
Anti-Inflammatory, Antioxidant, and WAT/BAT-Conversion Stimulation Induced by Novel PPAR Ligands: Results from Ex Vivo and In Vitro Studies


 ,
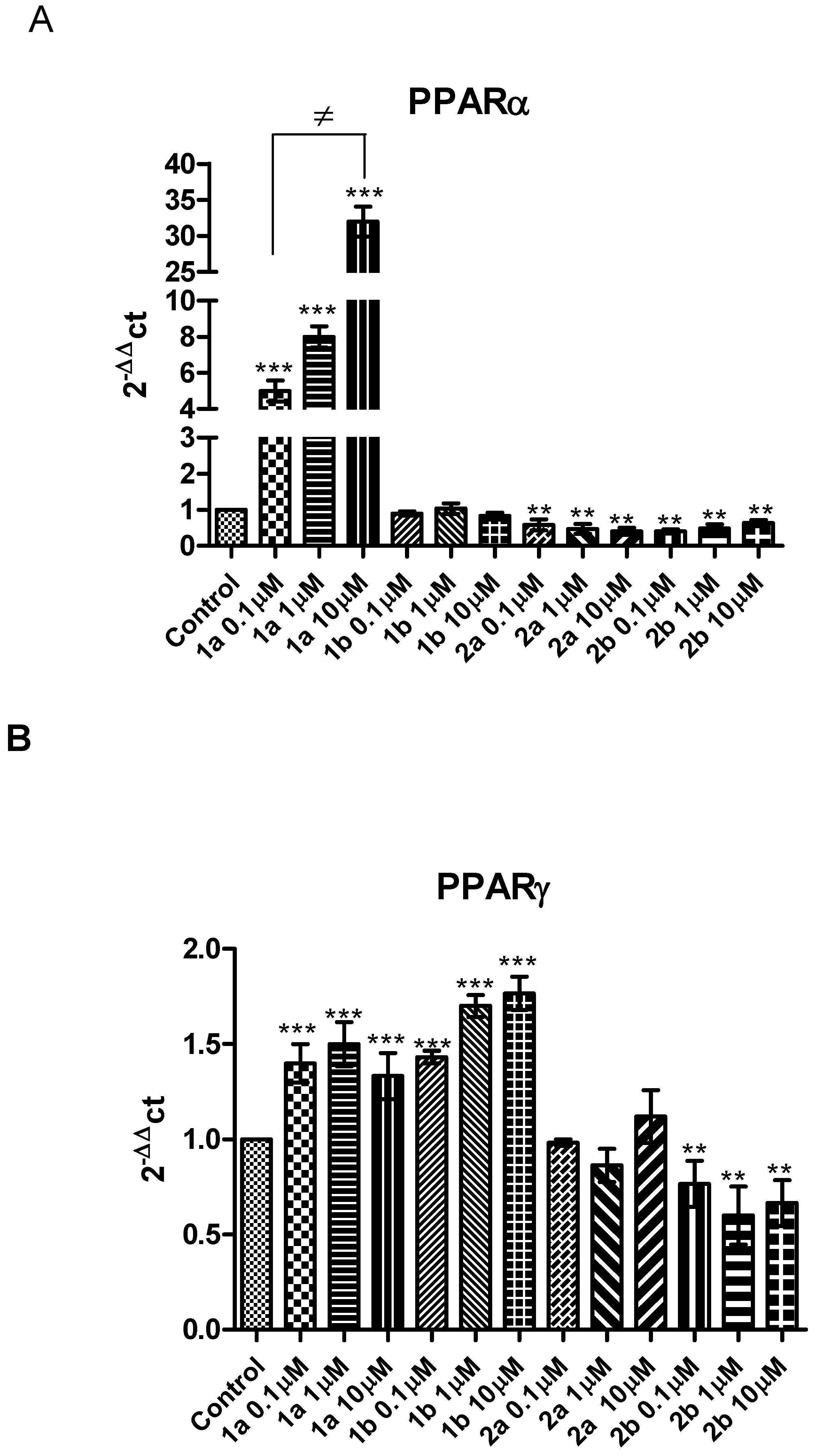
,  ,
,  ,
,  ,
, 
 ,
,  ,
,  ,
,  ,
, 
 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
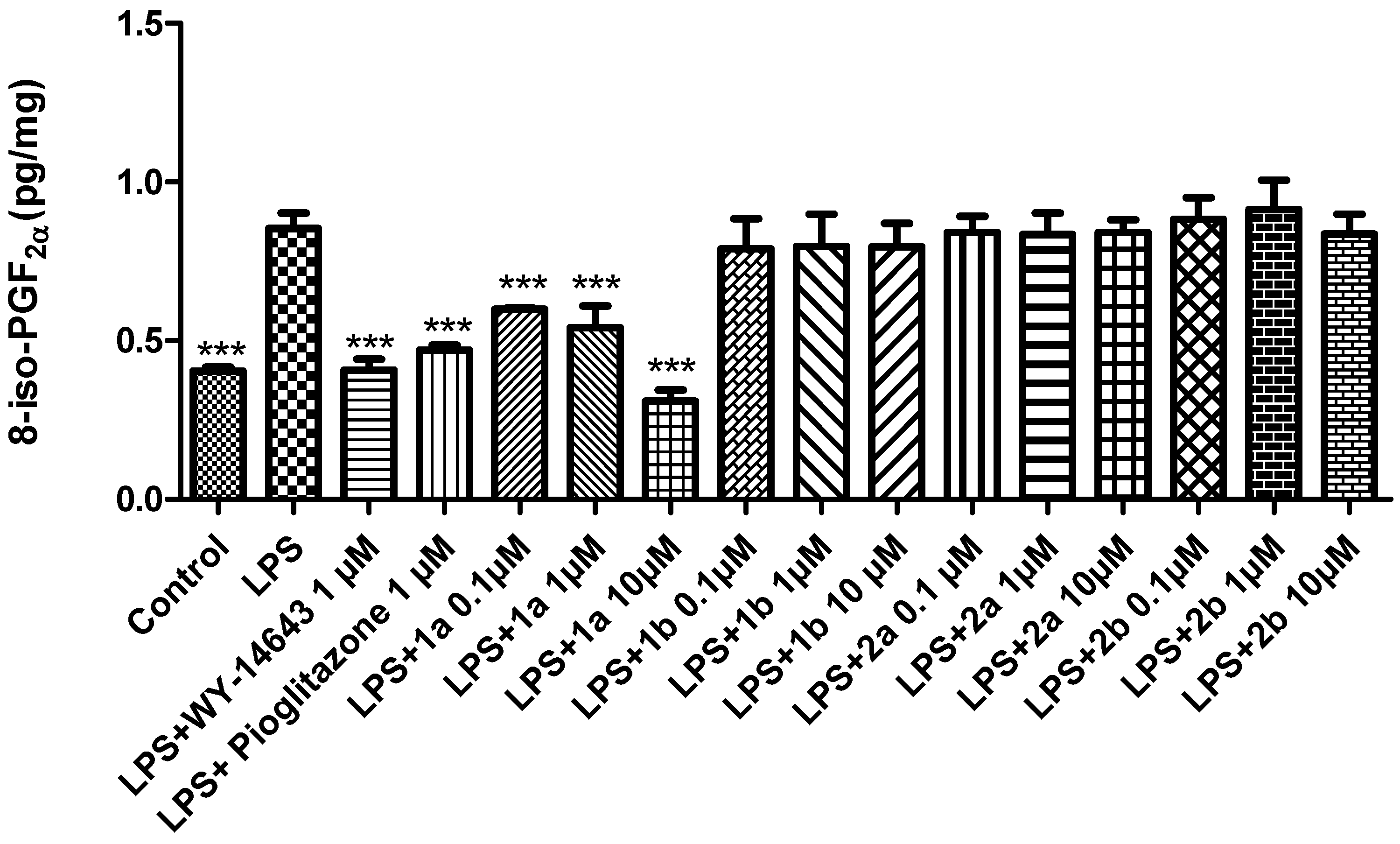{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
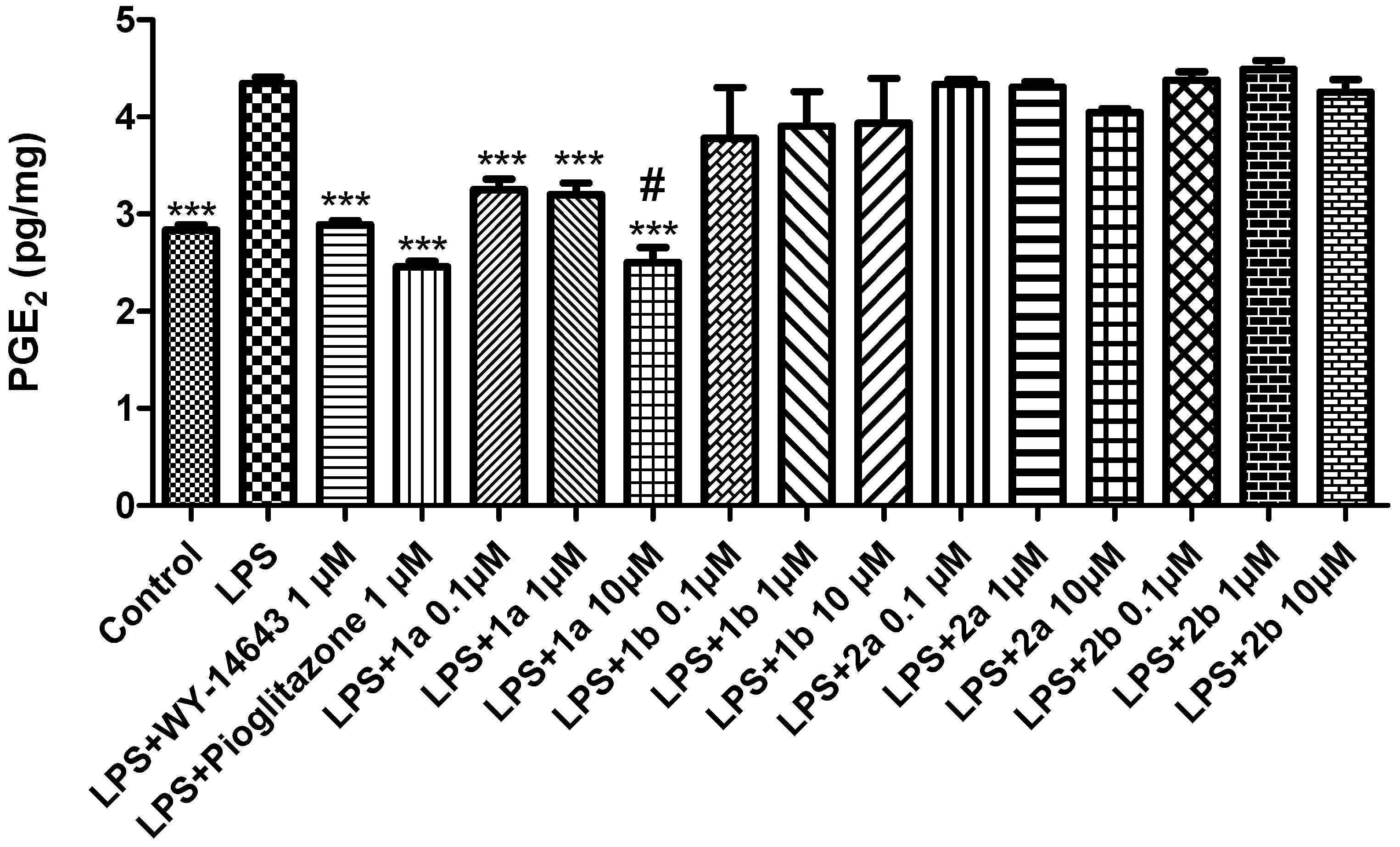
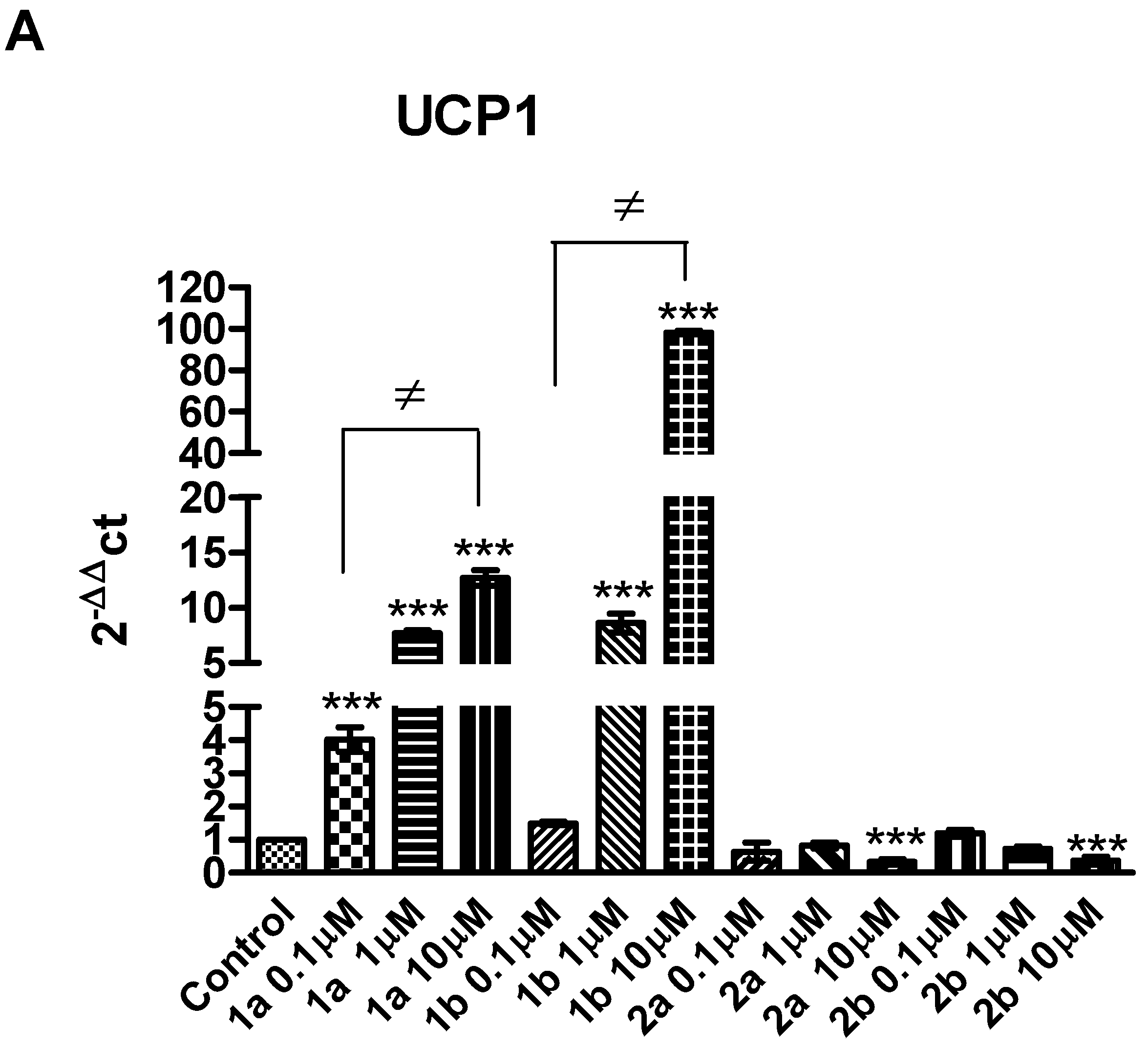
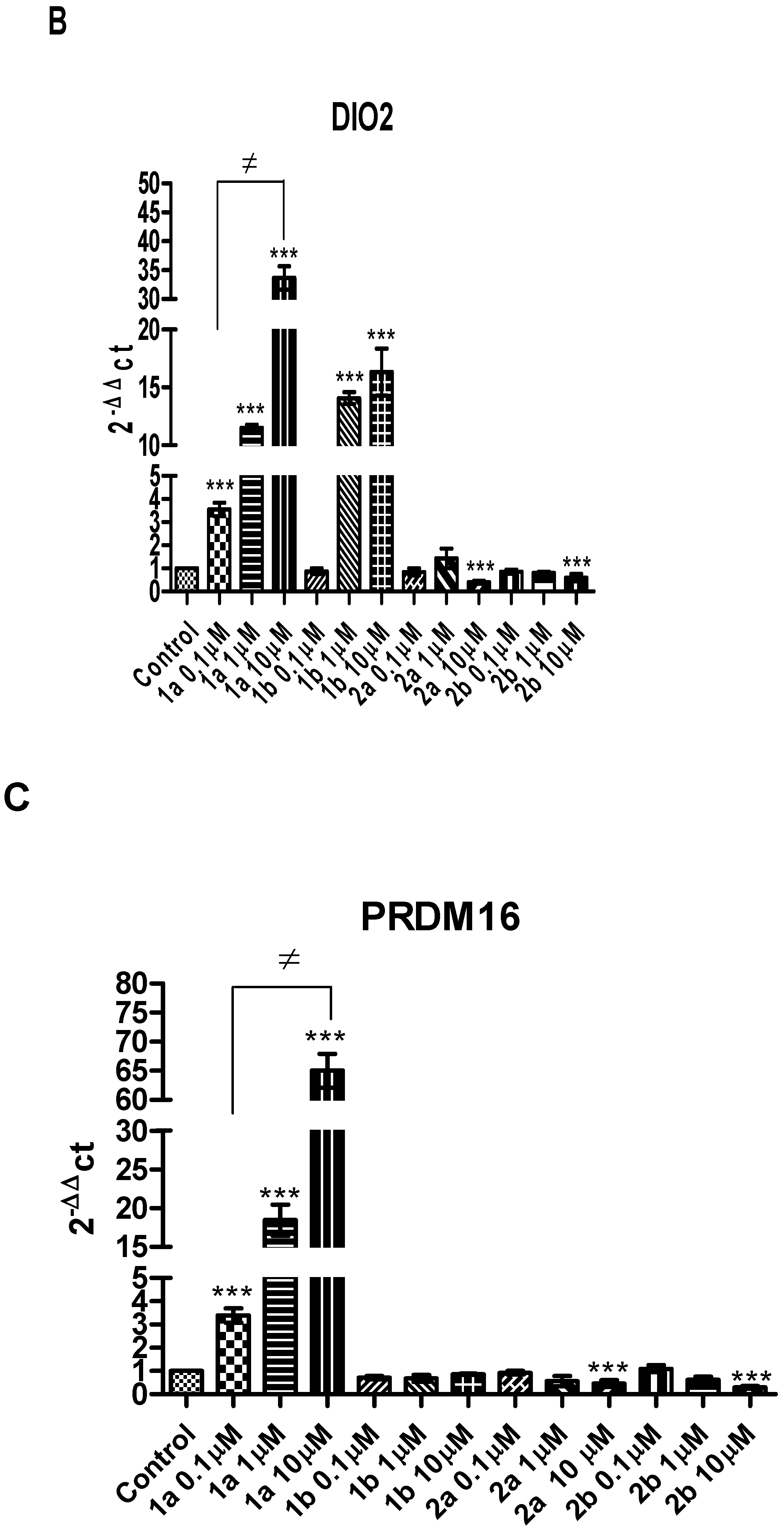
2. Results and Discussion
3. Materials and Methods
3.1. Chemistry
3.2. Cell Viability Assay
3.3. Ex Vivo Studies
3.4. Adipocyte Culture
3.5. PCR Assay
3.6. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Delerive, P.; Fruchart, J.C.; Staels, B. Peroxisome proliferator-activated receptors in inflammation control. J. Endocrinol. 2001, 169, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Moraes, L.A.; Piqueras, L.; Bishop-Bailey, D. Peroxisome proliferator-activated receptors and inflammation. Pharmacol. Ther. 2006, 110, 371–385. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Yang, Q. Peroxisome-proliferator-activated receptors regulate redox signaling in the cardiovascular system. World J. Cardiol. 2013, 5, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Laganà, A.S.; Vitale, S.G.; Nigro, A.; Sofo, V.; Salmeri, F.M.; Rossetti, P.; Rapisarda, A.M.C.; La Vignera, S.; Condorelli, R.A.; Rizzo, G.; et al. Pleiotropic actions of peroxisome proliferator-activated receptors (PPARs) in dysregulated metabolic homeostasis, inflammation and cancer: Current evidence and future perspectives. Int. J. Mol. Sci. 2016, 17, 999. [Google Scholar] [CrossRef] [PubMed]
- Li, T.T.; Tan, T.B.; Hou, H.Q.; Zhao, X.Y. Changes in peroxisome proliferatoractivated receptor alpha target gene expression in peripheral blood mononuclear cells associated with non-alcoholic fatty liver disease. Lipids Health Dis. 2018, 17, 256. [Google Scholar] [CrossRef] [PubMed]
- Delerive, P.; De Bosscher, K.; Besnard, S.; Vanden Berghe, W.; Peters, J.M.; Gonzalez, F.J.; Fruchart, J.C.; Tedgui, A.; Haegeman, G.; Staels, B. Peroxisome proliferators-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J. Biol. Chem. 1999, 274, 32048–32054. [Google Scholar] [CrossRef]
- Staels, B.; Koenig, W.; Habib, A.; Merval, R.; Lebret, M.; Torra, I.P.; Delerive, P.; Fadel, A.; Chinetti, G.; Fruchart, J.C.; et al. Activation of human aortic smooth-muscle cells is inhibited by PPARalpha but not by PPARgamma activators. Nature. 1998, 393, 790–793. [Google Scholar] [CrossRef]
- Devchand, P.R.; Keller, H.; Peters, J.M.; Vazquez, M.; Gonzalez, F.J.; Wahli, W. The PPARalpha-leukotriene B4 pathway to inflammation control. Nature. 1996, 384, 39–43. [Google Scholar] [CrossRef]
- Cuzzocrea, S.; Di Paola, R.; Mazzon, E.; Genovese, T.; Muià, C.; Centorrino, T.; Caputi, A.P. Role of endogenous and exogenous ligands for the peroxisome proliferators activated receptors alpha (PPAR-alpha) in the development of inflammatory bowel disease in mice. Lab. Investig. 2004, 84, 1643–1654. [Google Scholar] [CrossRef]
- Ziouzenkova, O.; Perrey, S.; Asatryan, L.; Hwang, J.; MacNaul, K.L.; Moller, D.E.; Rader, D.J.; Sevanian, A.; Zechner, R.; Hoefler, G.; et al. Lipolysis of triglyceride-rich lipoproteins generates PPAR ligands: Evidence for an antiinflammatory role for lipoprotein lipase. Proc. Natl. Acad. Sci. USA 2003, 100, 2730–2735. [Google Scholar] [CrossRef]
- Han, C.Y.; Chiba, T.; Campbell, J.S.; Fausto, N.; Chaisson, M.; Orasanu, G.; Plutzky, J.; Chait, A. Reciprocal and coordinate regulation of serum amyloid A versus apolipoprotein A-I and paraoxonase-1 by inflammation in murine hepatocytes. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1806–1813. [Google Scholar] [CrossRef] [PubMed]
- Stienstra, R.; Duval, C.; Müller, M.; Kersten, S. PPARs, Obesity, and Inflammation. PPAR Res. 2007, 2007, 95974. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Vela, M.E.; Torres, N.; Tovar, A.R. White adipose tissue as endocrine organ and its role in obesity. Arch. Med. Res. 2008, 39, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Giordano, A.; Frontini, A.; Cinti, S. Convertible visceral fat as a therapeutic target to curb obesity. Nat. Rev. Drug Discov. 2016, 15, 405–424. [Google Scholar] [CrossRef]
- Olefsky, J.M.; Glass, C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 2010, 72, 219–246. [Google Scholar] [CrossRef]
- Razina, A.O.; Runenko, S.D.; Achkasov, E.E. Obesity: Current global and russian trends. Vestn. Ross. Akad. Med. Nauk. 2016, 2, 154–159. [Google Scholar] [CrossRef]
- Budny, A.; Grochowski, C.; Kozłowski, P.; Kolak, A.; Kamińska, M.; Budny, B.; Abramiuk, M.; Burdan, F. Obesity as a tumour development triggering factor. Ann. Agric. Environ. Med. 2019, 26, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Singer-Englar, T.; Barlow, G.; Mathur, R. Obesity, diabetes, and the gut microbiome: An updated review. Expert Rev. Gastroenterol. Hepatol. 2019, 13, 3–15. [Google Scholar] [CrossRef]
- Hondares, E.; Rosell, M.; Díaz-Delfín, J.; Olmos, Y.; Monsalve, M.; Iglesias, R.; Villarroya, F.; Giralt, M. Peroxisome proliferator-activated receptor α (PPARα) induces PPARγ coactivator 1α (PGC-1α) gene expression and contributes to thermogenic activation of brown fat: Involvement of PRDM16. J. Biol. Chem. 2011, 286, 43112–43122. [Google Scholar] [CrossRef]
- Barbera, M.J.; Schluter, A.; Pedraza, N.; Iglesias, R.; Villarroya, F.; Giralt, M. Peroxisome proliferator-activated receptor alpha activates transcription of the brown fat uncoupling protein-1 gene. A link between regulation of the thermogenic and lipid oxidation pathways in the brown fat cell. J. Biol. Chem. 2001, 276, 1486–1493. [Google Scholar] [CrossRef]
- Lo, K.A.; Sun, L. Turning WAT into BAT: A review on regulators controlling the browning of white adipocytes. Biosci. Rep. 2013, 33, e00065. [Google Scholar] [CrossRef] [PubMed]
- Grozovsky, R.; Ribich, S.; Rosene, M.L.; Mulcahey, M.A.; Huang, S.A.; Patti, M.E.; Bianco, A.C.; Kim, B.W. Type 2 deiodinase expression is induced by peroxisomal proliferator-activated receptor-gamma agonists in skeletal myocytes. Endocrinology 2009, 150, 1976–1983. [Google Scholar] [CrossRef] [PubMed]
- Senn, L.; Costa, A.M.; Avallone, R.; Socała, K.; Wlaź, P.; Biagini, G. Is the peroxisome proliferator-activated receptor gamma a putative target for epilepsy treatment? Current evidence and future perspectives. Pharmacol. Ther. 2023, 241, 108316. [Google Scholar] [CrossRef]
- Lucchi, C.; Costa, A.M.; Giordano, C.; Curia, G.; Piat, M.; Leo, G.; Vinet, J.; Brunel, L.; Fehrentz, J.A.; Martinez, J.; et al. Involvement of PPARγ in the Anticonvulsant Activity of EP-80317, a Ghrelin Receptor Antagonist. Front. Pharmacol. 2017, 8, 676. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.M.; Russo, F.; Senn, L.; Ibatici, D.; Cannazza, G.; Biagini, G. Antiseizure Effects of Cannabidiol Leading to Increased Peroxisome Proliferator-Activated Receptor Gamma Levels in the Hippocampal CA3 Subfield of Epileptic Rats. Pharmaceuticals 2022, 15, 495. [Google Scholar] [CrossRef]
- Patel, C.; Wyne, K.L.; McGuire, D.K. Thiazolidinediones, peripheral oedema and congestive heart failure: What is the evidence? Diab. Vasc. Dis. Res. 2005, 2, 61–66. [Google Scholar] [CrossRef]
- Peraza, M.A.; Burdick, A.D.; Marin, H.E.; Gonzalez, F.J.; Peters, J.M. The toxicology of ligands for peroxisome proliferator-activated receptors (PPAR). Toxicol. Sci. 2006, 90, 269–295. [Google Scholar] [CrossRef]
- Tang, W.H.; Maroo, A. PPARgamma agonists: Safety issues in heart failure. Diabetes Obes. Metab. 2007, 9, 447–454. [Google Scholar] [CrossRef]
- Shearer, B.G.; Billin, A.N. The next generation of PPAR drugs: Do we have the tools to find them? Biochim. Biophys. Acta 2007, 1771, 1082–1093. [Google Scholar] [CrossRef]
- He, H.; Tao, H.; Xiong, H.; Duan, S.Z.; McGowan, F.X., Jr.; Mortensen, R.M.; Balschi, J.A. Rosiglitazone causes cardiotoxicity via peroxisome proliferatoractivated receptor gamma-independent mitochondrial oxidative stress in mouse hearts. Toxicol. Sci. 2014, 138, 468–481. [Google Scholar] [CrossRef]
- Hong, F.; Xu, P.; Zhai, Y. The Opportunities and Challenges of Peroxisome Proliferator-Activated Receptors Ligands in Clinical Drug Discovery and Development. Int. J. Mol. Sci. 2018, 19, 2189. [Google Scholar] [CrossRef]
- Frkic, R.L.; Marshall, A.C.; Blayo, A.L.; Pukala, T.L.; Kamenecka, T.M.; Griffin, P.R.; Bruning, J.B. PPARγ in Complex with an Antagonist and Inverse Agonist: A Tumble and Trap Mechanism of the Activation Helix. iSciences 2018, 5, 69–79. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, B.; Giancristofaro, A.; Ammazzalorso, A.; D’Angelo, A.; Fantacuzzi, M.; Giampietro, L.; Maccallini, C.; Petruzzelli, M.; Amoroso, R. Discovery of gemfibrozil analogues that activate PPARα and enhance the expression of gene CPT1A involved in fatty acid catabolism. Eur. J. Med. Chem. 2011, 46, 5218–5224. [Google Scholar] [CrossRef] [PubMed]
- Ammazzalorso, A.; Giancristofaro, A.; D’Angelo, A.; De Filippis, B.; Fantacuzzi, M.; Giampietro, L.; Maccallini, C.; Amoroso, R. Benzothiazole-based N-(phenylsulfonyl)amides as a novel family of PPARα antagonists. Bioorg. Med. Chem. Lett. 2011, 21, 4869–4872. [Google Scholar] [CrossRef] [PubMed]
- Giampietro, L.; D’Angelo, A.; Giancristofaro, A.; Ammazzalorso, A.; De Filippis, B.; Fantacuzzi, M.; Linciano, P.; Maccallini, C.; Amoroso, R. Synthesis and structure–activity relationships of fibrate-based analogues inside PPARs. Bioorg. Med. Chem. Lett. 2012, 22, 7662–7666. [Google Scholar] [CrossRef]
- Giampietro, L.; Ammazzalorso, A.; Amoroso, R.; De Filippis, B. Development of fibrates as important scaffold in medicinal chemistry. Chem. Med. Chem. 2019, 14, 1051–1066. [Google Scholar] [CrossRef]
- De Filippis, B.; Linciano, P.; Ammazzalorso, A.; Di Giovanni, C.; Fantacuzzi, M.; Giampietro, L.; Laghezza, A.; Maccallini, C.; Tortorella, P.; Lavecchia, A.; et al. Structural development studies of PPARs ligands based on tyrosine scaffold. Eur. J. Med. Chem. 2015, 89, 817–825. [Google Scholar] [CrossRef]
- Xu, H.E.; Lambert, M.H.; Montana, V.G.; Plunket, K.D.; Moore, L.B.; Collins, J.L.; Oplinger, J.A.; Kliewer, S.A.; Gampe, R.T., Jr.; McKee, D.D.; et al. Structural determinants of ligand binding selectivity between the peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 13919–13924. [Google Scholar] [CrossRef]
- Ammazzalorso, A.; Carrieri, A.; Verginelli, F.; Bruno, I.; Carbonara, G.; D’Angelo, A.; De Filippis, B.; Fantacuzzi, M.; Florio, R.; Fracchiolla, G.; et al. Synthesis, in vitro evaluation, and molecular modeling investigation of benzenesulfonimide peroxisome proliferator-activated receptors α antagonists. Eur. J. Med. Chem. 2016, 114, 191–200. [Google Scholar] [CrossRef]
- Ammazzalorso, A.; De Lellis, L.; Florio, R.; Bruno, I.; De Filippis, B.; Fantacuzzi, M.; Giampietro, L.; Maccallini, C.; Perconti, S.; Verginelli, F.; et al. Cytotoxic effect of a family of peroxisome proliferator-activated receptor antagonists in colorectal and pancreatic cancer lines. Chem. Biol. Drug Des. 2017, 90, 1029–1035. [Google Scholar] [CrossRef]
- Recinella, L.; Chiavaroli, A.; Orlando, G.; Menghini, L.; Ferrante, C.; Di Cesare Mannelli, L.; Ghelardini, C.; Brunetti, L.; Leone, S. Protective effects induced by two polyphenolic liquid complexes from olive (Olea europaea, mainly Cultivar Coratina) pressing juice in rat isolated tissues challenged with LPS. Molecules 2019, 24, 3002. [Google Scholar] [CrossRef] [PubMed]
- Leone, S.; Chiavaroli, A.; Recinella, L.; Orlando, G.; Ferrante, C.; Marconi, G.D.; Gasparo, I.; Bitto, A.; Salvatori, R.; Brunetti, L. Increased pain and inflammatory sensitivity in growth hormone-releasing hormone (GHRH) knockout mice. Prostaglandins Other Lipid Mediat. 2019, 144, 106362. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Liu, Z.; Sun, H.; Huang, Y.; Albrecht, E.; Zhao, R.; Yang, X. Lipopolysaccharide challenge significantly influences lipid metabolism and proteome of white adipose tissue in growing pigs. Lipids Health Dis. 2015, 14, 68. [Google Scholar] [CrossRef]
- Recinella, L.; Chiavaroli, A.; Orlando, G.; Ferrante, C.; Marconi, G.D.; Gesmundo, I.; Granata, R.; Cai, R.; Sha, W.; Schally, A.V.; et al. Antinflammatory, antioxidant, and behavioral effects induced by administration of growth hormone-releasing hormone analogs in mice. Sci. Rep. 2020, 10, 4850. [Google Scholar] [CrossRef] [PubMed]
- Recinella, L.; Chiavaroli, A.; Di Valerio, V.; Veschi, S.; Orlando, G.; Ferrante, C.; Gesmundo, I.; Granata, R.; Cai, R.; Sha, W.; et al. Protective effects of growth hormone-releasing hormone analogs in DSS-induced colitis in mice. Sci. Rep. 2021, 11, 2530. [Google Scholar] [CrossRef]
- Kannan, N.; Guruvayoorappan, C. Protective effect of Bauhinia tomentosa on acetic acid induced ulcerative colitis by regulating antioxidant and inflammatory mediators. Int. Immunopharmacol. 2013, 16, 57–66. [Google Scholar] [CrossRef]
- Kotoh, K.; Kato, M.; Kohjima, M.; Tanaka, M.; Miyazaki, M.; Nakamura, K.; Enjoji, M.; Nakamuta, M.; Takayanagi, R. Lactate dehydrogenase production in hepatocytes is increased at an early stage of acute liver failure. Exp. Ther. Med. 2011, 2, 195–199. [Google Scholar] [CrossRef]
- Faiola, B.; Falls, J.G.; Peterson, R.A.; Bordelon, N.R.; Brodie, T.A.; Cummings, C.A.; Romach, E.H.; Miller, R.T. PPAR alpha, more than PPAR delta, mediates the hepatic and skeletal muscle alterations induced by the PPAR agonist GW0742. Toxicol. Sci. 2008, 105, 384–394. [Google Scholar] [CrossRef]
- Morán-Salvador, E.; Titos, E.; Rius, B.; González-Périz, A.; García-Alonso, V.; López-Vicario, C.; Miquel, R.; Barak, Y.; Arroyo, V.; Clària, J. Cell-specific PPARγ deficiency establishes anti-inflammatory and anti-fibrogenic properties for this nuclear receptor in non-parenchymal liver cells. J. Hepatol. 2013, 59, 1045–1053. [Google Scholar] [CrossRef]
- Berlanga, A.; Guiu-Jurado, E.; Porras, J.A.; Auguet, T. Molecular pathways in non-alcoholic fatty liver disease. Clin. Exp. Gastroenterol. 2014, 7, 221–239. [Google Scholar]
- Schaefer, M.B.; Pose, A.; Ott, J.; Hecker, M.; Behnk, A.; Schulz, R.; Weissmann, N.; Günther, A.; Seeger, W.; Mayer, K. Peroxisome proliferator-activated receptor-alpha reduces inflammation and vascular leakage in a murine model of acute lung injury. Eur. Respir. J. 2008, 32, 1344–1353. [Google Scholar] [CrossRef] [PubMed]
- Yoon, E.K.; Lee, W.K.; Lee, J.H.; Yu, S.M.; Hwang, S.G.; Kim, S.J. ERK-1/-2 and p38 kinase oppositely regulate 15-deoxy-delta(12,14)-prostaglandinJ(2)-Induced PPAR-gamma activation that mediates dedifferentiation but not cyclooxygenase-2 expression in articular chondrocytes. J. Korean Med. Sci. 2007, 22, 1015–1121. [Google Scholar] [CrossRef] [PubMed]
- Basu, S. F2-Isoprostanes in human health and diseases: From molecular mechanisms to clinical implications. Antioxid. Redox Sign. 2008, 10, 1405–1434. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Tsuboyama-Kasaoka, N.; Nakatani, T.; Ishii, M.; Tsutsumi, S.; Aburatani, H.; Ezaki, O. Fish oil feeding alters liver gene expressions to defend against PPARalpha activation and ROS production. Am. J. Physiol. Gasterointest. Liver Physiol. 2002, 282, G338–G348. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Hamm, A.C.; Bonaus, M.; Jacob, A.; Jaekel, J.; Schorle, H.; Pankratz, M.J.; Katzenberger, J.D. Starvation response in mouse liver shows strong correlation with life-span-prolonging processes. Physiol. Genom. 2004, 17, 230–244. [Google Scholar] [CrossRef] [PubMed]
- Nedergaard, J.; Cannon, B. UCP1 mRNA does not produce heat. Biochim. Biophys. Acta 2013, 1831, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Cousin, B.; Cinti, S.; Morroni, M.; Raimbault, S.; Ricquier, D.; Pénicaud, L.; Casteilla, L. Occurrence of brown adipocytes in rat white adipose tissue: Molecular and morphological characterization. J. Cell Sci. 1992, 103, 931–942. [Google Scholar] [CrossRef]
- Nedergaard, J.; Cannon, B. The changed metabolic world with human brown adipose tissue: Therapeutic visions. Cell Metab. 2010, 11, 268–272. [Google Scholar] [CrossRef]
- Seale, P.; Kajimura, S.; Yang, W.; Chin, S.; Rohas, L.M.; Uldry, M.; Tavernier, G.; Langin, D.; Spiegelman, B.M. Transcriptional control of brown fat determination by PRDM16. Cell Metab. 2007, 6, 38–54. [Google Scholar] [CrossRef]
- Harms, M.; Seale, P. Brown and beige fat: Development, function and therapeutic potential. Nat. Med. 2013, 19, 1252–1263. [Google Scholar] [CrossRef]
- Seale, P.; Bjork, B.; Yang, W.; Kajimura, S.; Chin, S.; Kuang, S.; Scimè, A.; Devarakonda, S.; Conroe, H.M.; Erdjument-Bromage, H.; et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature 2008, 454, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Weiner, J.; Kranz, M.; Klöting, N.; Kunath, A.; Steinhoff, K.; Rijntjes, E.; Köhrle, J.; Zeisig, V.; Hankir, M.; Gebhardt, C.; et al. Thyroid hormone status defines brown adipose tissue activity and browning of white adipose tissues in mice. Sci. Rep. 2016, 6, 38124. [Google Scholar] [CrossRef] [PubMed]
- Fukui, Y.; Masui, S.; Osada, S.; Umesono, K.; Motojima, K. A new thiazolidinedione, NC-2100, which is a weak PPAR-gamma activator, exhibits potent antidiabetic effects and induces uncoupling protein 1 in white adipose tissue of KKAy obese mice. Diabetes 2000, 49, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Kelly, L.J.; Vicario, P.P.; Thompson, G.M.; Candelore, M.R.; Doebber, T.W.; Ventre, J.; Wu, M.S.; Meurer, R.; Forrest, M.J.; Conner, M.W.; et al. Peroxisome proliferator-activated receptors γ and α mediate in vivo regulation of uncoupling protein (UCP-1, UCP-2, UCP-3) gene expression. Endocrinology 1998, 139, 4920–4927. [Google Scholar] [CrossRef]
- Seale, P.; Conroe, H.M.; Estall, J.; Kajimura, S.; Frontini, A.; Ishibashi, J.; Cohen, P.; Cinti, S.; Spiegelman, B.M. Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J. Clin. Investig. 2011, 121, 96–105. [Google Scholar] [CrossRef]
- Veschi, S.; De Lellis, L.; Florio, R.; Lanuti, P.; Massucci, A.; Tinari, N.; De Tursi, M.; di Sebastiano, P.; Marchisio, M.; Natoli, C.; et al. Effects of repurposed drug candidates nitroxoline and nelfinavir as single agents or in combination with erlotinib in pancreatic cancer cells. J. Exp. Clin. Cancer Res. 2018, 37, 236. [Google Scholar] [CrossRef]
- Recinella, L.; Chiavaroli, A.; Masciulli, F.; Fraschetti, C.; Filippi, A.; Cesa, S.; Cairone, F.; Gorica, E.; De Leo, M.; Braca, A.; et al. Protective Effects Induced by a Hydroalcoholic Allium sativum Extract in Isolated Mouse Heart. Nutrients 2021, 13, 2332. [Google Scholar] [CrossRef]
- Recinella, L.; Gorica, E.; Chiavaroli, A.; Fraschetti, C.; Filippi, A.; Cesa, S.; Cairone, F.; Martelli, A.; Calderone, V.; Veschi, S.; et al. Anti-Inflammatory and Antioxidant Effects Induced by Allium sativum L. Extracts on an Ex Vivo Experimental Model of Ulcerative Colitis. Foods 2022, 11, 3559. [Google Scholar]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Recinella, L.; De Filippis, B.; Libero, M.L.; Ammazzalorso, A.; Chiavaroli, A.; Orlando, G.; Ferrante, C.; Giampietro, L.; Veschi, S.; Cama, A.; et al. Anti-Inflammatory, Antioxidant, and WAT/BAT-Conversion Stimulation Induced by Novel PPAR Ligands: Results from Ex Vivo and In Vitro Studies. Pharmaceuticals 2023, 16, 346. https://doi.org/10.3390/ph16030346
Recinella L, De Filippis B, Libero ML, Ammazzalorso A, Chiavaroli A, Orlando G, Ferrante C, Giampietro L, Veschi S, Cama A, et al. Anti-Inflammatory, Antioxidant, and WAT/BAT-Conversion Stimulation Induced by Novel PPAR Ligands: Results from Ex Vivo and In Vitro Studies. Pharmaceuticals. 2023; 16(3):346. https://doi.org/10.3390/ph16030346
Chicago/Turabian StyleRecinella, Lucia, Barbara De Filippis, Maria Loreta Libero, Alessandra Ammazzalorso, Annalisa Chiavaroli, Giustino Orlando, Claudio Ferrante, Letizia Giampietro, Serena Veschi, Alessandro Cama, and et al. 2023. "Anti-Inflammatory, Antioxidant, and WAT/BAT-Conversion Stimulation Induced by Novel PPAR Ligands: Results from Ex Vivo and In Vitro Studies" Pharmaceuticals 16, no. 3: 346. https://doi.org/10.3390/ph16030346
APA StyleRecinella, L., De Filippis, B., Libero, M. L., Ammazzalorso, A., Chiavaroli, A., Orlando, G., Ferrante, C., Giampietro, L., Veschi, S., Cama, A., Mannino, F., Gasparo, I., Bitto, A., Amoroso, R., Brunetti, L., & Leone, S. (2023). Anti-Inflammatory, Antioxidant, and WAT/BAT-Conversion Stimulation Induced by Novel PPAR Ligands: Results from Ex Vivo and In Vitro Studies. Pharmaceuticals, 16(3), 346. https://doi.org/10.3390/ph16030346