
Direct-Acting Antivirals and Host-Targeting Approaches against Enterovirus B Infections: Recent Advances

 , ,
, ,  ,
,  and
and 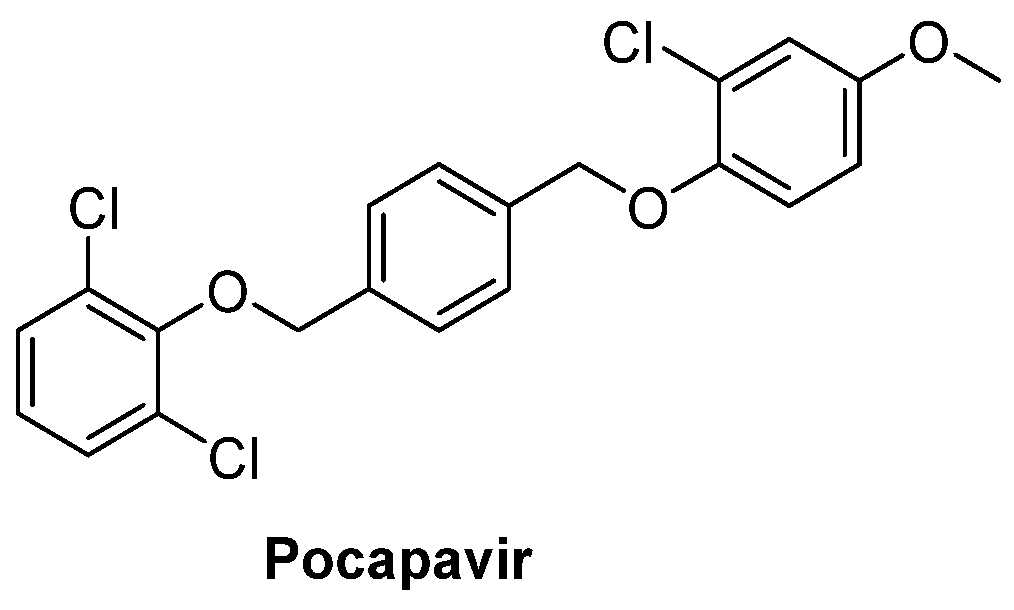{kind=link}
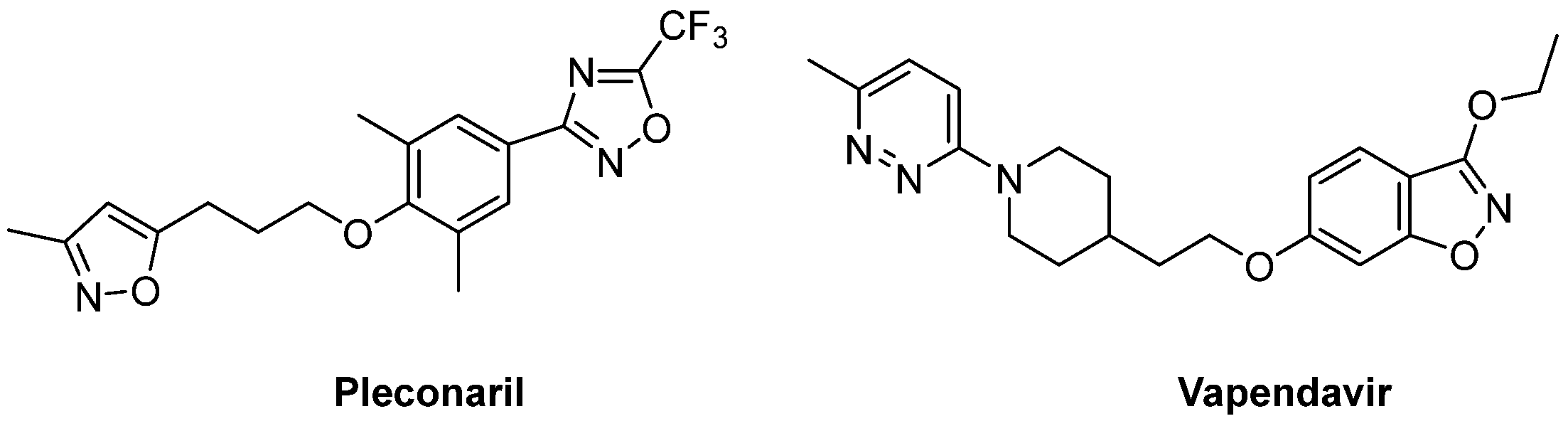{kind=link}
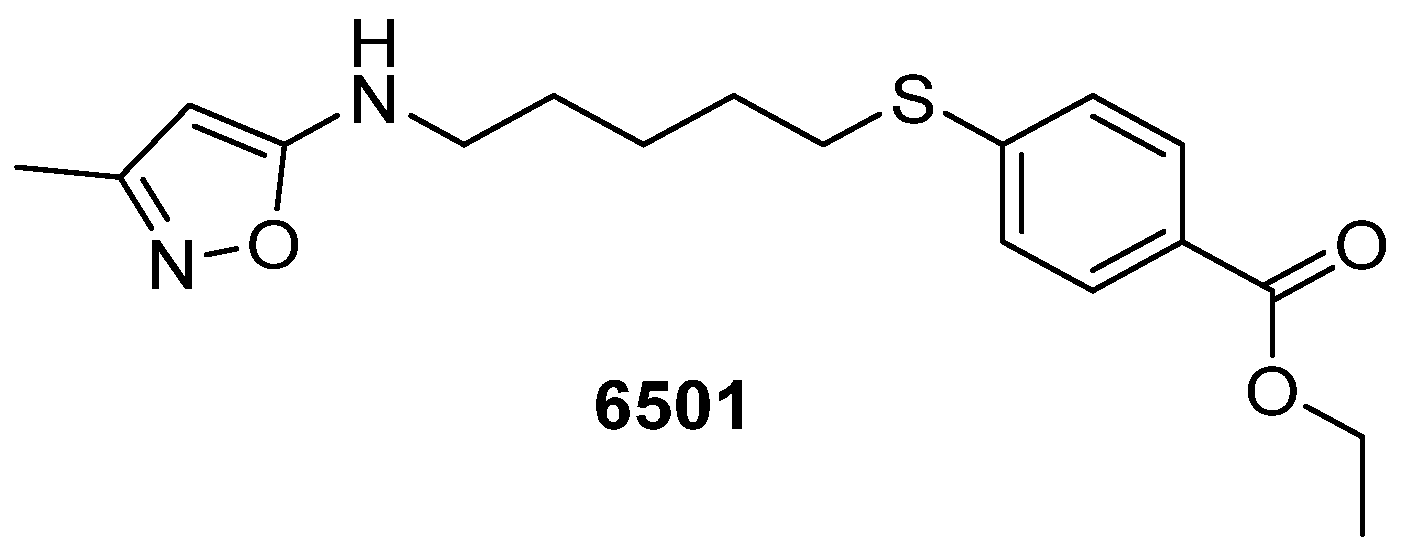{kind=link}
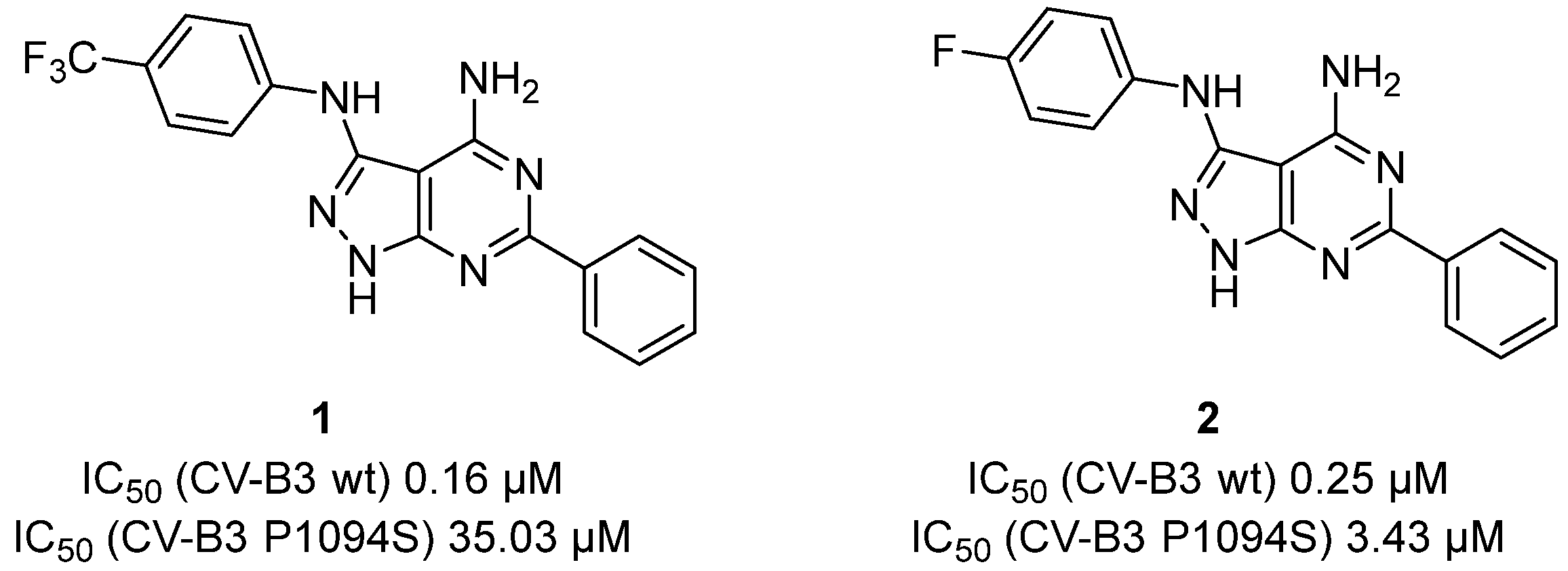{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Enterovirus Classification and Pathogenesis
1.2. Life Cycle of Enteroviruses
1.3. Antivirals against Enterovirus Infections
2. Direct-Acting Antivirals
2.1. Capsid Binders
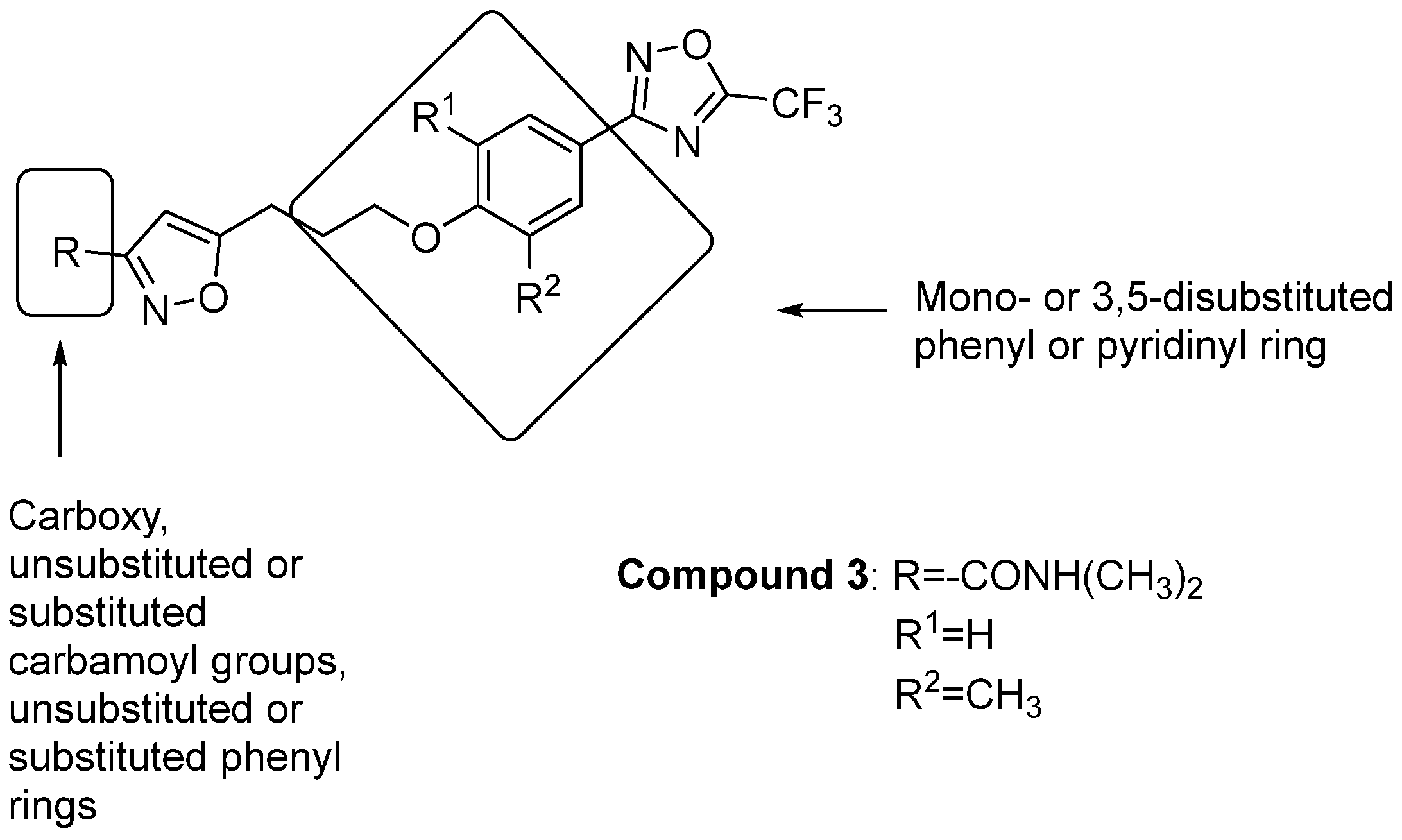
2.1.1. Antivirals Targeting the VP1 Hydrophobic Pocket
2.1.2. Antivirals Targeting the VP1-VP3 Interprotomer Binding Pocket
2.2. Non-Structural Protein Inhibitors
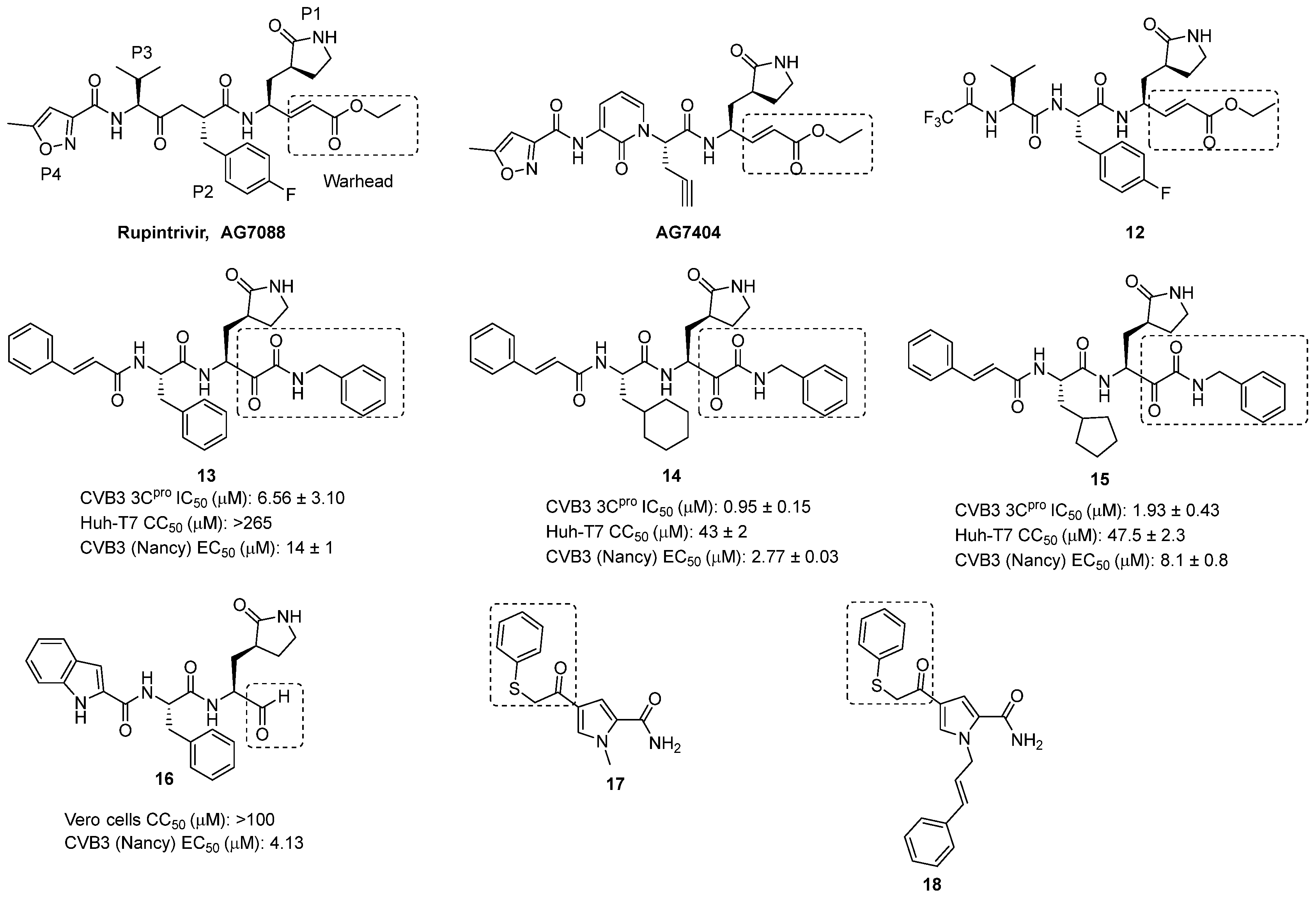
2.2.1. 3C Protease Inhibitors
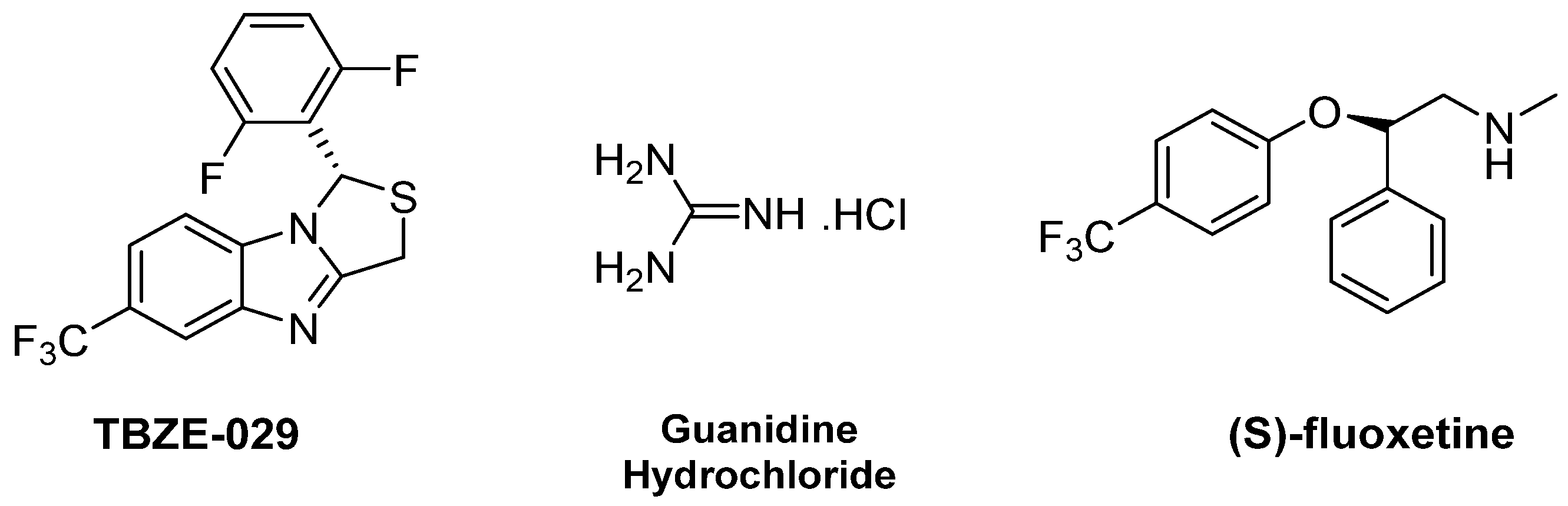
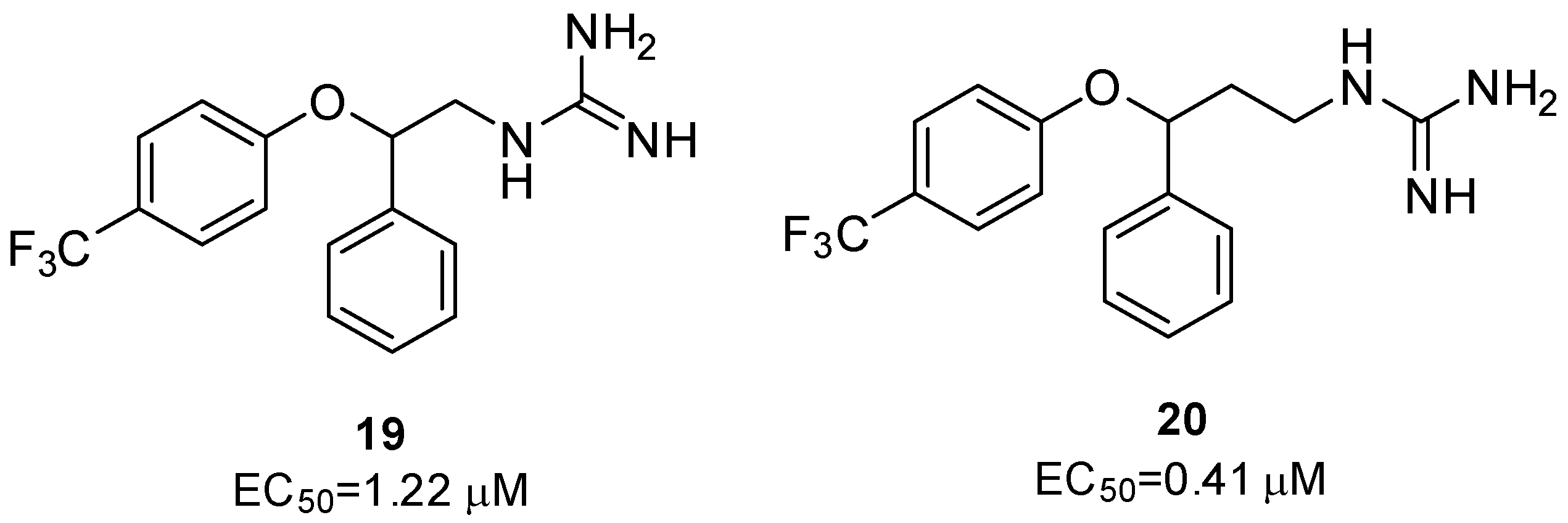
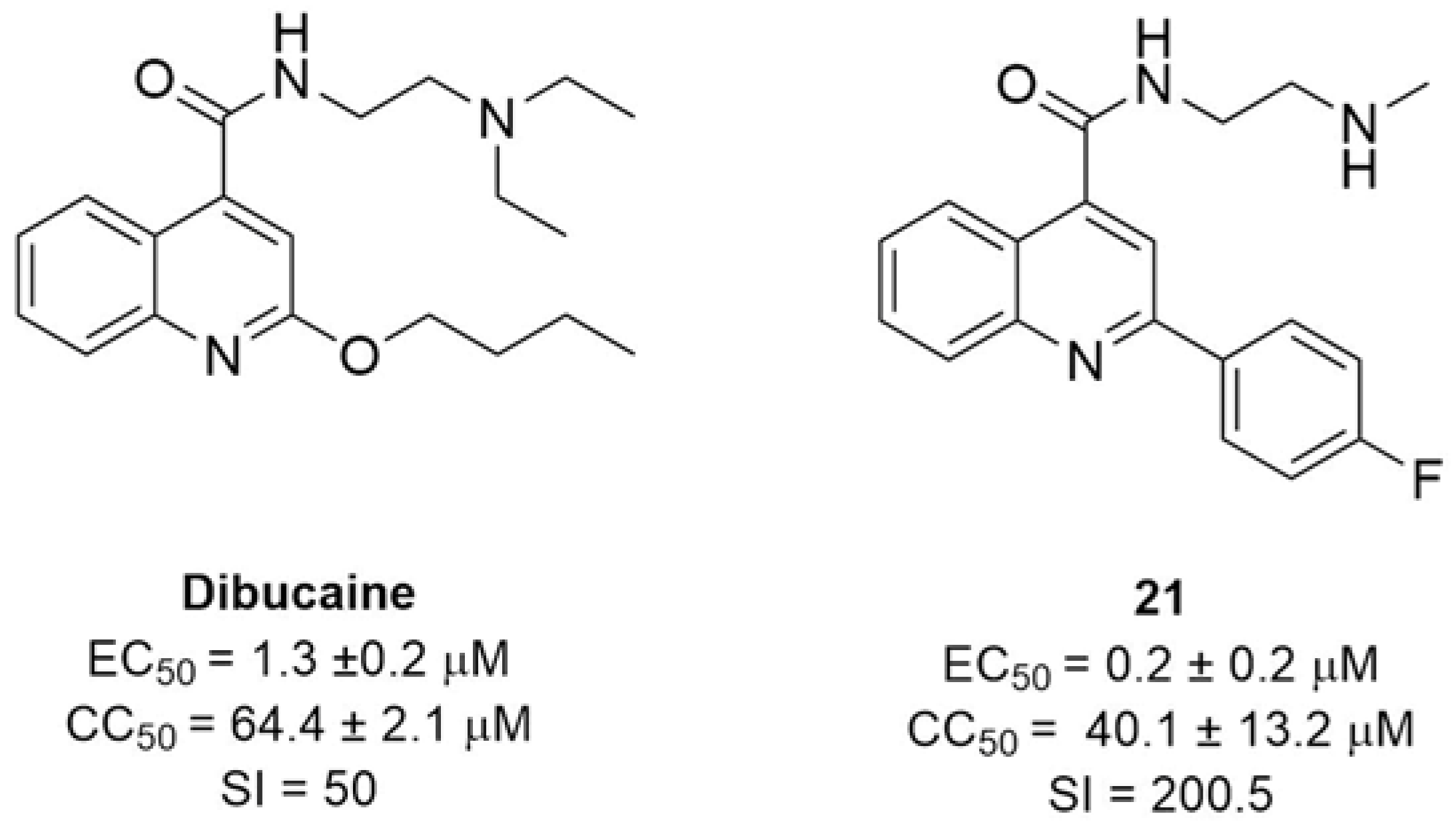
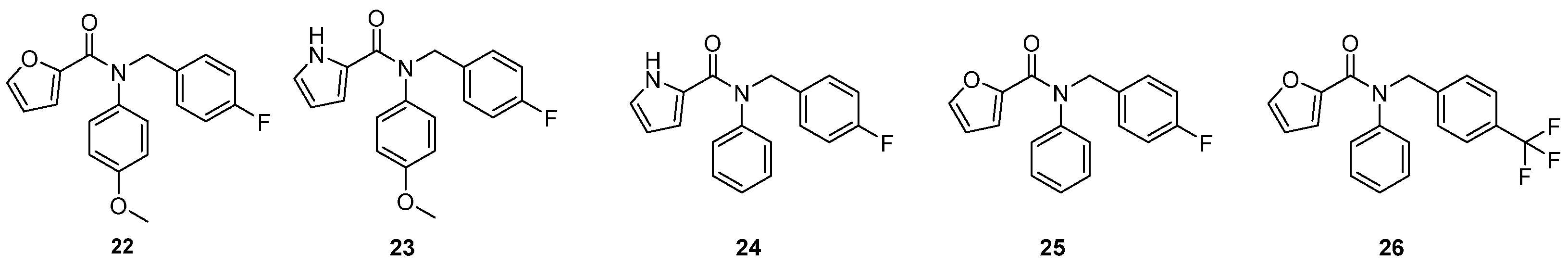
2.2.2. 2C Protease Inhibitors
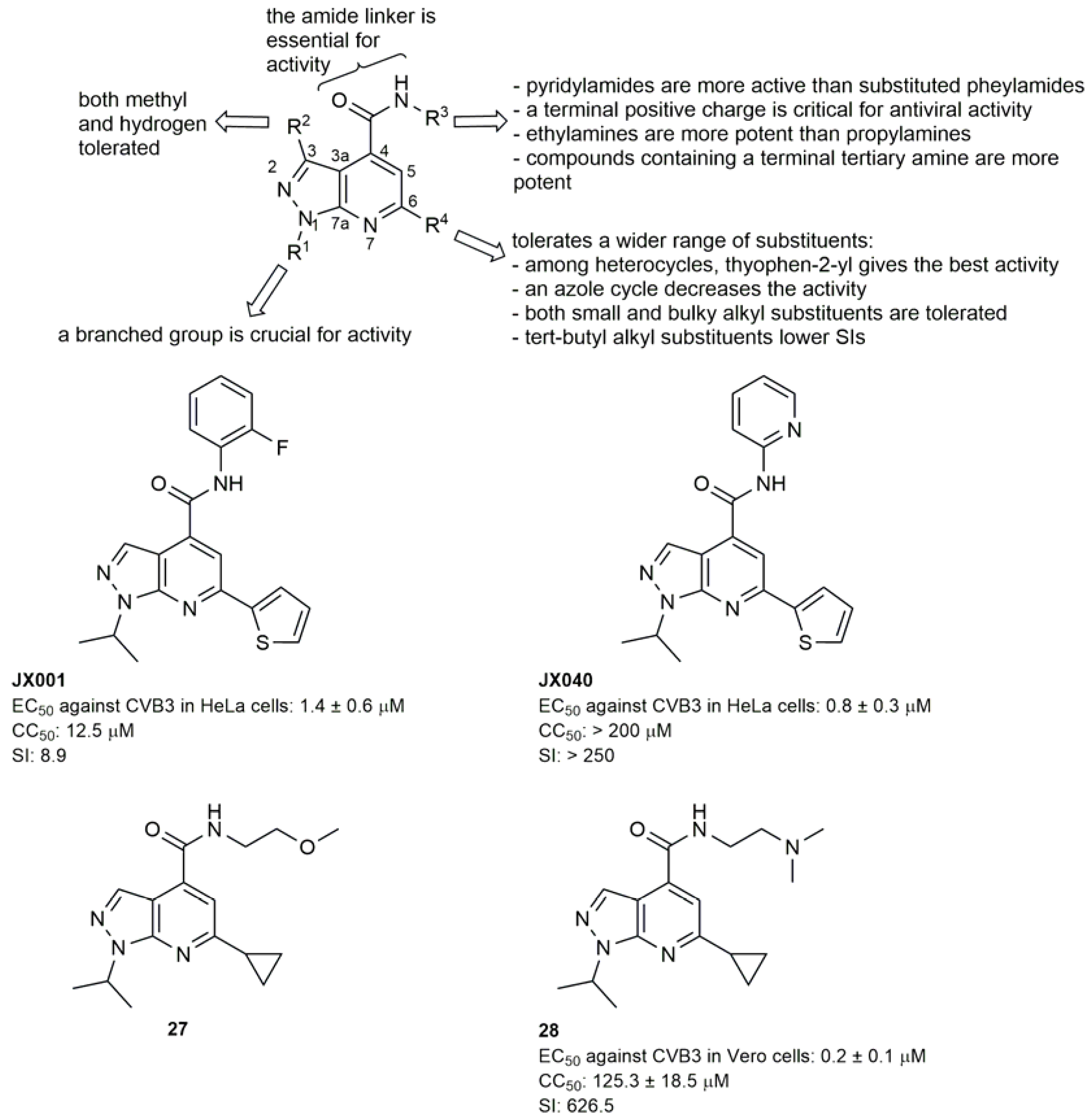
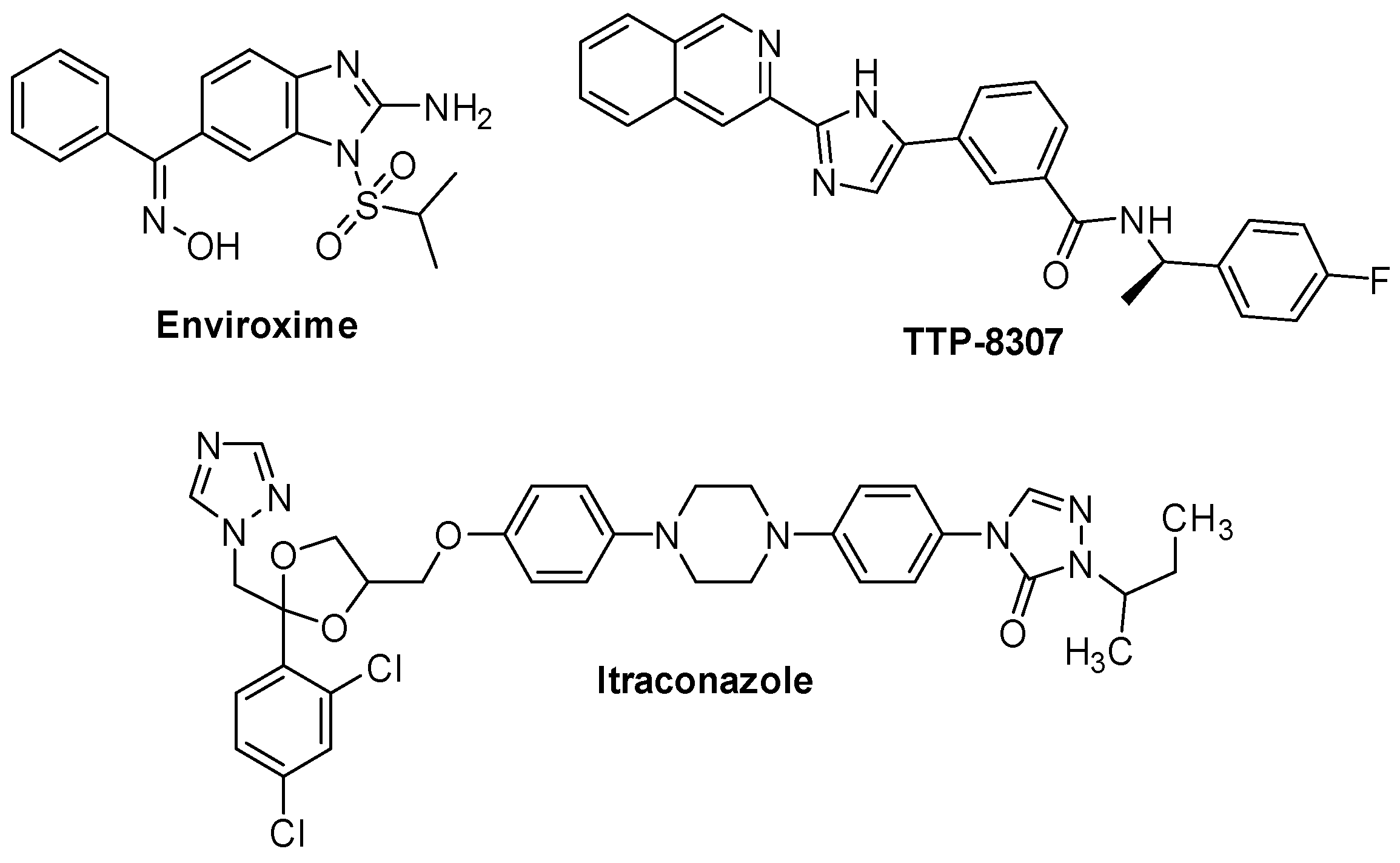
2.2.3. 3A Protein Inhibitors
2.2.4. 3Dpol Inhibitors
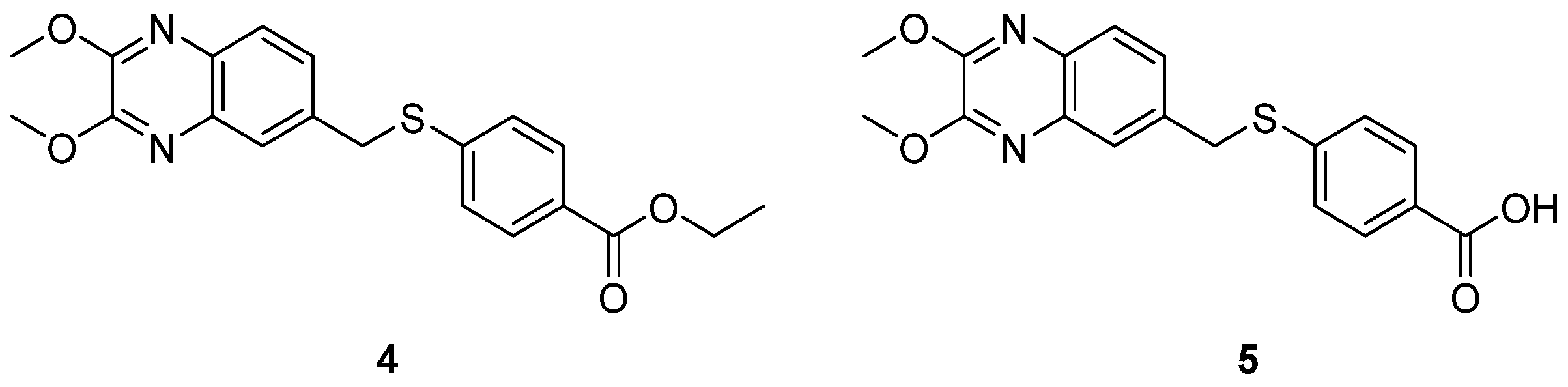
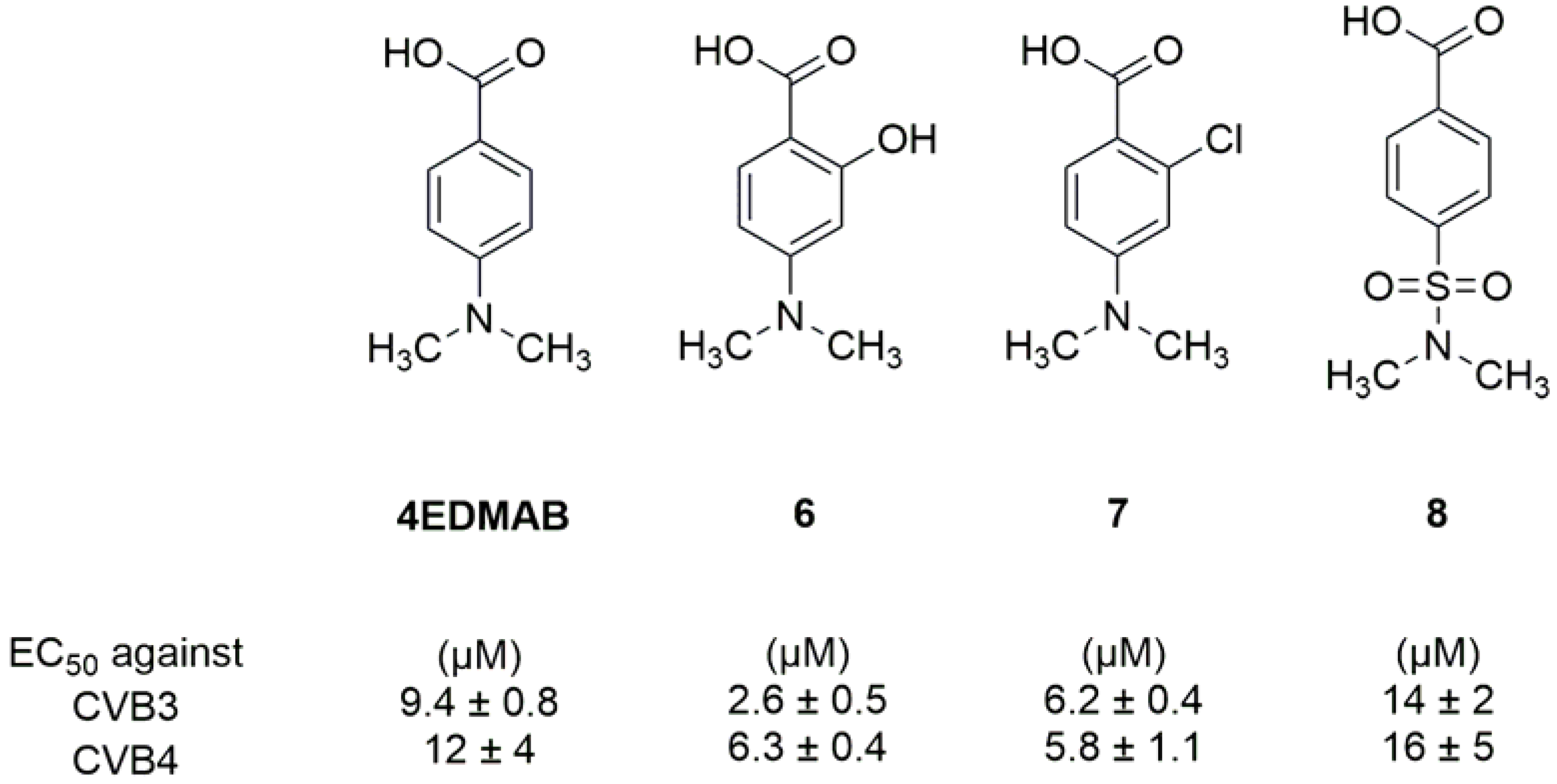
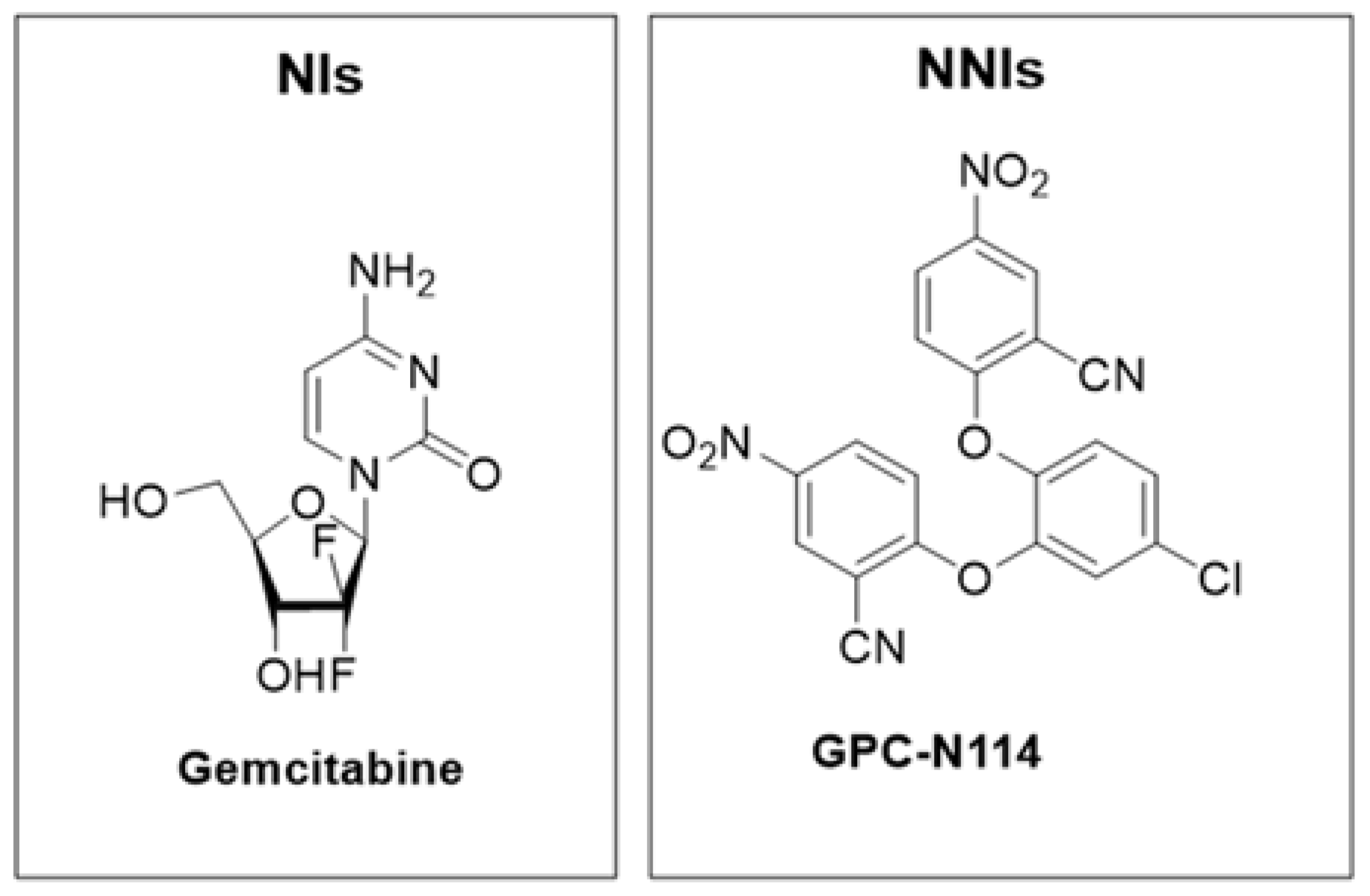
2.3. Inhibitors of the Host Factors
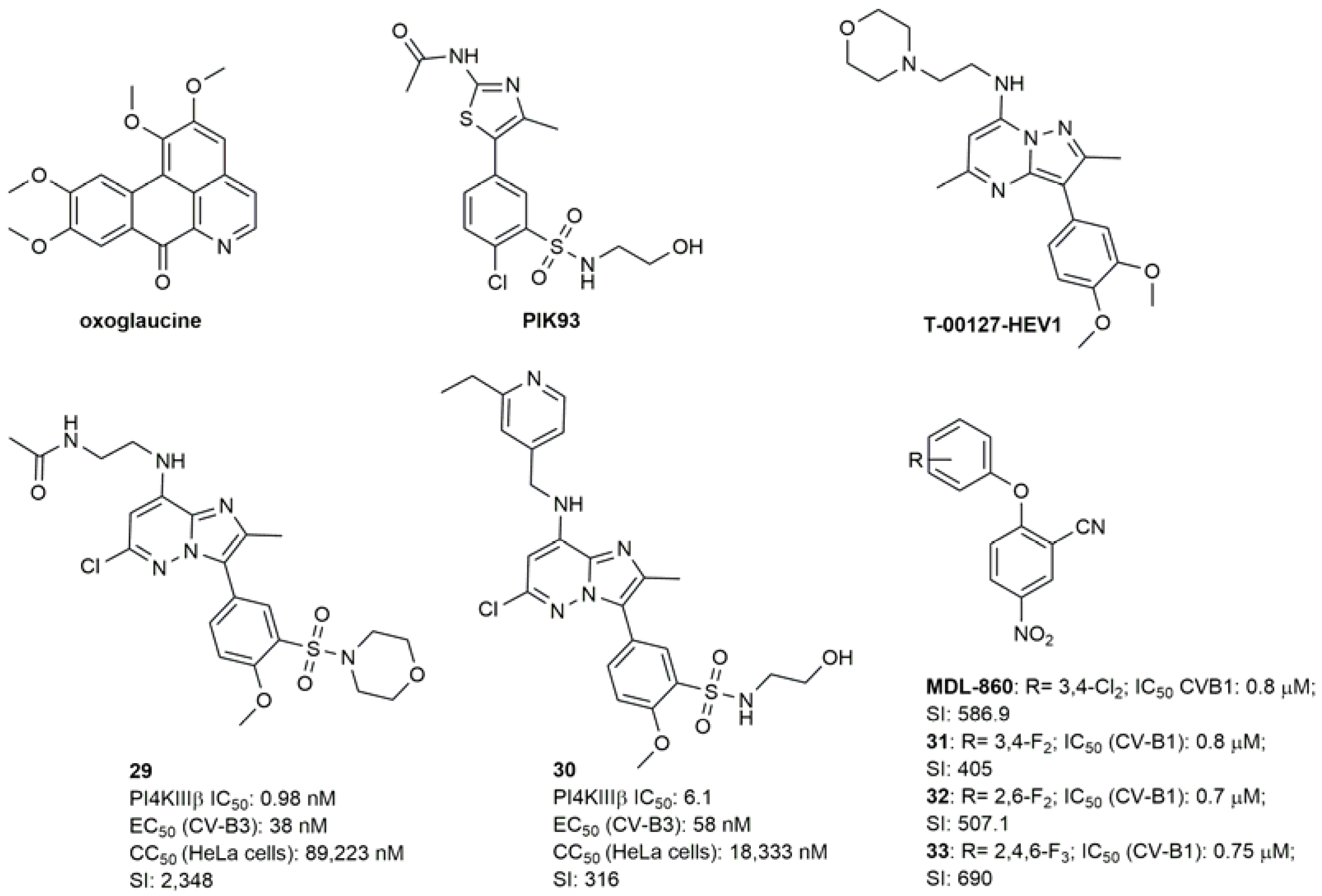
Inhibitors of PI4KIIIβ-PI4P-OSBP Pathway
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Anasir, M.I.; Zarif, F.; Poh, C.L. Antivirals Blocking Entry of Enteroviruses and Therapeutic Potential. J. Biomed. Sci. 2021, 28, 10. [Google Scholar] [CrossRef] [PubMed]
- Zell, R. Picornaviridae-the Ever-Growing Virus Family. Arch. Virol. 2018, 163, 299–317. [Google Scholar] [CrossRef] [PubMed]
- Baggen, J.; Thibaut, H.J.; Strating, J.R.P.M.; van Kuppeveld, F.J.M. The Life Cycle of Non-Polio Enteroviruses and How to Target It. Nat. Rev. Microbiol. 2018, 16, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Nekoua, M.P.; Alidjinou, E.K.; Hober, D. Persistent Coxsackievirus B Infection and Pathogenesis of Type 1 Diabetes Mellitus. Nat. Rev. Endocrinol. 2022, 18, 503–516. [Google Scholar] [CrossRef]
- Alhazmi, A.; Nekoua, M.P.; Mercier, A.; Vergez, I.; Sane, F.; Alidjinou, E.K.; Hober, D. Combating Coxsackievirus B Infections. Rev. Med. Virol. 2022, 33, e2406. [Google Scholar] [CrossRef]
- Laajala, M.; Reshamwala, D.; Marjomäki, V. Therapeutic Targets for Enterovirus Infections. Expert. Opin. Ther. Targets 2020, 24, 745–757. [Google Scholar] [CrossRef]
- Egorova, A.; Ekins, S.; Schmidtke, M.; Makarov, V. Back to the Future: Advances in Development of Broad-Spectrum Capsid-Binding Inhibitors of Enteroviruses. Eur. J. Med. Chem. 2019, 178, 606–622. [Google Scholar] [CrossRef]
- Epstein, S.; Thakkar, R.; Fong, K.T.; Ng, J.; Bearden, D.R.; Mishra, N.; Thakur, K.T.; Riley, C.S. Compassionate-Use Pocapavir and Immunoglobulin Therapy for Treatment of Rituximab-Associated Enterovirus Meningoencephalitis. J. Neurovirol. 2022, 28, 329–334. [Google Scholar] [CrossRef]
- Torres-Torres, S.; Myers, A.L.; Klatte, J.M.; Rhoden, E.E.; Oberste, M.S.; Collett, M.S.; McCulloh, R.J. First Use of Investigational Antiviral Drug Pocapavir (v-073) for Treating Neonatal Enteroviral Sepsis. Pediatr. Infect. Dis. J. 2015, 34, 52–54. [Google Scholar] [CrossRef]
- Wyllie, T. P011 An Experimental Treatment for Enteroviral Sepsis. Arch. Dis. Child. 2019, 104, e2. [Google Scholar] [CrossRef]
- Bauer, L.; Lyoo, H.; van der Schaar, H.M.; Strating, J.R.; van Kuppeveld, F.J. Direct-Acting Antivirals and Host-Targeting Strategies to Combat Enterovirus Infections. Curr. Opin. Virol. 2017, 24, 1–8. [Google Scholar] [CrossRef]
- Ren, P.; Zheng, Y.; Wang, W.; Hong, L.; Delpeyroux, F.; Arenzana-Seisdedos, F.; Altmeyer, R. Suramin Interacts with the Positively Charged Region Surrounding the 5-Fold Axis of the EV-A71 Capsid and Inhibits Multiple Enterovirus A. Sci. Rep. 2017, 7, 42902. [Google Scholar] [CrossRef] [Green Version]
- Reshamwala, D.; Shroff, S.; Sheik Amamuddy, O.; Laquintana, V.; Denora, N.; Zacheo, A.; Lampinen, V.; Hytonen, V.P.; Tastan Bishop, Ö.; Krol, S.; et al. Polyphenols Epigallocatechin Gallate and Resveratrol, and Polyphenol-Functionalized Nanoparticles Prevent Enterovirus Infection through Clustering and Stabilization of the Viruses. Pharmaceutics 2021, 13, 1182. [Google Scholar] [CrossRef]
- Bernard, A.; Lacroix, C.; Cabiddu, M.G.; Neyts, J.; Leyssen, P.; Pompei, R. Exploration of the Anti-Enterovirus Activity of a Series of Pleconaril/Pirodavir-like Compounds. Antivir. Chem. Chemother. 2015, 24, 56–61. [Google Scholar] [CrossRef]
- Egorova, A.; Kazakova, E.; Jahn, B.; Ekins, S.; Makarov, V.; Schmidtke, M. Novel Pleconaril Derivatives: Influence of Substituents in the Isoxazole and Phenyl Rings on the Antiviral Activity against Enteroviruses. Eur. J. Med. Chem. 2020, 188, 112007. [Google Scholar] [CrossRef]
- Makarov, V.A.; Braun, H.; Richter, M.; Riabova, O.B.; Kirchmair, J.; Kazakova, E.S.; Seidel, N.; Wutzler, P.; Schmidtke, M. Pyrazolopyrimidines: Potent Inhibitors Targeting the Capsid of Rhino- and Enteroviruses. ChemMedChem 2015, 10, 1629–1634. [Google Scholar] [CrossRef] [Green Version]
- Carta, A.; Sanna, G.; Briguglio, I.; Madeddu, S.; Vitale, G.; Piras, S.; Corona, P.; Peana, A.T.; Laurini, E.; Fermeglia, M.; et al. Quinoxaline Derivatives as New Inhibitors of Coxsackievirus B5. Eur. J. Med. Chem. 2018, 145, 559–569. [Google Scholar] [CrossRef]
- Madeddu, S.; Ibba, R.; Sanna, G.; Piras, S.; Riu, F.; Marongiu, A.; Ambrosino, A.; Caria, P.; Onnis, V.; Franci, G.; et al. Human Enterovirus B: Selective Inhibition by Quinoxaline Derivatives and Bioinformatic RNA-Motif Identification as New Targets. Pharmaceuticals 2022, 15, 181. [Google Scholar] [CrossRef]
- Ma, Y.; Abdelnabi, R.; Delang, L.; Froeyen, M.; Luyten, W.; Neyts, J.; Mirabelli, C. New Class of Early-Stage Enterovirus Inhibitors with a Novel Mechanism of Action. Antivir. Res. 2017, 147, 67–74. [Google Scholar] [CrossRef]
- Abdelnabi, R.; Geraets, J.A.; Ma, Y.; Mirabelli, C.; Flatt, J.W.; Domanska, A.; Delang, L.; Jochmans, D.; Kumar, T.A.; Jayaprakash, V.; et al. A Novel Druggable Interprotomer Pocket in the Capsid of Rhino- and Enteroviruses. PLoS Biol. 2019, 17, e3000281. [Google Scholar] [CrossRef]
- Shetnev, A.A.; Volobueva, A.S.; Panova, V.A.; Zarubaev, V.V.; Baykov, S.V. Design of 4-Substituted Sulfonamidobenzoic Acid Derivatives Targeting Coxsackievirus B3. Life 2022, 12, 1832. [Google Scholar] [CrossRef] [PubMed]
- Hayden, F.G.; Turner, R.B.; Gwaltney, J.M.; Chi-Burris, K.; Gersten, M.; Hsyu, P.; Patick, A.K.; Smith, G.J.; Zalman, L.S. Phase II, Randomized, Double-Blind, Placebo-Controlled Studies of Ruprintrivir Nasal Spray 2-Percent Suspension for Prevention and Treatment of Experimentally Induced Rhinovirus Colds in Healthy Volunteers. Antimicrob. Agents Chemother. 2003, 47, 3907–3916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patick, A.K.; Brothers, M.A.; Maldonado, F.; Binford, S.; Maldonado, O.; Fuhrman, S.; Petersen, A.; Smith, G.J.; Zalman, L.S.; Burns-Naas, L.A.; et al. In Vitro Antiviral Activity and Single-Dose Pharmacokinetics in Humans of a Novel, Orally Bioavailable Inhibitor of Human Rhinovirus 3C Protease. Antimicrob. Agents Chemother. 2005, 49, 2267–2275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ianevski, A.; Zusinaite, E.; Tenson, T.; Oksenych, V.; Wang, W.; Afset, J.E.; Bjørås, M.; Kainov, D.E. Novel Synergistic Anti-Enteroviral Drug Combinations. Viruses 2022, 14, 1866. [Google Scholar] [CrossRef]
- Kankam, M.K.; Burns, J.M.; Collett, M.S.; Corrado, M.L.; Hincks, J.R. A Phase 1 Study of the Safety, Tolerability, and Pharmacokinetics of Single and Multiple Oral Doses of V-7404 in Healthy Adult Volunteers. Antimicrob. Agents Chemother. 2021, 65, e0102921. [Google Scholar] [CrossRef]
- Rhoden, E.; Liu, H.-M.; Wang-Chern, S.-W.; Oberste, M.S. Anti-Poliovirus Activity of Protease Inhibitor AG-7404, and Assessment of in Vitro Activity in Combination with Antiviral Capsid Inhibitor Compounds. Antivir. Res. 2013, 98, 186–191. [Google Scholar] [CrossRef]
- van der Linden, L.; Ulferts, R.; Nabuurs, S.B.; Kusov, Y.; Liu, H.; George, S.; Lacroix, C.; Goris, N.; Lefebvre, D.; Lanke, K.H.W.; et al. Application of a Cell-Based Protease Assay for Testing Inhibitors of Picornavirus 3C Proteases. Antivir. Res. 2014, 103, 17–24. [Google Scholar] [CrossRef]
- Tan, Y.W.; Ang, M.J.Y.; Lau, Q.Y.; Poulsen, A.; Ng, F.M.; Then, S.W.; Peng, J.; Hill, J.; Hong, W.J.; Chia, C.S.B.; et al. Antiviral Activities of Peptide-Based Covalent Inhibitors of the Enterovirus 71 3C Protease. Sci. Rep. 2016, 6, 33663. [Google Scholar] [CrossRef] [Green Version]
- Marjomäki, V.; Kalander, K.; Hellman, M.; Permi, P. Enteroviruses and Coronaviruses: Similarities and Therapeutic Targets. Expert. Opin. Ther. Targets 2021, 25, 479–489. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Kusov, Y.; Nian, Y.; Ma, Q.; Wang, J.; von Brunn, A.; Leyssen, P.; Lanko, K.; Neyts, J.; et al. α-Ketoamides as Broad-Spectrum Inhibitors of Coronavirus and Enterovirus Replication: Structure-Based Design, Synthesis, and Activity Assessment. J. Med. Chem. 2020, 63, 4562–4578. [Google Scholar] [CrossRef]
- Dai, W.; Jochmans, D.; Xie, H.; Yang, H.; Li, J.; Su, H.; Chang, D.; Wang, J.; Peng, J.; Zhu, L.; et al. Design, Synthesis, and Biological Evaluation of Peptidomimetic Aldehydes as Broad-Spectrum Inhibitors against Enterovirus and SARS-CoV-2. J. Med. Chem. 2022, 65, 2794–2808. [Google Scholar] [CrossRef]
- Schulz, R.; Atef, A.; Becker, D.; Gottschalk, F.; Tauber, C.; Wagner, S.; Arkona, C.; Abdel-Hafez, A.A.; Farag, H.H.; Rademann, J.; et al. Phenylthiomethyl Ketone-Based Fragments Show Selective and Irreversible Inhibition of Enteroviral 3C Proteases. J. Med. Chem. 2018, 61, 1218–1230. [Google Scholar] [CrossRef]
- Kim, B.-K.; Ko, H.; Jeon, E.-S.; Ju, E.-S.; Jeong, L.S.; Kim, Y.-C. 2,3,4-Trihydroxybenzyl-Hydrazide Analogues as Novel Potent Coxsackievirus B3 3C Protease Inhibitors. Eur. J. Med. Chem. 2016, 120, 202–216. [Google Scholar] [CrossRef]
- Bauer, L.; Manganaro, R.; Zonsics, B.; Strating, J.R.P.M.; El Kazzi, P.; Lorenzo Lopez, M.; Ulferts, R.; van Hoey, C.; Maté, M.J.; Langer, T.; et al. Fluoxetine Inhibits Enterovirus Replication by Targeting the Viral 2C Protein in a Stereospecific Manner. ACS Infect. Dis. 2019, 5, 1609–1623. [Google Scholar] [CrossRef] [Green Version]
- Hurdiss, D.L.; El Kazzi, P.; Bauer, L.; Papageorgiou, N.; Ferron, F.P.; Donselaar, T.; van Vliet, A.L.W.; Shamorkina, T.M.; Snijder, J.; Canard, B.; et al. Fluoxetine Targets an Allosteric Site in the Enterovirus 2C AAA+ ATPase and Stabilizes a Ring-Shaped Hexameric Complex. Sci. Adv. 2022, 8, eabj7615. [Google Scholar] [CrossRef]
- De Palma, A.M.; Heggermont, W.; Lanke, K.; Coutard, B.; Bergmann, M.; Monforte, A.-M.; Canard, B.; De Clercq, E.; Chimirri, A.; Pürstinger, G.; et al. The Thiazolobenzimidazole TBZE-029 Inhibits Enterovirus Replication by Targeting a Short Region Immediately Downstream from Motif C in the Nonstructural Protein 2C. J. Virol. 2008, 82, 4720–4730. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Zhi, C.; Li, H.; Huang, D.; Fan, Q.; Cui, J.; Liang, C. Umifenovir Effectively Inhibits IL-10 Dependent Persistent Coxsackie B4 Virus Infection. Antivir. Res. 2017, 141, 165–173. [Google Scholar] [CrossRef]
- Zhao, Y.; Yan, K.; Wang, Y.; Cai, J.; Wei, L.; Li, S.; Xu, W.; Li, M. Lithium Chloride Confers Protection against Viral Myocarditis via Suppression of Coxsackievirus B3 Virus Replication. Microb. Pathog. 2020, 144, 104169. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, S.; Chen, Y.; Wang, Y.; Wang, T.; Wo, X.; Dong, Y.; Zhang, J.; Xu, W.; Qu, C.; et al. N-Acetyl Cysteine Effectively Alleviates Coxsackievirus B-Induced Myocarditis through Suppressing Viral Replication and Inflammatory Response. Antivir. Res. 2020, 179, 104699. [Google Scholar] [CrossRef]
- Wu, S.; Wang, H.-Q.; Guo, T.-T.; Li, Y.-H. Luteolin Inhibits CVB3 Replication through Inhibiting Inflammation. J. Asian Nat. Prod. Res. 2020, 22, 762–773. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, G.; Jia, J.; Zhong, J.; Yan, R.; Lin, X.; Zheng, K.; Zhu, Q. In-Vitro Antiviral Activity of Doxepin Hydrochloride against Group B Coxsackievirus. Virus Res. 2022, 317, 198816. [Google Scholar] [CrossRef]
- Zuo, J.; Quinn, K.K.; Kye, S.; Cooper, P.; Damoiseaux, R.; Krogstad, P. Fluoxetine Is a Potent Inhibitor of Coxsackie virus Replication. Antimicrob. Agents Chemother. 2012, 56, 4838–4844. [Google Scholar] [CrossRef] [Green Version]
- Manganaro, R.; Zonsics, B.; Bauer, L.; Lorenzo Lopez, M.; Donselaar, T.; Zwaagstra, M.; Saporito, F.; Ferla, S.; Strating, J.R.P.M.; Coutard, B.; et al. Synthesis and Antiviral Effect of Novel Fluoxetine Analogues as Enterovirus 2C Inhibitors. Antivir. Res. 2020, 178, 104781. [Google Scholar] [CrossRef]
- Ulferts, R.; de Boer, S.M.; van der Linden, L.; Bauer, L.; Lyoo, H.R.; Maté, M.J.; Lichière, J.; Canard, B.; Lelieveld, D.; Omta, W.; et al. Screening of a Library of FDA-Approved Drugs Identifies Several Enterovirus Replication Inhibitors That Target Viral Protein 2C. Antimicrob. Agents Chemother. 2016, 60, 2627–2638. [Google Scholar] [CrossRef] [Green Version]
- Musharrafieh, R.; Kitamura, N.; Hu, Y.; Wang, J. Development of Broad-Spectrum Enterovirus Antivirals Based on Quinoline Scaffold. Bioorg. Chem. 2020, 101, 103981. [Google Scholar] [CrossRef]
- Zuo, J.; Kye, S.; Quinn, K.K.; Cooper, P.; Damoiseaux, R.; Krogstad, P. Discovery of Structurally Diverse Small-Molecule Compounds with Broad Antiviral Activity against Enteroviruses. Antimicrob. Agents Chemother. 2015, 60, 1615–1626. [Google Scholar] [CrossRef] [Green Version]
- Bauer, L.; Manganaro, R.; Zonsics, B.; Hurdiss, D.L.; Zwaagstra, M.; Donselaar, T.; Welter, N.G.E.; van Kleef, R.G.D.M.; Lopez, M.L.; Bevilacqua, F.; et al. Rational Design of Highly Potent Broad-Spectrum Enterovirus Inhibitors Targeting the Nonstructural Protein 2C. PLoS Biol. 2020, 18, e3000904. [Google Scholar] [CrossRef]
- Xing, Y.; Zuo, J.; Krogstad, P.; Jung, M.E. Synthesis and Structure-Activity Relationship (SAR) Studies of Novel Pyrazolopyridine Derivatives as Inhibitors of Enterovirus Replication. J. Med. Chem. 2018, 61, 1688–1703. [Google Scholar] [CrossRef]
- Hu, Y.; Kitamura, N.; Musharrafieh, R.; Wang, J. Discovery of Potent and Broad-Spectrum Pyrazolopyridine-Containing Antivirals against Enteroviruses D68, A71, and Coxsackievirus B3 by Targeting the Viral 2C Protein. J. Med. Chem. 2021, 64, 8755–8774. [Google Scholar] [CrossRef]
- Belvisi, L.; D’Andrea, L.D.; Jiménez, M.A. Editorial: Peptides Targeting Protein-Protein Interactions: Methods and Applications. Front. Mol. Biosci. 2021, 8, 780106. [Google Scholar] [CrossRef]
- Fang, Y.; Wang, C.; Wang, C.; Yang, R.; Bai, P.; Zhang, X.-Y.; Kong, J.; Yin, L.; Qiu, Y.; Zhou, X. Antiviral Peptides Targeting the Helicase Activity of Enterovirus Nonstructural Protein 2C. J. Virol. 2021, 95, e02324-20. [Google Scholar] [CrossRef]
- Strating, J.R.P.M.; van der Linden, L.; Albulescu, L.; Bigay, J.; Arita, M.; Delang, L.; Leyssen, P.; van der Schaar, H.M.; Lanke, K.H.W.; Thibaut, H.J.; et al. Itraconazole Inhibits Enterovirus Replication by Targeting the Oxysterol-Binding Protein. Cell. Rep. 2015, 10, 600–615. [Google Scholar] [CrossRef] [Green Version]
- Rattanakomol, P.; Srimanote, P.; Tongtawe, P.; Khantisitthiporn, O.; Supasorn, O.; Thanongsaksrikul, J. Host Neuronal PRSS3 Interacts with Enterovirus A71 3A Protein and Its Role in Viral Replication. Sci. Rep. 2022, 12, 12846. [Google Scholar] [CrossRef]
- Heinz, B.A.; Vance, L.M. The Antiviral Compound Enviroxime Targets the 3A Coding Region of Rhinovirus and Poliovirus. J. Virol. 1995, 69, 4189–4197. [Google Scholar] [CrossRef] [Green Version]
- Phillpotts, R.J.; Jones, R.W.; Delong, D.C.; Reed, S.E.; Wallace, J.; Tyrrell, D.A. The Activity of Enviroxime against Rhinovirus Infection in Man. Lancet 1981, 1, 1342–1344. [Google Scholar] [CrossRef]
- De Palma, A.M.; Thibaut, H.J.; van der Linden, L.; Lanke, K.; Heggermont, W.; Ireland, S.; Andrews, R.; Arimilli, M.; Al-Tel, T.H.; De Clercq, E.; et al. Mutations in the Nonstructural Protein 3A Confer Resistance to the Novel Enterovirus Replication Inhibitor TTP-8307. Antimicrob. Agents Chemother. 2009, 53, 1850–1857. [Google Scholar] [CrossRef] [Green Version]
- Albulescu, L.; Bigay, J.; Biswas, B.; Weber-Boyvat, M.; Dorobantu, C.M.; Delang, L.; van der Schaar, H.M.; Jung, Y.-S.; Neyts, J.; Olkkonen, V.M.; et al. Uncovering Oxysterol-Binding Protein (OSBP) as a Target of the Anti-Enteroviral Compound TTP-8307. Antivir. Res. 2017, 140, 37–44. [Google Scholar] [CrossRef]
- Gao, Q.; Yuan, S.; Zhang, C.; Wang, Y.; Wang, Y.; He, G.; Zhang, S.; Altmeyer, R.; Zou, G. Discovery of Itraconazole with Broad-Spectrum in Vitro Antienterovirus Activity That Targets Nonstructural Protein 3A. Antimicrob. Agents Chemother. 2015, 59, 2654–2665. [Google Scholar] [CrossRef] [Green Version]
- van der Linden, L.; Wolthers, K.C.; van Kuppeveld, F.J.M. Replication and Inhibitors of Enteroviruses and Parechoviruses. Viruses 2015, 7, 4529–4562. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.; Kim, C.; Kim, D.; Song, J.-H.; Choi, M.; Choi, K.; Kang, M.; Lee, K.; Kim, H.S.; Shin, J.S.; et al. Synergistic Antiviral Activity of Gemcitabine and Ribavirin against Enteroviruses. Antivir. Res. 2015, 124, 1–10. [Google Scholar] [CrossRef]
- Lee, K.; Kim, D.-E.; Jang, K.-S.; Kim, S.-J.; Cho, S.; Kim, C. Gemcitabine, a Broad-Spectrum Antiviral Drug, Suppresses Enterovirus Infections through Innate Immunity Induced by the Inhibition of Pyrimidine Biosynthesis and Nucleotide Depletion. Oncotarget 2017, 8, 115315–115325. [Google Scholar] [CrossRef] [PubMed]
- van der Linden, L.; Vives-Adrián, L.; Selisko, B.; Ferrer-Orta, C.; Liu, X.; Lanke, K.; Ulferts, R.; De Palma, A.M.; Tanchis, F.; Goris, N.; et al. The RNA Template Channel of the RNA-Dependent RNA Polymerase as a Target for Development of Antiviral Therapy of Multiple Genera within a Virus Family. PLoS Pathog. 2015, 11, e1004733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lind, K.; Svedin, E.; Domsgen, E.; Kapell, S.; Laitinen, O.H.; Moll, M.; Flodström-Tullberg, M. Coxsackievirus Counters the Host Innate Immune Response by Blocking Type III Interferon Expression. J. Gen. Virol. 2016, 97, 1368–1380. [Google Scholar] [CrossRef] [PubMed]
- Cheung, N.N.; Lai, K.K.; Dai, J.; Kok, K.H.; Chen, H.; Chan, K.-H.; Yuen, K.-Y.; Kao, R.Y.T. Broad-Spectrum Inhibition of Common Respiratory RNA Viruses by a Pyrimidine Synthesis Inhibitor with Involvement of the Host Antiviral Response. J. Gen. Virol. 2017, 98, 946–954. [Google Scholar] [CrossRef]
- Huan, C.; Qu, X.; Li, Z. Host Restrictive Factors Are the Emerging Storm Troopers Against Enterovirus: A Mini-Review. Front. Immunol. 2022, 13, 910780. [Google Scholar] [CrossRef]
- Ji, X.; Li, Z. Medicinal Chemistry Strategies toward Host Targeting Antiviral Agents. Med. Res. Rev. 2020, 40, 1519–1557. [Google Scholar] [CrossRef]
- Owino, C.O.; Chu, J.J.H. Recent Advances on the Role of Host Factors during Non-Poliovirus Enteroviral Infections. J. Biomed. Sci. 2019, 26, 47. [Google Scholar] [CrossRef] [Green Version]
- Peischard, S.; Ho, H.T.; Theiss, C.; Strutz-Seebohm, N.; Seebohm, G. A Kidnapping Story: How Coxsackievirus B3 and Its Host Cell Interact. Cell Physiol. Biochem. 2019, 53, 121–140. [Google Scholar] [CrossRef]
- Duyvesteyn, H.M.E.; Ren, J.; Walter, T.S.; Fry, E.E.; Stuart, D.I. Glutathione Facilitates Enterovirus Assembly by Binding at a Druggable Pocket. Commun. Biol. 2020, 3, 9. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.-C.; Liu, Y.; Wang, C.; Strauss, M.; Rehage, N.; Chen, Y.-H.; Altan-Bonnet, N.; Hogle, J.; Wimmer, E.; Mueller, S.; et al. An Interaction between Glutathione and the Capsid Is Required for the Morphogenesis of C-Cluster Enteroviruses. PLoS Pathog. 2014, 10, e1004052. [Google Scholar] [CrossRef]
- Moghimi, S.; Viktorova, E.; Zimina, A.; Szul, T.; Sztul, E.; Belov, G.A. Enterovirus Infection Induces Massive Recruitment of All Isoforms of Small Cellular Arf GTPases to the Replication Organelles. J. Virol. 2020, 95, e01629-20. [Google Scholar] [CrossRef] [PubMed]
- Thibaut, H.J.; van der Linden, L.; Jiang, P.; Thys, B.; Canela, M.-D.; Aguado, L.; Rombaut, B.; Wimmer, E.; Paul, A.; Pérez-Pérez, M.-J.; et al. Binding of Glutathione to Enterovirus Capsids Is Essential for Virion Morphogenesis. PLoS Pathog. 2014, 10, e1004039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seizer, P.; Klingel, K.; Sauter, M.; Westermann, D.; Ochmann, C.; Schönberger, T.; Schleicher, R.; Stellos, K.; Schmidt, E.-M.; Borst, O.; et al. Cyclophilin A Affects Inflammation, Virus Elimination and Myocardial Fibrosis in Coxsackievirus B3-Induced Myocarditis. J. Mol Cell Cardiol. 2012, 53, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Geller, R.; Vignuzzi, M.; Andino, R.; Frydman, J. Evolutionary Constraints on Chaperone-Mediated Folding Provide an Antiviral Approach Refractory to Development of Drug Resistance. Genes Dev. 2007, 21, 195–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanke, K.H.W.; van der Schaar, H.M.; Belov, G.A.; Feng, Q.; Duijsings, D.; Jackson, C.L.; Ehrenfeld, E.; van Kuppeveld, F.J.M. GBF1, a Guanine Nucleotide Exchange Factor for Arf, Is Crucial for Coxsackievirus B3 RNA Replication. J. Virol. 2009, 83, 11940–11949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Linden, L.; van der Schaar, H.M.; Lanke, K.H.W.; Neyts, J.; van Kuppeveld, F.J.M. Differential Effects of the Putative GBF1 Inhibitors Golgicide A and AG1478 on Enterovirus Replication. J. Virol. 2010, 84, 7535–7542. [Google Scholar] [CrossRef] [Green Version]
- Ferlin, J.; Farhat, R.; Belouzard, S.; Cocquerel, L.; Bertin, A.; Hober, D.; Dubuisson, J.; Rouillé, Y. Investigation of the Role of GBF1 in the Replication of Positive-Sense Single-Stranded RNA Viruses. J. Gen. Virol. 2018, 99, 1086–1096. [Google Scholar] [CrossRef]
- Martínez, J.L.; Arias, C.F. Role of the Guanine Nucleotide Exchange Factor GBF1 in the Replication of RNA Viruses. Viruses 2020, 12, 682. [Google Scholar] [CrossRef]
- Viktorova, E.G.; Nchoutmboube, J.A.; Ford-Siltz, L.A.; Iverson, E.; Belov, G.A. Phospholipid Synthesis Fueled by Lipid Droplets Drives the Structural Development of Poliovirus Replication Organelles. PLoS Pathog. 2018, 14, e1007280. [Google Scholar] [CrossRef] [Green Version]
- Quaranta, P.; Lottini, G.; Chesi, G.; Contrafatto, F.; Russotto, R.; Macera, L.; Lai, M.; Spezia, P.G.; Brai, A.; Botta, M.; et al. DDX3 Inhibitors Show Antiviral Activity against Positive-Sense Single-Stranded RNA Viruses but Not against Negative-Sense Single-Stranded RNA Viruses: The Coxsackie B Model. Antivir. Res. 2020, 178, 104750. [Google Scholar] [CrossRef]
- Laufman, O.; Perrino, J.; Andino, R. Viral Generated Inter-Organelle Contacts Redirect Lipid Flux for Genome Replication. Cell 2019, 178, 275–289. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, M.; Cheng, A.; Wen, X.; Ou, X.; Mao, S.; Gao, Q.; Sun, D.; Jia, R.; Yang, Q.; et al. Enterovirus Replication Organelles and Inhibitors of Their Formation. Front. Microbiol. 2020, 11, 1817. [Google Scholar] [CrossRef] [PubMed]
- van der Schaar, H.M.; van der Linden, L.; Lanke, K.H.W.; Strating, J.R.P.M.; Pürstinger, G.; de Vries, E.; de Haan, C.A.M.; Neyts, J.; van Kuppeveld, F.J.M. Coxsackievirus Mutants That Can Bypass Host Factor PI4KIIIβ and the Need for High Levels of PI4P Lipids for Replication. Cell Res. 2012, 22, 1576–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arita, M.; Kojima, H.; Nagano, T.; Okabe, T.; Wakita, T.; Shimizu, H. Phosphatidylinositol 4-Kinase III Beta Is a Target of Enviroxime-like Compounds for Antipoliovirus Activity. J. Virol. 2011, 85, 2364–2372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arita, M.; Philipov, S.; Galabov, A.S. Phosphatidylinositol 4-Kinase III Beta Is the Target of Oxoglaucine and Pachypodol (Ro 09-0179) for Their Anti-Poliovirus Activities, and Is Located at Upstream of the Target Step of Brefeldin A. Microbiol. Immunol. 2015, 59, 338–347. [Google Scholar] [CrossRef]
- Šála, M.; Kögler, M.; Plačková, P.; Mejdrová, I.; Hřebabecký, H.; Procházková, E.; Strunin, D.; Lee, G.; Birkus, G.; Weber, J.; et al. Purine Analogs as Phosphatidylinositol 4-Kinase IIIβ Inhibitors. Bioorg. Med. Chem Lett. 2016, 26, 2706–2712. [Google Scholar] [CrossRef]
- Mejdrová, I.; Chalupská, D.; Plačková, P.; Müller, C.; Šála, M.; Klíma, M.; Baumlová, A.; Hřebabecký, H.; Procházková, E.; Dejmek, M.; et al. Rational Design of Novel Highly Potent and Selective Phosphatidylinositol 4-Kinase IIIβ (PI4KB) Inhibitors as Broad-Spectrum Antiviral Agents and Tools for Chemical Biology. J. Med. Chem. 2017, 60, 100–118. [Google Scholar] [CrossRef]
- Arita, M.; Dobrikov, G.; Pürstinger, G.; Galabov, A.S. Allosteric Regulation of Phosphatidylinositol 4-Kinase III Beta by an Antipicornavirus Compound MDL-860. ACS Infect. Dis. 2017, 3, 585–594. [Google Scholar] [CrossRef]
- Dobrikov, G.M.; Slavchev, I.; Nikolova, I.; Stoyanova, A.; Nikolova, N.; Mukova, L.; Nikolova, R.; Shivachev, B.; Galabov, A.S. Synthesis and Anti-Enterovirus Activity of New Analogues of MDL-860. Bioorg. Med. Chem. Lett. 2017, 27, 4540–4543. [Google Scholar] [CrossRef]
- Nikolova, I.; Slavchev, I.; Ravutsov, M.; Dangalov, M.; Nikolova, Y.; Zagranyarska, I.; Stoyanova, A.; Nikolova, N.; Mukova, L.; Grozdanov, P.; et al. Anti-Enteroviral Activity of New MDL-860 Analogues: Synthesis, in Vitro/in Vivo Studies and QSAR Analysis. Bioorg. Chem. 2019, 85, 487–497. [Google Scholar] [CrossRef]
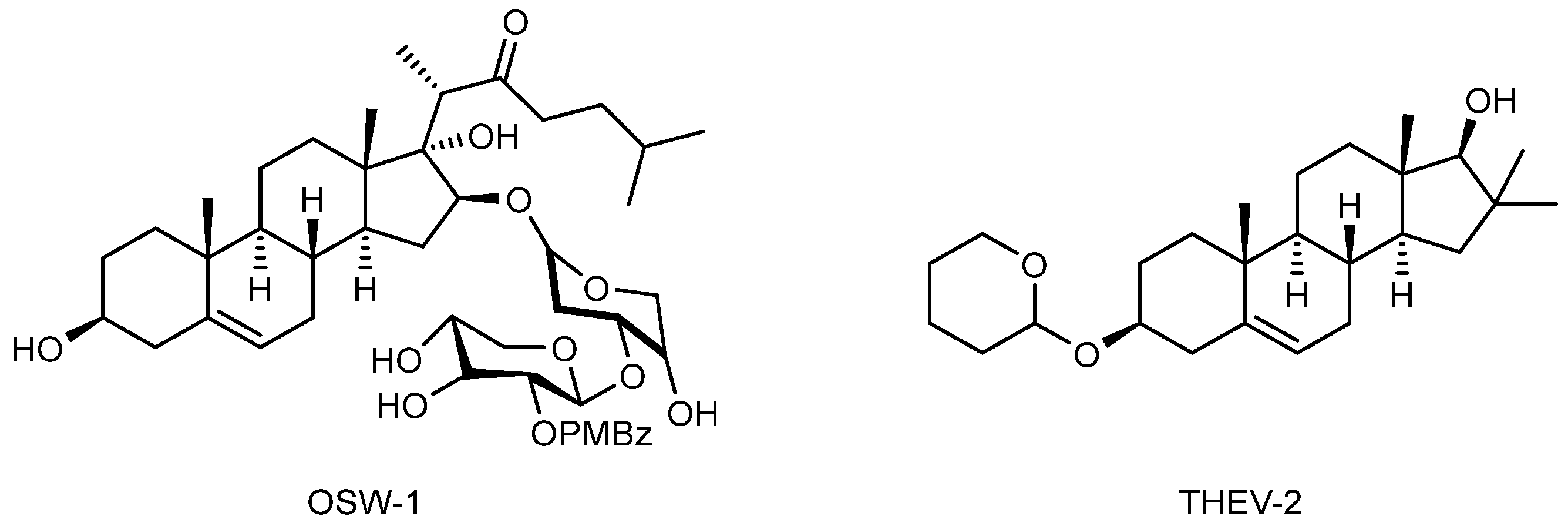
- Albulescu, L.; Strating, J.R.P.M.; Thibaut, H.J.; van der Linden, L.; Shair, M.D.; Neyts, J.; van Kuppeveld, F.J.M. Broad-Range Inhibition of Enterovirus Replication by OSW-1, a Natural Compound Targeting OSBP. Antivir. Res. 2015, 117, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Arita, M.; Kojima, H.; Nagano, T.; Okabe, T.; Wakita, T.; Shimizu, H. Oxysterol-Binding Protein Family I Is the Target of Minor Enviroxime-like Compounds. J. Virol. 2013, 87, 4252–4260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, B.L.; Severance, Z.C.; Bensen, R.C.; Le-McClain, A.T.; Malinky, C.A.; Mettenbrink, E.M.; Nuñez, J.I.; Reddig, W.J.; Blewett, E.L.; Burgett, A.W.G. Differing Activities of Oxysterol-Binding Protein (OSBP) Targeting Anti-Viral Compounds. Antivir. Res. 2019, 170, 104548. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.L.; Severance, Z.C.; Bensen, R.C.; Le, A.T.; Kothapalli, N.R.; Nuñez, J.I.; Ma, H.; Wu, S.; Standke, S.J.; Yang, Z.; et al. Transient Compound Treatment Induces a Multigenerational Reduction of Oxysterol-Binding Protein (OSBP) Levels and Prophylactic Antiviral Activity. ACS Chem. Biol. 2019, 14, 276–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-S.; Choi, H.-J.; Song, J.-H.; Ko, H.-J.; Yoon, K.; Seong, J.-M. Antiviral Activity of Itraconazole against Echovirus 30 Infection In Vitro. Osong. Public Health Res. Perspect. 2017, 8, 318–324. [Google Scholar] [CrossRef]
- Policastro, L.R.; Dolci, I.; Godoy, A.S.; Silva Júnior, J.V.J.; Ruiz, U.E.A.; Santos, I.A.; Jardim, A.C.G.; Samby, K.; Burrows, J.N.; Wells, T.N.C.; et al. The Antifungal Itraconazole Is a Potent Inhibitor of Chikungunya Virus Replication. Viruses 2022, 14, 1351. [Google Scholar] [CrossRef]
- Van Damme, E.; De Meyer, S.; Bojkova, D.; Ciesek, S.; Cinatl, J.; De Jonghe, S.; Jochmans, D.; Leyssen, P.; Buyck, C.; Neyts, J.; et al. In Vitro Activity of Itraconazole against SARS-CoV-2. J. Med. Virol. 2021, 93, 4454–4460. [Google Scholar] [CrossRef]
- Bauer, L.; Ferla, S.; Head, S.A.; Bhat, S.; Pasunooti, K.K.; Shi, W.Q.; Albulescu, L.; Liu, J.O.; Brancale, A.; van Kuppeveld, F.J.M.; et al. Structure-Activity Relationship Study of Itraconazole, a Broad-Range Inhibitor of Picornavirus Replication That Targets Oxysterol-Binding Protein (OSBP). Antivir. Res. 2018, 156, 55–63. [Google Scholar] [CrossRef]
- Wang, J.; Hu, Y.; Zheng, M. Enterovirus A71 Antivirals: Past, Present, and Future. Acta Pharm. Sin. B 2022, 12, 1542–1566. [Google Scholar] [CrossRef]


















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tammaro, C.; Guida, M.; Appetecchia, F.; Biava, M.; Consalvi, S.; Poce, G. Direct-Acting Antivirals and Host-Targeting Approaches against Enterovirus B Infections: Recent Advances. Pharmaceuticals 2023, 16, 203. https://doi.org/10.3390/ph16020203
Tammaro C, Guida M, Appetecchia F, Biava M, Consalvi S, Poce G. Direct-Acting Antivirals and Host-Targeting Approaches against Enterovirus B Infections: Recent Advances. Pharmaceuticals. 2023; 16(2):203. https://doi.org/10.3390/ph16020203
Chicago/Turabian StyleTammaro, Chiara, Michela Guida, Federico Appetecchia, Mariangela Biava, Sara Consalvi, and Giovanna Poce. 2023. "Direct-Acting Antivirals and Host-Targeting Approaches against Enterovirus B Infections: Recent Advances" Pharmaceuticals 16, no. 2: 203. https://doi.org/10.3390/ph16020203
APA StyleTammaro, C., Guida, M., Appetecchia, F., Biava, M., Consalvi, S., & Poce, G. (2023). Direct-Acting Antivirals and Host-Targeting Approaches against Enterovirus B Infections: Recent Advances. Pharmaceuticals, 16(2), 203. https://doi.org/10.3390/ph16020203