
30th Annual GP2A Medicinal Chemistry Conference

 , ,
, ,  , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- Best Presentation by a PhD Student: Magdalena Wojciechowski, University of Münster, Institute of Pharmaceutical and Medicinal Chemistry. Prize sponsored by the European Journal of Medicinal Chemistry.
- Runner-Up Presentation by a PhD Student: Christina Kosch, Helmholtz Institute of Pharmaceutical Research Saarland (HIPS). Prize sponsored by RSC Medicinal Chemistry.
- Best Presentation by a Postdoctoral Researcher: Dr. Manuela Jörg, Medicinal Chemistry, Monash University. Prize sponsored by Almac Group.
- Runner-Up Presentation by a Postdoctoral Researcher: Dr. Mariia Nesterkina, Helmholtz Institute of Pharmaceutical Research Saarland (HIPS). Prize sponsored by Pharmaceuticals.
- Best Poster Presentations: Julian Breidenbach (Rheinische Friedrich-Wilhelms-Universität Bonn), Hugo Bloux (CERMN, University of Caen), Aline Renata Pavan (UNESP, Brazil), Rhys Francis (The University of Nottingham), Nikolina Stipaničev (Trinity College Dublin). Prizes sponsored by the European Journal of Medicinal Chemistry, European Journal of Medicinal Chemistry, RSC Medicinal Chemistry, ThermoFisher, and CEM.
2. Keynote Lectures
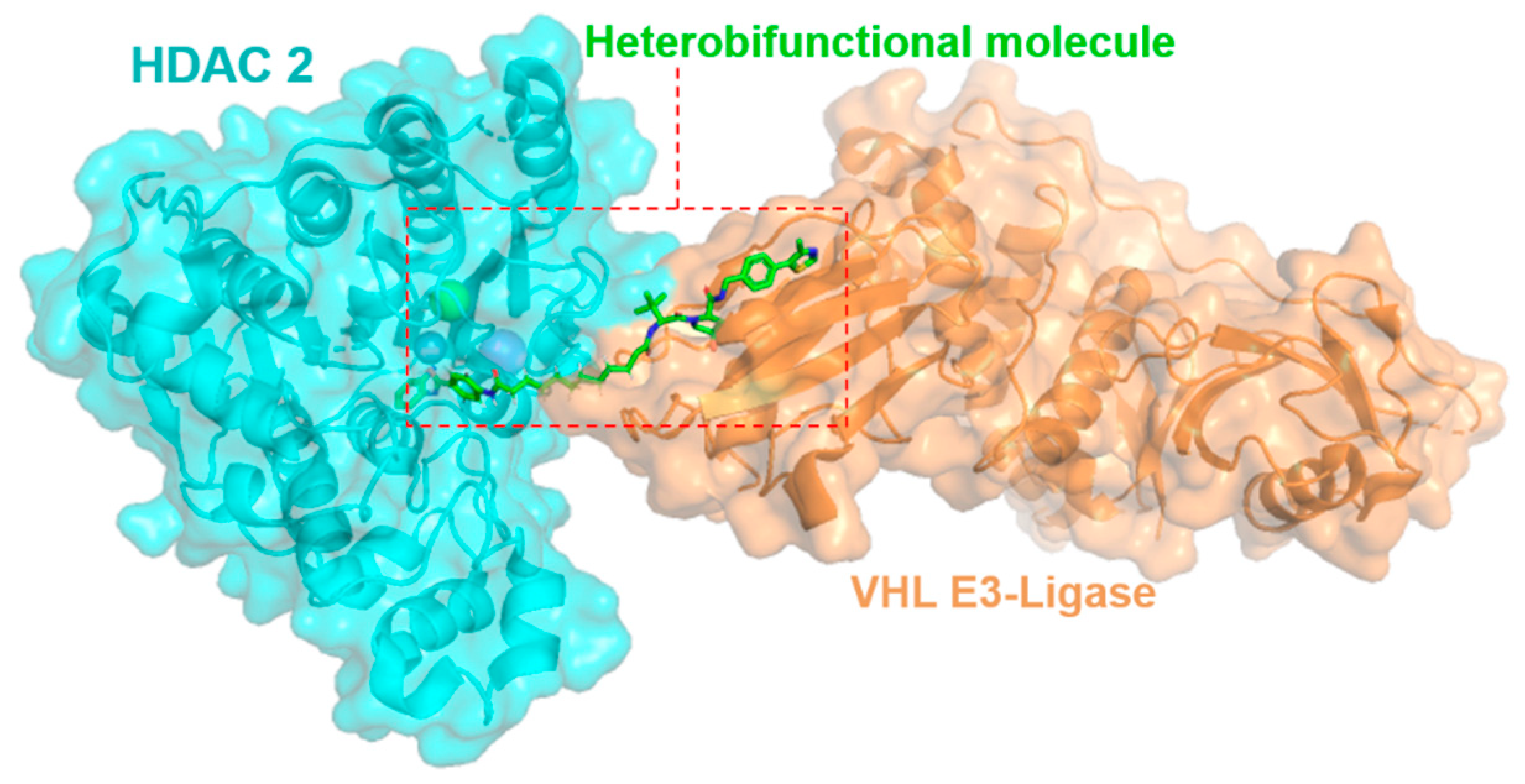
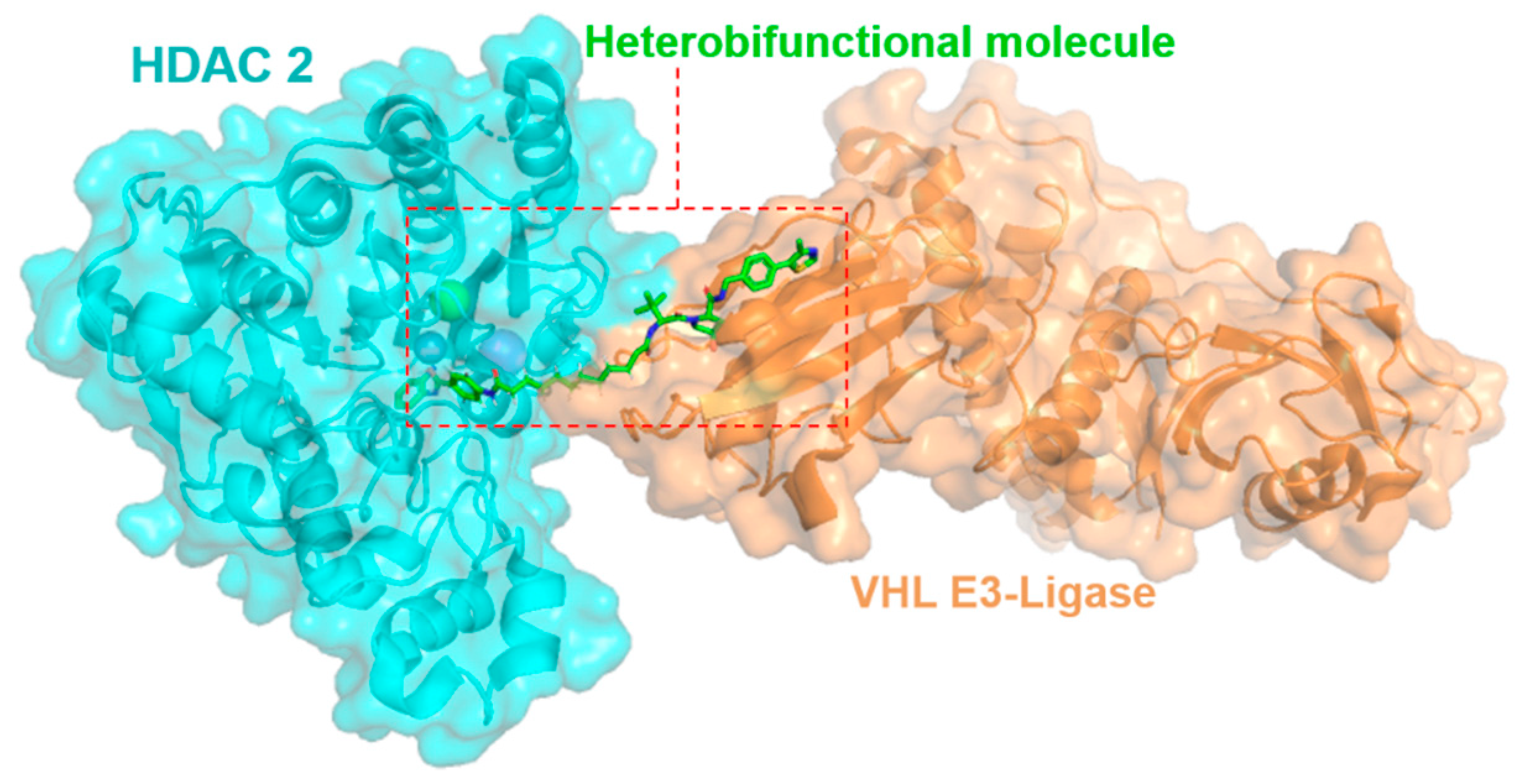
2.1. Exploring Heterobifunctional Molecules and Class-I Histone Deacetylase Enzymes in Complexes
2.2. Targeting the Protein Kinases’ Cysteinome: Design of Covalent MPS1/TTK and BMX Inhibitors
2.3. A Tricycle Journey to In Vivo Degraders of the Oncogenic Transcription Factor BCL6
2.4. IDE and ERAP Inhibitors Discovered by KTGS and Their Use in Therapeutics
2.5. Structure-Affinity Relationship Study of a Potential Therapeutic Peptide for the Treatment of MELAS
2.6. New Strategies for the Identification and Control of Disease Relevant Protein-Protein Interactions
2.7. Addressing Unusual Anti-Infective Targets
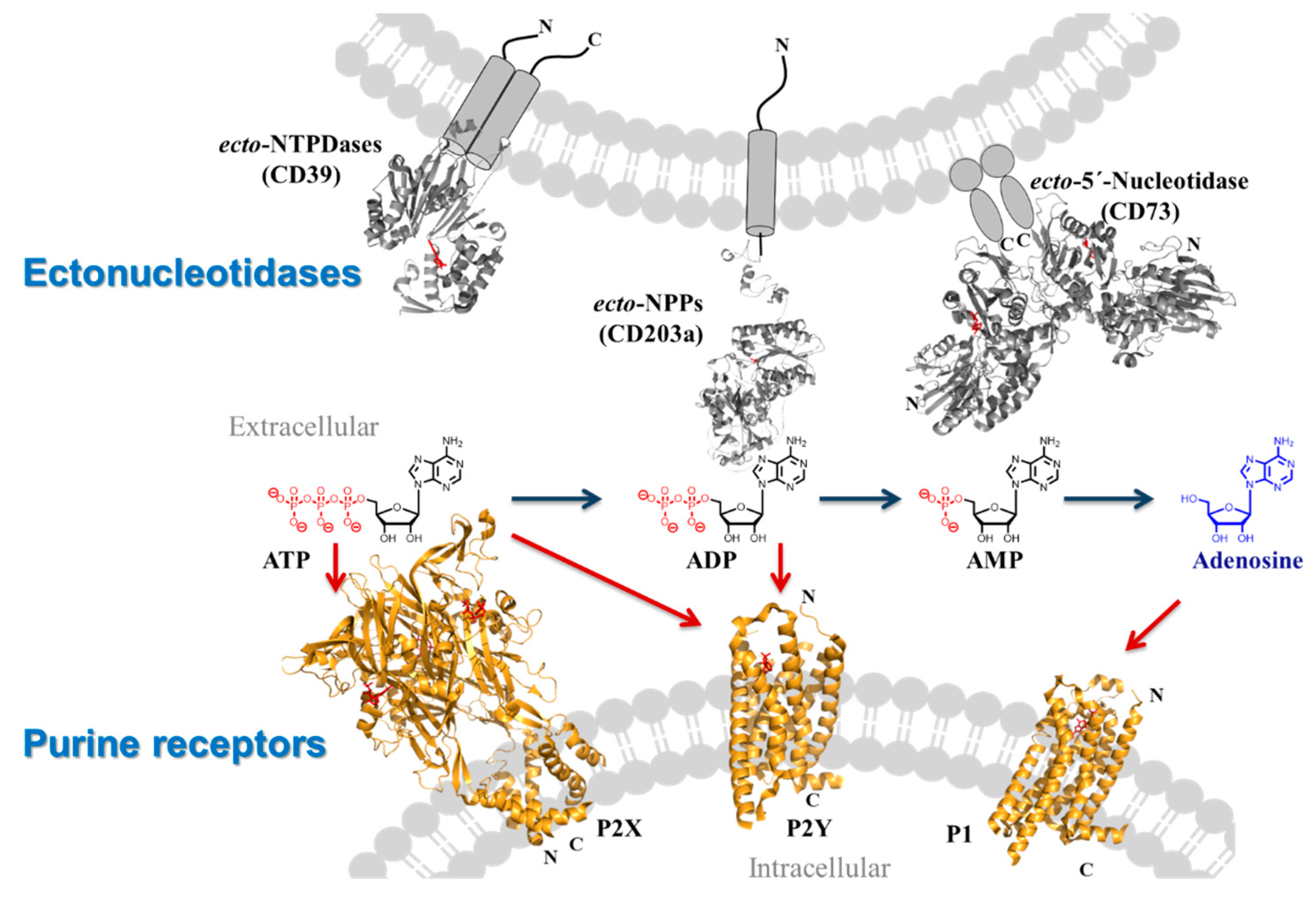
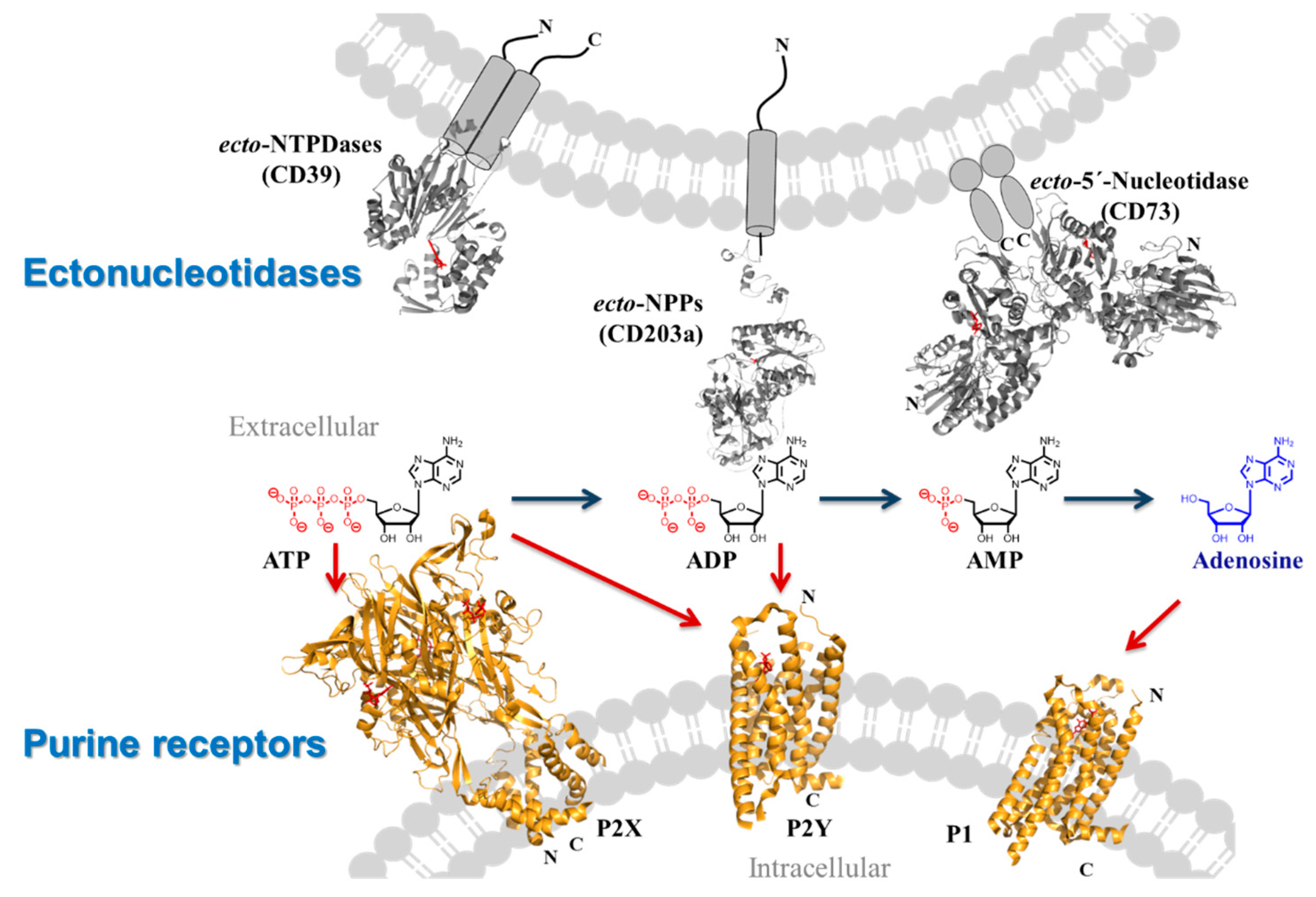
2.8. Targeting Membrane Proteins Involved in Purinergic Signalling–Important Players in Inflammation, Immunity and Cancer
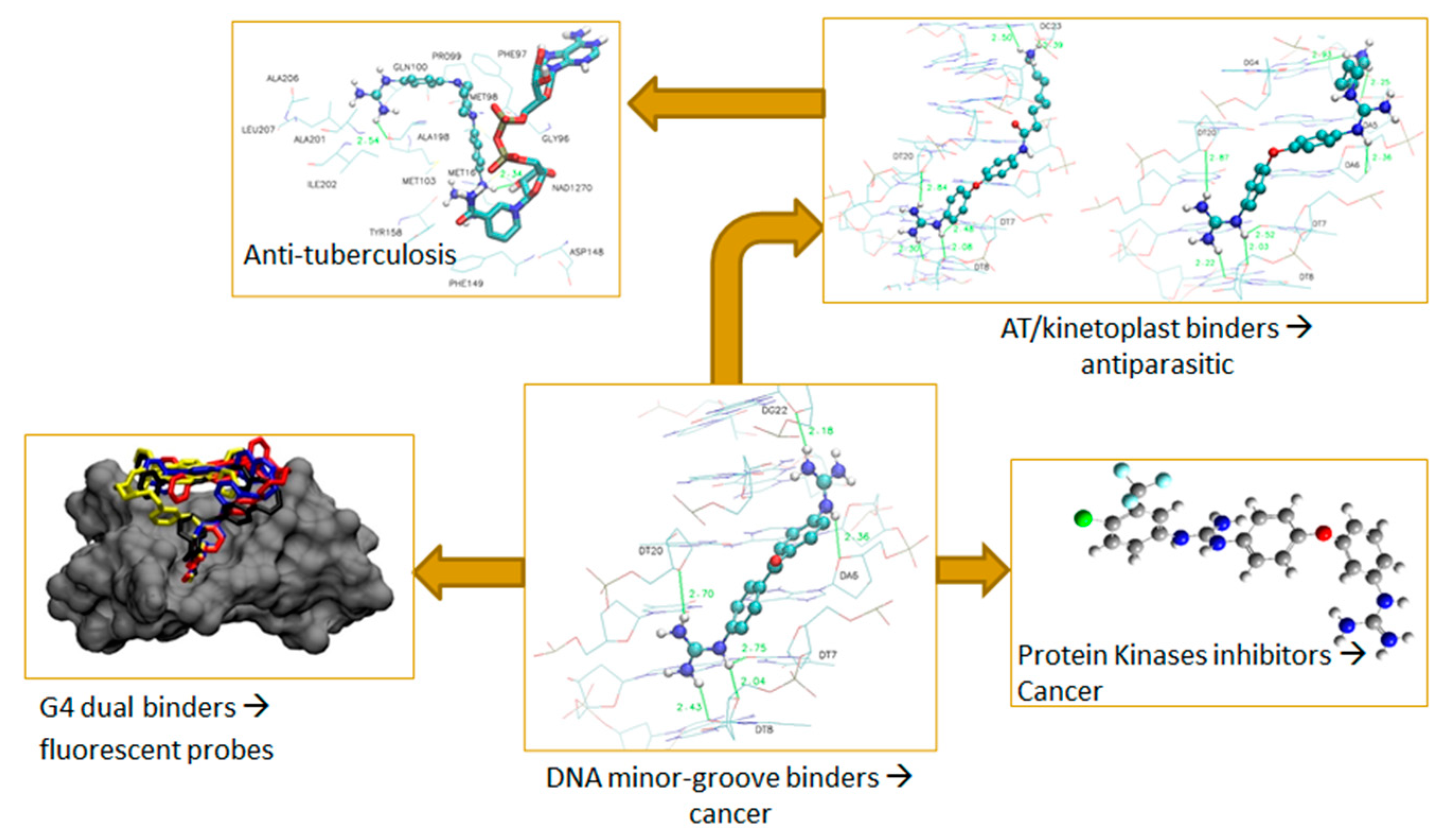
2.9. Where Targeting DNA Can Take You…
3. Early Career Researcher Presentations
3.1. Identification and Structure-Activity Relationship Profiling of Positive Allosteric Modulators Targeting Muscarinic Acetylcholine Receptors
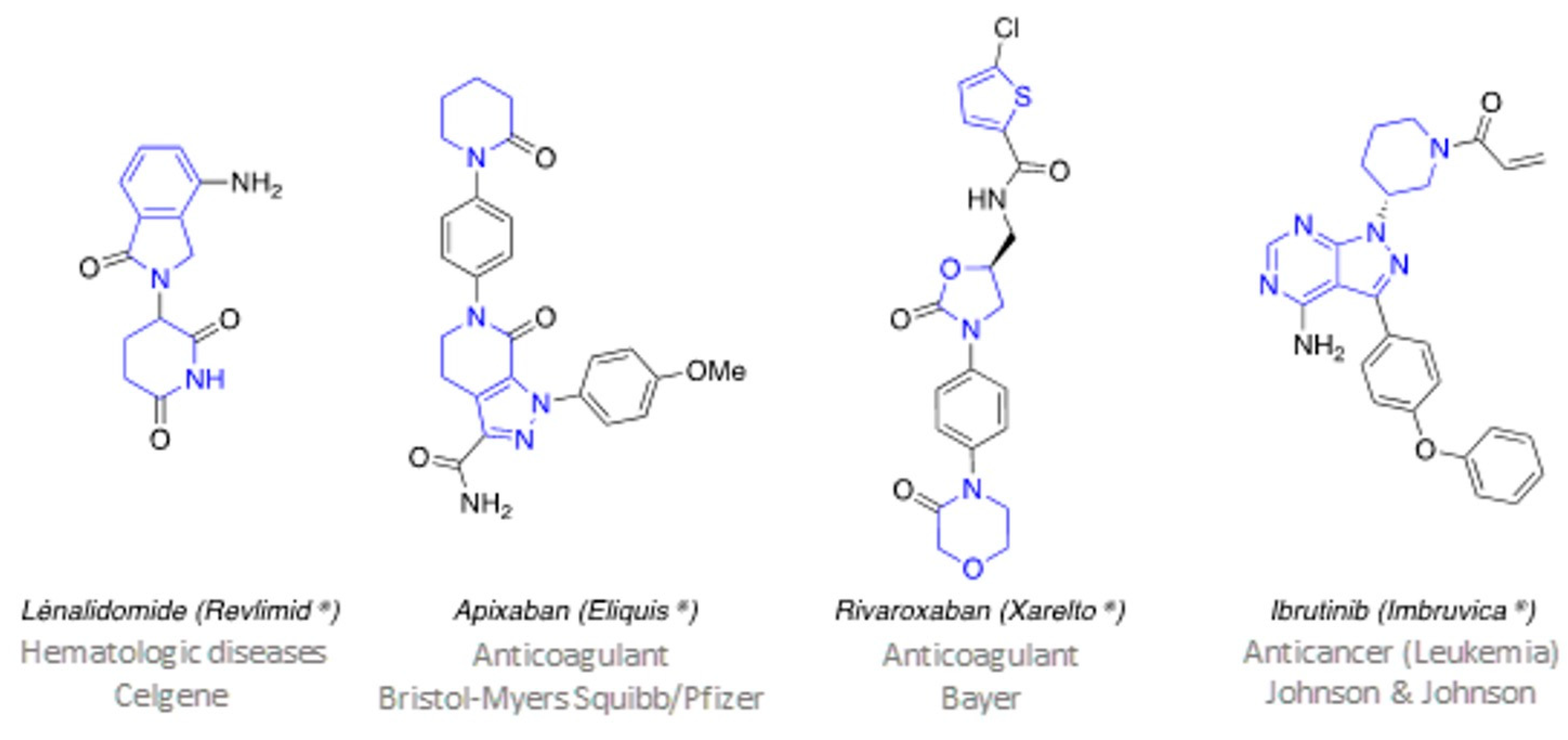
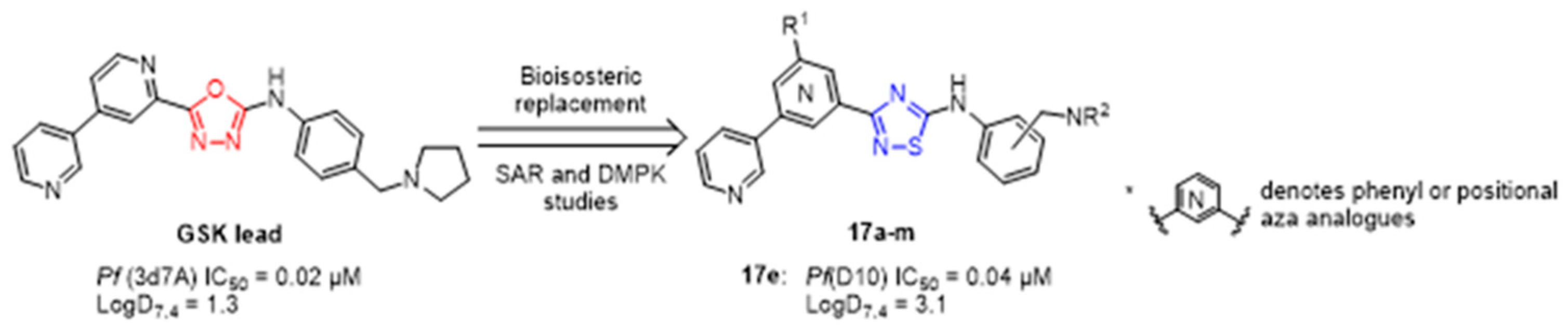
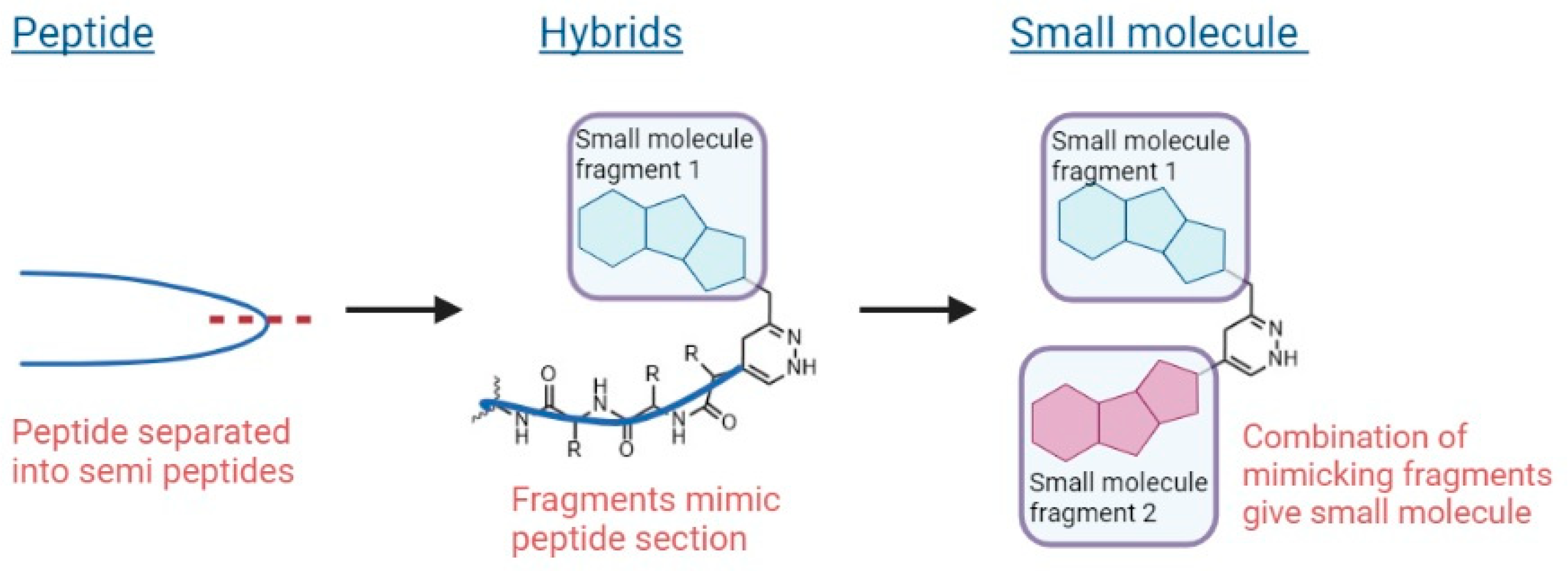
3.2. New Aza-Heterocyclic Structures for Feeding the Drug Space in Medicinal Chemistry
3.3. A novel Polycerasoidol Analogue with Pan-PPAR Agonism, Anti-Inflammatory Effects and Amelioration of Metabolic Abnormalities in Ob/Ob Mice
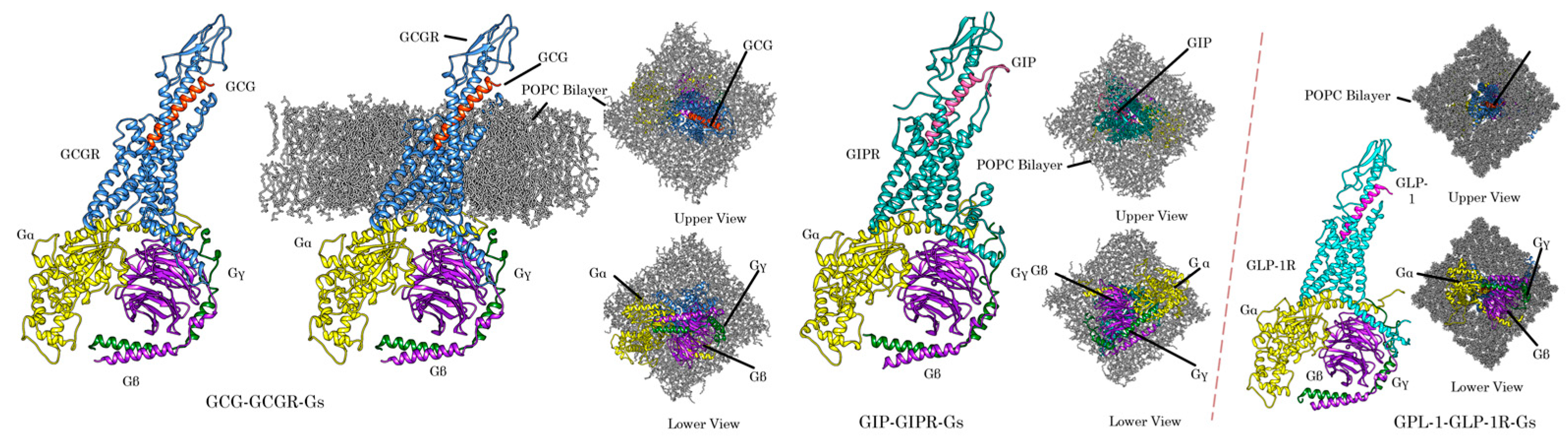
3.4. Triagonist Peptide Designed to Activate Incretin/Glucagon System
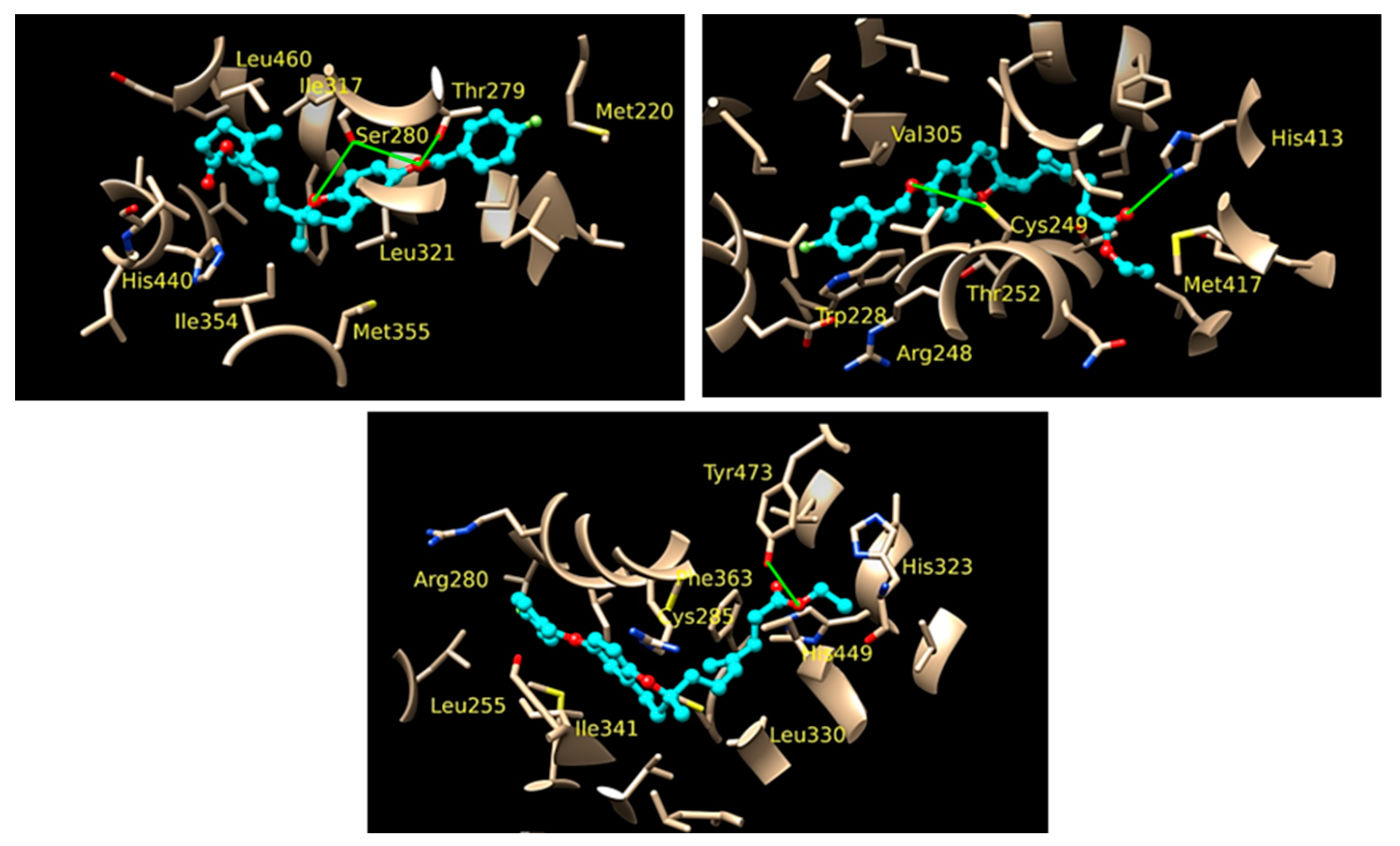
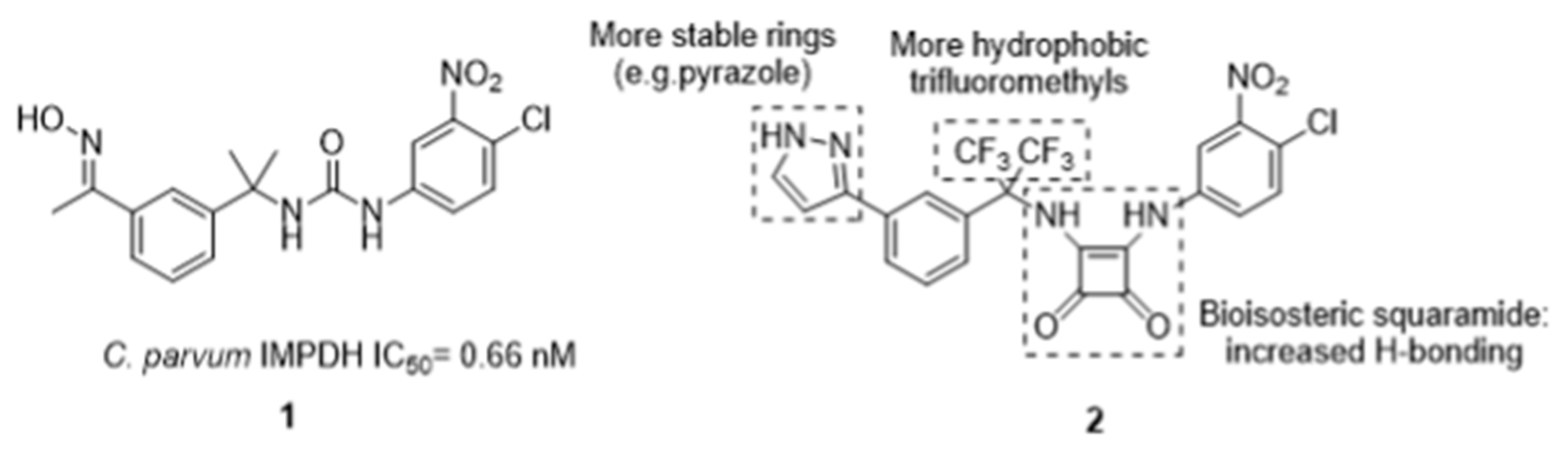
3.5. Design and Synthesis of Novel Drugs for the Treatment of Cryptosporidiosis
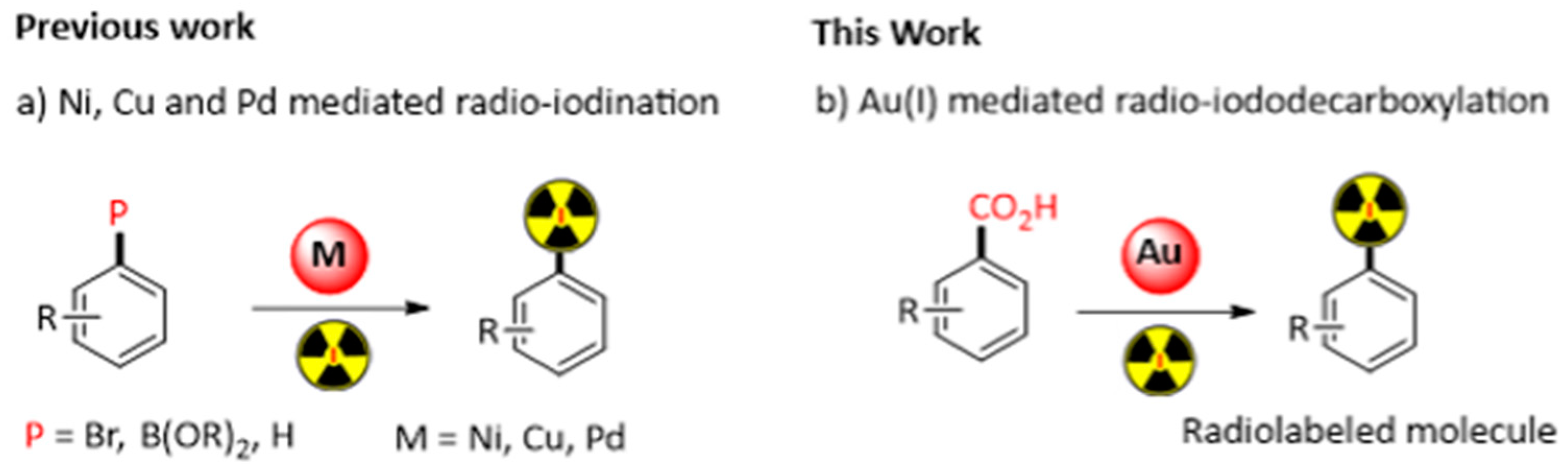
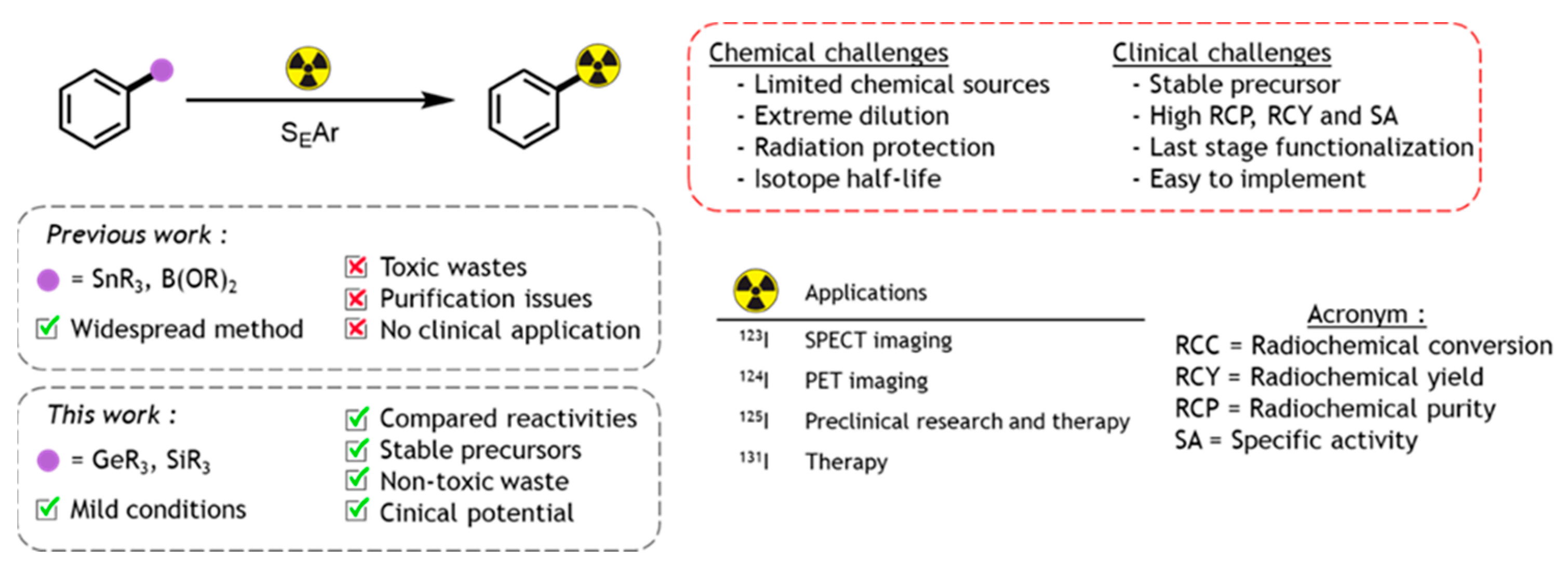
3.6. Gold(I) Mediated Radio-Iododecarboxylation: Application in Nuclear Medicine
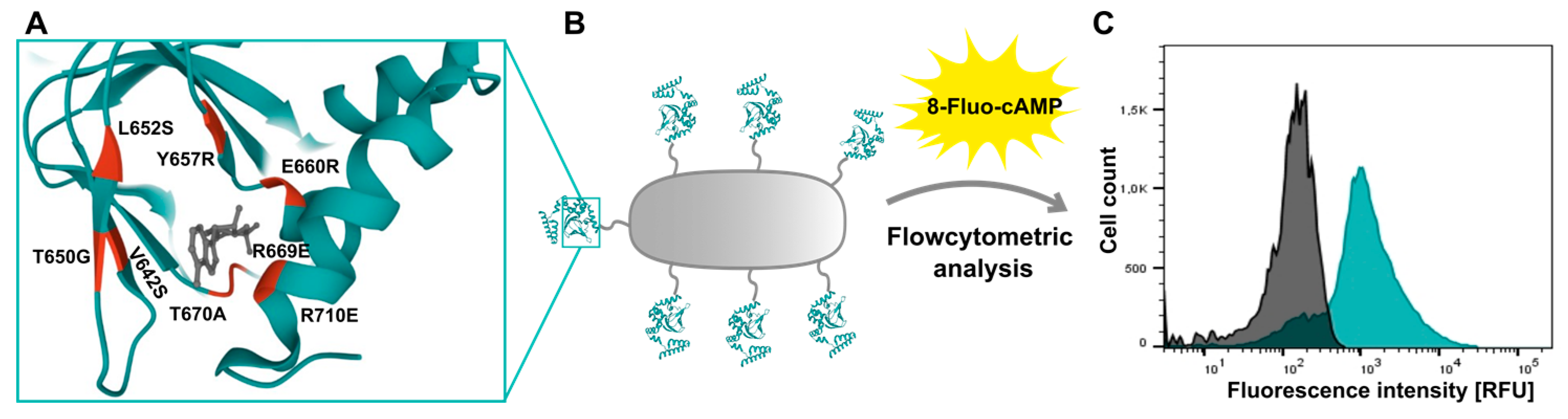
3.7. Mutational Analysis of the Surface Displayed HCN4 C-Linker-CNBD: A Flow Cytometry Based Ligand Binding Approach
3.8. Hit-to-Lead Optimization of a Novel RSV Small Molecule Entry Inhibitor
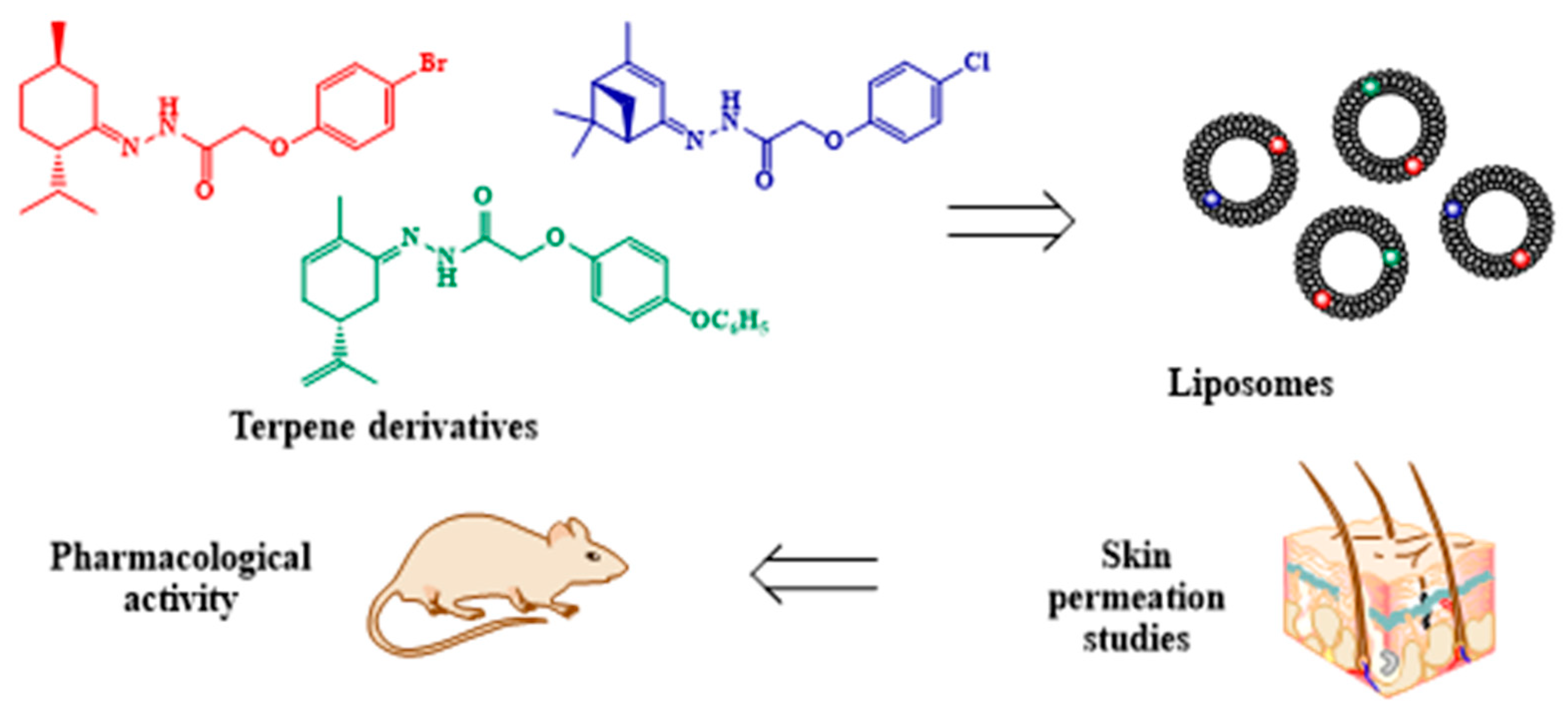
3.9. Terpenoid Hydrazones as Biomembrane Penetration Enhancers with Multi-Target Activity
3.10. Aminobenzofuran-Containing Analogues of Proximicins B and C Exhibit Higher Antiproliferative Aactivity against Glioblastoma Cells Compared to Temozolomide in In vitro Cellular Assays
4. Poster Presentations
4.1. Self-Assemblies of Azacitidine Prodrugs: A Promising Strategy of Treatment for Myelodysplastic Syndromes and Acute Myeloid Leukemia
4.2. Development of Novel Therapeutic Agents to Enhance Insulin Secretion
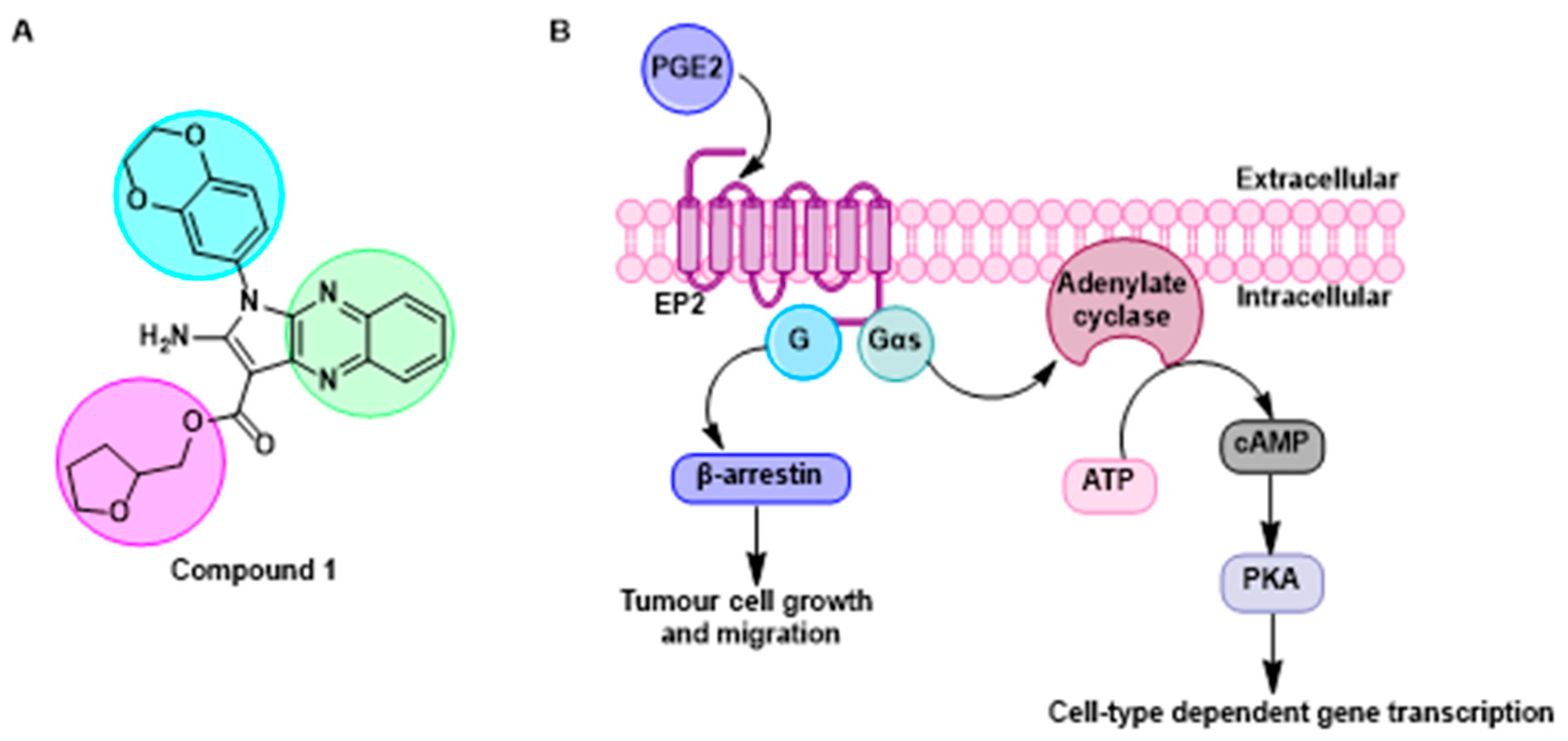
4.3. Design and Synthesis of Novel Fluorescent Allosteric Modulators for the Prostaglandin EP2 Receptor
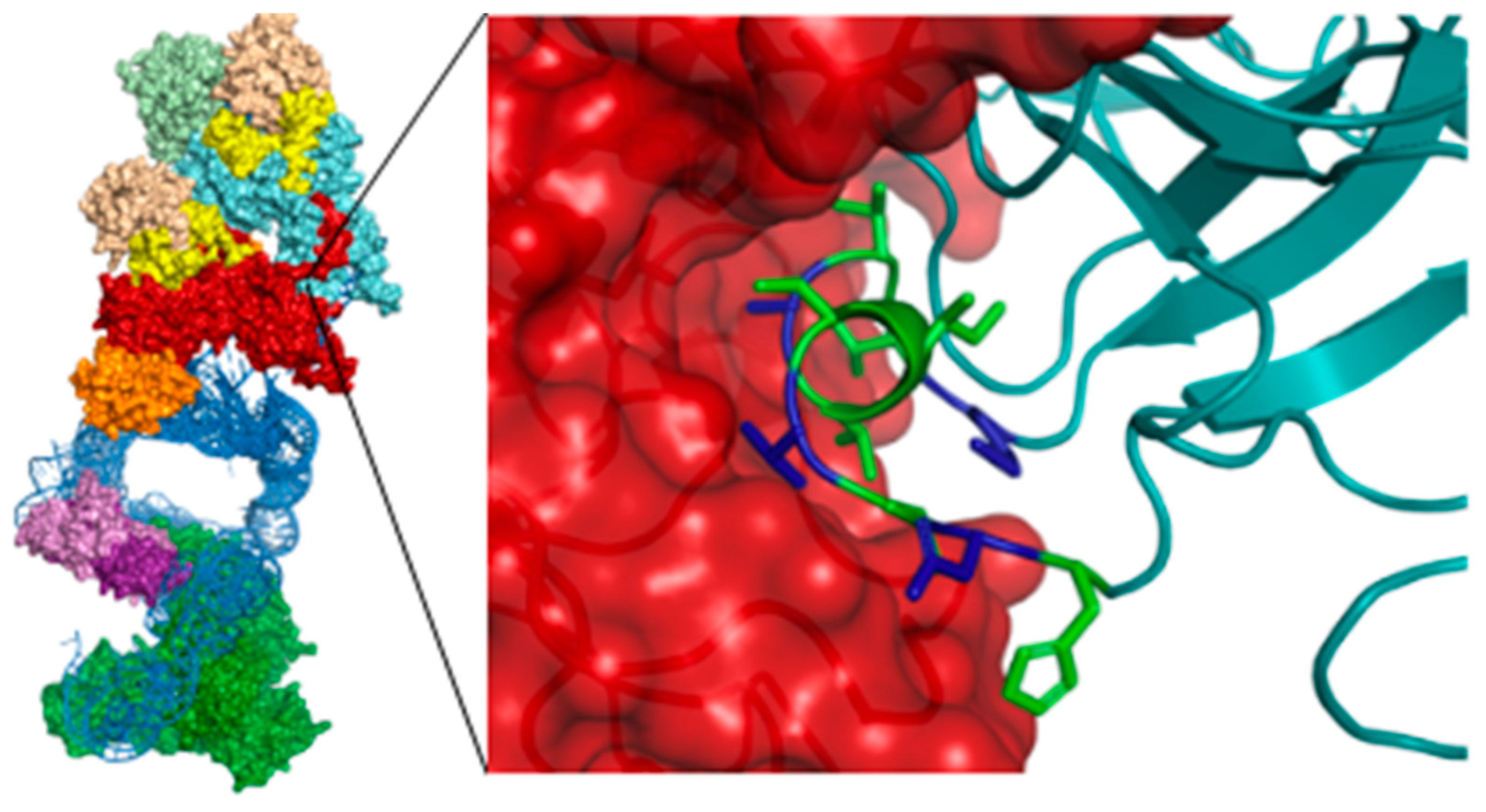
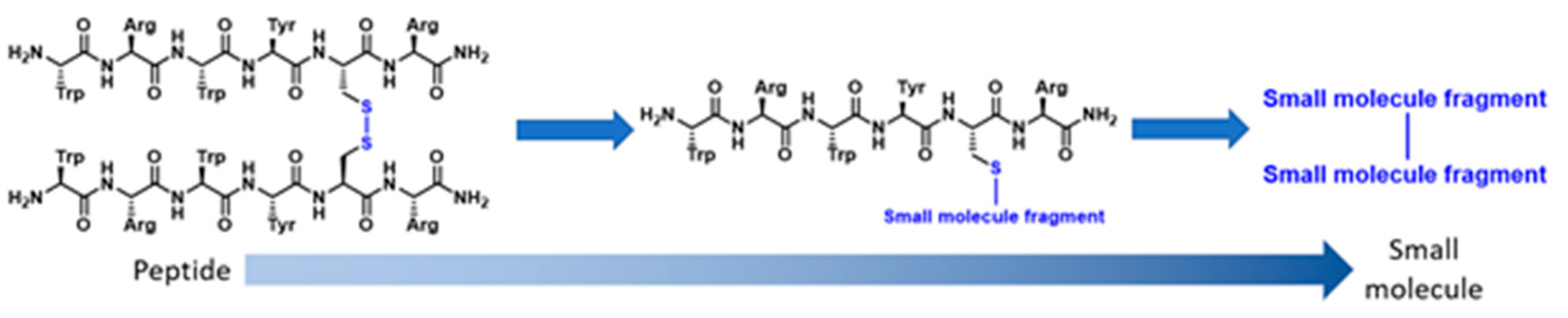
4.4. Removing Cancer’s Immortality: The Design and Synthesis of Linear and Stapled Peptides Targeting the Dyskerin-Dyskerin PPI in Telomerase
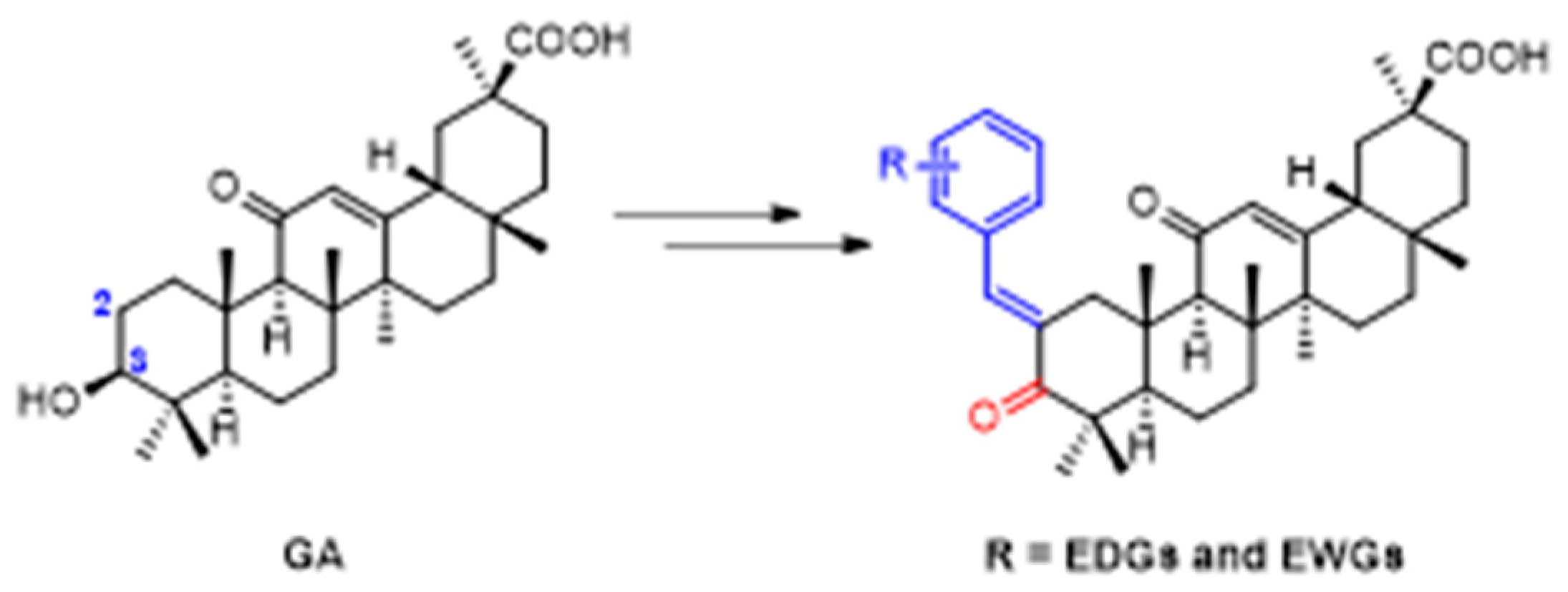
4.5. The Synthesis of 18β-Glycyrrhetinic Acid Derivatives which have Increased Antibacterial Activity against Staphylococcus Aureus
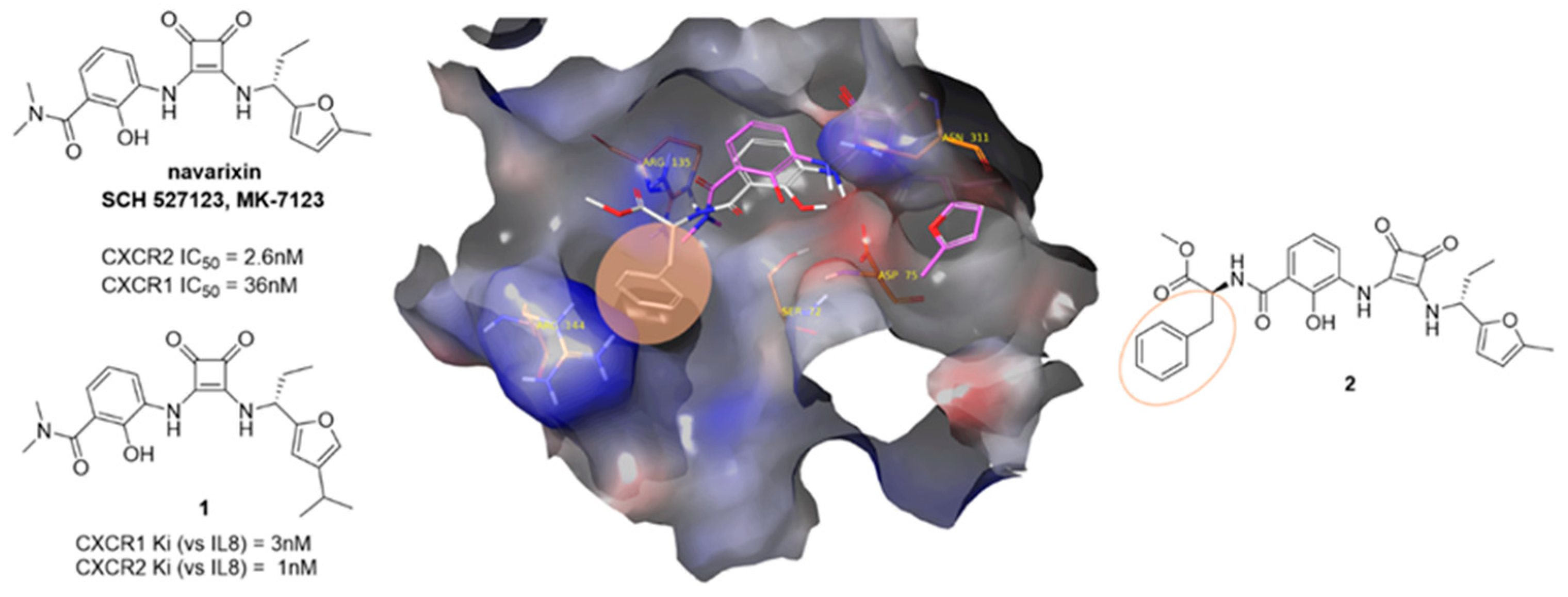
4.6. Exploration of Intracellular Binding Pockets of CXC Chemokine Receptors for Allosteric Modulation
4.7. Base Mediated Radio-Iodination of Arenes from Organo-Silanes and-Germanes as Radiolabelling Precursors
4.8. A Marine Fungal Vanadium Haloperoxidase as an Alternative for Bromination of Electron-Rich Substrates
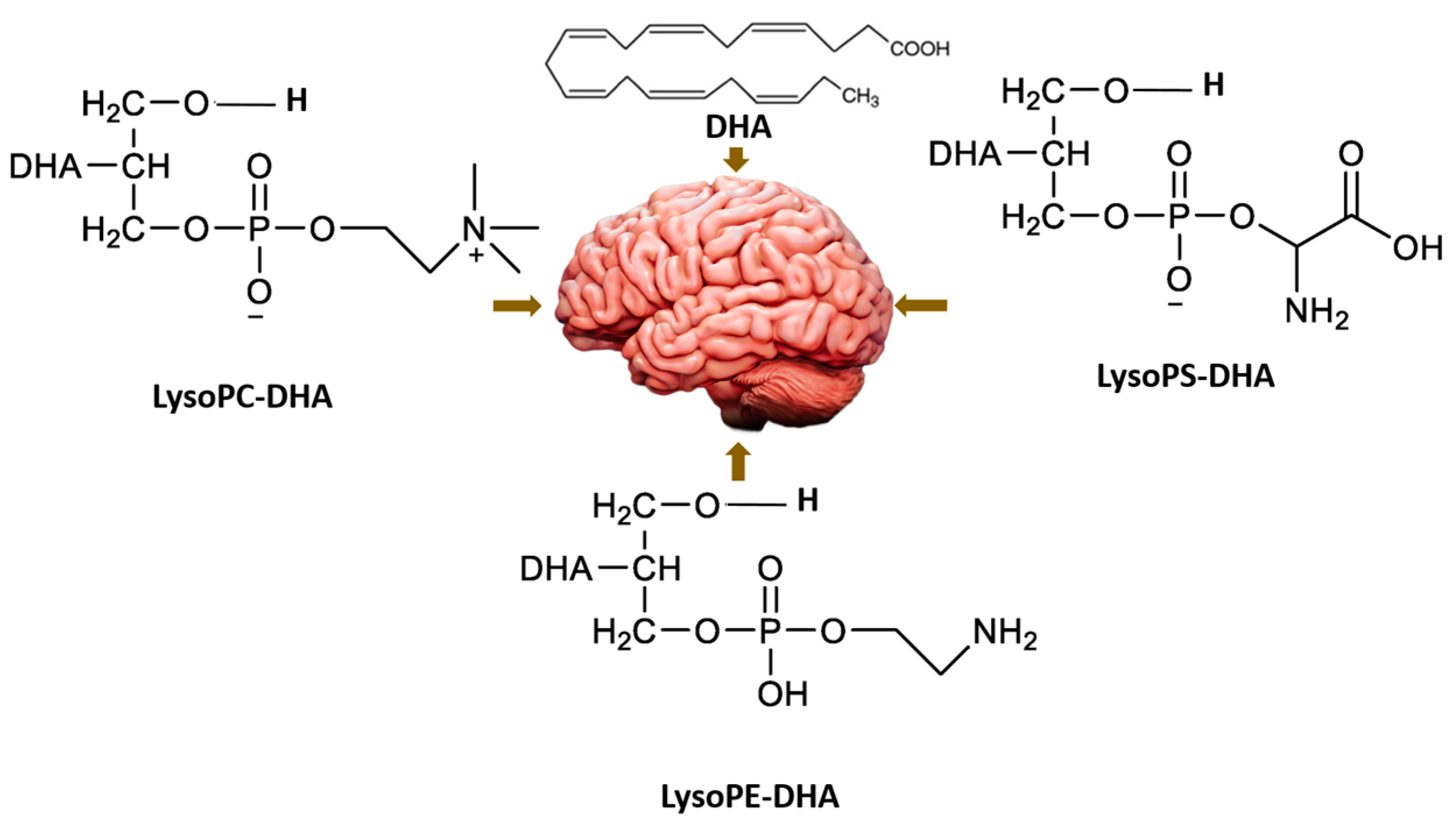
4.9. Emerging Role of Phospholipids and Lysophospholipids for Improving Brain Docosahexaenoic Acid as Potential Preventive and Therapeutic Strategies for Neurological Diseases
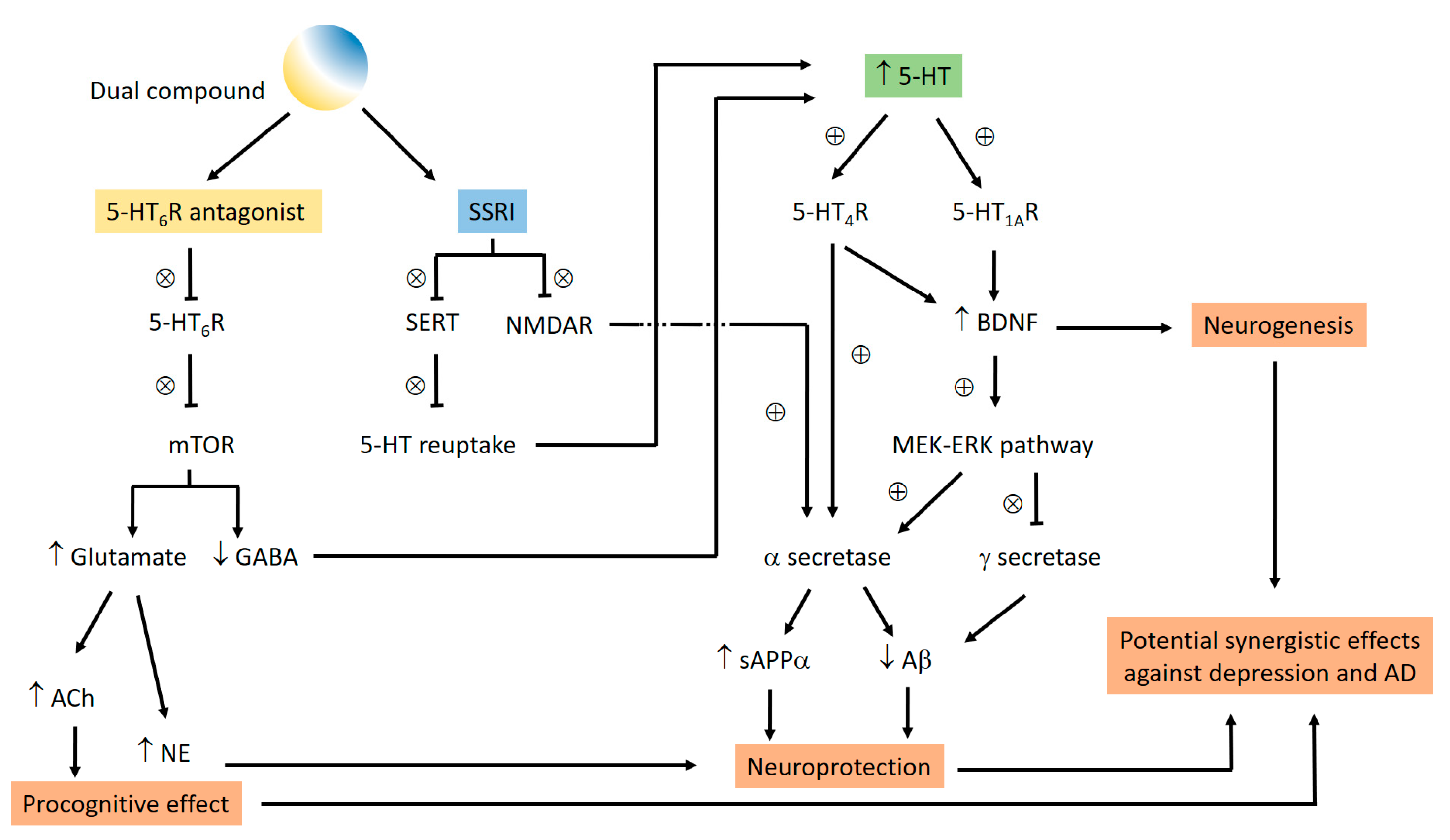
4.10. Playing the Serotoninergic Piano against Alzheimer’s Disease
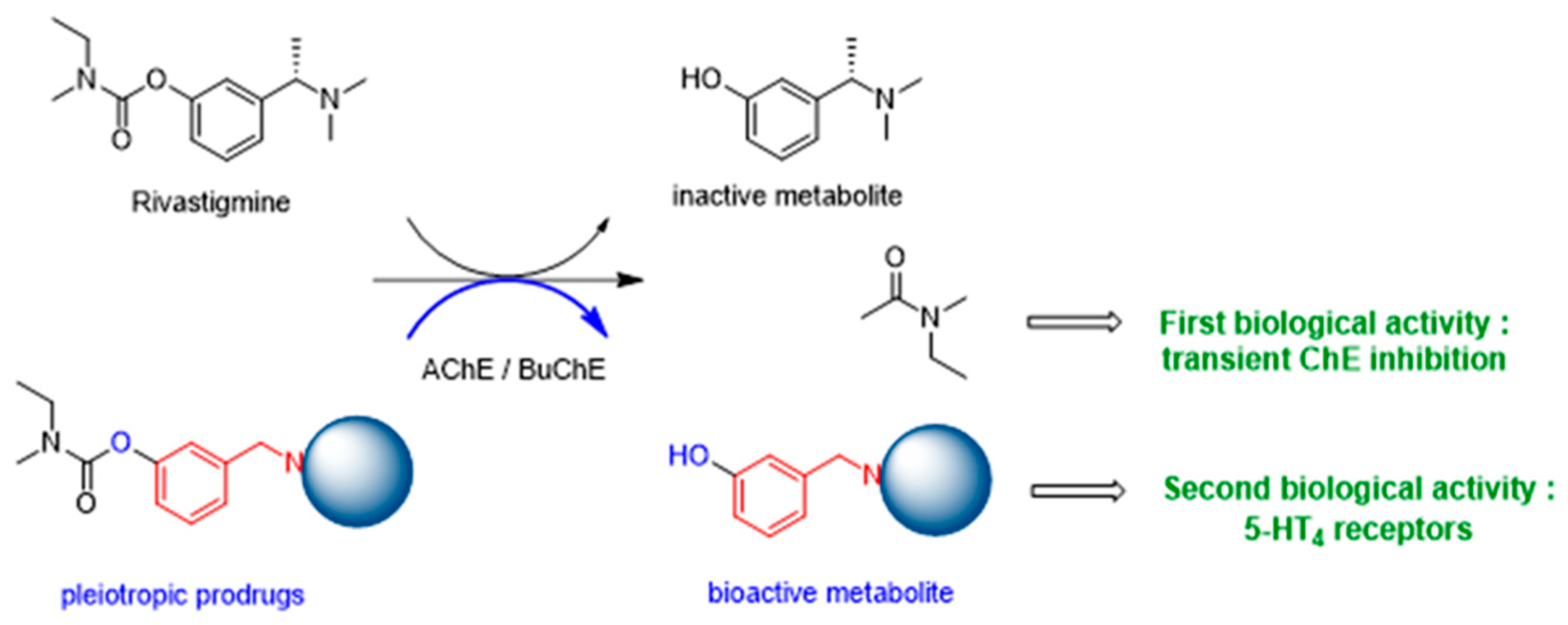
4.11. Innovative Pleiotropic Prodrugs with Potential Therapeutic Interest in Down Syndrome and Alzheimer’s Disease
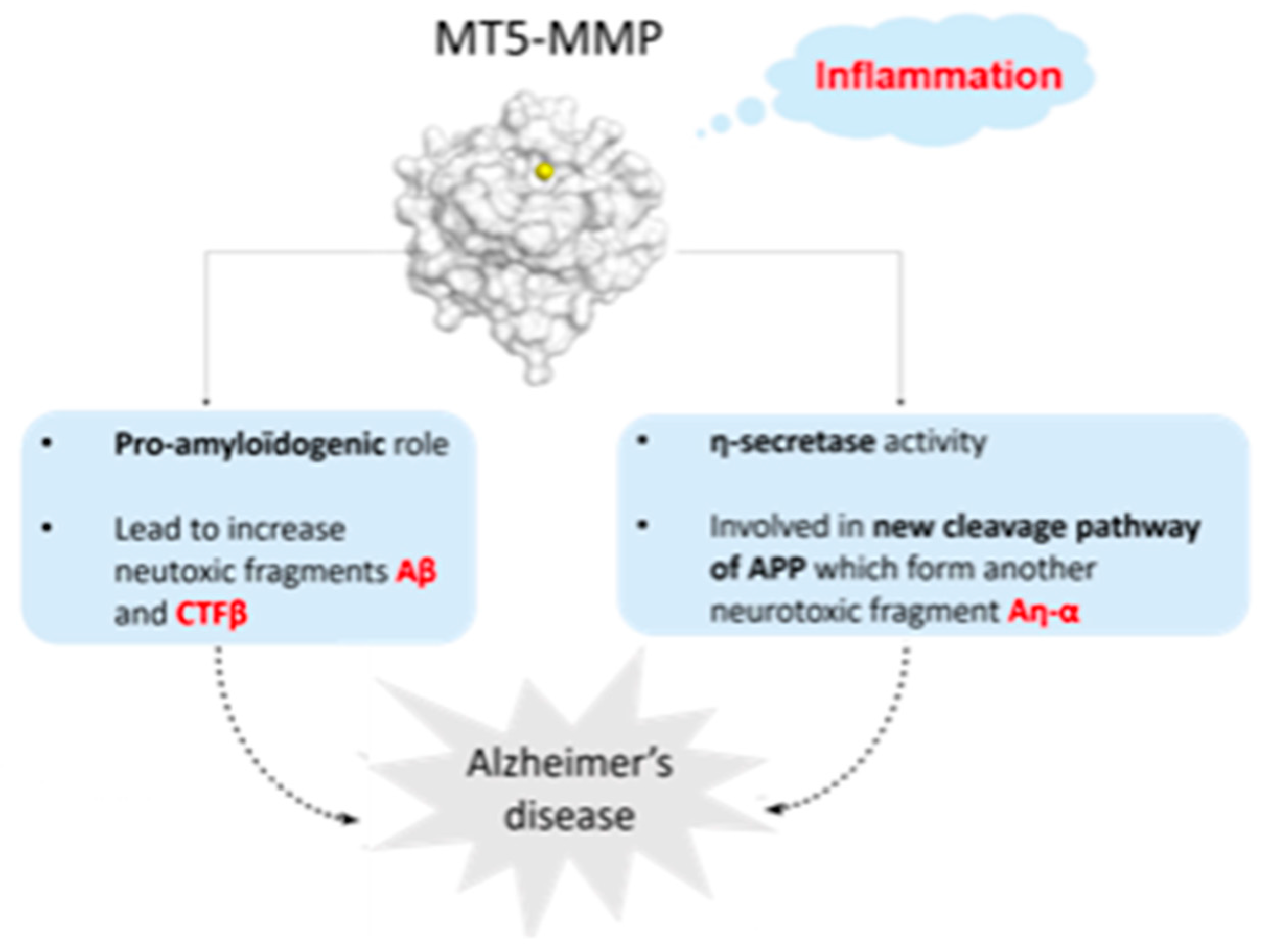
4.12. Drug Design and Biological Evaluation of MT5-MMP (Membrane-Type 5 Matrix MetalloProteinase) Inhibitors for a New Potential Treatment of Alzheimer’s Disease
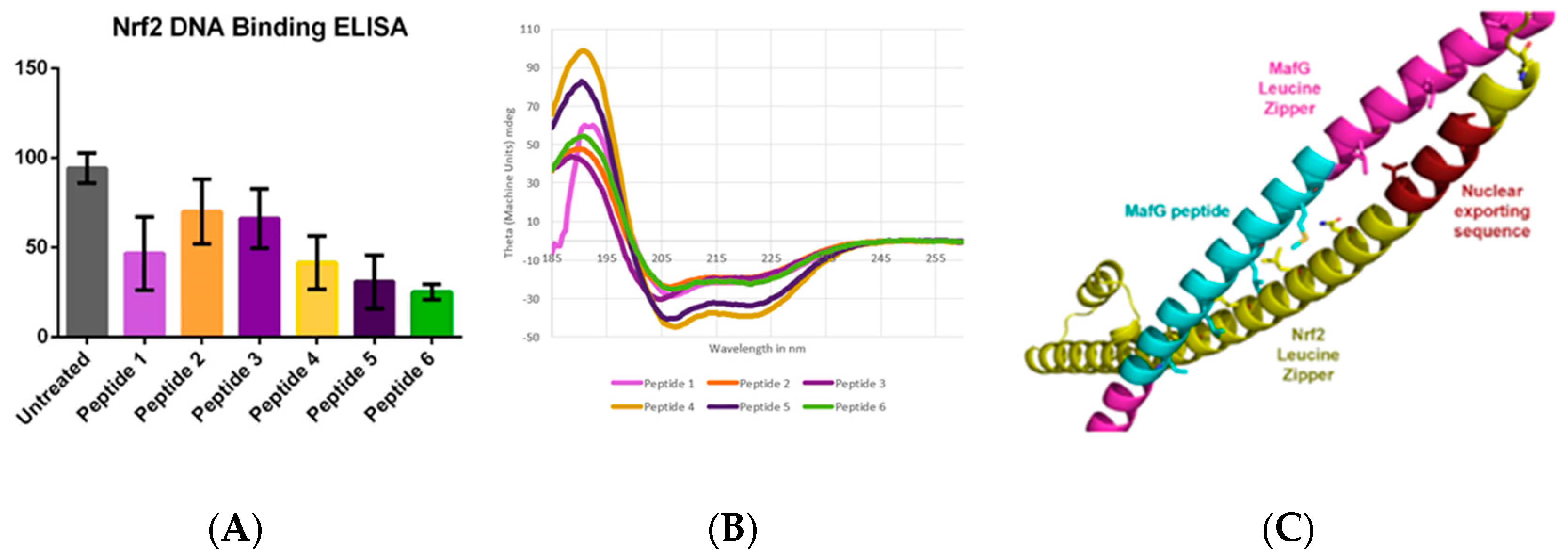
4.13. Controlling Coiled Coils: Harnessing Transcription Factors with Protein-Protein Interactions
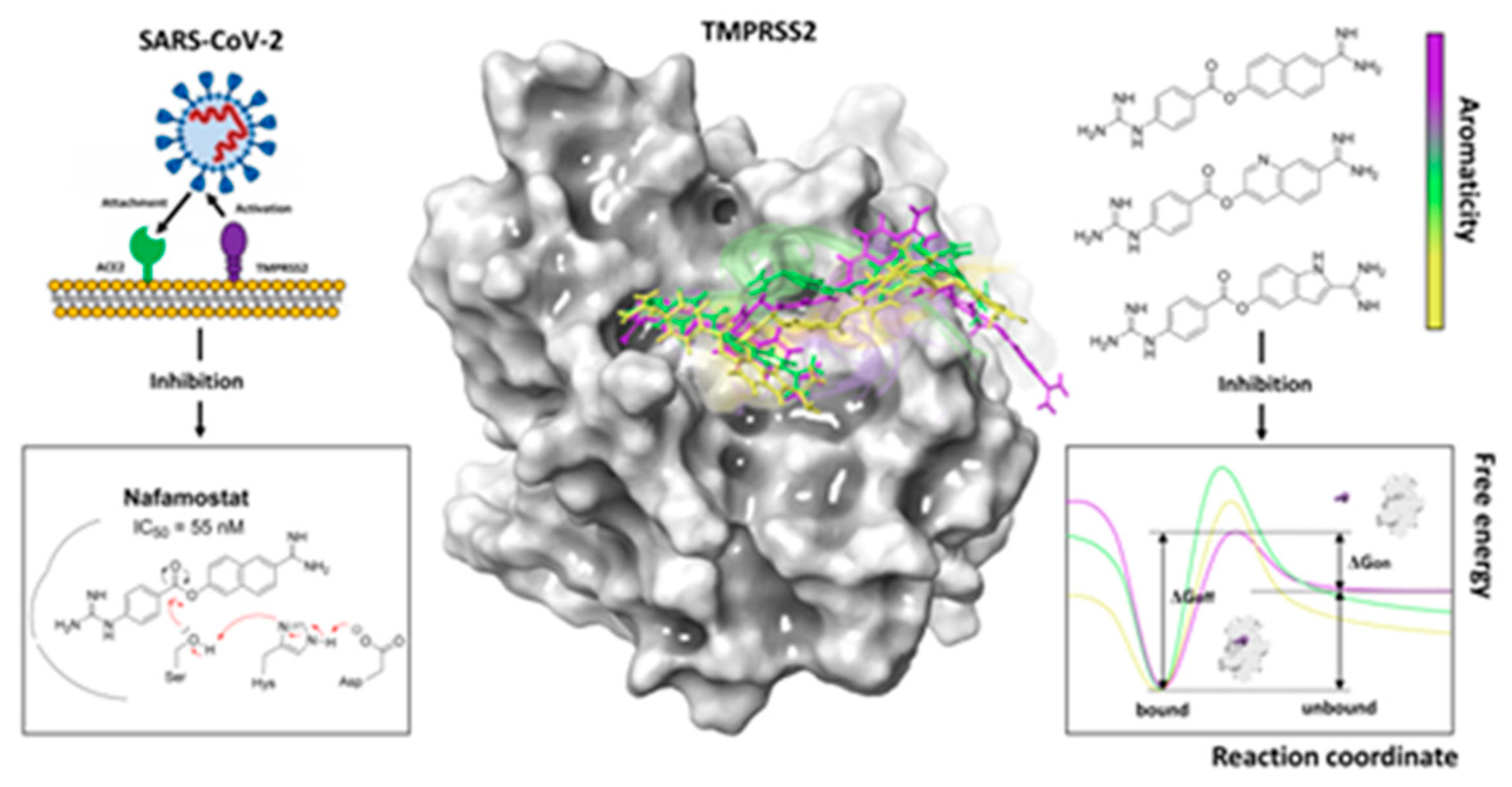
4.14. Influence of Aromaticity on the Covalent Inhibition of Nafamostat Derivatives as Potential SARS-CoV-2 Antiviral Agents
4.15. Identification of Heterocyclic Aminothiadiazoles as Analogues of Fast Acting 1,3,4-Oxadiazole Based Antimalarials
4.16. Design and Synthesis of 12-Thiazole Abietanes as Selective Inhibitors of the Human Metabolic Serine Hydrolase hABHD16A
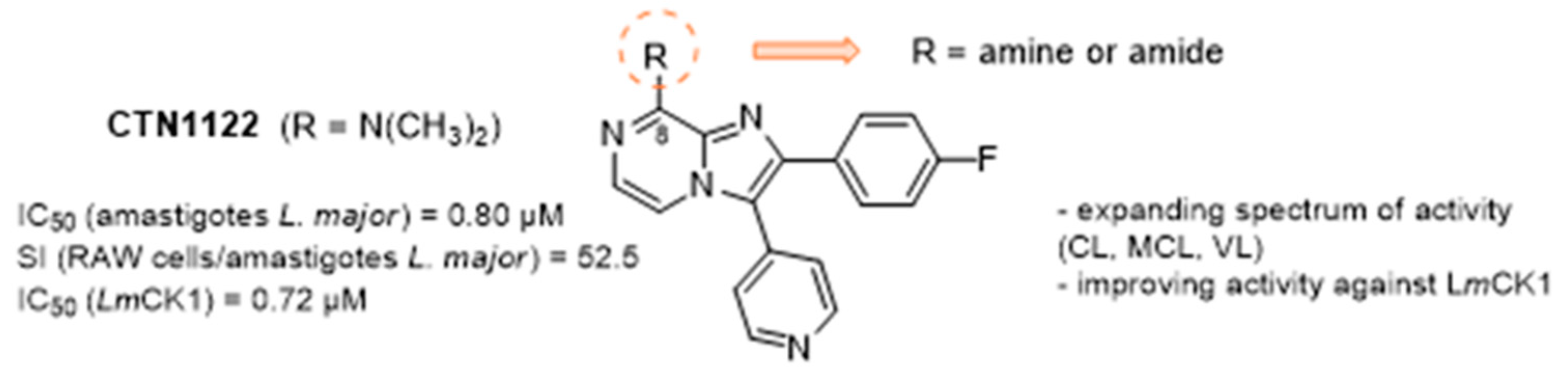
4.17. 8-Substituted 2,3-Diarylimidazo [1,2-a]Pyrazines as Casein Kinase 1 Inhibitors Endowed with Antileishmanial Activity
4.18. 2-Prenylated Benzopyrans with PPARα/γ Agonist Activity as Potential Drugs for Metabolic Syndrome
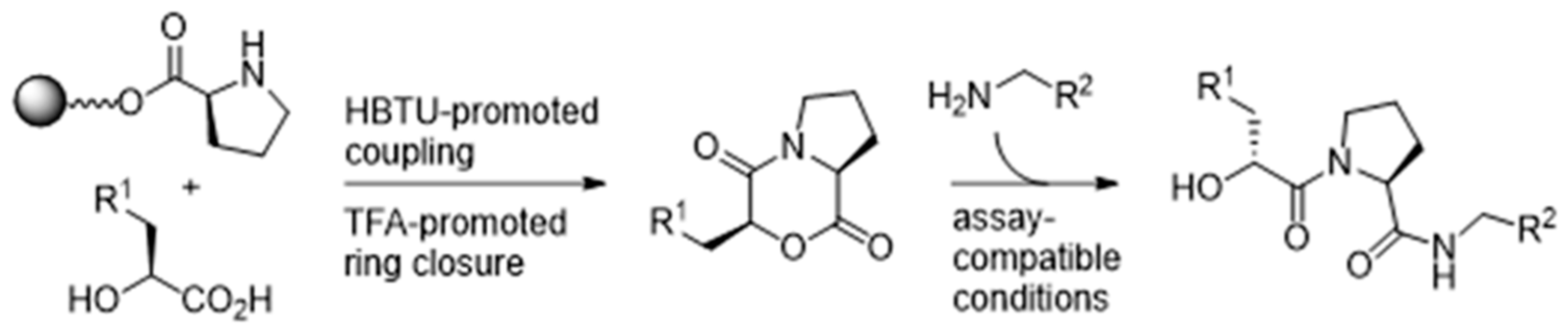
4.19. A Novel Approach to Traceless Generation of Serine Protease Inhibitors under Bioassay-Compatible Conditions
4.20. PROTAC Technology: A New Opportunity to Target Proteins Overexpressed in Chemoresistant Ovarian Cancer
4.21. Novel Fluorescent Porphyrin Probes to Target G-Quadruplexes
4.22. Synthesis and Physicochemical Properties of Amino Acid Prodrugs of the Radiosensitser Pyrazinib
4.23. The Synthesis of DSA Analogues for the Treatment of Cancer
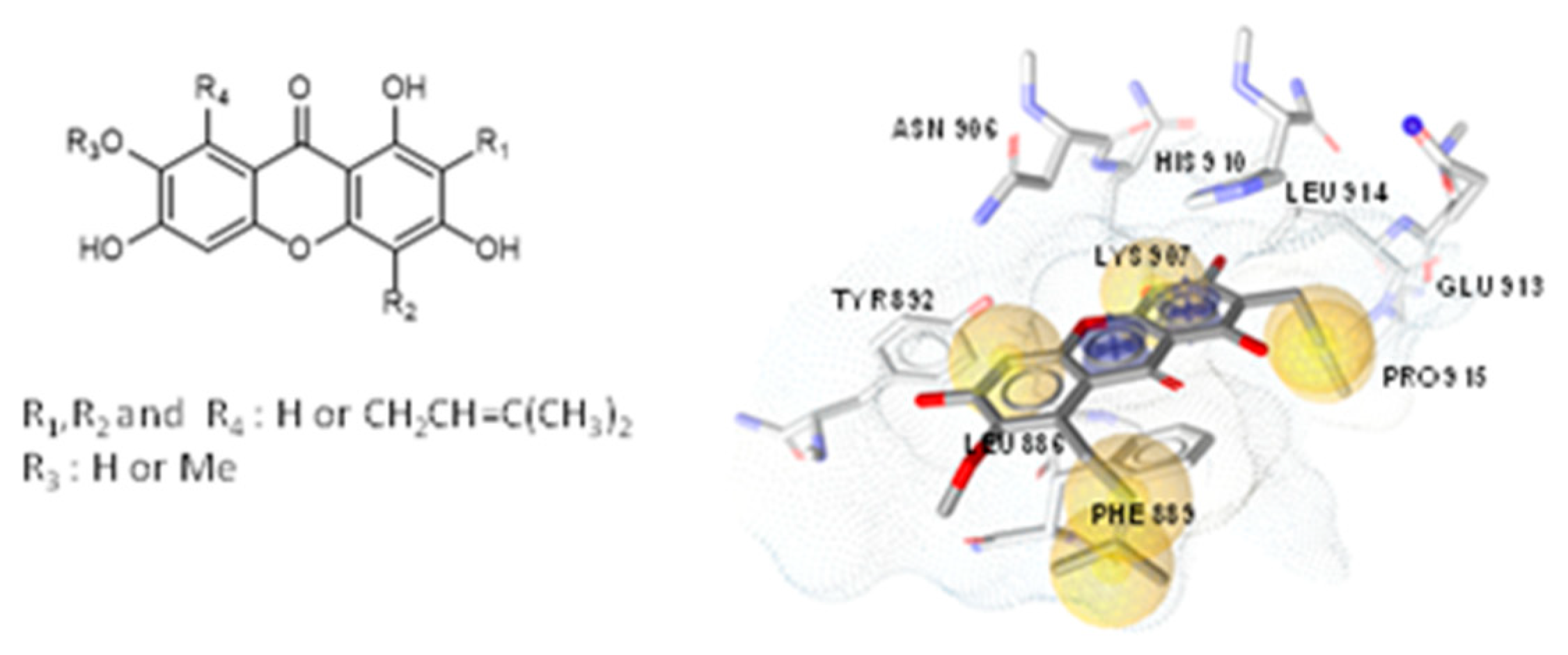
4.24. Synthesis of Prenylated Xanthones as Potential Fungal UPR Pathway Inhibitors
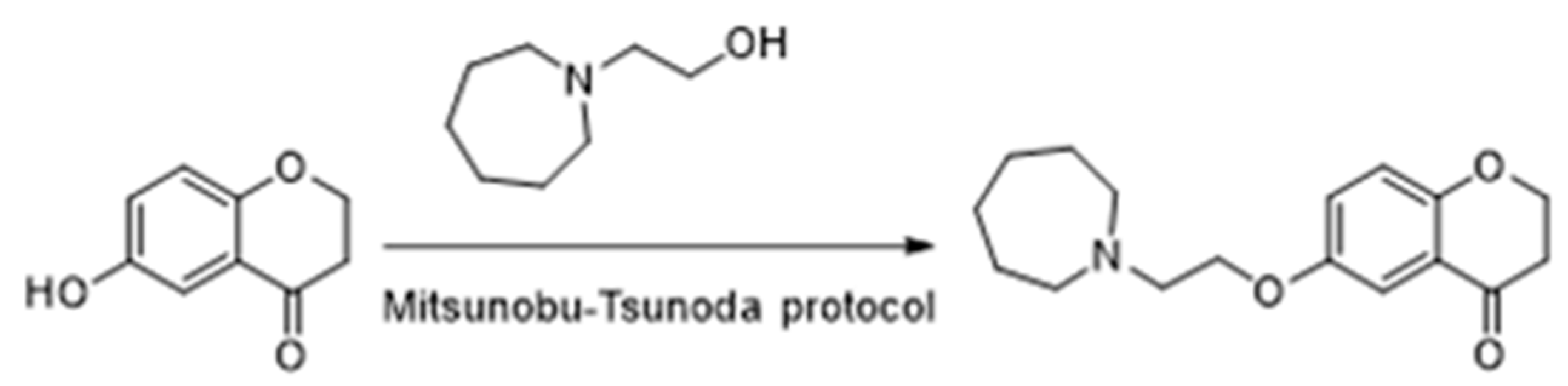
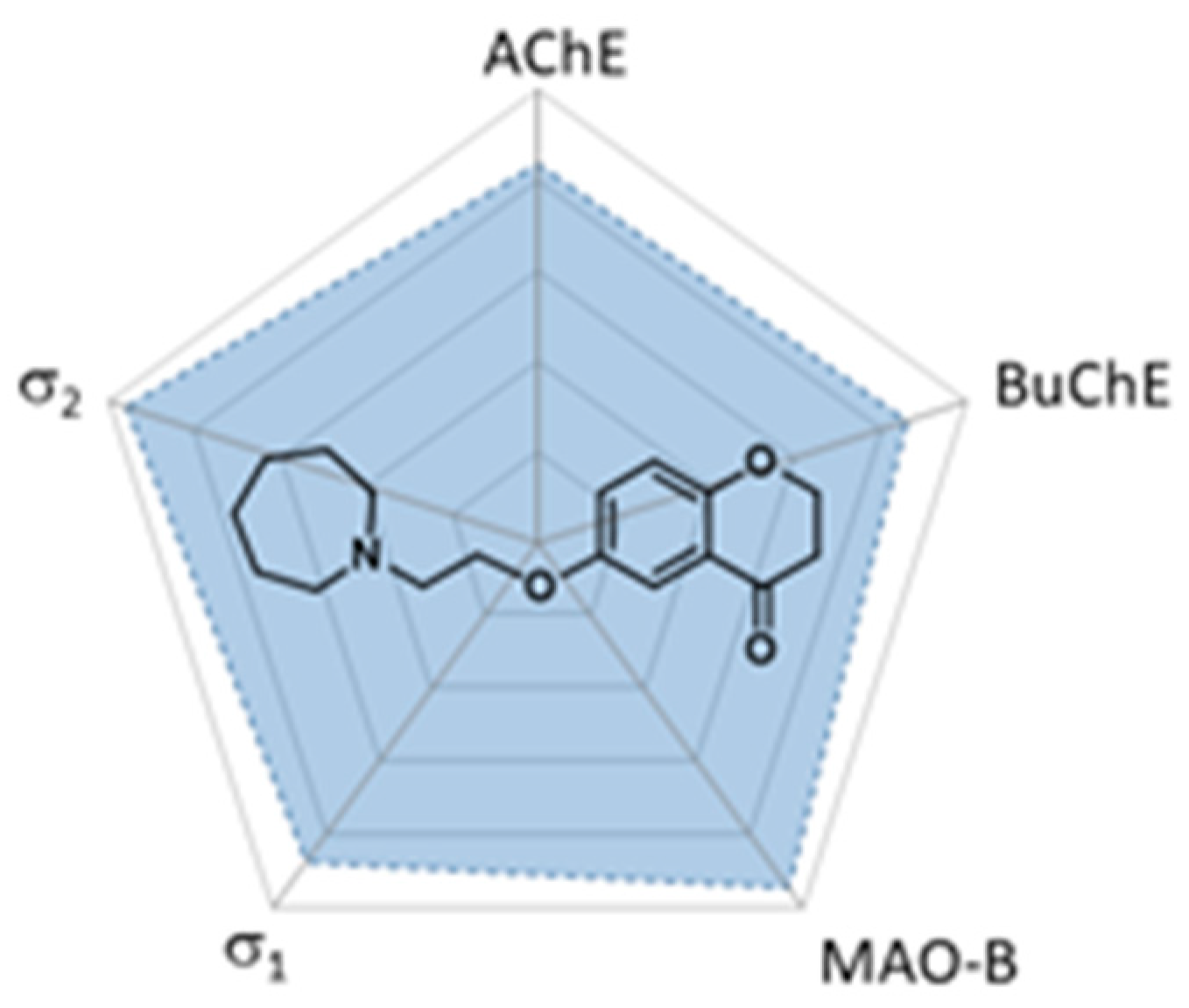
4.25. Chromanones as a Privileged Scaffold for Multineurotarget Anti-Alzheimer Agents
4.26. Design, Synthesis, and Enzymatic Evaluation of New HDAC 1 and 2 Orto-Aminobenzamides Inhibitors
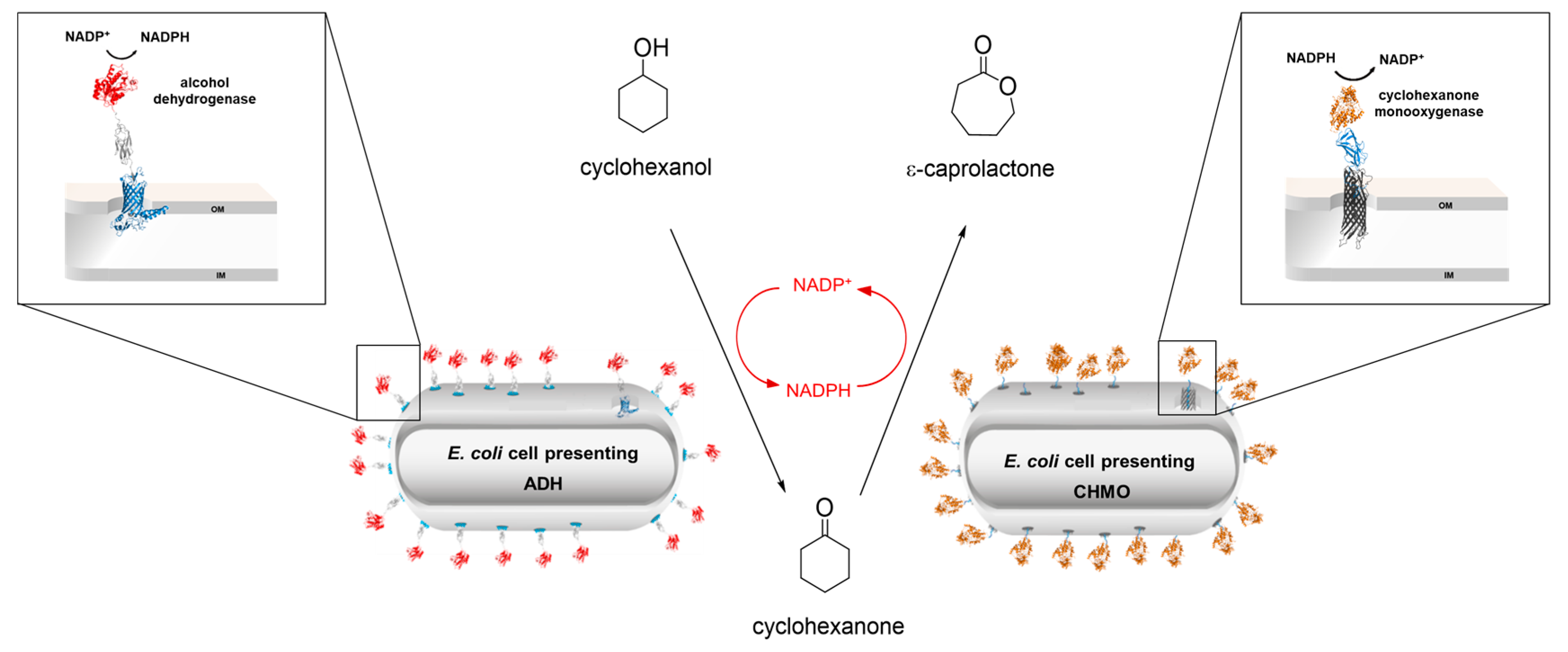
4.27. Enzyme Cascade Converting Cyclohexanol into ε-Caprolactone Coupled with NADPH Recycling Using Surface Displayed Alcohol Dehydrogenase and Cyclohexanone Monooxygenase on E. coli
4.28. Determination and Comparison of Kinetic Parameters for CK2 Enzyme Variants CK2α, CK2α’, CK2α2β2 and CK2α’2β2 in a Universal Buffer
4.29. Can We Expect Novel Chemical Structures from Highly Studied Fungal Strains? The Case Study of Penicillium Chrysogenum
4.30. Taking Back Control of the Immune System
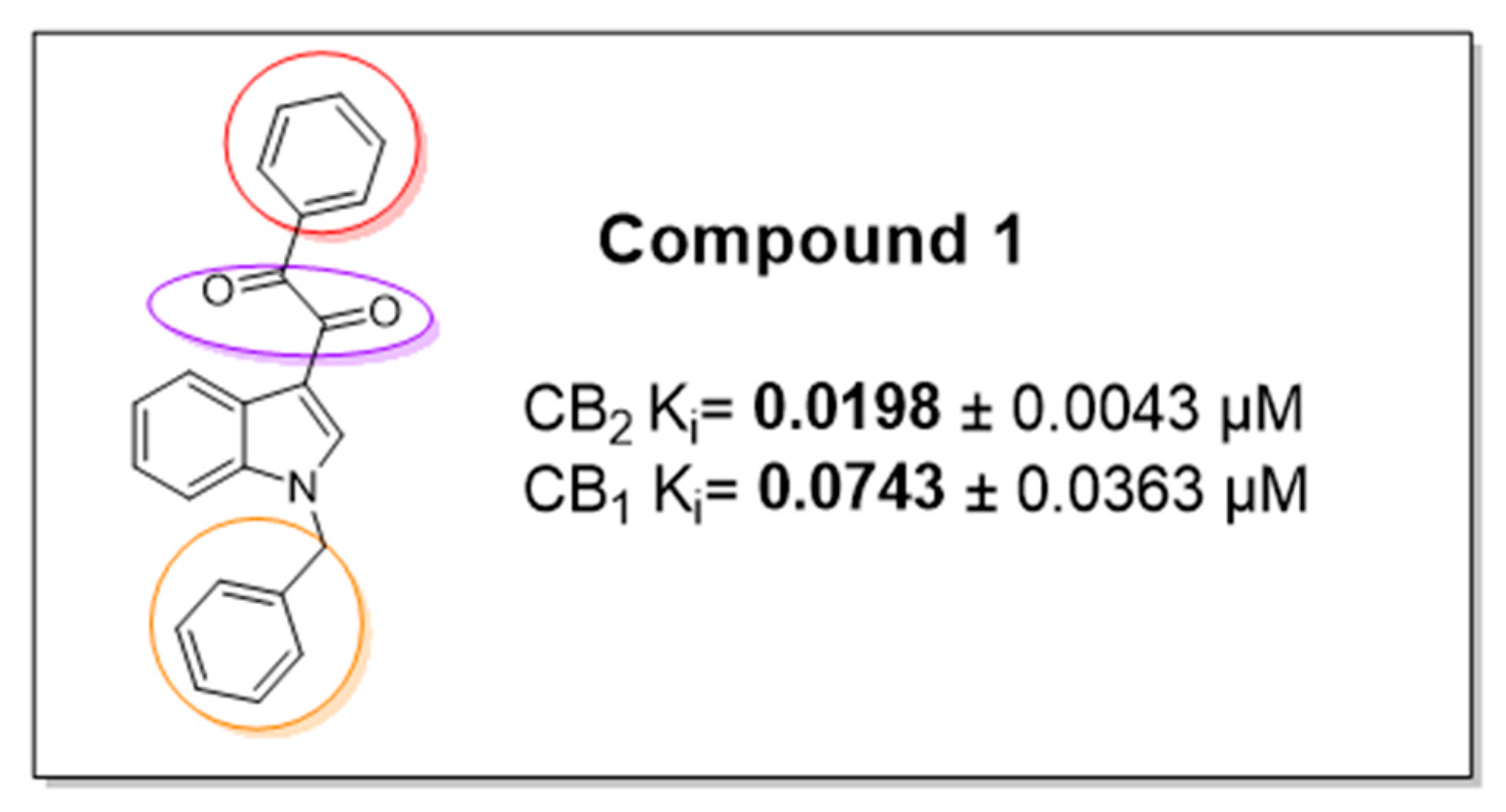
4.31. Discovery, Synthesis and Biological Evaluation of Indolyl-Diketone Derivatives as Cannabinoid Receptor Agonists
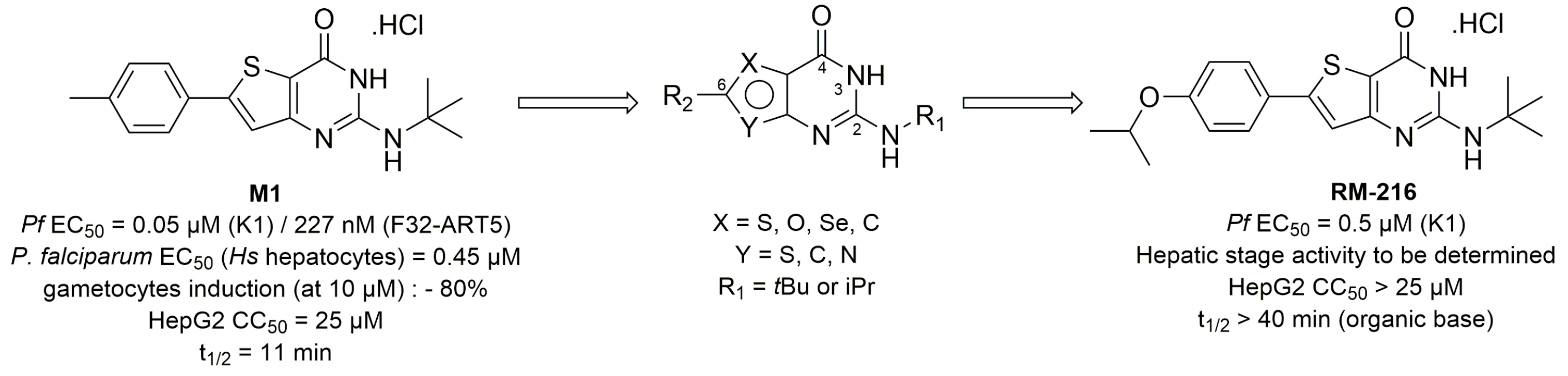
4.32. C6-Modulation and Scaffold Hopping of Theinopyrimidinone Antiplasmodial Hit with Multi-Stage Activity
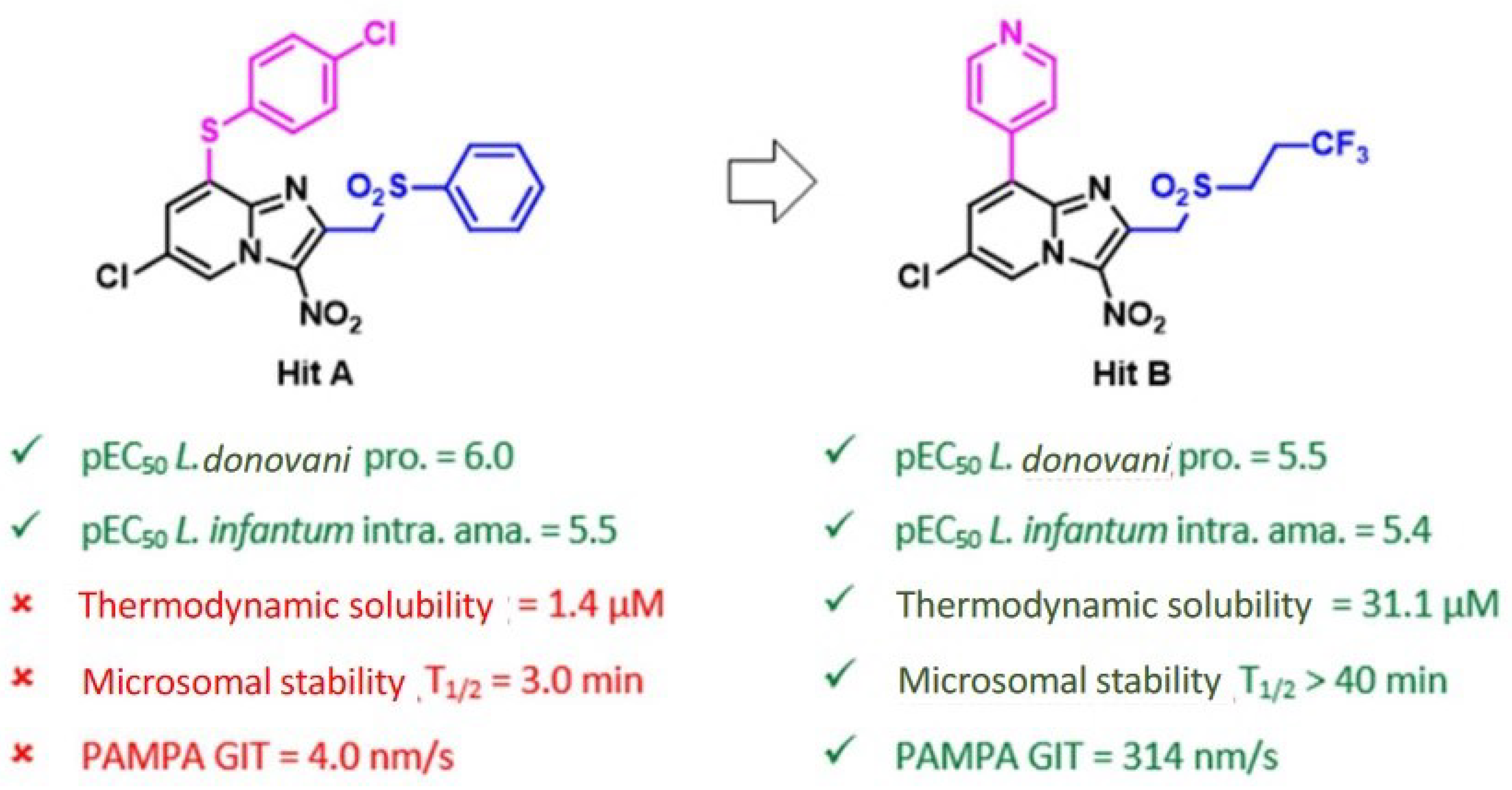
4.33. Improving the Druglikeness of a Nitroimidazopyridine Antileishmanial hit
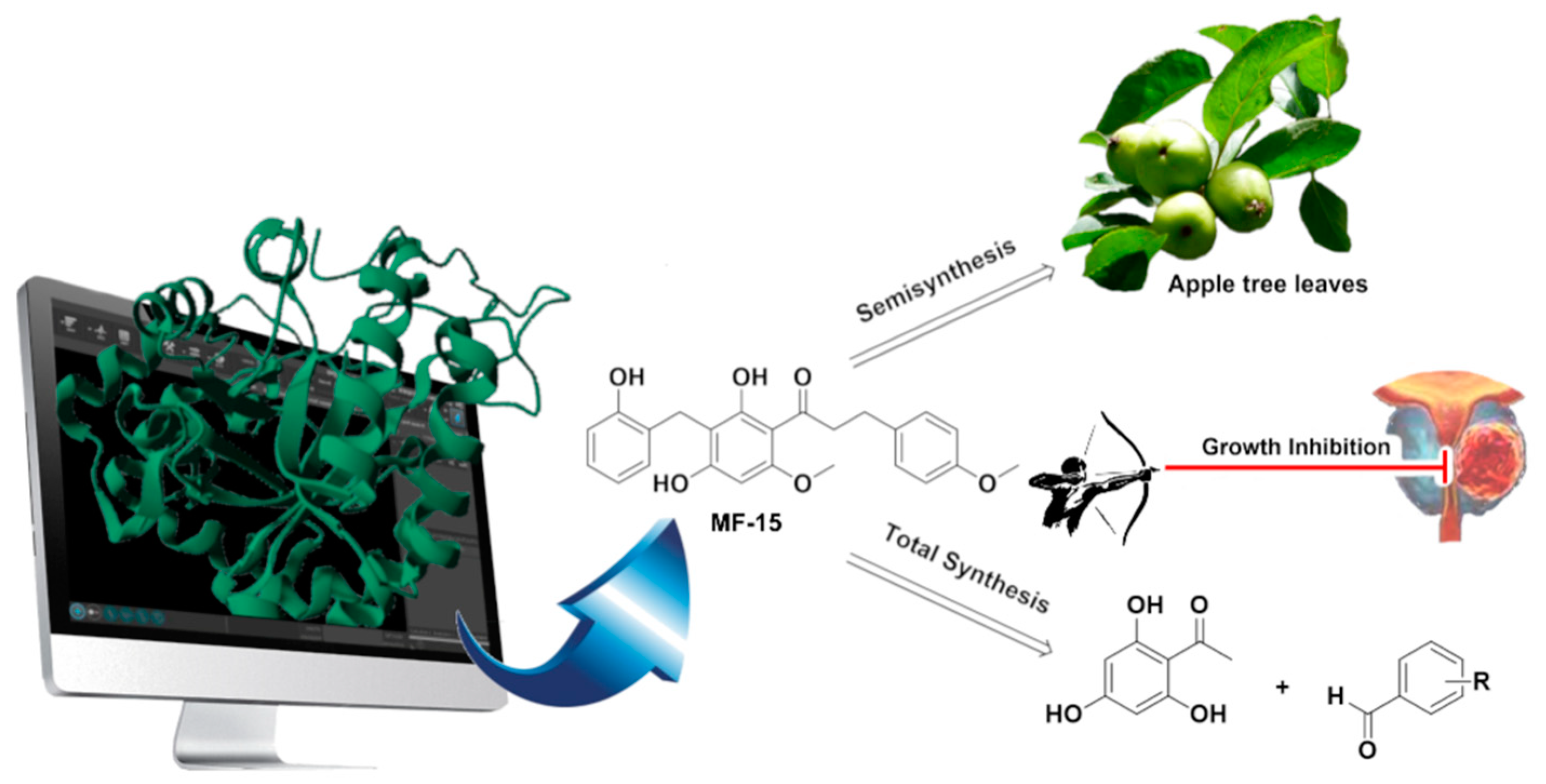
4.34. Novel (Semi-)Synthetic Phloretin-Based Inhibitors of AKR1C3 Overexpressed (Castration-Resistant) Prostate Cancer
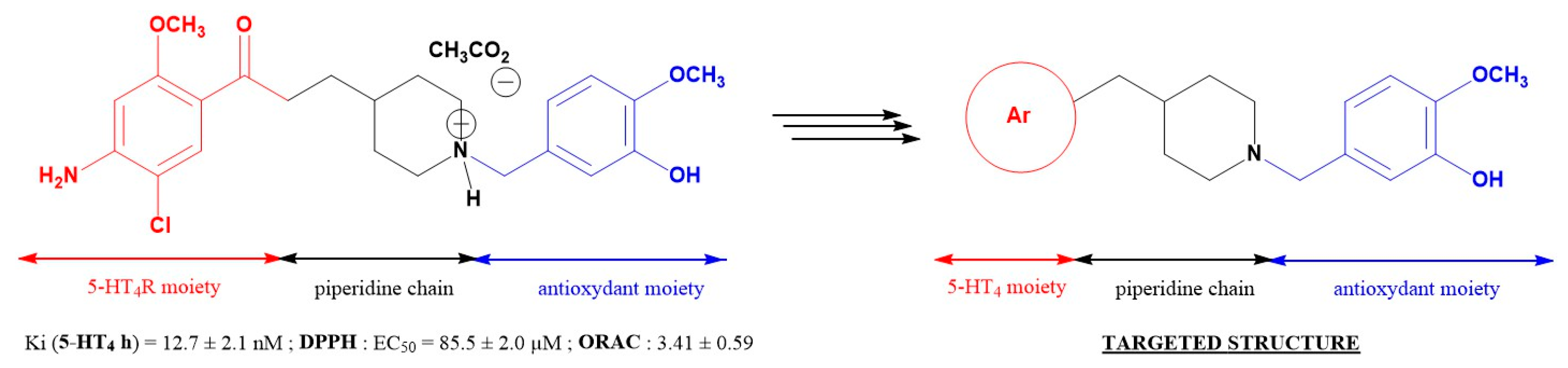
4.35. Novel Pleiotropic Compounds Inhibiting 5-HT4 Receptors with Antioxidant Properties for the Treatment of Alzheimer’s Disease
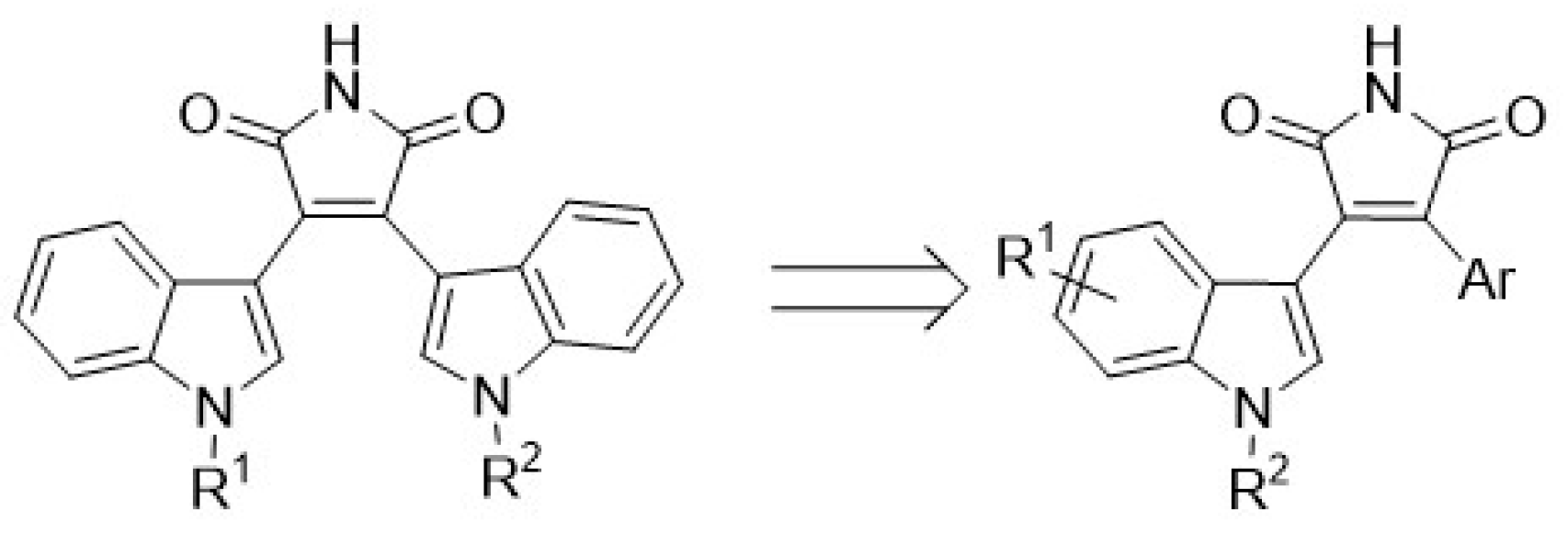
4.36. Bisindolylmaleimide Based Heterocyclic Compounds Are Involved in Fluconazole Susceptibility Restoration towards Candida albicans Strains
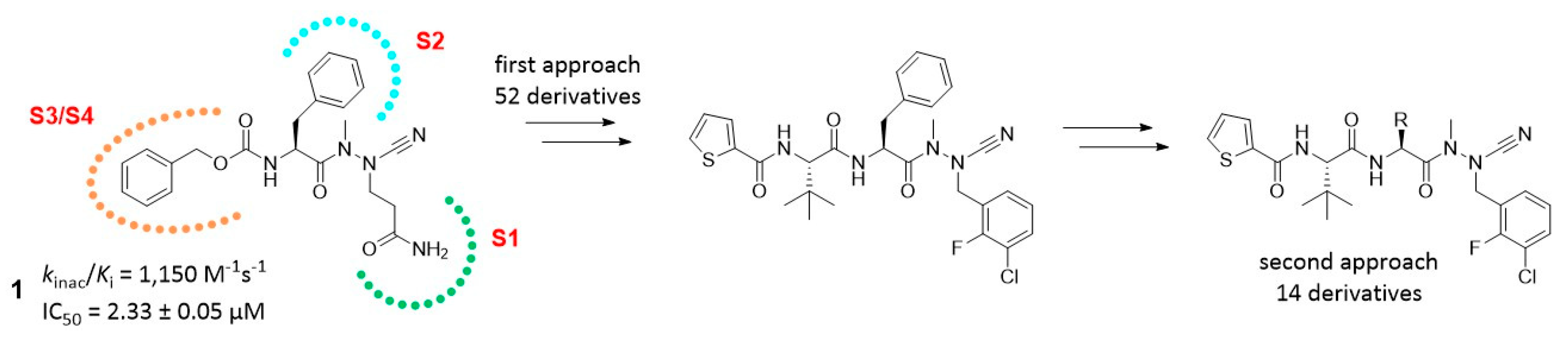
4.37. Structure-Inhibition Relationships with Regard to the S2 Site of SARS-CoV-2 Mpro
4.38. Peptides and Small Molecules Stabilise DNA Four-Way Junctions
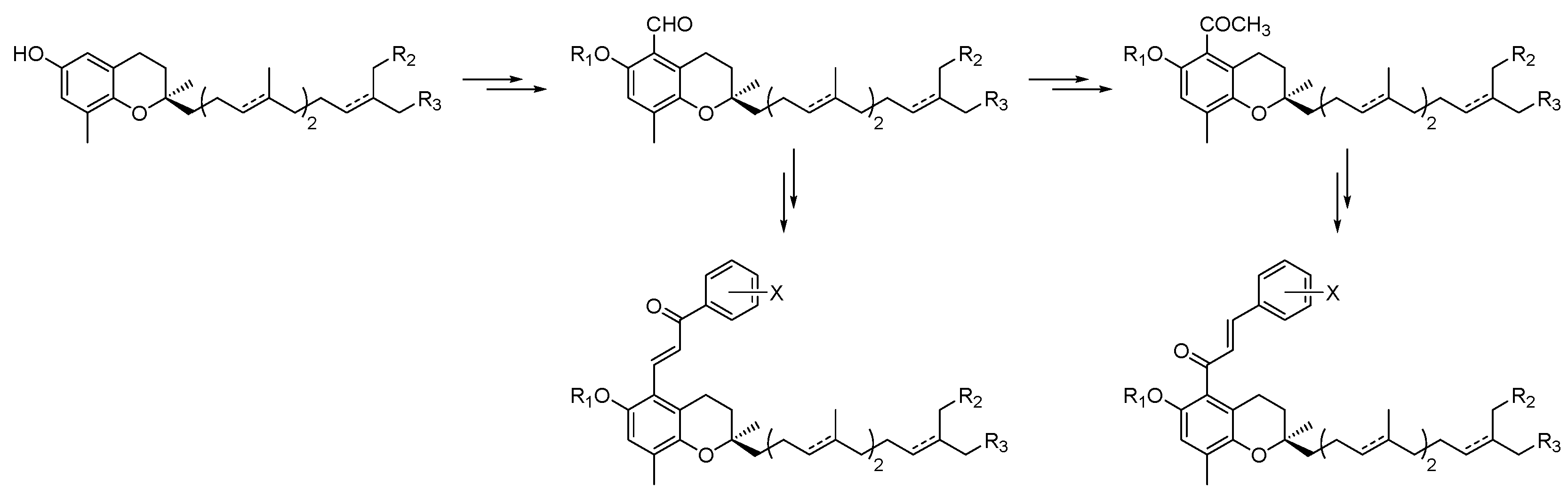
4.39. Anti-Inflammatory Potential of Chalcones in the Vitamin E Series
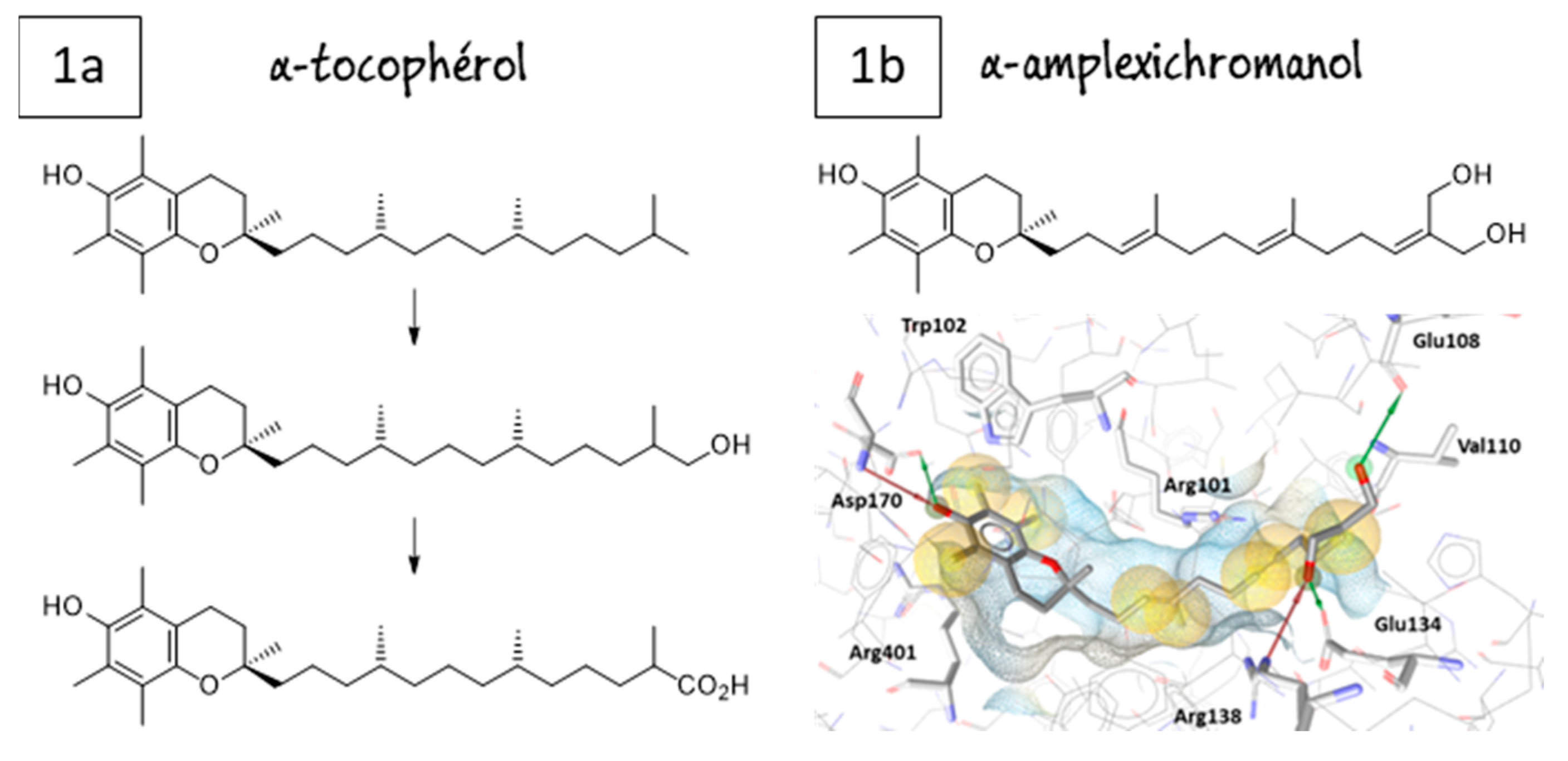
4.40. α-Amplexichromanol as a Highly Potent 5-LOX Inhibitor
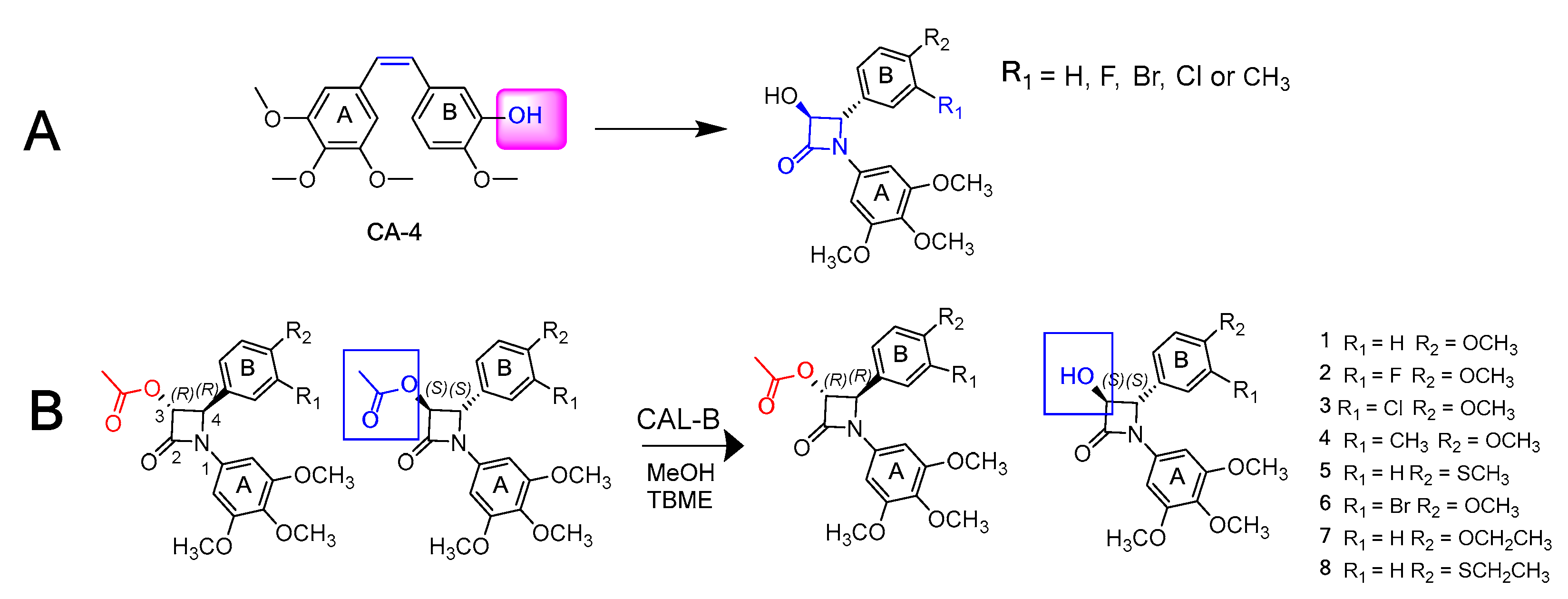
4.41. A Comparison of Chiral Diastereomeric Versus Kinetic Enzymatic Resolution for Enantioseparation of Microtubule Depolymerising Beta-Lactams
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yoon, S.; Eom, G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med. J. 2016, 52, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Millard, C.J.; Watson, P.J.; Fairall, L.; Schwabe, J.W.R. Targeting Class I Histone Deacetylases in a “Complex” Environment. Trends Pharmacol. Sci. 2017, 38, 363–377. [Google Scholar] [CrossRef]
- Smalley, J.P.; Cowley, S.M.; Hodgkinson, J.T. Bifunctional HDAC Therapeutics: One Drug to Rule Them All? Molecules 2020, 25, 4394. [Google Scholar] [CrossRef]
- Smalley, J.P.; Adams, G.E.; Milard, C.J.; Song, Y.; Norris, J.K.S.; Schwabe, J.W.R.; Cowley, S.M.; Hodgkinson, J.T. PROTAC-mediated degration of class I histone deacetylase enzymes in corepressor complexes. Chem. Commun. 2020, 56, 4476–4479. [Google Scholar] [CrossRef]
- Cowley, S.M.; Hodgkinson, J.; Schwabe, J.; Cross, J.; Smalley, J.; Grace, A.; Millard, C. HDAC Degrdaer. WIPO PCT Application Number WO2021GB50156, 2021. [Google Scholar]
- Smalley, J.P.; Baker, I.M.; Pytel, W.A.; Lin, L.-Y.; Bowman, K.J.; Schwabe, J.W.R.; Cowley, S.M.; Hodgkinson, J.T. Optimization of Class I Histone Deacetylase PROTACs Reveals that HDAC1/2 Degradation is Critical to Induce Apoptosis and Cell Arrest in Cancer Cells. J. Med. Chem. 2022, 65, 5642–5659. [Google Scholar] [CrossRef]
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schiöth, H.B. Trends in kinase drug discovery: Targets, indications and inhibitor design. Nat. Rev. Drug. Discov. 2021, 20, 839–861. [Google Scholar] [CrossRef]
- Chaikuad, A.; Koch, P.; Laufer, S.A.; Knapp, S. The cysteinome of protein kinases as a target in drug development. Angew. Chem. Int. Ed. 2018, 57, 4372–4385. [Google Scholar] [CrossRef]
- Hillebrand, L.; Gehringer, M. Never Gonna Give You Up–Current Developments in Covalent Protein Kinase Inhibitors. CHIMIA 2022, 76, 435–447. [Google Scholar] [CrossRef]
- Serafim, R.A.M.; da Silva Santiago, A.; Schwalm, M.P.; Hu, Z.; Dos Reis, C.V.; Takarada, J.E.; Mezzomo, P.; Massirer, K.B.; Kudolo, M.; Gerstenecker, S.; et al. Development of the first covalent monopolar spindle kinase 1 (MPS1/TTK) Inhibitor. J. Med. Chem. 2022, 65, 3173–3192. [Google Scholar]
- Forster, M.; Liang, X.J.; Schröder, M.; Gerstenecker, S.; Chaikuad, A.; Knapp, S.; Laufer, S.; Gehringer, M. Discovery of a Novel Class of Covalent Dual Inhibitors Targeting the Protein Kinases BMX and BTK. Int. J. Mol. Sci. 2020, 21, 9269. [Google Scholar] [CrossRef]
- Bellenie, B.R.; Cheung, K.J.; Varela, A.; Pierrat, O.A.; Collie, G.W.; Box, G.M.; Bright, M.D.; Gowan, S.; Hayes, A.; Rodrigues, M.J.; et al. Achieving in Vivo Target Depletion through the Discovery and Optimization of Benzimidazolone BCL6 Degraders. J. Med. Chem. 2020, 63, 4047–4068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, O.A.; Cheung, K.M.J.; Brennan, A.; Lloyd, M.G.; Rodrigues, M.J.; Pierrat, O.A.; Collie, G.W.; Le Bihan, Y.V.; Huckvale, R.; Harnden, A.C.; et al. Optimizing shape complementarity enables the discovery of potent tricyclic BCL6 inhibitors. J. Med. Chem. 2022, 65, 8169–8190. [Google Scholar] [CrossRef] [PubMed]
- Huckvale, R.; Harnden, A.C.; Cheung, K.M.J.; Pierrat, O.A.; Talbot, R.; Box, G.M.; Henley, A.T.; de Haven Brandon, A.K.; Hallsworth, A.E.; Bright, M.D.; et al. Improved binding affinity and pharmacokinetics enable sustained degradation of BCL6 in vivo. J. Med. Chem. 2022, 65, 8191–8207. [Google Scholar] [CrossRef] [PubMed]
- Mabonga, L.; Kappo, A.P. Protein-protein interaction modulators: Advances, successes and remaining challenges. Biophys. Rev. 2019, 11, 559–581. [Google Scholar] [CrossRef] [PubMed]
- Beekman, A.M.; O’Connell, M.A.; Howell, L.A. Peptide-Directed Binding for the Discovery of Modulators of α-Helix-Mediated Protein-Protein Interactions: Proof-of-Concept Studies with the Apoptosis Regulator Mcl-1. Angew. Chemie 2017, 56, 10446–10450. [Google Scholar] [CrossRef] [Green Version]
- Beekman, A.M.; Cominetti, M.M.D.; Walpole, S.J.; Prabhu, S.; O’Connell, M.A.; Angulo, J.; Searcey, M. Identification of selective protein-protein interaction inhibitors using efficient in silico peptide-directed ligand design. Chem. Sci. 2019, 10, 4502–4508. [Google Scholar] [CrossRef] [Green Version]
- Howell, L.A.; Beekman, A.M. In silico peptide-directed ligand design complements experimental peptide-directed binding for protein–protein interaction modulator discovery. RSC Chem. Biol. 2021, 2, 215–219. [Google Scholar] [CrossRef]
- Miethke, M.; Pieroni, M.; Weber, T.; Brönstrup, M.; Hammann, P.; Halby, L.; Arimondo, P.B.; Glaser, P.; Aigle, B.; Bode, H.B.; et al. Towards the sustainable discovery and development of new antibiotics. Nat. Rev. Chem. 2021, 5, 726. [Google Scholar] [CrossRef]
- Bousis, S.; Setyawati, I.; Diamanti, E.; Slotboom, D.J.; Hirsch, A.K. Energy-coupling factor transporters as novel antimicrobial targets. Adv. Ther. 2019, 2, 1800066. [Google Scholar] [CrossRef] [Green Version]
- Kiefer, A.F.; Bousis, S.; Hamed, M.M.; Diamanti, E.; Haupenthal, J.; Hirsch, A.K. Structure-Guided Optimization of Small-Molecule Folate Uptake Inhibitors Targeting the Energy-Coupling Factor Transporters. J. Med. Chem. 2022, 65, 8869–8880. [Google Scholar] [CrossRef]
- Bousis, S.; Winkler, S.; Haupenthal, J.; Fulco, F.; Diamanti, E.; Hirsch, A.K.H. An Efficient Way to Screen Inhibitors of Energy-Coupling Factor (ECF) Transporters in a Bacterial Uptake Assay. Int. J. Mol. Sci. 2022, 23, 2637. [Google Scholar] [CrossRef]
- Mancini, F.; Unver, M.Y.; Elgaher, W.A.; Jumde, V.R.; Alhayek, A.; Lukat, P.; Herrmann, J.; Witte, M.D.; Köck, M.; Blankenfeldt, W.; et al. Protein-Templated Hit Identification through an Ugi Four-Component Reaction. Chem. Eur. J. 2020, 26, 14585. [Google Scholar] [CrossRef] [PubMed]
- Kaya, C.; Walter, I.; Alhayek, A.; Shafiei, R.; Jézéquel, G.; Andreas, A.; Konstantinović, J.; Schönauer, E.; Sikandar, A.; Haupenthal, J.; et al. Structure-Based Design of α-Substituted Mercaptoacetamides as Inhibitors of the Virulence Factor LasB from Pseudomonas aeruginosa. ACS Infect. Dis. 2022, 8, 1010. [Google Scholar] [CrossRef]
- Kaya, C.; Walter, I.; Yahiaoui, S.; Sikandar, A.; Alhayek, A.; Konstantinović, J.; Kany, A.M.; Haupenthal, J.; Köhnke, J.; Hartmann, R.W.; et al. Substrate-inspired fragment merging and growing affords efficacious LasB inhibitors. Angew. Chem. Int. Ed. 2021, 61, e202112295. [Google Scholar] [CrossRef] [PubMed]
- Nagle, P.S.; Rodriguez, F.; Kahvedzic, A.; Quinn, S.J.; Rozas, I. Asymmetrical diaromatic guanidinium/2-aminoimidazolinium derivatives: Synthesis and DNA affinity. J. Med. Chem. 2009, 52, 7113–7121. [Google Scholar] [CrossRef] [PubMed]
- Nagle, P.; Quinn, S.J.; Kelly, J.M.; O’Donovan, D.H.; Khan, A.R.; Rodriguez, F.; Nguyen, B.; Wilson, W.D.; Rozas, I. Understanding the DNA binding of novel non-symmetrical guanidinium/2-aminoimidazolinium derivatives. Org. Biomol. Chem. 2010, 8, 5558–5567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagle, P.S.; Rodriguez, F.; Nguyen, B.; Wilson, W.D.; Rozas, I. High DNA affinity of a series of peptide linked diaromatic guanidinium-like derivatives. J. Med. Chem. 2012, 55, 4397–4406. [Google Scholar] [CrossRef] [PubMed]
- Nagle, P.S.; McKeever, C.; Rodriguez, F.; Nguyen, B.; Wilson, W.D.; Rozas, I. Unexpected DNA affinity and sequence selectivity through core rigidity in guanidinium-based minor groove binders. J. Med. Chem. 2014, 57, 7663–7672. [Google Scholar] [CrossRef] [PubMed]
- Grover, J.; Trujillo, C.; Saad, M.; Emandi, G.; Stipaničev, N.; Bernhard, S.S.; Guédin, A.; Mergny, J.L.; Senge, M.O.; Rozas, I. Dual-binding conjugates of diaromatic guanidines and porphyrins for recognition of G-quadruplexes. Org. Biomol. Chem. 2020, 18, 5617–5624. [Google Scholar] [CrossRef]
- Jörg, M.; Khajehali, E.; van der Westhuizen, E.T.; Choy, K.H.C.; Shackleford, D.M.; Tobin, A.B.; Sexton, P.M.; Valant, C.; Capuano, B.; Christopoulos, A.; et al. Development of novel 4-arylpyridin-2-one and 6-arylpyrimidin-4-one positive allosteric modulators of the M1 muscarinic acetylcholine receptor. ChemMedChem 2021, 16, 216–233. [Google Scholar] [CrossRef]
- Jörg, M.; van der Westhuizen, E.T.; Khajehali, E.; Burger, W.A.; White, J.M.; Choy, K.H.; Tobin, A.B.; Sexton, P.M.; Valant, C.; Capuano, B.; et al. 6-Phenylpyrimidin-4-ones as positive allosteric modulators at the M1 mAChR: The determinants of allosteric activity. ACS Chem. Neurosci. 2019, 10, 1099–1114. [Google Scholar] [CrossRef] [PubMed]
- Van der Westhuizen, E.T.; Spathis, A.; Khajehali, E.; Jörg, M.; Mistry, S.N.; Capuano, B.; Tobin, A.B.; Sexton, P.M.; Scammells, P.J.; Valant, C.; et al. Assessment of the molecular mechanisms of action of novel 4-Phenylpyridine-2-one and 6-Phenylpyrimidin-4-one allosteric modulators at the M1 muscarinic acetylcholine receptors. Mol. Pharm. 2018, 94, 770–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khajehali, E.; Valant, C.; Jörg, M.; Tobin, A.B.; Conn, P.J.; Lindsley, C.W.; Sexton, P.M.; Scammells, P.J.; Christopoulos, A. Probing the binding site of novel selective positive allosteric modulators at the M1 muscarinic acetylcholine receptor. Biochem. Pharmacol. 2018, 154, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Dallagnol, J.C.; Khajehali, E.; Van Der Westhuizen, E.T.; Jörg, M.; Valant, C.; Gonçalves, A.G.; Capuano, B.; Christopoulos, A.; Scammells, P.J. Synthesis and pharmacological evaluation of heterocyclic carboxamides: Positive allosteric modulators of the M1 muscarinic acetylcholine receptor with weak agonist activity and diverse modulatory profiles. J. Med. Chem. 2018, 61, 2875–2894. [Google Scholar] [CrossRef] [PubMed]
- Mistry, S.N.; Lim, H.; Jörg, M.; Capuano, B.; Christopoulos, A.; Lane, J.R.; Scammells, P.J. Novel fused arylpyrimidinone based allosteric modulators of the M1 muscarinic acetylcholine receptor. ACS Chem. Neurosci 2016, 7, 647–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mistry, S.N.; Jörg, M.; Lim, H.; Vinh, N.B.; Sexton, P.M.; Capuano, B.; Christopoulos, A.; Lane, J.R.; Scammells, P.J. 4-Phenylpyridin-2-one derivatives: A novel class of positive allosteric modulator of the M1 muscarinic acetylcholine receptor. J. Med. Chem. 2016, 59, 388–409. [Google Scholar] [CrossRef] [Green Version]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257. [Google Scholar] [CrossRef]
- Nathubhai, A.; Haikarainen, T.; Koivunen, J.; Murthy, S.; Koumanov, F.; Lloyd, M.D.; Holman, G.D.; Pihlajaniemi, T.; Tosh, D.; Lehtiö, L.; et al. Highly potent and isoform selective dual site binding Tankyrase/Wnt signaling inhibitors that increase cellular glucose uptake and have antiproliferative activity. J. Med. Chem. 2017, 60, 814–820. [Google Scholar] [CrossRef] [Green Version]
- Marques, P.; Villarroel-Vicente, C.; Collado, A.; García, A.; Vila, L.; Duplan, I.; Hennuyer, N.; Garibotto, F.; Enriz, R.D.; Dacquet, C.; et al. Anti-inflammatory effects and improved metabolic derangements in ob/ob mice by a newly synthesized prenylated benzopyran with pan-PPAR activity. Pharmacol. Res. 2023, 187, 106638. [Google Scholar] [CrossRef]
- Müller, T.D.; Blüher, M.; Tschöp, M.H.; DiMarchi, R.D. Anti-obesity drug discovery: Advances and challenges. Nat. Rev. Drug Discov. 2022, 21, 201–223. [Google Scholar] [CrossRef]
- Pickford, P.; Lucey, M.; Rujan, R.M.; McGlone, E.R.; Bitsi, S.; Ashford, F.B.; Corrêa, I.R.; Hodson, D.J.; Tomas, A.; Deganutti, G.; et al. Partial agonism improves the anti-hyperglycaemic efficacy of an oxyntomodulin-derived GLP-1R/GCGR co-agonist. Mol. Metab. 2021, 51, 101242. [Google Scholar] [CrossRef] [PubMed]
- Checkley, W.; White, J. A review of the global burden, novel diagnostics, therapeutics, and vaccine 795 targets for cryptosporidium. Lancet Infect. Dis. 2015, 15, 85–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudou, M.; Cleary, E.; ÓhAiseadha, C.; Garvey, P.; McKeown, P.; O’Dwyer, J.; Hynds, P. Spatiotemporal epidemiology of cryptosporidiosis in the Republic of Ireland, 2008–2017: Development of a space–time “cluster recurrence” index. BMC Infect. Dis. 2021, 21, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Striepen, B.; Pruijssers, A.J.; Huang, J.; Li, C.; Gubbels, M.J.; Umejiego, N.N.; Hedstrom, L.; Kissinger, J.C. Gene transfer in the evolution of parasite nucleotide biosynthesis. Proc. Natl. Acad. Sci. USA 2004, 101, 3154–3159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorla, S.K.; Kavitha, M.; Zhang, M.; Liu, X.; Sharling, L.; Gollapalli, D.R.; Striepen, B.; Hedstrom, L.; Cuny, G.D. Selective and potent urea inhibitors of Cryptosporidium parvum inosine 5′-monophosphate dehydrogenase. J. Med. Chem. 2012, 55, 7759–7771. [Google Scholar] [CrossRef] [Green Version]
- Wilbur, D.S. Radiohalogenation of proteins: An overview of radionuclides, labeling methods and reagents for conjugate labeling. Bioconjug. Chem. 1992, 3, 433–470. [Google Scholar] [CrossRef]
- Volkert, W.A.; Hoffman, T.J. Therapeutic radiopharmaceuticals. Chem. Rev. 1999, 99, 2269–2292. [Google Scholar] [CrossRef]
- Pimlott, S.L.; Sutherland, A. Molecular tracers for the PET and SPECT imaging of disease. Chem. Soc. Rev. 2011, 40, 149–162. [Google Scholar] [CrossRef]
- Adam, M.J.; Wilbur, D.S. Radiohalogens for imaging and therapy. Chem. Soc. Rev. 2005, 34, 153–163. [Google Scholar] [CrossRef]
- Dubost, E.; McErlain, H.; Babin, V.; Sutherland, A.; Cailly, T. Recent advances in synthetic methods for radioiodination. J. Org. Chem. 2020, 85, 8300–8310. [Google Scholar] [CrossRef]
- Cornella, J.; Rosillo-Lopez, M.; Larrosa, I. A novel mode of reactivity for gold (I): The decarboxylative activation of (hetero) aromatic carboxylic acids. Adv. Synth. Catal. 2011, 353, 1359–1366. [Google Scholar] [CrossRef]
- Wahl-Schott, C.; Biel, M. HCN channels: Structure, cellular regulation and physiological function. Cell Mol. Life Sci. 2009, 66, 470–494. [Google Scholar] [CrossRef] [PubMed]
- Jose, J.; Meyer, T.F. The autodisplay story, from discovery to biotechnical and biomedical applications. Microbiol. Mol. Biol. Rev. 2007, 71, 600–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nickelsen, A.; Jose, J. Label-free flow cytometry-based enzyme inhibitor identification. Anal. Chim. Acta 2021, 1179, 338826. [Google Scholar] [CrossRef]
- Ng, L.C.T.; Zhuang, M.; Van Petegem, F.; Li, Y.X.; Accili, E.A. Binding and structural asymmetry governs ligand sensitivity in a cyclic nucleotide–gated ion channel. J. Gen. Physiol. 2019, 151, 1190–1212. [Google Scholar] [CrossRef] [Green Version]
- Falsey, A.R.; Hennessey, P.A.; Formica, M.A.; Cox, C.; Walsh, E.E. Respiratory syncytial virus infection in elderly and high-risk adults. NEJM 2005, 352, 1749–1759. [Google Scholar] [CrossRef]
- Nair, H.; Nokes, D.J.; Gessner, B.D.; Dherani, M.; Madhi, S.A.; Singleton, R.J.; O’Brien, K.L.; Roca, A.; Wright, P.F.; Bruce, N.; et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: A systematic review and meta-analysis. Lancet 2010, 375, 1545–1555. [Google Scholar] [CrossRef] [Green Version]
- Haber, N. Respiratory syncytial virus infection in elderly adults. Med. Mal. Infect. 2018, 48, 377–382. [Google Scholar] [CrossRef]
- Behzadi, M.A.; Leyva-Grado, V.H. Overview of current therapeutics and novel candidates against influenza, respiratory syncytial virus, and Middle East respiratory syndrome coronavirus infections. Front. Microbiol. 2019, 10, 1327. [Google Scholar] [CrossRef] [Green Version]
- Mejias, A.; Ramilo, O. New options in the treatment of respiratory syncytial virus disease. J. Infect. 2015, 71, S80–S87. [Google Scholar] [CrossRef]
- Resch, B. Product review on the monoclonal antibody palivizumab for prevention of respiratory syncytial virus infection. Hum. Vaccin. Immunother. 2017, 9, 2138–2149. Available online: https://www.tandfonline.com/doi/full/10.1080/21645515.2017.1337614 (accessed on 16 January 2023). [CrossRef] [PubMed] [Green Version]
- Nesterkina, M.; Smola, S.; Rusakova, N.; Kravchenko, I. Terpenoid Hydrazones as Biomembrane Penetration Enhancers: FT-IR Spectroscopy and Fluorescence Probe Studies. Molecules 2021, 27, 206. [Google Scholar] [CrossRef] [PubMed]
- Nesterkina, M.; Muratov, V.; Ognichenko, L.; Kravchenko, I.; Kuz’min, V. Quantitative structure–activity relationship study on prolonged anticonvulsant activity of terpene derivatives in pentylenetetrazole test. Open Chemistry 2021, 19, 1184–1192. [Google Scholar] [CrossRef]
- Subramani, R.; Aalbersberg, W. Marine actinomycetes: An ongoing source of novel bioactive metabolites. Microbiol. Res. 2012, 167, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Goodfellow, M.; Brown, R.; Ahmed, L.; Pathom-Aree, W.; Bull, A.T.; Jones, A.L.; Stach, J.E.; Zucchi, T.D.; Zhang, L.; Wang, J. Verrucosispora fiedleri sp. nov., an actinomycete isolated from a fjord sediment which synthesizes proximicins. Antonie van Leeuwenhoek 2013, 103, 493–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, K.; Keller, S.; Wolter, F.E.; Röglin, L.; Beil, W.; Seitz, O.; Nicholson, G.; Bruntner, C.; Riedlinger, J.; Fiedler, H.P.; et al. Proximicins A, B, and C-Antitumor furan analogues of netropsin from the marine actinomycete Verrucosispora induce upregulation of p53 and the cyclin kinase inhibitor p21. Angew. Chem. Int. Ed. 2008, 47, 3258–3261. [Google Scholar] [CrossRef] [PubMed]
- Brucoli, F.; Natoli, A.; Marimuthu, P.; Borrello, M.T.; Stapleton, P.; Gibbons, S.; Schätzlein, A. Efficient synthesis and biological evaluation of proximicins A, B and C. Bioorg. Med. Chem. 2012, 20, 2019–2024. [Google Scholar] [CrossRef]
- Hong, M.; He, G. The 2016 revision to the World Health Organization classification of myelodysplastic syndromes. J. Transl. Intern. Med. 2017, 5, 139–143. [Google Scholar] [CrossRef] [Green Version]
- Baroud, M.; Lepeltier, E.; El-Makhour, Y.; Lautram, N.; Bejaud, J.; Thepot, S.; Duval, O. Azacitidine Omega-3 Self-Assemblies: Synthesis, Characterization, and Potent Applications for Myelodysplastic Syndromes. Pharmaceuticals 2021, 14, 1317. [Google Scholar] [CrossRef]
- Peramo, A.; Mura, S.; Semen, O. Yesylevskyy; Bruno Cardey; Dunja Sobot; Stephanie Denis; Christophe Ramseyer; Didier Desmaële; Patrick Couvreur. Squalene versus cholesterol: Which is the best nanocarrier for the delivery to cells of the anticancer drug gemcitabine? C. R. Chim. 2018, 21, 974–986. Available online: https://www.sciencedirect.com/science/article/pii/S1631074818300584?via%3Dihub (accessed on 16 January 2023). [CrossRef]
- Dheer, D.; Nicolas, J.; Shankar, R. Cathepsin-sensitive nanoscale drug delivery systems for cancer therapy and other diseases. Adv. Drug. Deliv. Rev. 2019, 151–152, 130–151. Available online: https://www.sciencedirect.com/science/article/pii/S0169409X19300134?via%3Dihub (accessed on 16 January 2023). [CrossRef] [PubMed] [Green Version]
- Aynalem, S.B.; Zeleke, A.J. Prevalence of Diabetes Mellitus and Its Risk Factors among Individuals Aged 15 Years and Above in Mizan-Aman Town, Southwest Ethiopia, 2016: A Cross Sectional Study. Int. J. Endocrinol. 2018, 9317987. [Google Scholar] [CrossRef] [PubMed]
- Micheal, E.S.; Covic, L.; Kuliopulos, A. Trace amine-associated receptor 1 (TAAR1) promotes anti-diabetic signaling in insulin-secreting cells. J. Biol. Chem. 2019, 294, 4401–4411. [Google Scholar] [CrossRef] [PubMed]
- Cripps, M.; Bagnati, M.; Jones, T.A.; Ogunkolade, B.W.; Sayers, S.R.; Caton, P.W.; Hanna, K.; Billacura, M.P.; Fair, K.; Nelson, C.; et al. Identification of a subset of trace amine-associated receptors and ligands as potential modulators of insulin secretion. Biochem. Pharmacol. 2020, 171, 113685. [Google Scholar] [CrossRef] [PubMed]
- Galley, G.; Stalder, H.; Goergler, A.; Hoener, M.C.; Norcross, R.D. Optimisation of imidazole compounds as selective TAAR1 agonists: Discovery of RO5073012. Bioorg. Med. Chem. Lett. 2012, 22, 5244–5248. [Google Scholar] [CrossRef]
- Bradaia, A.; Trube, G.; Stalder, H.; Norcross, R.D.; Ozmen, L.; Wettstein, J.G.; Pinard, A.; Buchy, D.; Gassmann, M.; Hoener, M.C.; et al. The selective antagonist EPPTB reveals TAAR1-mediated regulatory mechanisms in dopaminergic neurons of the mesolimbic system. Proc. Natl. Acad. Sci. USA. 2009, 106, 20081–20086. [Google Scholar] [CrossRef] [Green Version]
- Revel, F.G.; Moreau, J.L.; Gainetdinov, R.R.; Bradaia, A.; Sotnikova, T.D.; Mory, R.; Durkin, S.; Zbinden, K.G.; Norcross, R.; Meyer, C.A.; et al. TAAR1 activation modulates monoaminergic neurotransmission, preventing hyperdopaminergic and hypoglutamatergic activity. Proc. Natl. Acad. Sci. USA 2011, 108, 8485–8490. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.; Amaradhi, R.; Ganesh, T.; Dingledine, R. An agonist dependent allosteric antagonist of prostaglandin EP2 receptors. ACS Chem. Neurosci. 2020, 11, 1436–1446. [Google Scholar] [CrossRef]
- Sun, X.; Li, Q. Prostaglandin EP2 receptor: Novel therapeutic target for human cancers (Review). Int. J. Mol. Med. 2018, 42, 1203–1214. [Google Scholar] [CrossRef] [Green Version]
- Forselles, K.A.; Root, J.; Clarke, T.; Davey, D.; Aughton, K.; Dack, K.; Pullen, N. In vitro and in vivo characterization of PF-04418948, a novel, potent and selective prostaglandin EP2 receptor antagonist. Br. J. Pharmacol. 2011, 164, 1847–1856. [Google Scholar] [CrossRef] [Green Version]
- Ghanim, G.E.; Fountain, A.J.; van Roon, A.-M.M.; Rangan, R.; Das, R.; Collins, K.; Nguyen, T.H.D. Structure of human telomerase holoenzyme with bound telomeric DNA. Nature 2021, 593, 449–453. [Google Scholar] [CrossRef]
- Oyama, K.; Kawada-Matsuo, M.; Oogai, Y.; Hayashi, T.; Nakamura, N.; Komatsuzawa, H. Antibacterial Effects of Glycyrrhetinic Acid and Its Derivatives on Staphylococcus aureus. PLoS ONE 2016, 11, e0165831. [Google Scholar] [CrossRef] [Green Version]
- Kowalska, A.; Kalinowska-Lis, U. 18 β -Glycyrrhetinic acid: Its core biological properties and dermatological applications. Int. J. Cosmet. Sci. 2019, 41, 325–331. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhu, Q.; Zhong, Y.; Cui, X.; Jiang, Z.; Wu, P.; Zheng, X.; Zhang, K.; Zhao, S. Synthesis, anti-microbial and anti-inflammatory activities of 18β-glycyrrhetinic acid derivatives. Bioorg. Chem. 2020, 101, 103985. [Google Scholar] [CrossRef]
- Li, X.; Liu, Y.; Wang, N.; Liu, Y.; Wang, S.; Wang, H.; Li, A.; Ren, S. Synthesis and discovery of 18β-glycyrrhetinic acid derivatives inhibiting cancer stem cell properties in ovarian cancer cells. RSC Adv. 2019, 9, 27294–27304. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Tu, B.; Liang, J.; Guo, S.; Cao, N.; Chen, S.; Luo, Z.; Li, J.; Zheng, W.; Tang, X.; et al. Synthesis and biological evaluation of pentacyclic triterpenoid derivatives as potential novel antibacterial agents. Bioorganic Chem. 2021, 109, 104692. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Cheng, Y.; Ma, X.-L.; Wei, Y.-Q.; Wei, X.-W. Potential roles and targeted therapy of the CXCLs/CXCR2 axis in cancer and inflammatory diseases. BBA Rev. Cancer 2019, 1871, 289–312. [Google Scholar] [CrossRef]
- Dwyer, M.P.; Yu, Y. CXCR2 modulators: A patent review (2009–2013). Expert Opin. Ther. Pat. 2014, 24, 519–534. [Google Scholar] [CrossRef]
- Rennard, S.I.; Dale, D.C.; Donohue, J.F.; Kanniess, F.; Magnussen, H.; Sutherland, E.R.; Watz, H.; Lu, S.; Stryszak, P.; Rosenberg, E.; et al. CXCR2 Antagonist MK-7123. A Phase 2 Proof-of-Concept Trial for Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2015, 191, 1001–1011. [Google Scholar] [CrossRef]
- Salchow, K.; Bond, M.; Evans, S.; Press, N.; Charlton, S.; Hunt, P.; Bradley, M. A common intracellular allosteric binding site for antagonists of the CXCR2 receptor. Br. J. Pharmacol. 2010, 159, 1429–1439. [Google Scholar] [CrossRef] [Green Version]
- Chao, J.; Taveras, A.G.; Chao, J.; Aki, C.; Dwyer, M.; Yu, Y.; Purakkattle, B.; Rindgen, D.; Jakway, J.; Hipkin, W.; et al. C (4)-alkyl substituted furanyl cyclobutenediones as potent, orally bioavailable CXCR2 and CXCR1 receptor antagonists. Bioorg. Med. Chem. Lett. 2007, 17, 3778–3783. [Google Scholar] [CrossRef]
- McKinnie, S.M.; Miles, Z.D.; Moore, B.S. Characterization and Biochemical Assays of Streptomyces Vanadium-Dependent Chloroperoxidases. Methods Enzymol. 2018, 604, 405–424. [Google Scholar]
- Agarwal, V.; Miles, Z.D.; Winter, J.M.; Eustáquio, A.S.; El Gamal, A.A.; Moore, B.S. Enzymatic Halogenation and Dehalogenation Reactions: Pervasive and Mechanistically Diverse. Chem. Rev. 2017, 117, 5619–5674. [Google Scholar] [CrossRef] [Green Version]
- Leblanc, C.; Vilter, H.; Fournier, J.-B.; Delage, L.; Potin, P.; Rebuffet, E.; Michel, G.; Solari, P.; Feiters, M.; Czjzek, M. Vanadium haloperoxidases: From the discovery 30 years ago to X-ray crystallographic and V K-edge absorption spectroscopic studies. Coord. Chem. Rev. 2015, 301–302, 134–146. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Lv, J.; Zou, J.; Tian, F.; Shang, Z. Halogen–water–hydrogen bridges in biomolecules. J. Struct. Biol. 2010, 169, 172–182. [Google Scholar] [CrossRef]
- Xu, Z.; Yang, Z.; Liu, Y.; Lu, Y.; Chen, K.; Zhu, W. Halogen Bond: Its Role beyond Drug–Target Binding Affinity for Drug Discovery and Development. J. Chem. Inf. Model. 2014, 54, 69–78. [Google Scholar] [CrossRef]
- Hachem, M.; Nacir, H. Emerging Role of Phospholipids and Lysophospholipids for Improving Brain Docosahexaenoic Acid as Potential Preventive and Therapeutic Strategies for Neurological Diseases. Int. J. Mol. Sci. 2022, 23, 3969. [Google Scholar] [CrossRef]
- Lott, I.T.; Head, E. Dementia in Down syndrome: Unique insights for Alzheimer disease research. Nat. Rev. Neurol. 2019, 15, 135–147. [Google Scholar] [CrossRef]
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-target-Directed Ligands To Combat Neurodegenerative Diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef]
- Eady, N.; Sheehan, R.; Rantell, K.; Sinai, A.; Bernal, J.; Bohnen, I.; Bonell, S.; Courtenay, K.; Dodd, K.; Gazizova, D.; et al. Impact of cholinesterase inhibitors or memantine on survival in adults with Down syndrome and dementia: Clinical cohort study. Br. J. Psychiatry 2018, 212, 155–160. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Pintus, F.; Di Petrillo, A.; Medda, R.; Caria, P.; Matos, M.J.; Viña, D.; Pieroni, E.; Delogu, F.; Era, B.; et al. Novel 2-pheynlbenzofuran derivatives as selective butyrylcholinesterase inhibitors for Alzheimer’s disease. Sci. Rep. 2018, 8, 4424. [Google Scholar] [CrossRef] [Green Version]
- Kandiah, N.; Pai, M.-C.; Senanarong, V.; Looi, I.; Ampil, E.; Park, K.W.; Karanam, A.K.; Christopher, S. Rivastigmine: The advantages of dual inhibition of acetylcholinesterase and butyrylcholinesterase and its role in subcortical vascular dementia and Parkinson’s disease dementia. Clin. Interv. Aging 2017, 12, 697–707. [Google Scholar] [CrossRef] [Green Version]
- Toublet, F.-X.; Lecoutey, C.; Lalut, J.; Hatat, B.; Davis, A.; Since, M.; Corvaisier, S.; Freret, T.; Santos, J.S.D.O.; Claeysen, S.; et al. Inhibiting Acetylcholinesterase to Activate Pleiotropic Prodrugs with Therapeutic Interest in Alzheimer’s Disease. Molecules 2019, 24, 2786. [Google Scholar] [CrossRef] [Green Version]
- Baranger, K.; Marchalant, Y.; Bonnet, A.E.; Crouzin, N.; Carrete, A.; Paumier, J.-M.; Py, N.A.; Bernard, A.; Bauer, C.; Charrat, E.; et al. MT5-MMP is a new pro-amyloidogenic proteinase that promotes amyloid pathology and cognitive decline in a transgenic mouse model of Alzheimer’s disease. Cell Mol. Life Sci. 2016, 73, 217–236. [Google Scholar] [CrossRef] [Green Version]
- Zipfel, P.; Rochais, C.; Baranger, K.; Rivera, S.; Dallemagne, P. Matrix Metalloproteinases as New Targets in Alzheimer’s Disease: Opportunities and Challenges. J. Med. Chem. 2020, 63, 10705–10725. [Google Scholar] [CrossRef]
- Hirotsu, Y.; Katsuoka, F.; Funayama, R.; Nagashima, T.; Nishida, Y.; Nakayama, K.; Engel, J.D.; Yamamoto, M. Nrf2–MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res. 2012, 40, 10228–10239. [Google Scholar] [CrossRef] [Green Version]
- Zimta, A.-A.; Cenariu, D.; Irimie, A.; Magdo, L.; Nabavi, S.M.; Atanasov, A.G.; Berindan-Neagoe, I. The Role of Nrf2 Activity in Cancer Development and Progression. Cancers 2019, 11, 1755. [Google Scholar] [CrossRef] [Green Version]
- Lapenta, F.; Aupič, J.; Strmšek, Ž.; Jerala, R. Coiled coil protein origami: From modular design principles towards biotechnological applications. Chemical Society Reviews 2018, 47, 3530–3542. [Google Scholar] [CrossRef] [Green Version]
- Truebestein, L.; Leonard, T.A. Coiled-coils: The long and short of it. Bioessays 2016, 38, 903–916. [Google Scholar] [CrossRef] [Green Version]
- López-García, P.; de Araujo, A.D.; Bergues-Pupo, A.E.; Tunn, I.; Fairlie, D.P.; Blank, K.G. Fortified coiled coils: Enhancing mechanical stability with lactam or metal staples. Angewandte Chemie 2021, 60, 232–236. [Google Scholar] [CrossRef]
- Alquraishi, M. AlphaFold at CASP13. Bioinformatics 2019, 35, 4862–4865. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Shrimp, J.H.; Kales, S.C.; Sanderson, P.E.; Simeonov, A.; Shen, M.; Hall, M.D. An Enzymatic TMPRSS2 Assay for Assessment of Clinical Candidates and Discovery of Inhibitors as Potential Treatment of COVID-19. ACS Pharmacol. Transl. Sci. 2020, 3, 997–1007. [Google Scholar] [CrossRef]
- WHO. World Malaria Report; World Health Organization: Geneva, Switzerland, 2021; pp. 1–263.
- Sandoval, E.; Lafuente-Monasterio, M.J.; Almela, M.J.; Castañeda, P.; Díaz, M.B.J.; Martínez-Martínez, M.S.; Vidal, J.; Angulo-Barturen, I.; Bamborough, P.; Burrows, J.; et al. The Discovery of Novel Antimalarial Aminoxadiazoles as a Promising Nonendoperoxide Scaffold. F. J. Med. Chem. 2017, 60, 6880–6896. [Google Scholar] [CrossRef]
- Gao, Y.-J.; Ji, R.-R. Chemokines, neuronal–glial interactions, and central processing of neuropathic pain. Pharmacol. Ther. 2010, 126, 56–68. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, Y.; Zhang, L.; Cheng, J.K.; Ji, R.R. Cytokine mechanisms of central sensitization: Distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J. Neurosci. 2008, 28, 5189–5194. [Google Scholar] [CrossRef] [Green Version]
- Kamat, S.S.; Camara, K.; Parsons, W.H.; Chen, D.-H.; Dix, M.M.; Bird, T.D.; Howell, A.R.; Cravatt, B.F. Immunomodulatory lysophosphatidylserines are regulated by ABHD16A and ABHD12 interplay. Nat. Chem. Biol. 2015, 11, 164–171. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-Y. A neuroinflammation emerging target. Nat. Chem. Biol. 2015, 11, 99–100. [Google Scholar] [CrossRef]
- Ahonen, T.J.; Savinainen, J.R.; Yli-Kauhaluoma, J.; Kalso, E.; Laitinen, J.T.; Moreira, V.M. Discovery of 12-Thiazole Abietanes as Selective Inhibitors of the Human Metabolic Serine Hydrolase hABHD16A. ACS Med. Chem. Lett. 2018, 9, 1269–1273. [Google Scholar] [CrossRef]
- WHO. Leishmaniasis. Available online: https://www.who.int/en/news-room/fact-sheets/detail/leishmaniasis (accessed on 30 May 2022).
- Sangshetti, J.N.; Khan, F.A.K.; Kulkarni, A.A.; Arote, R.; Patil, R.H. Antileishmanial drug discovery: Comprehensive review of the last 10 years. RSC Adv. 2015, 5, 32376–32415. [Google Scholar] [CrossRef]
- Marchand, P.; Bazin, M.-A.; Pagniez, F.; Rivière, G.; Bodero, L.; Marhadour, S.; Nourrisson, M.-R.; Picot, C.; Ruchaud, S.; Bach, S.; et al. Synthesis, antileishmanial activity and cytotoxicity of 2,3-diaryl- and 2,3,8-trisubstituted imidazo[1,2-a]pyrazines. Eur. J. Med. Chem. 2015, 103, 381–395. [Google Scholar] [CrossRef]
- Durieu, E.; Prina, E.; Leclercq, O.; Oumata, N.; Gaboriaud-Kolar, N.; Vougogiannopoulou, K.; Aulner, N.; Defontaine, A.; No, J.H.; Ruchaud, S.; et al. From Drug Screening to Target Deconvolution: A Target-Based Drug Discovery Pipeline Using Leishmania Casein Kinase 1 Isoform 2 To Identify Compounds with Antileishmanial Activity. Antimicrob. Agents Chemother. 2016, 60, 2822–2833. [Google Scholar] [CrossRef] [Green Version]
- Bazin, M.A.; Cojean, S.; Pagniez, F.; Bernadat, G.; Cavé, C.; Ourliac-Garnier, I.; Nourrisson, M.R.; Morgado, C.; Picot, C.; Leclercq, O.; et al. In vitro identification of imidazo [1,2-a] pyrazine-based antileishmanial agents and evaluation of L. major casein kinase 1 inhibition. Eur. J. Med. Chem. 2021, 210, 112956. [Google Scholar] [CrossRef]
- Bermejo, A.; Collado, A.; Barrachina, I.; Marqués, P.; El Aouad, N.; Franck, X.; Garibotto, F.; Dacquet, C.; Caignard, D.H.; Suvire, F.D.; et al. Polycerasoidol, a natural prenylated benzopyran with a dual PPARα/PPARγ agonist activity and anti-inflammatory effect. J. Nat. Prod. 2019, 82, 1802–1812. [Google Scholar] [CrossRef]
- García, A.; Vila, L.; Marín, P.; Bernabeu, Á.; Villarroel-Vicente, C.; Hennuyer, N.; Staels, B.; Franck, X.; Figadère, B.; Cabedo, N.; et al. Synthesis of 2-Prenylated Alkoxylated Benzopyrans by Horner–Wadsworth–Emmons Olefination with PPARα/γ Agonist Activity. ACS Med. Chem. Lett 2021, 12, 1783–1786. [Google Scholar] [CrossRef]
- Ramström, O.; Lehn, J.-M. Drug discovery by dynamic combinatorial libraries. Nat. Rev. Drug Discov. 2002, 1, 26–36. [Google Scholar] [CrossRef]
- Steinmetzer, T.; Baum, B.; Biela, A.; Klebe, G.; Nowak, G.; Bucha, E. Beyond Heparinization: Design of Highly Potent Thrombin Inhibitors Suitable for Surface Coupling. ChemMedChem 2012, 7, 1965–1973. [Google Scholar] [CrossRef]
- Vu, L.P.; Gütschow, M. Diketomorpholines: Synthetic Accessibility and Utilization. ACS Omega 2022, 7(1), 48–54. [Google Scholar] [CrossRef]
- Vu, L.P.; Zyulina, M.; Hingst, A.; Schnakenburg, G.; Gütschow, M. Combinatorial assembly, traceless generation and in situ evaluation of inhibitors for therapeutically relevant serine proteases. Bioorg. Chem. 2022, 121, 105676. [Google Scholar] [CrossRef]
- Brotin, E.; Meryet-Figuière, M.; Simonin, K.; Duval, R.E.; Villedieu, M.; Leroy-Dudal, J.; Saison-Behmoaras, E.; Gauduchon, P.; Denoyelle, C.; Poulain, L. Bcl-XL and MCL-1 constitute pertinent targets in ovarian carcinoma and their concomitant inhibition is sufficient to induce apoptosis. Int. J. Cancer 2010, 126, 885–895. [Google Scholar] [PubMed]
- Simonin, K.; N’Diaye, M.; Lheureux, S.; Loussouarn, C.; Dutoit, S.; Briand, M.; Giffard, F.; Brotin, E.; Blanc-Fournier, C.; Poulain, L. Platinum compounds sensitize ovarian carcinoma cells to ABT-737 by modulation of the Mcl-1/Noxa axis. Apoptosis 2013, 18, 492–508. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA. 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gloaguen, C.; Voisin-Chiret, A.S.; Sopkova-de Oliveira Santos, J.; Fogha, J.; Gautier, F.; De Giorgi, M.; Burzicki, G.; Perato, S.; Pétigny-Lechartier, C.; Simonin-Le Jeune, K.; et al. First evidence that oligopyridines, α-helix foldamers, inhibit Mcl-1 and sensitize ovarian carcinoma cells to Bcl-xL-targeting strategies. J. Med. Chem. 2015, 58, 1644–1668. [Google Scholar] [CrossRef]
- Guedeney, N.; Cornu, M.; Schwalen, F.; Kieffer, C.; Voisin-Chiret, A.S. PROTAC technology: A new drug design for chemical biology with many challenges in drug discovery. Drug Discov. Today 2023, 1, 103395. [Google Scholar] [CrossRef]
- Burge, S.; Parkinson, G.N.; Hazel, P.; Todd, A.K.; Neidle, S. Quadruplex DNA: Sequence, topology and structure. Nucleic Acids Res. 2006, 34, 5402–5415. [Google Scholar] [CrossRef] [Green Version]
- Ou, T.-M.; Lu, Y.-J.; Tan, J.-H.; Huang, Z.-S.; Wong, K.-Y.; Gu, L.-Q. G-Quadruplexes: Targets in Anticancer Drug Design. ChemMedChem 2008, 3, 690–713. [Google Scholar] [CrossRef]
- Rozas, I.; Senge, M.O. A two-pronged attack on DNA: Targeting guanine quadruplexes with nonplanar porphyrins and DNA-binding small molecules. Future Med. Chem. 2016, 8, 609–612. [Google Scholar] [CrossRef] [Green Version]
- Buckley, A.M.; Dunne, M.R.; Lynam-Lennon, N.; Kennedy, S.A.; Cannon, A.; Reynolds, A.L.; Maher, S.G.; Reynolds, J.V.; Kennedy, B.N.; O’Sullivan, J. Pyrazinib (P3), [(E)-2-(2-Pyrazin-2-yl-vinyl)-phenol], a small molecule pyrazine compound enhances radiosensitivity in oesophageal adenocarcinoma. Cancer Lett. 2019, 447, 115–129. [Google Scholar] [CrossRef]
- Pysz, I.; Jackson, P.J.M.; Thurston, D.E. Chapter 1: Introduction to Antibody–Drug Conjugates (ADCs). In Drug Discovery: Cytotoxic Payloads for Antibody-Drug Conjugates; Royal Society of Chemistry: London, UK, 2019. [Google Scholar]
- Thurston, D.E.; Jackson, P.J. Cytotoxic Payloads for Antibody-Drug Conjugates; Royal Society of Chemistry: London, UK, 2019; pp. 185–191. [Google Scholar]
- Pillow, T.H.; Tercel, M. Chapter 11: Duocarmycin–PBD Dimers as Antibody–Drug Conjugate (ADC) Payloads. In Drug Discovery: Cytotoxic Payloads for Antibody-Drug Conjugates; Royal Society of Chemistry: London, UK, 2019. [Google Scholar]
- Adams, C.J.; Kopp, M.C.; Larburu, N.; Nowak, P.R.; Ali, M.M.U. Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator IRE1. Front. Mol. Biosci. 2019, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Joubert, A.; Simoneau, P.; Campion, C.; Bataillé-Simoneau, N.; Iacomi-Vasilescu, B.; Poupard, P.; François, J.M.; Georgeault, S.; Sellier, E.; Guillemette, T. Impact of the unfolded protein response on the pathogenicity of the necrotrophic fungus Alternaria brassicicola. Mol. Microbiol. 2011, 79, 1305–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelly, B.-S.; Thomas, C.; Thomas, G.; Anne-Marie, L.R.; Pascal, R.-P.; Philippe, S.; Guillaume, V. Compositions and Methods for Controlling Phytopathogenic Infections. European Patent Application EP21306671, 30 November 2021. [Google Scholar]
- León, R.; Garcia, A.G.; Marco-Contelles, J. Recent advances in the multitarget-directed ligands approach for the treatment of Alzheimer’s disease. J. Med. Res. Rev. 2013, 33, 139–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swamy, K.C.K.; Kumar, N.N.B.; Balaraman, E.; Kumar, K.V.P.P. Mitsunobu and Related Reactions: Advances and Applications. Chem. Rev. 2009, 109, 2551–2651. [Google Scholar] [PubMed]
- Nowakowska, Z.; Kędzia, B.; Schroeder, G. Synthesis, physicochemical properties and antimicrobial evaluation of new (E)-chalcones. Eur. J. Med. Chem. 2008, 43, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Lemke, C.; Christmann, J.; Jiafei, Y.; Alonso, J.M.; Serrano, E.; Chioua, M.; Ismaili, L.; Martínez-Grau, M.A.; Beadle, C.D.; Vetman, T.; et al. Chromenones as Multineurotargeting Inhibitors of Human Enzymes. ACS Omega 2019, 4, 22161–22168. [Google Scholar] [CrossRef] [Green Version]
- Mertens, M.D.; Hinz, S.; Müller, C.E.; Gütschow, M. Alkynyl–coumarinyl ethers as MAO-B inhibitors. Bioorg. Med. Chem. 2014, 22, 1916–1928. [Google Scholar] [CrossRef] [PubMed]
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation and Methylation of Histones and their Possible Role in the Regulation of RNA Synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef] [Green Version]
- Johnstone, R.W. Histone-deacetylase inhibitors: Novel drugs for the treatment of cancer. Nat. Rev. Drug Discov. 2002, 1, 287–299. [Google Scholar] [CrossRef]
- Eckslager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef] [Green Version]
- Clouthier, C.M.; Pelletier, J.N. Expanding the organic toolbox: A guide to integrating biocatalysis in synthesis. Chem. Soc. Rec. 2012, 41, 1585–1605. [Google Scholar] [CrossRef]
- Schüürmann, J.; Quehl, P.; Festel, G.; Jose, J. Bacterial whole-cell biocatalysts by surface display of enzymes: Toward industrial application. Appl. Microbiol. Biotechnol. 2014, 98, 8031–8046. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Furtmann, C.; Lenz, F.; Srinivasamurthy, V.; Bornscheuer, U.T.; Jose, J. Enzyme cascade converting cyclohexanol into ε-caprolactone coupled with NADPH recycling using surface displayed alcohol dehydrogenase and cyclohexanone monooxygenase on E. coli. Microbiol. Biotechnol. 2022, 15, 2235–2249. [Google Scholar] [CrossRef] [PubMed]
- Borgo, C.; D’amore, C.; Sarno, S.; Salvi, M.; Ruzzene, M. Protein kinase CK2: A potential therapeutic target for diverse human diseases. Signal Transduct. Target Ther. 2021, 6, 183. [Google Scholar] [CrossRef] [PubMed]
- Niefind, K.; Guerra, B.; Ermakowa, I.; Issinger, O. Crystal structure of human protein kinase CK2: Insights into basic properties of the CK2 holoenzyme. Embo. J. 2001, 20, 5320–5331. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui-Jain, A.; Drygin, D.; Streiner, N.; Chua, P.; Pierre, F.; O’Brien, S.E.; Bliesath, J.; Omori, M.; Huser, N.; Ho, C.; et al. CX-4945, an Orally Bioavailable Selective Inhibitor of Protein Kinase CK2, Inhibits Prosurvival and Angiogenic Signaling and Exhibits Antitumor Efficacy. Cancer Res. 2010, 70, 10288–10298. [Google Scholar] [CrossRef] [Green Version]
- Sarno, S.; Reddy, H.; Meggio, F.; Ruzzene, M.; Davies, S.P.; Donella-Deana, A.; Shugar, D.; Pinna, L.A. Selectivity of 4, 5, 6, 7-tetrabromobenzotriazole, an ATP site-directed inhibitor of protein kinase CK2 (‘casein kinase-2’). FEBS Lett. 2001, 496, 44–48. [Google Scholar] [CrossRef]
- Farkona, S.; Diamandis, E.P.; Blasutig, I.M. Cancer immunotheraphy: The beginning of the end of cancer? BMC Med. 2016, 14, 73. [Google Scholar] [CrossRef] [Green Version]
- Magiera-Mularz, K.; Skalniak, L.; Zak, K.M.; Musielak, B.; Rudzinska-Szostak, E.; Berlicki, Ł.; Kocik, J.; Grudnik, P.; Sala, D.; Zarganes-Tzitzikas, T.; et al. Bioactive macrocyclic inhibitors of the PD-1/PD-L1 immune checkpoint. Angew. Chem. Int. 2017, 56, 13732–13735. [Google Scholar] [CrossRef]
- Sunshine, J.; Taube, M.J. Pd-1/pd-l1 inhibitors. Curr. Opin. Pharmacol. 2015, 23, 32–38. [Google Scholar] [CrossRef] [Green Version]
- Pacher, P.; Bátkai, S.; Kunos, G. The Endocannabinoid System as an Emerging Target of Pharmacotherapy. Pharmacol. Rev. 2006, 58, 389–462. [Google Scholar] [CrossRef] [Green Version]
- Schoeder, C.T.; Hess, C.; Madea, B.; Meiler, J.; Müller, C.E. Pharmacological evaluation of new constituents of “Spice”: Synthetic cannabinoids based on indole, indazole, benzimidazole and carbazole scaffolds. Forensic Toxicol. 2018, 36, 385–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taliani, S.; Trincavelli, M.L.; Cosimelli, B.; Laneri, S.; Severi, E.; Barresi, E.; Pugliesi, I.; Daniele, S.; Giacomelli, C.; Greco, G.; et al. Modulation of A2B adenosine receptor by 1-Benzyl-3-ketoindole derivatives. Eur. J. Med. Chem. 2013, 69, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Ouji, M.; Augereau, J.M.; Paloque, L.; Benoit-Vical, F. Plasmodium falciparum resistance to artemisinin-based combination therapies: A sword of Damocles in the path toward malaria elimination. Parasite 2018, 25, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, A.; Suzanne, P.; Lancelot, J.-C.; Verhaeghe, P.; Lesnard, A.; Basmaciyan, L.; Hutter, S.; Laget, M.; Dumètre, A.; Paloque, L.; et al. Discovery of New Thienopyrimidinone Derivatives Displaying Antimalarial Properties toward Both Erythrocytic and Hepatic Stages of Plasmodium. Eur. J. Med. Chem. 2015, 95, 16–28. [Google Scholar] [CrossRef]
- Bosson-Vanga, H.; Primas, N.; Franetich, J.-F.; Lavazec, C.; Gomez, L.; Ashraf, K.; Tefit, M.; Soulard, V.; Dereuddre-Bosquet, N.; Le Grand, R.; et al. A New Thienopyrimidinone Chemotype Shows Multistage Activity against Plasmodium falciparum, Including Artemisinin-Resistant Parasites. Microbiol. Spectr. 2021, 9, e00274-21. [Google Scholar] [CrossRef]
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef]
- Fersing, C.; Basmaciyan, L.; Boudot, C.; Pedron, J.; Hutter, S.; Cohen, A.; Caroline, D.; Primas, N.; Laget, M.; Casanova, M.; et al. Nongenotoxic 3-Nitroimidazo[1,2-a]Pyridines Are NTR1 Substrates That Display Potent in Vitro Antileishmanial Activity. ACS Med. Chem. Lett. 2019, 10, 34–39. [Google Scholar] [CrossRef] [Green Version]
- American Cancer Society. Key Statistics for Prostate Cancer. Available online: https://www.cancer.org/cancer/prostate-cancer/about/key-statistics.html (accessed on 20 August 2022).
- Adeniji, A.O.; Chen, M.; Penning, T.M. AKR1C3 as a target in castrate resistant prostate cancer. J. Steroid Biochem. Mol. Biol. 2013, 137, 136–149. [Google Scholar] [CrossRef] [Green Version]
- Kafka, M.; Mayr, F.; Temml, V.; Möller, G.; Adamski, J.; Höfer, J.; Schwaiger, S.; Heidegger, I.; Matuszczak, B.; Schuster, D.; et al. Dual Inhibitory Action of a Novel AKR1C3 Inhibitor on Both Full-Length AR and the Variant AR-V7 in Enzalutamide Resistant Metastatic Castration Resistant Prostate Cancer. Cancers 2020, 12, 2092. [Google Scholar] [CrossRef]
- Prince, M.J.; Wimo, A.; Guerchet, M.M.; Ali, G.C.; Wu, Y.T.; Prina, M. World Alzheimer Report 2015-The Global Impact of Dementia: An analysis of prevalence, incidence, cost and trends; Alzheimer’s Disease International: London, UK, 2015; Volume 87. [Google Scholar]
- Rosini, M.; Simoni, E.; Milelli, A.; Minarini, A.; Melchiorre, C. Oxidative stress in Alzheimer’s disease: Are we connecting the dots? J. Med. Chem. 2014, 57, 2821–2831. [Google Scholar] [CrossRef]
- Lanthier, C.; Payan, H.; Liparulo, I.; Hatat, B.; Lecoutey, C.; Since, M.; Davis, A.; Bergamini, C.; Claeysen, S.; Dallemagne, P.; et al. Novel multi target-directed ligands targeting 5-HT4 receptors with in cellulose antioxidant properties as promising leads in Alzheimer’s disease. Eur. J. Med. Chem. 2019, 182, 111596. [Google Scholar] [CrossRef]
- Monge, R.A.; Román, E.; Nombela, C.; Pla, J. The MAP kinase signal transduction network in Candida albicans. J. Microbiol. 2006, 152, 905–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, B.B.; Mylonakis, E. Our Paths Might Cross: The Role of the Fungal Cell Wall Integrity Pathway in Stress Response and Cross Talk with Other Stress Response Pathways. Eukaryot Cell 2009, 8, 1616–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaFayette, S.L.; Collins, C.; Zaas, A.K.; Schell, W.A.; Betancourt-Quiroz, M.; Gunatilaka, A.A.L.; Perfect, J.R.; Cowen, L.E. PKC Signaling Regulates Drug Resistance of the Fungal Pathogen Candida albicans via Circuitry Comprised of Mkc1, Calcineurin, and Hsp90. PLoS Pathog. 2010, 6, e1001069. [Google Scholar] [CrossRef] [Green Version]
- Perfect, J.R. The antifungal pipeline: A reality check. Nat. Rev. Drug Discov. 2017, 16, 603–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morio, F.; Jensen, R.H.; Le Pape, P.; Arendrup, M.C. Molecular basis of antifungal drug resistance in yeasts. Int. J. Antimicrob. Agents. 2017, 50, 599–606. [Google Scholar] [CrossRef]
- Liu, N.; Tu, J.; Dong, G.; Wang, Y.; Sheng, C. Emerging New Targets for the Treatment of Resistant Fungal Infections. J. Med. Chem. 2018, 61, 5484–5511. [Google Scholar] [CrossRef]
- Dao, V.H.; Ourliac-Garnier, I.; Bazin, M.-A.; Jacquot, C.; Baratte, B.; Ruchaud, S.; Bach, S.; Grovel, O.; Le Pape, P.; Marchand, P. Benzofuro[3,2-d]pyrimidines inspired from cercosporamide CaPkc1 inhibitor: Synthesis and evaluation of fluconazole susceptibility restoration. Bioorg. Med. Chem. Lett. 2018, 28, 2250–2255. [Google Scholar] [CrossRef]
- Winfield, H.J.; Cahill, M.M.; O’Shea, K.D.; Pierce, L.T.; Robert, T.; Ruchaud, S.; Bach, S.; Marchand, P.; McCarthy, F.O. Synthesis and anticancer activity of novel bisindolylhydroxymaleimide derivatives with potent GSK-3 kinase inhibition. Bioorg. Med. Chem. 2018, 26, 4209–4224. [Google Scholar] [CrossRef]
- Vangeel, L.; Chiu, W.; De Jonghe, S.; Maes, P.; Slechten, B.; Raymenants, J.; André, E.; Leyssen, P.; Neyts, J.; Jochmans, D. Remdesivir, Molnupiravir and Nirmatrelvir remain active against SARS-CoV-2 Omicron and other variants of concern. Antivir. Res. 2022, 198, 105252. [Google Scholar] [CrossRef]
- Breidenbach, J.; Lemke, C.; Pillaiyar, T.; Schäkel, L.; Al Hamwi, G.; Diett, M.; Gedschold, R.; Geiger, N.; Lopez, V.; Mirza, S.; et al. Targeting the Main Protease of SARS-CoV-2: From the Establishment of High Throughput Screening to the Design of Tailored Inhibitors. Angew. Chem. Int. Ed. 2021, 60, 10423–10429. [Google Scholar] [CrossRef] [PubMed]
- Howell, L.A.; Searcey, M. Targeting Higher-Order DNA: Beyond the G-Quadruplex. ChemBioChem. 2009, 10(13), 2139–2143. [Google Scholar] [CrossRef] [PubMed]
- Eichman, B.; Ortiz-Lombardía, M.; Aymamí, J.; Coll, M.; Ho, P.S. The Inherent Properties of DNA Four-way Junctions: Comparing the Crystal Structures of Holliday Junctions. J. Mol. Biol. 2002, 320, 1037–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boldt, J.L.; Pinilla, C.; Segall, A.M. Reversible Inhibitors of λ Integrase-mediated Recombination Efficiently Trap Holliday Junction Intermediates and Form the Basis of a Novel Assay for Junction Resolution. J. Biol. Chem. 2004, 279, 3472–3483. [Google Scholar] [CrossRef] [Green Version]
- Gunderson, C.W.; Segall, A.M. DNA repair, a novel antibacterial target: Holliday junction-trapping peptides induce DNA damage and chromosome segregation defects. Mol. Microbiol. 2006, 59, 1129–1148. [Google Scholar] [CrossRef]
- Weiss, U. Inflammation. Nature 2008, 454, 427. [Google Scholar] [CrossRef] [Green Version]
- Koeberle, A.; Werz, O. Natural products as inhibitors of prostaglandin E2 and pro-inflammatory 5-lipoxygenase-derived lipid mediator biosynthesis. Biotechnol. Adv. 2018, 36, 1709–1723. [Google Scholar] [CrossRef]
- Alsabil, K.; Viault, G.; Suor-Cherer, S.; Helesbeux, J.J.; Merza, J.; Dumontet, V.; Peña-Rodriguez, L.M.; Richomme, P.; Séraphin, D. Efficient ortho-formylation in vitamin E series, application to the semi-synthesis of natural 5-and 7-formyl-δ-tocotrienols revealing an unprecedented 5-bromo-7-formyl exchange. Tetrahedron 2017, 73, 6863–6870. [Google Scholar] [CrossRef]
- Mesmin, B.; Antonny, B. Vitamin Currency in a Lipid Exchange Market. Science 2013, 340, 1051–1052. [Google Scholar] [CrossRef]
- Pein, H.; Ville, A.; Pace, S.; Temml, V.; Garscha, U.; Raasch, M.; Alsabil, K.; Viault, G.; Dinh, C.-P.; Guilet, D.; et al. Endogenous metabolites of vitamin E limit inflammation by targeting 5-lipoxygenase. Nat. Commun. 2018, 9, 3834. [Google Scholar] [CrossRef] [Green Version]
- Neukirch, K.; Alsabil, K.; Dinh, C.P.; Bilancia, R.; Raasch, M.; Ville, A.; Cerqua, I.; Viault, G.; Bréard, D.; Pace, S.; et al. Exploration of long-chain vitamin E metabolites for the discovery of a highly potent, orally effective, and metabolically stable 5-LOX inhibitor that limits inflammation. J. Med. Chem. 2021, 64, 11496–11526. [Google Scholar] [CrossRef] [PubMed]
- Malebari, A.M.; Greene, L.M.; Nathwani, S.M.; Fayne, D.; O’Boyle, N.M.; Wang, S.; Twamley, B.; Zisterer, D.M.; Meegan, M.J. β-Lactam analogues of combretastatin A-4 prevent metabolic inactivation by glucuronidation in chemoresistant HT-29 colon cancer cells. Eur. J. Med. Chem. 2017, 130, 261–285. [Google Scholar] [CrossRef] [PubMed]
- Malebari, A.M.; Fayne, D.; Nathwani, S.M.; O’Connell, F.; Noorani, S.; Twamley, B.; O’Boyle, N.M.; O’Sullivan, J.; Zisterer, D.M.; Meegan, M.J. β-Lactams with antiproliferative and antiapoptotic activity in breast and chemoresistant colon cancer cells. Eur J Med. Chem. 2020, 189, 112050. [Google Scholar] [CrossRef] [PubMed]
- McLoughlin, E.C.; O’Brien, J.E.; Trujillo, C.; Meegan, M.J.; O’Boyle, N.M. Application of 2D EXSY and qNMR Spectroscopy for Diastereomeric Excess Determination Following Chiral Resolution of β-Lactams. ChemistryOpen 2022, e202200119. [Google Scholar] [CrossRef] [PubMed]











































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Boyle, N.M.; Helesbeux, J.-J.; Meegan, M.J.; Sasse, A.; O’Shaughnessy, E.; Qaisar, A.; Clancy, A.; McCarthy, F.; Marchand, P. 30th Annual GP2A Medicinal Chemistry Conference. Pharmaceuticals 2023, 16, 432. https://doi.org/10.3390/ph16030432
O’Boyle NM, Helesbeux J-J, Meegan MJ, Sasse A, O’Shaughnessy E, Qaisar A, Clancy A, McCarthy F, Marchand P. 30th Annual GP2A Medicinal Chemistry Conference. Pharmaceuticals. 2023; 16(3):432. https://doi.org/10.3390/ph16030432
Chicago/Turabian StyleO’Boyle, Niamh M., Jean-Jacques Helesbeux, Mary J. Meegan, Astrid Sasse, Elizabeth O’Shaughnessy, Alina Qaisar, Aoife Clancy, Florence McCarthy, and Pascal Marchand. 2023. "30th Annual GP2A Medicinal Chemistry Conference" Pharmaceuticals 16, no. 3: 432. https://doi.org/10.3390/ph16030432