
Pyrazole-Enriched Cationic Nanoparticles Induced Early- and Late-Stage Apoptosis in Neuroblastoma Cells at Sub-Micromolar Concentrations

 ,
,  , ,
, ,  and
and 
Abstract
1. Introduction
1.1. Why BBB4-G4K NPs and CB1H-P7 NPs?
1.2. The Rational of the Present Work
2. Results and Discussion
2.1. Cytotoxicity of BBB4-G4K on NB Cells
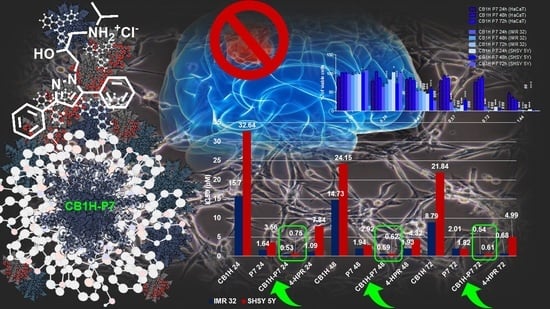
2.2. Cytotoxicity of CB1H-P7 NPs on NB Cells
2.2.1. Cytotoxicity of CB1H-P7 NPs on IMR 32 NB Cells
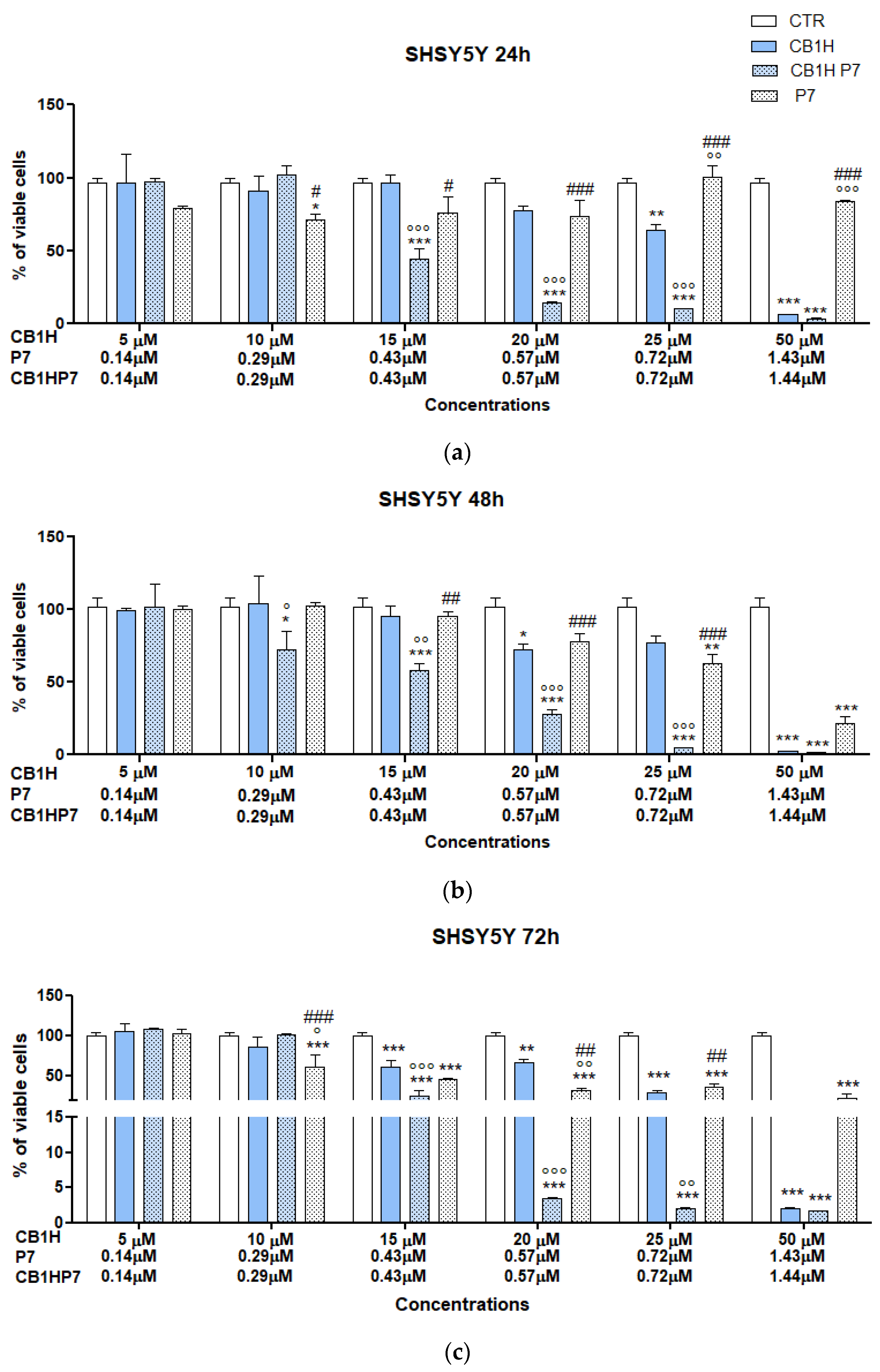
2.2.2. Cytotoxicity of CB1H-P7 NPs on SHSY 5Y NB Cells
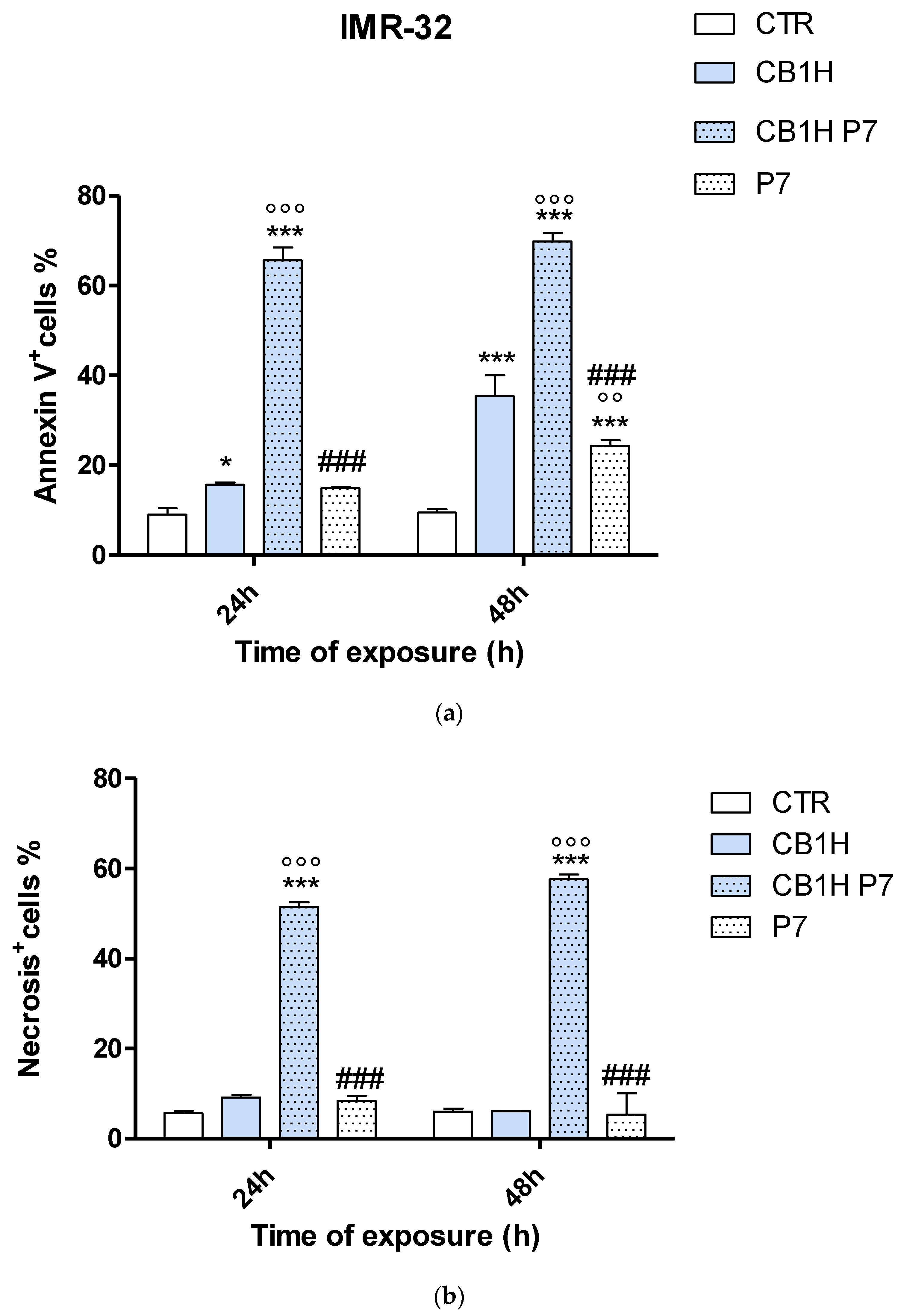
2.3. Effect of CB1H, P7 and CB1H-P7 NPs on Apoptosis and Necrosis of IMR-32 and SHSY 5Y Cells
3. Materials and Methods
3.1. In Vitro Biological Experiments
3.1.1. Cell Line Neuroblastoma
3.1.2. Cell Proliferation Assay
3.1.3. Analysis of Apoptosis and Necrosis by Annexin-V and 7-AAD Staining
3.1.4. Selectivity Index
3.2. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brodeur, G.M.; Castleberry, R.P. Neuroblastoma. In Principles and Practice of Paediatric Oncology; Pizzo, P.A., Poplack, D.G., Eds.; J. B. Lippincott & Co.: Philadelphia, PA, USA, 1997; pp. 761–797. [Google Scholar]
- Franks, L.M.; Bollen, A.; Seeger, R.C.; Stram, D.O.; Matthay, K.K. Neuroblastoma in adults and adolescents: An indolent course with poor survival. Cancer 1997, 10, 2028–2035. [Google Scholar] [CrossRef]
- Dembowska-Bagińska, B.; Brozyna, A.; Perek, D.; Krenska, A.; Stypińska, M.; Drogosiewicz, M.; Perek-Polnik, M.; Rutynowska-Pronicka, O. Cancer among adolescents 15–19 years of age. Own experience. Med. Wieku Rozw. 2006, 10, 725–735. [Google Scholar]
- Johnson, K.J.; Puumala, S.E.; Soler, J.T.; Spector, L.G. Perinatal characteristics and risk of neuroblastoma. Int. J. Cancer 2008, 123, 1166–1172. [Google Scholar] [CrossRef]
- Alfei, S.; Marengo, B.; Zuccari, G.; Turrini, F.; Domenicotti, C. Dendrimer Nanodevices and Gallic Acid as Novel Strategies to Fight Chemoresistance in Neuroblastoma Cells. Nanomaterials 2020, 10, 1243. [Google Scholar] [CrossRef]
- Maris, J.M.; Hogarty, M.D.; Bagatell, R.; Cohn, S.L. Neuroblastoma. Lancet 2007, 369, 2106–2120. [Google Scholar] [CrossRef]
- Leone, G.; Voso, M.T.; Sica, S.; Morosetti, R.; Pagano, L. Therapy related leukemias: Susceptibility, prevention and treatment. Leuk. Lymphoma 2001, 41, 255–276. [Google Scholar] [CrossRef]
- Haupt, R.; Fears, T.R.; Heise, A.; Gadner, H.; Loiacono, G.; De Terlizzi, M.; Tucker, M.A. Risk of secondary leukemia after treatment with etoposide (VP-16) for Langerhans’cell histiocytosis in Italian and Austrian-German populations. Int. J. Cancer 1997, 71, 9–13. [Google Scholar] [CrossRef]
- Bernardini, S.; Bellincampi, L.; Ballerini, S.; Ranalli, M.; Pastore, A.; Cortese, C.; Federici, G. Role of GST P1-1 in mediating the effect of etoposide on human neuroblastoma cell line Sh-Sy5y. J. Cell. Biochem. 2002, 86, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Alfei, S.; Marengo, B.; Valenti, G.E.; Domenicotti, C. Synthesis of Polystyrene-Based Cationic Nanomaterials with Pro-Oxidant Cytotoxic Activity on Etoposide-Resistant Neuroblastoma Cells. Nanomaterials 2021, 11, 977. [Google Scholar] [CrossRef] [PubMed]
- Alfei, S.; Schito, A.M. Positively Charged Polymers as Promising Devices against Multidrug Resistant Gram-Negative Bacteria: A Review. Polymers 2020, 12, 1195. [Google Scholar] [CrossRef]
- Alfei, S.; Piatti, G.; Caviglia, D.; Schito, A.M. Synthesis, Characterization and Bactericidal Activity of a 4-Ammoniumbuthylstyrene-Based Random Copolymer. Polymers 2021, 13, 1140. [Google Scholar] [CrossRef] [PubMed]
- Alfei, S.; Schito, A.M. From Nanobiotechnology, Positively Charged Biomimetic Dendrimers as Novel Antibacterial Agents: A Review. Nanomaterials 2020, 10, 2022. [Google Scholar] [CrossRef]
- Gelman, M.A.; Weisblum, B.; Lynn, D.M.; Gellman, S.H. Biocidal activity of polystyrenes that are cationic by virtue of protonation. Org. Lett. 2004, 6, 557–560. [Google Scholar] [CrossRef]
- Palermo, E.; Kuroda, K. Chemical structure of cationic groups in amphiphilic polymethacrylates modulates the antimicrobial and hemolytic activities. Biomacromolecules 2009, 10, 1416–1428. [Google Scholar] [CrossRef]
- Büyükkiraz, E.M.; Kesmen, Z. Antimicrobial peptides (AMPs): A promising class of antimicrobial compounds. J. Appl. Microbiol. 2022, 132, 1573–1596. [Google Scholar] [CrossRef]
- Bellotto, O.; Semeraro, S.; Bandiera, A.; Tramer, F.; Pavan, N.; Marchesan, S. Polymer Conjugates of Antimicrobial Peptides (AMPs) with d-Amino Acids (d-aa): State of the Art and Future Opportunities. Pharmaceutics 2022, 14, 446. [Google Scholar] [CrossRef]
- Scorciapino, M.A.; Serra, I.; Manzo, G.; Rinaldi, A.C. Antimicrobial Dendrimeric Peptides: Structure, Activity and New Therapeutic Applications. Int. J. Mol. Sci. 2017, 18, 542. [Google Scholar] [CrossRef]
- Valenti, G.E.; Alfei, S.; Caviglia, D.; Domenicotti, C.; Marengo, B. Antimicrobial Peptides and Cationic Nanoparticles: A Broad-Spectrum Weapon to Fight Multi-Drug Resistance Not Only in Bacteria. Int. J. Mol. Sci. 2022, 23, 6108. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Tay, J.; Hedrick, J.; Yang, Y.Y. Synthetic macromolecules as therapeutics that overcome resistance in cancer and microbial infection. Biomaterials 2020, 252, 120078. [Google Scholar] [CrossRef]
- Schito, A.M.; Piatti, G.; Caviglia, D.; Zuccari, G.; Alfei, S. Broad-Spectrum Bactericidal Activity of a Synthetic Random Copolymer Based on 2-Methoxy-6-(4-Vinylbenzyloxy)-Benzylammonium Hydrochloride. Int. J. Mol. Sci. 2021, 22, 5021. [Google Scholar] [CrossRef] [PubMed]
- Alfei, S.; Brullo, C.; Caviglia, D.; Piatti, G.; Zorzoli, A.; Marimpietri, D.; Zuccari, G.; Schito, A.M. Pyrazole-Based Water-Soluble Dendrimer Nanoparticles as a Potential New Agent against Staphylococci. Biomedicines 2022, 10, 17. [Google Scholar] [CrossRef]
- Schito, A.M.; Caviglia, D.; Brullo, C.; Zorzoli, A.; Marimpietri, D.; Alfei, S. Enhanced Antibacterial Activity of a Cationic Macromolecule by Its Complexation with a Weakly Active Pyrazole Derivative. Biomedicines 2022, 10, 1607. [Google Scholar] [CrossRef]
- Alfei, S.; Brullo, C.; Caviglia, D.; Zuccari, G. Preparation and Physicochemical Characterization of Water-Soluble Pyrazole-Based Nanoparticles by Dendrimer Encapsulation of an Insoluble Bioactive Pyrazole Derivative. Nanomaterials 2021, 11, 2662. [Google Scholar] [CrossRef] [PubMed]
- Alfei, S.; Zuccari, G.; Caviglia, D.; Brullo, C. Synthesis and Characterization of Pyrazole-Enriched Cationic Nanoparticles as New Promising Antibacterial Agent by Mutual Cooperation. Nanomaterials 2022, 12, 1215. [Google Scholar] [CrossRef] [PubMed]
- Kunda, N.K. Antimicrobial peptides as novel therapeutics for non-small cell lung cancer. Drug Discov. Today 2020, 25, 238–247. [Google Scholar] [CrossRef]
- Giuliani, A. Antimicrobial peptides: An overview of a promising class of therapeutics. Cent. Eur. J. Biol. 2007, 2, 1–33. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, M.; Ericsson, A.C. Antimicrobial peptides: Potential application in liver cancer. Front. Microbiol. 2019, 10, 1257. [Google Scholar] [CrossRef]
- Desai, T.J.; Toombs, J.E.; Minna, J.D.; Brekken, R.A.; Udugamasooriya, D.G. Identification of lipid-phosphatidylserine (PS) as the target of unbiasedly selected cancer specific peptide-peptoid hybrid PPS1. Oncotarget 2016, 7, 30678–30690. [Google Scholar] [CrossRef]
- Baxter, A.A.; Lay, F.T.; Poon, I.K.H.; Kvansakul, M.; Hulett, M.D. Tumor cell membrane-targeting cationic antimicrobial peptides: Novel insights into mechanisms of action and therapeutic prospects. Cell. Mol. Life Sci. 2017, 74, 3809–3825. [Google Scholar] [CrossRef]
- Chuang, C.M.; Monie, A.; Wu, A.; Mao, C.P.; Hung, C.F. Treatment with ll-37 peptide enhances antitumor effects induced by cpg oligodeoxynucleotides against ovarian cancer. Hum. Gene Ther. 2009, 20, 303–313. [Google Scholar] [CrossRef]
- Okumura, K.; Itoh, A.; Isogai, E.; Hirose, K.; Hosokawa, Y.; Abiko, Y.; Shibata, T.; Hirata, M.; Isogai, H. C-Terminal domain of human cap18 antimicrobial peptide induces apoptosis in oral squamous cell carcinoma sas-h1 cells. Cancer Lett. 2004, 212, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Mader, J.S.; Mookherjee, N.; Hancock, R.E.; Bleackley, R.C. The human host defense peptide ll-37 induces apoptosis in a calpain- and apoptosis-inducing factor-dependent manner involving bax activity. Mol. Cancer Res. 2009, 7, 689–702. [Google Scholar] [CrossRef]
- Xhindoli, D.; Pacor, S.; Guida, F.; Antcheva, N.; Tossi, A. Native oligomerization determines the mode of action and biological activities of human cathelicidin ll-37. Biochem. J. 2014, 457, 263–275. [Google Scholar] [CrossRef]
- Cheng, M.; Ho, S.; Yoo, J.H.; Tran, D.H.; Bakirtzi, K.; Su, B.; Tran, D.H.; Kubota, Y.; Ichikawa, R.; Koon, H.W. Cathelicidin suppresses colon cancer development by inhibition of cancer associated fibroblasts. Clin. Exp. Gastroenterol. 2015, 8, 13–29. [Google Scholar] [PubMed]
- Xu, N.; Wang, Y.S.; Pan, W.B.; Xiao, B.; Wen, Y.J.; Chen, X.C.; Chen, L.J.; Deng, H.X.; You, J.; Kan, B.; et al. Human α-defensin-1 inhibits growth of human lung adenocarcinoma xenograft in nude mice. Mol. Cancer Ther. 2008, 7, 1588–1597. [Google Scholar] [CrossRef]
- Wang, Y.S.; Li, D.; Shi, H.S.; Wen, Y.J.; Yang, L.; Xu, N.; Chen, X.C.; Chen, X.; Chen, P.; Li, J.; et al. Intratumoral expression of mature human neutrophil peptide-1 mediates antitumor immunity in mice. Clin. Cancer Res. 2009, 15, 6901–6911. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.K.; Lay, F.T.; Poon, I.K.; Hinds, M.G.; Kvansakul, M.; Hulett, M.D. Human β-defensin 3 contains an oncolytic motif that binds pi(4,5)p2 to mediate tumour cell permeabilisation. Oncotarget 2016, 7, 2054–2069. [Google Scholar] [CrossRef]
- Eliassen, L.T.; Berge, G.; Leknessund, A.; Wikman, M.; Lindin, I.; Lokke, C.; Ponthan, F.; Johnsen, J.I.; Sveinbjornsson, B.; Kogner, P.; et al. The antimicrobial peptide, lactoferricin b, is cytotoxic to neuroblastoma cells In Vitro and inhibits xenograft growth In Vivo. Int. J. Cancer 2006, 119, 493–500. [Google Scholar] [CrossRef]
- Furlong, S.J.; Ridgway, N.D.; Hoskin, D.W. Modulation of ceramide metabolism in t-leukemia cell lines potentiates apoptosis induced by the cationic antimicrobial peptide bovine lactoferricin. Int. J. Oncol. 2008, 32, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Domingues, T.M.; Riske, K.A.; Miranda, A. Revealing the lytic mechanism of the antimicrobial peptide gomesin by observing giant unilamellar vesicles. Langmuir 2010, 26, 11077–11084. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, L.; Wu, Y.; Wang, L.; Ma, C.; Xi, X.; Bininda-Emonds, O.R.P.; Shaw, C.; Chen, T.; Zhou, M. Evaluation of the bioactivity of a mastoparan peptide from wasp venom and of its analogues designed through targeted engineering. Int. J. Biol. Sci. 2018, 14, 599–607. [Google Scholar] [CrossRef]
- Wu, J.M.; Jan, P.S.; Yu, H.C.; Haung, H.Y.; Fang, H.J.; Chang, Y.I.; Cheng, J.W.; Chen, H.M. Structure and function of a custom anticancer peptide, cb1a. Peptides 2009, 30, 839–848. [Google Scholar] [CrossRef]
- Li, X.; Shen, B.; Chen, Q.; Zhang, X.; Ye, Y.; Wang, F.; Zhang, X. Antitumor effects of cecropin b-lhrh’ on drug-resistant ovarian and endometrial cancer cells. BMC Cancer 2016, 16, 251. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.A.; Maloy, W.L.; Zasloff, M.; Jacob, L.S. Anticancer efficacy of magainin2 and analogue peptides. Cancer Res. 1993, 53, 3052–3057. [Google Scholar] [PubMed]
- Lehmann, J.; Retz, M.; Sidhu, S.S.; Suttmann, H.; Sell, M.; Paulsen, F.; Harder, J.; Unteregger, G.; Stockle, M. Antitumor activity of the antimicrobial peptide magainin ii against bladder cancer cell lines. Eur. Urol. 2006, 50, 141–147. [Google Scholar] [CrossRef]
- Shin, S.Y.; Kang, J.H.; Jang, S.Y.; Kim, Y.; Kim, K.L.; Hahm, K.S. Effects of the hinge region of cecropin a(1-8)-magainin 2(1-12), a synthetic antimicrobial peptide, on liposomes, bacterial and tumor cells. Biochim. Biophys. Acta 2000, 1463, 209–218. [Google Scholar] [CrossRef]
- Lee, H.S.; Park, C.B.; Kim, J.M.; Jang, S.A.; Park, I.Y.; Kim, M.S.; Cho, J.H.; Kim, S.C. Mechanism of anticancer activity of buforin iib, a histone h2a-derived peptide. Cancer Lett. 2008, 271, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Asoodeh, A.; Klonisch, T.; Halayko, A.J.; Kadkhoda, K.; Kroczak, T.J.; Gibson, S.B.; Booy, E.P.; Naderi-Manesh, H.; Los, M. Brevinin-2r(1) semi-selectively kills cancer cells by a distinct mechanism, which involves the lysosomal-mitochondrial death pathway. J. Cell. Mol. Med. 2008, 12, 1005–1022. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Lyu, P.; Xie, S.; Qin, H.; Pu, W.; Xu, H.; Chen, T.; Shaw, C.; Ge, L.; Kwok, H.F. Lfb: A novel antimicrobial brevinin-like peptide from the skin secretion of the fujian large headed frog, limnonectes fujianensi. Biomolecules 2019, 9, 242. [Google Scholar] [CrossRef]
- Liu, Y.; Du, Q.; Ma, C.; Xi, X.; Wang, L.; Zhou, M.; Burrows, J.F.; Chen, T.; Wang, H. Structure-Activity relationship of an antimicrobial peptide, phylloseptin-pha: Balance of hydrophobicity and charge determines the selectivity of bioactivities. Drug Des. Dev. Ther. 2019, 13, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, L.; Ma, C.; Zhang, Y.; Xi, X.; Wang, L.; Zhou, M.; Burrows, J.F.; Chen, T. A novel antimicrobial peptide, ranatuerin-2plx, showing therapeutic potential in inhibiting proliferation of cancer cells. Biosci. Rep. 2018, 38, BSR20180710. [Google Scholar] [CrossRef] [PubMed]
- Bartels, E.J.H.; Dekker, D.; Amiche, M. Dermaseptins, multifunctional antimicrobial peptides: A review of their pharmacology, effectivity, mechanism of action, and possible future directions. Front. Pharmacol. 2019, 10, 1421. [Google Scholar] [CrossRef]
- Dos Santos, C.; Hamadat, S.; Le Saux, K.; Newton, C.; Mazouni, M.; Zargarian, L.; Miro-Padovani, M.; Zadigue, P.; Delbe, J.; Hamma-Kourbali, Y.; et al. Studies of the antitumor mechanism of action of dermaseptin b2, a multifunctional cationic antimicrobial peptide, reveal a partial implication of cell surface glycosaminoglycans. PLoS ONE 2017, 12, e0182926. [Google Scholar] [CrossRef]
- Li, M.; Xi, X.; Ma, C.; Chen, X.; Zhou, M.; Burrows, J.F.; Chen, T.; Wang, L. A novel dermaseptin isolated from the skin secretion of phyllomedusa tarsius and its cationicity-enhanced analogue exhibiting effective antimicrobial and anti-proliferative activities. Biomolecules 2019, 9, 628. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.C.; Lin, L.C.; Tzen, J.T.; Chen, J.Y. Characteristics of the antitumor activities in tumor cells and modulation of the inflammatory response in raw264.7 cells of a novel antimicrobial peptide, chrysophsin-1, from the red sea bream (Chrysophrys major). Peptides 2011, 32, 900–910. [Google Scholar] [CrossRef] [PubMed]
- Anju, A.; Smitha, C.K.; Preetha, K.; Boobal, R.; Rosamma, P. Molecular characterization, recombinant expression and bioactivity profile of an antimicrobial peptide, ss-arasin from the indian mud crab, Scylla serrata. Fish Shellfish Immunol. 2019, 88, 352–358. [Google Scholar] [CrossRef]
- Hansen, I.K.O.; Isaksson, J.; Poth, A.G.; Hansen, K.O.; Andersen, A.J.C.; Richard, C.S.M.; Blencke, H.M.; Stensvag, K.; Craik, D.J.; Haug, T. Isolation and characterization of antimicrobial peptides with unusual disulfide connectivity from the colonial ascidian synoicum turgens. Mar. Drugs 2020, 18, 51. [Google Scholar] [CrossRef] [PubMed]
- Papo, N.; Seger, D.; Makovitzki, A.; Kalchenko, V.; Eshhar, Z.; Degani, H.; Shai, Y. Inhibition of tumor growth and elimination of multiple metastases in human prostate and breast xenografts by systemic inoculation of a host defense-like lytic peptide. Cancer Res. 2006, 66, 5371–5378. [Google Scholar] [CrossRef]
- Mai, J.C.; Mi, Z.; Kim, S.H.; Ng, B.; Robbins, P.D. A proapoptotic peptide for the treatment of solid tumors. Cancer Res. 2001, 61, 7709–7712. [Google Scholar]
- Haug, B.E.; Camilio, K.A.; Eliassen, L.T.; Stensen, W.; Svendsen, J.S.; Berg, K.; Mortensen, B.; Serin, G.; Mirjolet, J.F.; Bichat, F.; et al. Discovery of a 9-mer cationic peptide (ltx-315) as a potential first in class oncolytic peptide. J. Med. Chem. 2016, 59, 2918–2927. [Google Scholar] [CrossRef] [PubMed]
- Sveinbjornsson, B.; Camilio, K.A.; Haug, B.E.; Rekdal, O. Ltx-315: A first-in-class oncolytic peptide that reprograms the tumor microenvironment. Future Med. Chem. 2017, 9, 1339–1344. [Google Scholar] [CrossRef]
- Heulot, M.; Jacquier, N.; Aeby, S.; Le Roy, D.; Roger, T.; Trofimenko, E.; Barras, D.; Greub, G.; Widmann, C. The anticancer peptide tat-rasgap317-326 exerts broad antimicrobial activity. Front. Microbiol. 2017, 8, 994. [Google Scholar] [CrossRef] [PubMed]
- Zuccari, G.; Russo, E.; Villa, C.; Calcagno, E.; Mantero, E.; Zorzoli, A.; Marimpietri, D.; Marchitto, L.; Alfei, S. Preparation and Characterization of Amorphous Solid Dispersions for the Solubilization of Fenretinide. Pharmaceuticals 2023, 16, 388. [Google Scholar] [CrossRef]
- Kuz’Mina, U.S.; Raskil’Dina, G.Z.; Ishmetova, D.V.; Sakhabutdinova, G.N.; Dzhumaev, S.S.; Borisova, Y.G.; Vakhitova, Y.V.; Zlotskii, S.S. Cytotoxic Activity Against SH-SY5Y Neuroblastoma Cells of Heterocyclic Compounds Containing gem-Dichlorocyclopropane and/or 1,3-Dioxacycloalkane Fragments. Pharm. Chem. J. 2022, 55, 1293–1298. [Google Scholar] [CrossRef]
- Kelloff, G.J.; Crowell, J.A.; Boone, C.W.; Steele, V.E.; Lubet, R.A.; Greenwald, P.; Alberts, D.S.; Covey, J.M.; Doody, L.A.; Knapp, G.G.; et al. Clinical development plan: N-(4-hydroxyphenyl)retinamide. J. Cell. Biochem. Suppl. 1994, 20, 176–196. [Google Scholar] [PubMed]
- Reynolds, C.P.; Lemons, R.S. Retinoid therapy of childhood cancer. Hematol. Oncol. Clin. North Am. 2001, 15, 867–910. [Google Scholar] [CrossRef]
- Delia, D.; Aiello, A.; Lombardi, L.; Pelicci, P.G.; Grignani, F.; Grignani, F.; Formelli, F.; Menard, S.; Costa, A.; Veronesi, U.; et al. N-(4-Hydroxyphenyl)Retinamide Induces Apoptosis of Malignant Hemopoietic Cell Lines Including Those Unresponsive to Retinoic Acid1. Cancer Res. 1993, 53, 6036–6041. [Google Scholar] [PubMed]
- Kazmi, S.M.; Plante, R.K.; Visconti, V.; Lau, C.Y. Comparison of N-(4-hydroxyphenyl)retinamide and all-trans-retinoic acid in the regulation of retinoid receptor-mediated gene expression in human breast cancer cell lines. Cancer Res. 1996, 56, 1056–1062. [Google Scholar]
- Reynolds, C.P.; Wang, Y.; Melton, L.J.; Einhorn, P.A.; Slamon, D.J.; Maurer, B.J. Retinoic-Acid-Resistant Neuroblastoma Cell Lines Show Altered MYC Regulation and High Sensitivity to Fenretinide. Med. Pediatr. Oncol. 2000, 35, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, M.S.; Shao, Z.-M.; Li, X.-S.; Ordonez, J.V.; Conley, B.A.; Wu, S.; Dawson, M.I.; Han, Q.-X.; Chao, W.; Quick, T.; et al. N-(4-Hydroxyphenyl)Retinamide (4-HPR)-Mediated Biological Actions Involve Retinoid Receptor-Independent Pathways in Human Breast Carcinoma. Carcinogenesis 1995, 16, 2477–2486. [Google Scholar] [CrossRef]
- Supino, R.; Crosti, M.; Clerici, M.; Warlters, A.; Cleris, L.; Zunino, F.; Formelli, F. Induction of Apoptosis by Fenretinide (4HPR) in Human Ovarian Carcinoma Cells and Its Association with Retinoic Acid Receptor Expression. Int. J. Cancer 1996, 65, 491–497. [Google Scholar] [CrossRef]
- Patruno, I.; Thompson, D.; Dall’Angelo, S.; Windhorst, A.D.; Vugts, D.I.; Poot, A.J.; Mody, N.; Zanda, M. Design, Synthesis, Radiosynthesis and Biological Evaluation of Fenretinide Analogues as Anticancer and Metabolic Syndrome-Preventive Agents. ChemMedChem 2020, 15, 1579. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.M.; Di Pietrantonio, A.M.; Hsieh, T.C. Mechanism of fenretinide (4-HPR)-induced cell death. Apoptosis Int. J. Program. Cell Death 2001, 6, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Debatin, K.M. Caspase activation in cancer therapy. In Caspases—Their Role in Cell Death and Cell Survival; Los, M., Walczak, H., Eds.; Kluwer Academic Press: Amsterdam, Holland, 2002. [Google Scholar]
- Koshibu-Koizumi, J.; Akazawa, M.; Iwamoto, T.; Takasaki, M.; Mizuno, F.; Kobayashi, R.; Abe, A.; Tomoda, A.; Hamatake, M.; Ishida, R. Antitumor activity of a phenoxazine compound, 2-amino-4,4α-dihydro-4α,7-dimethyl-3H-phenoxazine-3-one against human B cell and T cell lymphoblastoid cell lines: Induction of mixed types of cell death, apoptosis, and necrosis. J. Cancer Res. Clin. Oncol. 2002, 128, 363–368. [Google Scholar] [CrossRef]
- Akao, Y.; Nakagawa, Y.; Akiyama, K. Arsenic trioxide induces apoptosis in neuroblastoma cell lines through the activation of caspase 3 in vitro. FEBS Lett. 1999, 455, 59–62. [Google Scholar] [CrossRef]
- Holub, J.L.; Qiu, Y.-Y.; Chu, F.; Madonna, M.B. The role of nerve growth factor in caspase-dependent apoptosis in human BE(2)C neuroblastoma. J. Pediatr. Surg. 2011, 46, 1191–1196. [Google Scholar] [CrossRef]
- Jang, M.H.; Shin, M.C.; Kang, I.S.; Baik, H.H.; Cho, Y.H.; Chu, J.P.; Kim, E.H.; Kim, C.J. Caffeine induces apoptosis in human neuroblastoma cell line SK-N-MC. J. Korean Med. Sci. 2020, 17, 674–678. [Google Scholar] [CrossRef]
- Fulda, S. Apoptosis pathways and neuroblastoma therapy. Curr. Pharm. Des. 2009, 15, 430–435. [Google Scholar] [CrossRef]
- Torkin, R.; Lavoie, J.F.; Kaplan, D.R.; Yeger, H. Induction of caspase-dependent, p53-mediated apoptosis by apigenin in human neuroblastoma. Mol. Cancer Ther. 2005, 4, 1–11. [Google Scholar] [CrossRef]
- Ihre, H.; Hult, A.; Fréchet, J.M.J.; Gitsov, I. Double-Stage Convergent Approach for the Synthesis of Functionalized Dendritic Aliphatic Polyesters Based on 2,2-Bis(hydroxymethyl)propionic Acid. Macromolecules 1998, 31, 4061–4068. [Google Scholar] [CrossRef]
- Ihre, H.; Padilla De Jesús, O.L.; Fréchet, J.M. Fast and convenient divergent synthesis of aliphatic ester dendrimers by anhydride coupling. J. Am. Chem. Soc. 2001, 123, 5908–5917. [Google Scholar] [CrossRef] [PubMed]
- Alfei, S.; Castellaro, S.; Taptue, G.B. Synthesis and NMR characterization of dendrimers based on 2, 2-bis-(hydroxymethyl)-propanoic acid (bis-HMPA) containing peripheral amino acid residues for gene transfection. Org. Commun. 2017, 10, 144–177. [Google Scholar] [CrossRef]
- Alfei, S.; Castellaro, S. Synthesis and characterization of polyester-based dendrimers containing peripheral arginine or mixed amino acids as potential vectors for gene and drug delivery. Macromol. Res. 2017, 25, 1172–1186. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AMP Name | Structure Class | Net Charge | Source | Tumor Target | Mechanism | Ref. |
|---|---|---|---|---|---|---|
| Cathelicidins LL37 hCAP18 | Unknown | 6 | Human | HTC/STC | MP/Apoptosis | [31,32,33,34,35] |
| α-Defensin-1 HNP-1 | Beta | 3 | Human | HTC/STC | Apoptosis | [36,37] |
| Human b-defensin-3 (hBD3) | Mixed | 11 | Human | HTC/STC | MP | [38] |
| Lactoferricin B (LfcinB) | Beta | 8 | Bovine | HTC/STC | MP/Apoptosis | [39,40] |
| Gomesin | Beta | 6 | Spider | STC | MP | [41] |
| Mastoparan-C (MP-C) | Helix | 4 | Venom | STC | Apoptosis | [42] |
| Cecropin B | Unknown | 8 | Silk moth | HTC/STC | MP/Apoptosis | [43,44] |
| Magainin 2 | Helix | 3 | Frog | HTC/STC | MP | [45,46] |
| CA-MA-2 | Helix | 8 | Hybrid | STC | MP | [47] |
| BuforinIIb | Unstructured | 7 | Frog | HTC/STC | Apoptosis | [48] |
| Brevenin-2R | Helix | 5 | Frog | STC | LDP | [49] |
| LFB | Unknown | 4 | Frog | STC | MP | [50] |
| Phylloseptin-PHa | Helix | 2 | Frog | STC | MP | [51] |
| Ranatuerin-2PLx | Helix | 2 | Frog | STC | Apoptosis | [52] |
| Dermaseptin-PS1 | Helix | 5 | Frog | STC/ICD | MP | [53,54] |
| Dermaseptin (DPT9) | Helix | 2 | Phyllomedusatarsius | STC | MP | [55] |
| chrysophsin-1 | Helix | 6 | Red sea bream | HTC/STC | MP | [56] |
| Ss-arasin | Bridge | 8 | Indian mud crab | STC | Uncharacterized | [57] |
| Turgencin A and B | Helix | 3 | Synoicum turgens | STC | Uncharacterized | [58] |
| D-K6L9 | Helix | 3 | Engineered | STC | MP | [59] |
| KLA | Unknown | 19 | Engineered | STC | MP | [60] |
| LTX-315 | Unknown | 5 | Engineered | HTC/STC | MP/ICD | [61,62] |
| TAT-RasGAP317-326 | Unknown | 8 | Engineered | STC | MP | [63] |
| Cells | Times (h) | CB1H (µM) | P7 NPs (µM) | CB1H-P7 NPs (µM) |
|---|---|---|---|---|
| IMR 32 | 24 | 25.45 | 1.16 | 0.47 |
| 48 | 24.55 | 1.70 | 0.43 | |
| 72 | 25.05 | 1.63 | 0.46 | |
| SHSY 5Y | 24 | 31.25 | 1.13 | 0.54 |
| 48 | 31.76 | 1.00 | 0.52 | |
| 72 | 24.79 | 0.61 | 0.47 | |
| HaCaT * | 24 | 57.30 | 2.10 | 1.50 |
| 48 | 42.69 | 2.38 | 1.42 | |
| 72 | 47.63 | 1.71 | 1.33 | |
| Selectivity Index | ||||
| IMR 32 | 24 | 2.3 | 1.8 | 3.2 |
| 48 | 1.7 | 1.4 | 3.3 | |
| 72 | 1.9 | 1.0 | 2.9 | |
| SHSY 5Y | 24 | 1.8 | 1.9 | 2.8 |
| 48 | 1.3 | 2.4 | 2.7 | |
| 72 | 1.9 | 2.8 | 2.8 | |
| Compound | Cells | Times | Sign. Viability Reduction (µM) a | Alive Cells (%) | IC50 (µM) |
|---|---|---|---|---|---|
| 4-HPR | IMR 32 | 24 h | 1 | 28.2 | 1.09 |
| 48 h | 1 | 39.3 | 1.93 | ||
| 72 h | 0.5 | 40.4 | 0.68 | ||
| SHSY-5Y | 24 h | 5 | 45.9 | 7.84 | |
| 48 h | 0.5 | 74.4 | 4.32 | ||
| 72h | 1 | 76.2 | 4.99 | ||
| CB1H-P7 | IMR 32 | 24 h | 0.43 | 51.5 | 0.47 |
| 48 h | 0.43 | 52.6 | 0.43 | ||
| 72 h | 0.43 | 50.8 | 0.46 | ||
| SHSY-5Y | 24 h | 0.43 | 44.9 | 0.54 | |
| 48 h | 0.43 | 58.0 | 0.52 | ||
| 72 h | 0.43 | 26.4 | 0.47 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zuccari, G.; Zorzoli, A.; Marimpietri, D.; Brullo, C.; Alfei, S. Pyrazole-Enriched Cationic Nanoparticles Induced Early- and Late-Stage Apoptosis in Neuroblastoma Cells at Sub-Micromolar Concentrations. Pharmaceuticals 2023, 16, 393. https://doi.org/10.3390/ph16030393
Zuccari G, Zorzoli A, Marimpietri D, Brullo C, Alfei S. Pyrazole-Enriched Cationic Nanoparticles Induced Early- and Late-Stage Apoptosis in Neuroblastoma Cells at Sub-Micromolar Concentrations. Pharmaceuticals. 2023; 16(3):393. https://doi.org/10.3390/ph16030393
Chicago/Turabian StyleZuccari, Guendalina, Alessia Zorzoli, Danilo Marimpietri, Chiara Brullo, and Silvana Alfei. 2023. "Pyrazole-Enriched Cationic Nanoparticles Induced Early- and Late-Stage Apoptosis in Neuroblastoma Cells at Sub-Micromolar Concentrations" Pharmaceuticals 16, no. 3: 393. https://doi.org/10.3390/ph16030393
APA StyleZuccari, G., Zorzoli, A., Marimpietri, D., Brullo, C., & Alfei, S. (2023). Pyrazole-Enriched Cationic Nanoparticles Induced Early- and Late-Stage Apoptosis in Neuroblastoma Cells at Sub-Micromolar Concentrations. Pharmaceuticals, 16(3), 393. https://doi.org/10.3390/ph16030393