
Targeting the IGF-Axis in Cultured Pediatric High-Grade Glioma Cells Inhibits Cell Cycle Progression and Survival
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
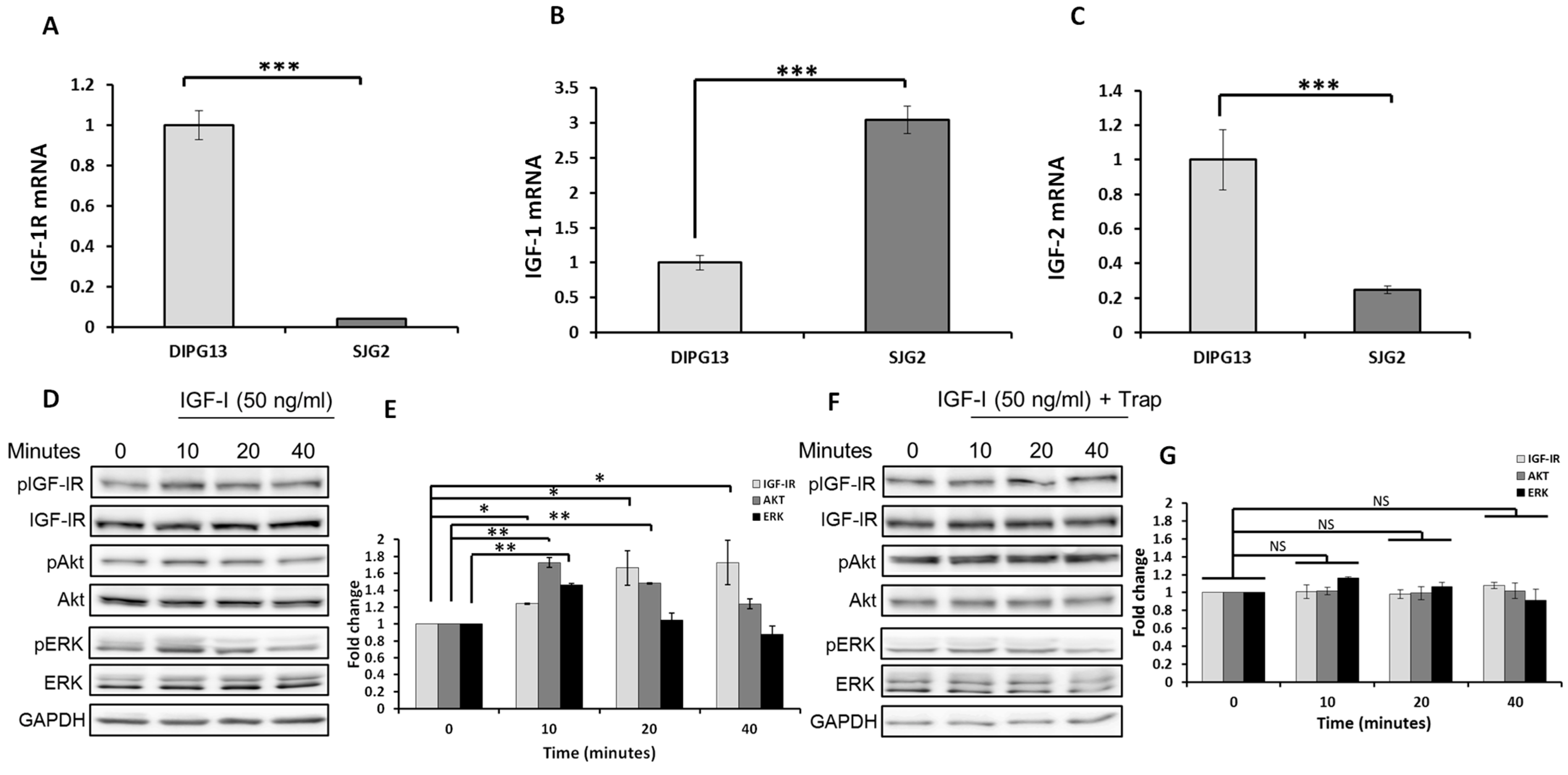
2.1. The IGF-1R Is Expressed in the pHGG Cells Selected, and Its Signaling Can Be Blocked by the IGF-Trap
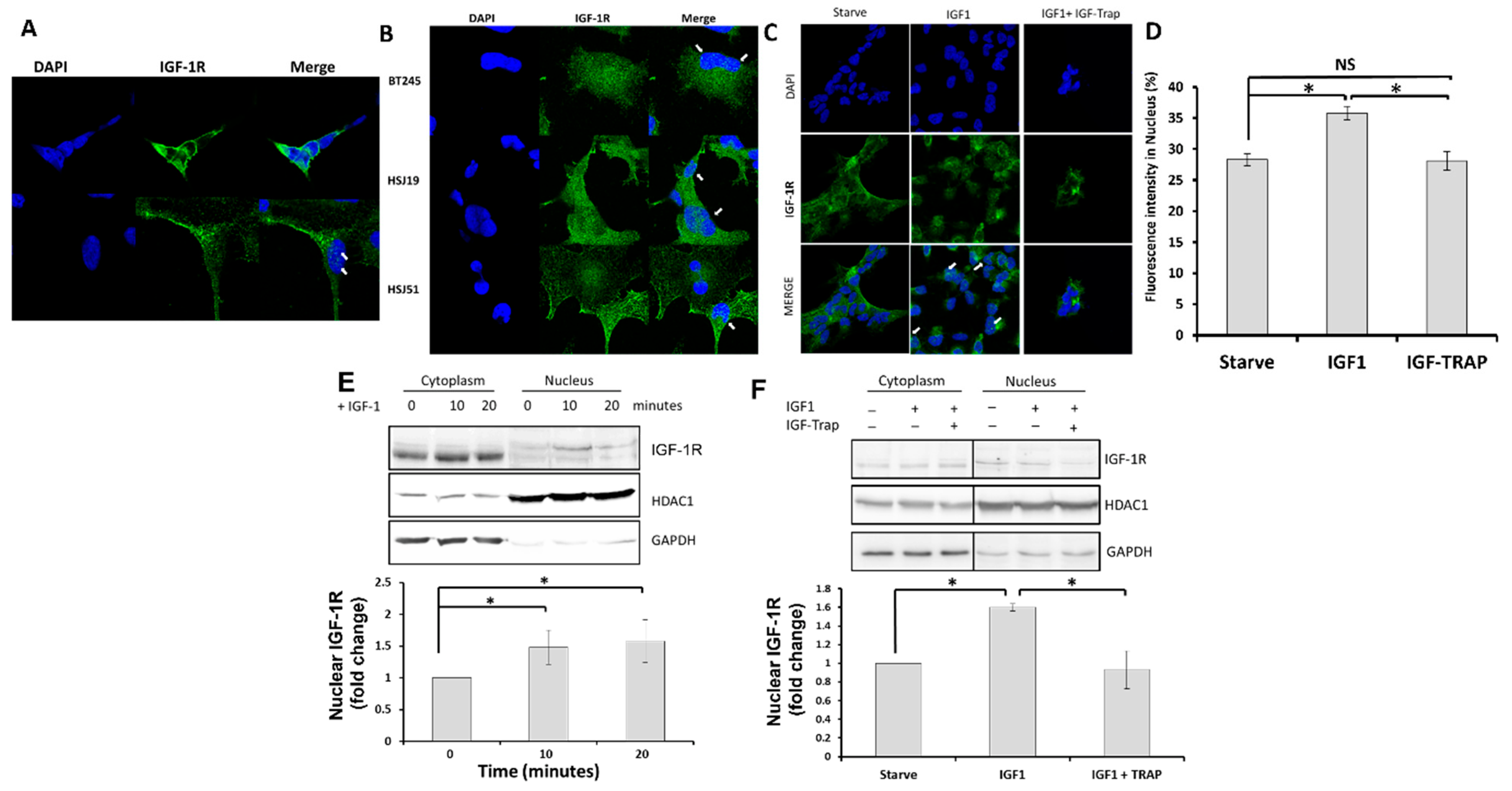
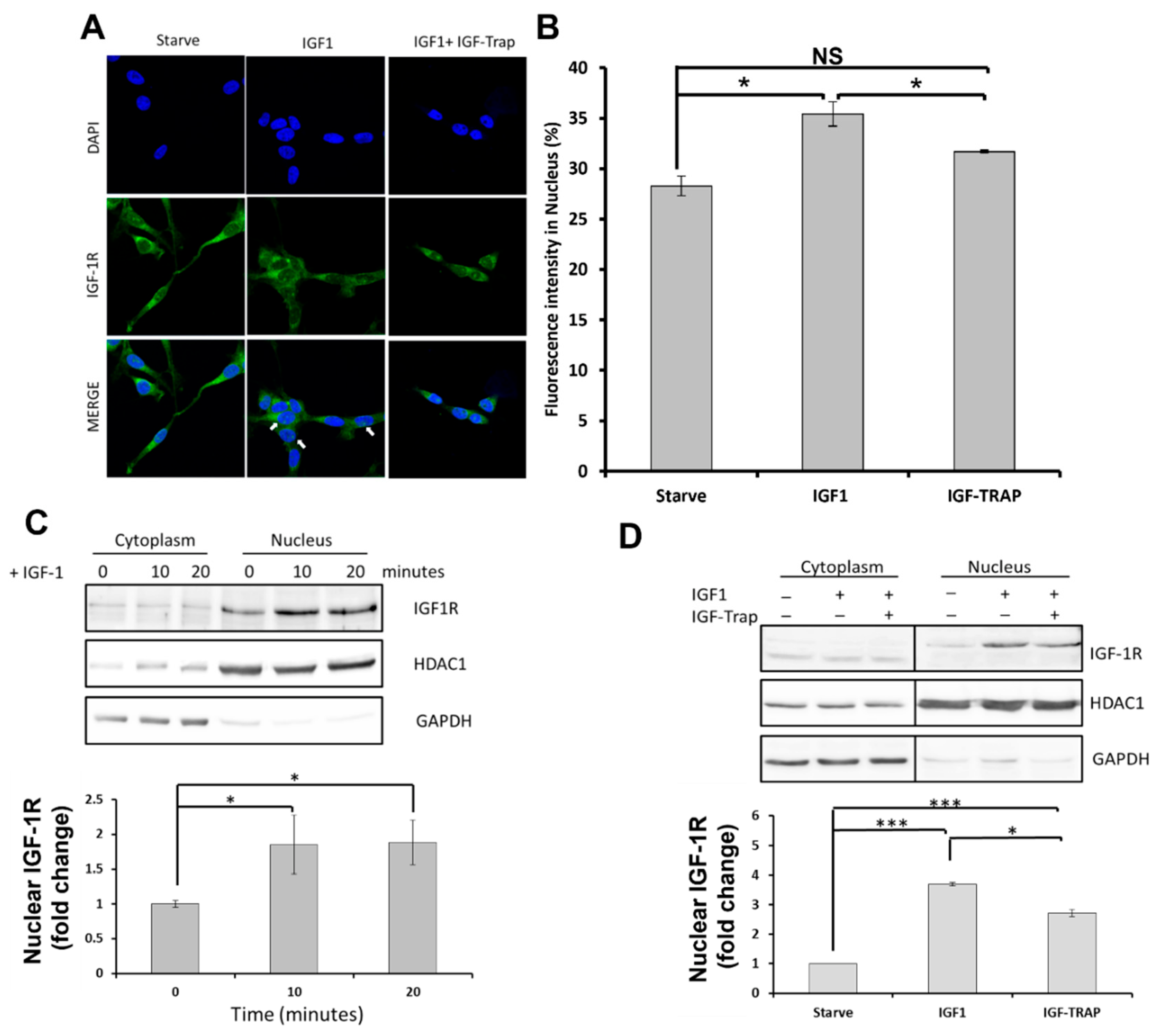
2.2. In Response to Ligand Binding, the IGF-1R Is also Transported to the Nucleus in pHGG Cells
Ligand Binding Increases Nuclear Levels of IGF-1R, and This Is Blocked by the IGF-Trap
2.3. Nuclear IGF-1R Activates Gene Transcription in DIPG13 Cells
Increased Cyclin D1 and IGF-1R Expression Is ERK-Activation Independent
2.4. Increased Cyclin D1 Expression in Response to IGF-1-Induced Nuclear Transport Is Cell Specific
2.5. IGF-1R Signaling Promotes Survival and Proliferation in Pediatric High-Grade Glioma Cells
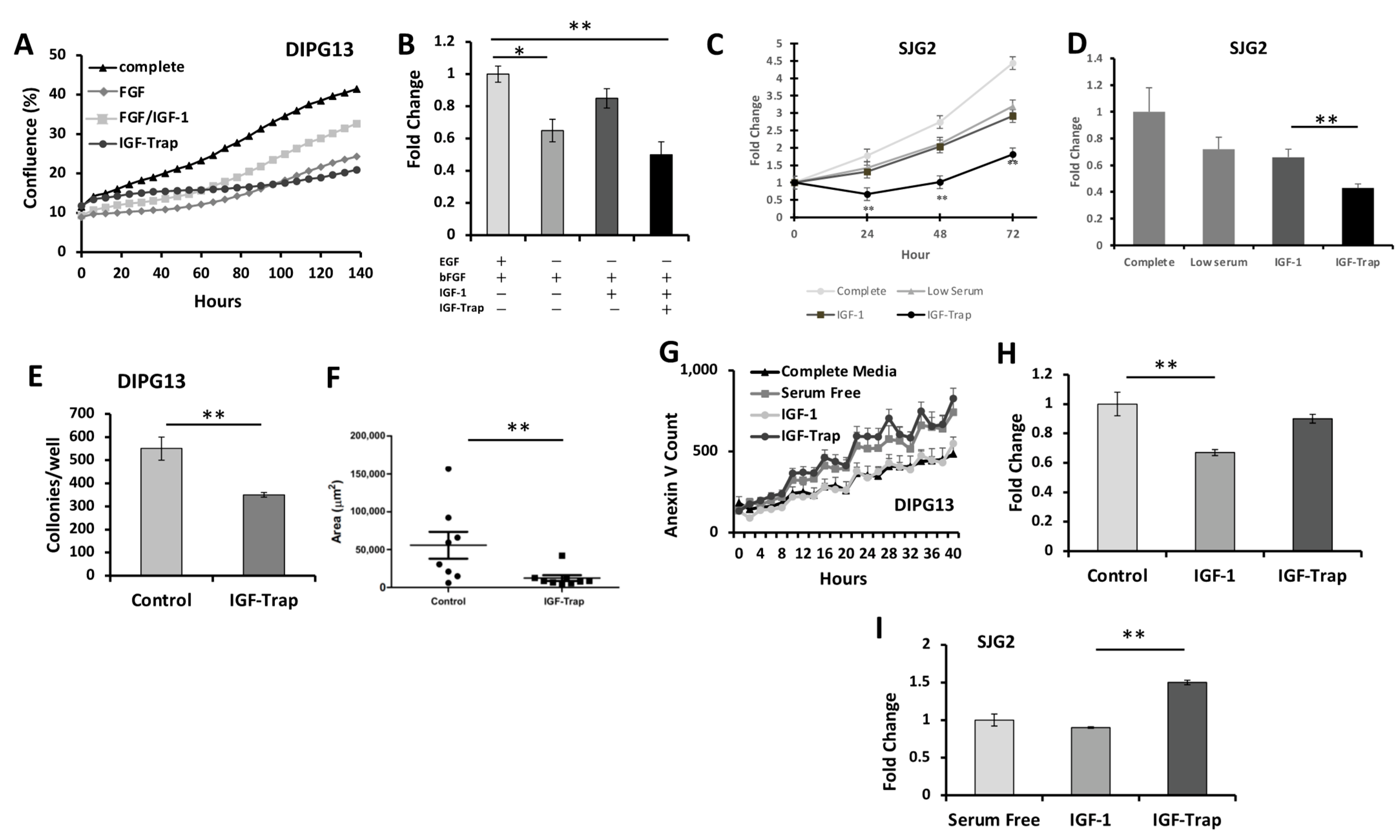
2.5.1. Increased Proliferation of pHGG in the Presence of IGF-1 Is Blocked by the IGF-Trap
2.5.2. IGF-Trap Can Reduce Colony Formation by DIPG13 Cells
2.5.3. The IGF-Trap Increases pHGG Apoptosis
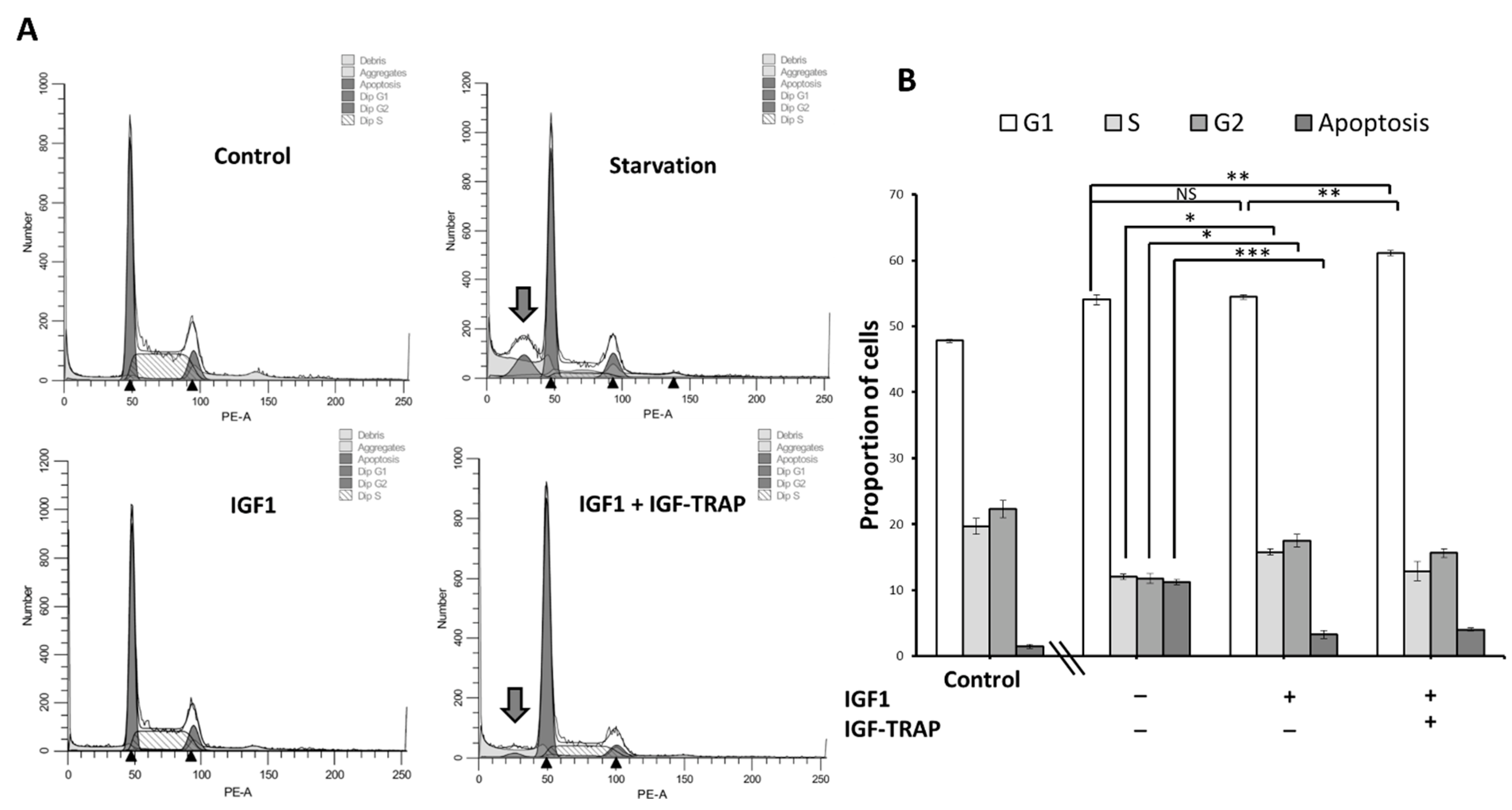
2.6. IGF-1 Increases Cell Cycle Progression in DIPG13 Cells, and This Is Blocked by the IGF-Trap
3. Discussion
4. Materials and Methods
5. Conclusions and Significance
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Cacciotti, C.; Fleming, A.; Ramaswamy, V. Advances in the molecular classification of pediatric brain tumors: A guide to the galaxy. J. Pathol. 2020, 251, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Chamdine, O.; Gajjar, A. Molecular characteristics of pediatric high-grade gliomas. CNS Oncol. 2014, 3, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Glod, J.; Rahme, G.; Kaur, H.; Raabe, E.H.; Hwang, E.I.; Israel, M.A. Pediatric brain tumors: Current knowledge and therapeutic opportunities. J. Pediatr. Hematol. 2016, 38, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Clément, F.; Martin, A.; Venara, M.; de Luján Calcagno, M.; Mathó, C.; Maglio, S.; Lombardi, M.G.; Bergadá, I.; Pennisi, P.A. Type 1 IGF receptor localization in paediatric gliomas: Significant association with WHO grading and clinical outcome. Horm. Cancer 2018, 9, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Wrigley, S.; Arafa, D.; Tropea, D. Insulin-like growth factor 1: At the crossroads of brain development and aging. Front. Cell. Neurosci. 2017, 11, 14. [Google Scholar] [CrossRef] [PubMed]
- D’Ercole, A.J.; Ye, P.; Calikoglu, A.S.; Gutierrez-Ospina, G. The role of the insulin-like growth factors in the central nervous system. Mol. Neurobiol. 1996, 13, 227–255. [Google Scholar] [CrossRef]
- Sehat, B.; Tofigh, A.; Lin, Y.; Trocmé, E.; Liljedahl, U.; Lagergren, J.; Larsson, O. SUMOylation mediates the nuclear translocation and signaling of the IGF-1 receptor. Sci. Signal. 2010, 3, ra10. [Google Scholar] [CrossRef]
- Warsito, D.; Sjöström, S.; Andersson, S.; Larsson, O.; Sehat, B. Nuclear IGF1R is a transcriptional co-activator of LEF1/TCF. EMBO Rep. 2012, 13, 244–250. [Google Scholar] [CrossRef]
- Wang, N.; Rayes, R.F.; Elahi, S.M.; Lu, Y.; Hancock, M.A.; Massie, B.; Rowe, G.E.; Aomari, H.; Hossain, S.; Durocher, Y.; et al. The IGF-Trap: Novel inhibitor of carcinoma growth and metastasis. Mol. Cancer Ther. 2015, 14, 982–993. [Google Scholar] [CrossRef]
- Vaniotis, G.; Moffett, S.; Sulea, T.; Wang, N.; Elahi, S.M.; Lessard, E.; Baardsnes, J.; Perrino, S.; Durocher, Y.; Frystyk, J.; et al. Enhanced anti-metastatic bioactivity of an IGF-TRAP re-engineered to improve physicochemical properties. Sci. Rep. 2018, 8, 17361. [Google Scholar] [CrossRef] [Green Version]
- Tsui, J.; Qi, S.; Perrino, S.; Leibovitch, M.; Brodt, P. Identification of a resistance mechanism to IGF-IR targeting in human triple negative MDA-MB-231 breast cancer cells. Biomolecules 2021, 11, 527. [Google Scholar] [CrossRef] [PubMed]
- Samani, A.A.; Nalbantoglu, J.; Brodt, P. Glioma cells with genetically engineered IGF-I receptor downregulation can persist in the brain in a dormant state. Front. Oncol. 2020, 10, 555945. [Google Scholar] [CrossRef] [PubMed]
- Samani, A.A.; Yakar, S.; LeRoith, D.; Brodt, P. The role of the IGF system in cancer growth and metastasis: Overview and recent insights. Endocr. Rev. 2007, 28, 20–47. [Google Scholar] [CrossRef] [PubMed]
- Simpson, A.D.; Soo, Y.W.J.; Rieunier, G.; Aleksic, T.; Ansorge, O.; Jones, C.; Macaulay, V.M. Type 1 IGF receptor associates with adverse outcome and cellular radioresistance in paediatric high-grade glioma. Br. J. Cancer 2019, 122, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Krug, B.; De Jay, N.; Harutyunyan, A.S.; Deshmukh, S.; Marchione, D.M.; Guilhamon, P.; Bertrand, K.C.; Mikael, L.G.; McConechy, M.K.; Chen, C.C.; et al. Pervasive H3K27 acetylation leads to ERV expression and a therapeutic vulnerability in H3K27M gliomas. Cancer Cell 2019, 35, 782–797.e8. [Google Scholar] [CrossRef]
- Harutyunyan, A.S.; Krug, B.; Chen, H.; Papillon-Cavanagh, S.; Zeinieh, M.; De Jay, N.; Deshmukh, S.; Chen, C.C.; Belle, J.; Mikael, L.G.; et al. H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nat. Commun. 2019, 10, 1262. [Google Scholar] [CrossRef] [PubMed]
- Mackay, A.; Burford, A.; Carvalho, D.; Izquierdo, E.; Fazal-Salom, J.; Taylor, K.R.; Bjerke, L.; Clarke, M.; Vinci, M.; Nandhabalan, M.; et al. Integrated molecular meta-analysis of 1000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell 2017, 32, 520–537.e5. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Bender, S.; Gronych, J.; Warnatz, H.-J.; Hutter, B.; Gröbner, S.; Ryzhova, M.; Pfaff, E.; Hovestadt, V.; Weinberg, F.; International Cancer Genome Consortium Pedbrain Tumor Project; et al. Recurrent MET fusion genes represent a drug target in pediatric glioblastoma. Nat. Med. 2016, 22, 1314–1320. [Google Scholar] [CrossRef]
- Joo, K.M.; Jin, J.; Kim, E.; Kim, K.H.; Kim, Y.; Kang, B.G.; Kang, Y.-J.; Lathia, J.D.; Cheong, K.H.; Song, P.H.; et al. MET signaling regulates glioblastoma stem cells. Cancer Res. 2012, 72, 3828–3838. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Li, A.; Glas, M.; Lal, B.; Ying, M.; Sang, Y.; Xia, S.; Trageser, D.; Guerrero-Cázares, H.; Eberhart, C.G.; et al. c-Met signaling induces a reprogramming network and supports the glioblastoma stem-like phenotype. Proc. Natl. Acad. Sci. USA 2011, 108, 9951–9956. [Google Scholar] [CrossRef]
- Saunders, V.C.; Lafitte, M.; Adrados, I.; Quereda, V.; Feurstein, D.; Ling, Y.; Fallahi, M.; Rosenberg, L.H.; Duckett, D.R. Identification of an EGFRvIII-JNK2-HGF/c-Met-Signaling axis required for intercellular crosstalk and glioblastoma multiforme cell invasion. Mol. Pharmacol. 2015, 88, 962–969. [Google Scholar] [CrossRef] [PubMed]
- Daniel, J.A.; Cooper, B.H.; Palvimo, J.J.; Zhang, F.P.; Brose, N.; Tirard, M. Analysis of SUMO1-conjugation at synapses. eLife 2017, 6, e26338. [Google Scholar] [CrossRef] [PubMed]
- Sarfstein, R.; Werner, H. Minireview: Nuclear insulin and insulin-like growth factor-1 receptors: A novel paradigm in signal transduction. Endocrinology 2013, 154, 1672–1679. [Google Scholar] [CrossRef] [PubMed]
- Brodt, P.; Fallavollita, L.; Khatib, A.M.; Samani, A.A.; Zhang, D. Cooperative regulation of the invasive and metastatic phenotypes by different domains of the type I insulin-like growth factor receptor beta subunit. J. Biol. Chem. 2001, 276, 33608–33615. [Google Scholar] [CrossRef]
- Aziz-Bose, R.; Monje, M. Diffuse intrinsic pontine glioma: Molecular landscape and emerging therapeutic targets. Curr. Opin. Oncol. 2019, 31, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Hoeman, C.; Shen, C.; Becher, O.J. CDK4/6 and PDGFRA signaling as therapeutic targets in diffuse intrinsic pontine glioma. Front. Oncol. 2018, 8, 191. [Google Scholar] [CrossRef]
- Bielen, A.; Perryman, L.; Box, G.M.; Valenti, M.; de Haven Brandon, A.; Martins, V.; Jury, A.; Popov, S.; Gowan, S.; Jeay, S.; et al. Enhanced efficacy of IGF1R inhibition in pediatric glioblastoma by combinatorial targeting of PDGFRalpha/beta. Mol. Cancer. Ther. 2011, 10, 1407–1418. [Google Scholar] [CrossRef] [PubMed]
- Ajeawung, N.F.; Wang, H.Y.; Gould, P.; Kamnasaran, D. Advances in molecular targets for the treatment of medulloblastomas. Clin. Investig. Med. 2012, 35, E246–E259. [Google Scholar] [CrossRef]
- Reiss, K. Insulin-like growth factor-I receptor—A potential therapeutic target in medulloblastomas. Expert Opin. Ther. Targets 2002, 6, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Burnier, J.V.; Wang, N.; Michel, R.P.; Hassanain, M.; Li, S.; Lu, Y.; Metrakos, P.; Antecka, E.; Burnier, M.N.; Ponton, A.; et al. Type IV collagen-initiated signals provide survival and growth cues required for liver metastasis. Oncogene 2011, 30, 3766–3783. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Brodt, P. Type 1 insulin-like growth factor regulates MT1-MMP synthesis and tumor invasion via PI 3-kinase/Akt signaling. Oncogene 2003, 22, 974–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.M.; Leibovitch, M.; Zeinieh, M.; Jabado, N.; Brodt, P. Targeting the IGF-Axis in Cultured Pediatric High-Grade Glioma Cells Inhibits Cell Cycle Progression and Survival. Pharmaceuticals 2023, 16, 297. https://doi.org/10.3390/ph16020297
Chen YM, Leibovitch M, Zeinieh M, Jabado N, Brodt P. Targeting the IGF-Axis in Cultured Pediatric High-Grade Glioma Cells Inhibits Cell Cycle Progression and Survival. Pharmaceuticals. 2023; 16(2):297. https://doi.org/10.3390/ph16020297
Chicago/Turabian StyleChen, Yinhsuan Michely, Matthew Leibovitch, Michele Zeinieh, Nada Jabado, and Pnina Brodt. 2023. "Targeting the IGF-Axis in Cultured Pediatric High-Grade Glioma Cells Inhibits Cell Cycle Progression and Survival" Pharmaceuticals 16, no. 2: 297. https://doi.org/10.3390/ph16020297