
- Guo, S.; Jiang, X.; Mao, B.; Li, Q.-X. he design, analysis and application of mouse clinical trials in oncology drug development. BMC Cancer 2019, 19, 718. [Google Scholar] [CrossRef]
- Cho, W.-K.; Spille, J.-H.; Hecht, M.; Lee, C.; Li, C.; Grube, V.; Cisse, I.I. Heterogeneity of paclitaxel distribution in different tumor models assessed by MALDI mass spectrometry imaging. Sci. Rep. 2016, 6, 39284. [Google Scholar] [CrossRef]
- Cho, W.K.; Spille, J.H.; Hecht, M.; Lee, C.; Li, C.; Grube, V.; Cisse, I.I. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 2018, 361, 412–415. [Google Scholar] [CrossRef]
- Mandal, R.; Becker, S.; Strebhardt, K. Targeting CDK9 for Anti-Cancer Therapeutics. Cancers 2021, 13, 2181. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.P. Cdk7: A kinase at the core of transcription and in the crosshairs of cancer drug discovery. Transcription 2019, 10, 47–56. [Google Scholar] [CrossRef]
- Gao, Y.; Volegova, M.; Nasholm, N.; Das, S.; Kwiatkowski, N.; Abraham, B.J.; Zhang, T.; Gray, N.S.; Gustafson, C.; Krajewska, M.; et al. Synergistic Anti-Tumor Effect of Combining Selective CDK7 and BRD4 Inhibition in Neuroblastoma. Front. Oncol. 2022, 11, 773186. [Google Scholar] [CrossRef] [PubMed]
- Boeva, V.; Louis-Brennetot, C.; Peltier, A.; Durand, S.; Pierre-Eugène, C.; Raynal, V.; Etchevers, H.C.; Thomas, S.; Lermine, A.; Daudigeos-Dubus, E.; et al. Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat. Genet. 2017, 49, 1408. [Google Scholar] [CrossRef] [PubMed]
- Durbin, A.D.; Zimmerman, M.W.; Dharia, N.V.; Abraham, B.J.; Iniguez, A.B.; Weichert-Leahey, N.; He, S.; Krill-Burger, J.M.; Root, D.E.; Vazquez, F.; et al. Selective gene dependencies in MYCN-amplified neuroblastoma include the core transcriptional regulatory circuitry. Nat. Genet. 2018, 50, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Puissant, A.; Frumm, S.M.; Alexe, G.; Bassil, C.F.; Qi, J.; Chanthery, Y.H.; Nekritz, E.A.; Zeid, R.; Gustafson, W.C.; Greninger, P.; et al. Targeting MYCN in Neuroblastoma by BET Bromodomain Inhibition. Cancer Discov. 2013, 3, 308–323. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chin, D.H.; Osman, I.; Porch, J.; Kim, H.; Buck, K.K.; Rodriguez, J.; Carapia, B.; Yan, D.; Moura, S.B.; Sperry, J.; et al. BET Bromodomain Degradation Disrupts Function but Not 3D Formation of RNA Pol2 Clusters. Pharmaceuticals 2023, 16, 199. https://doi.org/10.3390/ph16020199
Chin DH, Osman I, Porch J, Kim H, Buck KK, Rodriguez J, Carapia B, Yan D, Moura SB, Sperry J, et al. BET Bromodomain Degradation Disrupts Function but Not 3D Formation of RNA Pol2 Clusters. Pharmaceuticals. 2023; 16(2):199. https://doi.org/10.3390/ph16020199
Chicago/Turabian StyleChin, Diana H., Issra Osman, Jadon Porch, Hyunmin Kim, Kristen K. Buck, Javier Rodriguez, Bianca Carapia, Deborah Yan, Stela B. Moura, Jantzen Sperry, and et al. 2023. "BET Bromodomain Degradation Disrupts Function but Not 3D Formation of RNA Pol2 Clusters" Pharmaceuticals 16, no. 2: 199. https://doi.org/10.3390/ph16020199
APA StyleChin, D. H., Osman, I., Porch, J., Kim, H., Buck, K. K., Rodriguez, J., Carapia, B., Yan, D., Moura, S. B., Sperry, J., Nakashima, J., Altman, K., Altman, D., & Gryder, B. E. (2023). BET Bromodomain Degradation Disrupts Function but Not 3D Formation of RNA Pol2 Clusters. Pharmaceuticals, 16(2), 199. https://doi.org/10.3390/ph16020199