
Engineered Mesenchymal Stem Cells Over-Expressing BDNF Protect the Brain from Traumatic Brain Injury-Induced Neuronal Death, Neurological Deficits, and Cognitive Impairments

 ,
, 

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
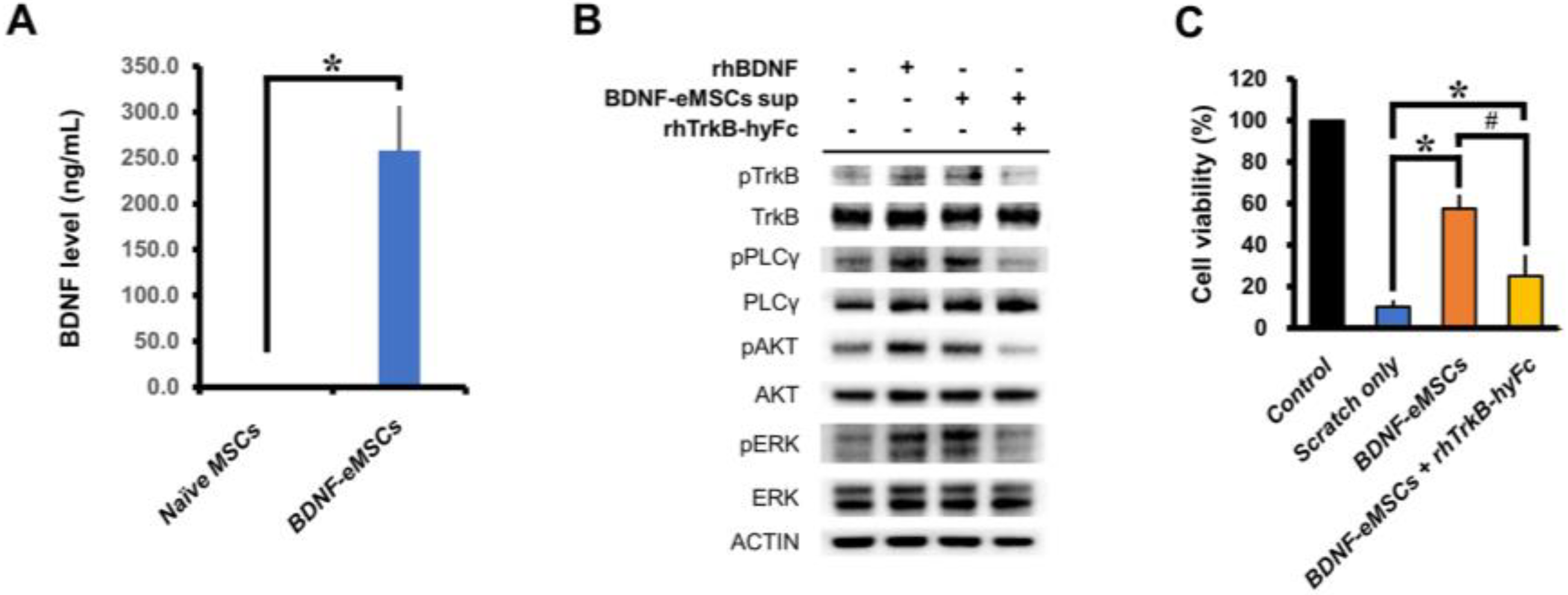
2.1. In Vitro Cellular Characterization and Neural Protection Effect of BDNF-eMSCs
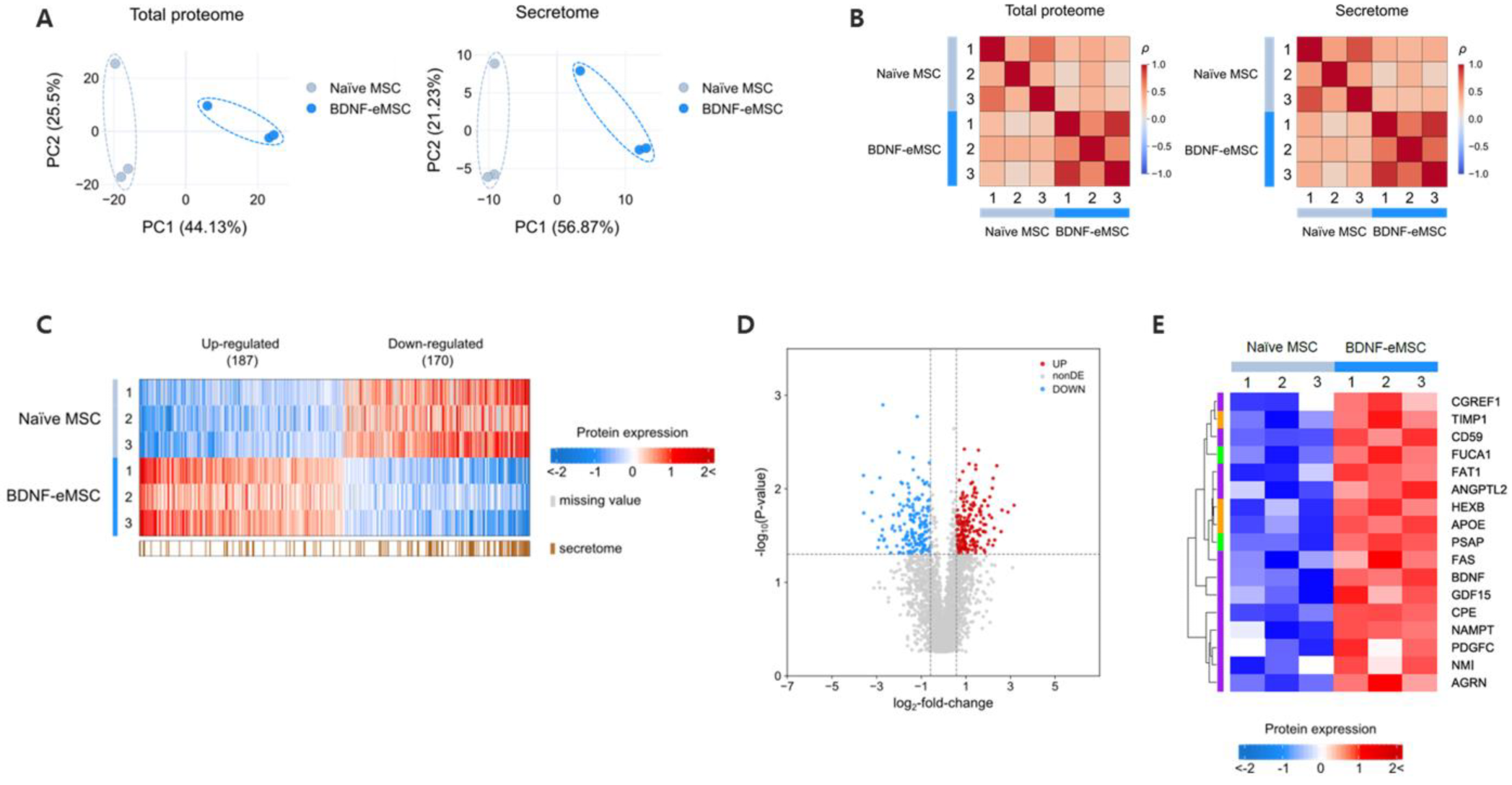
2.2. Proteomic Analysis of Supernatant of BDNF-eMSCs
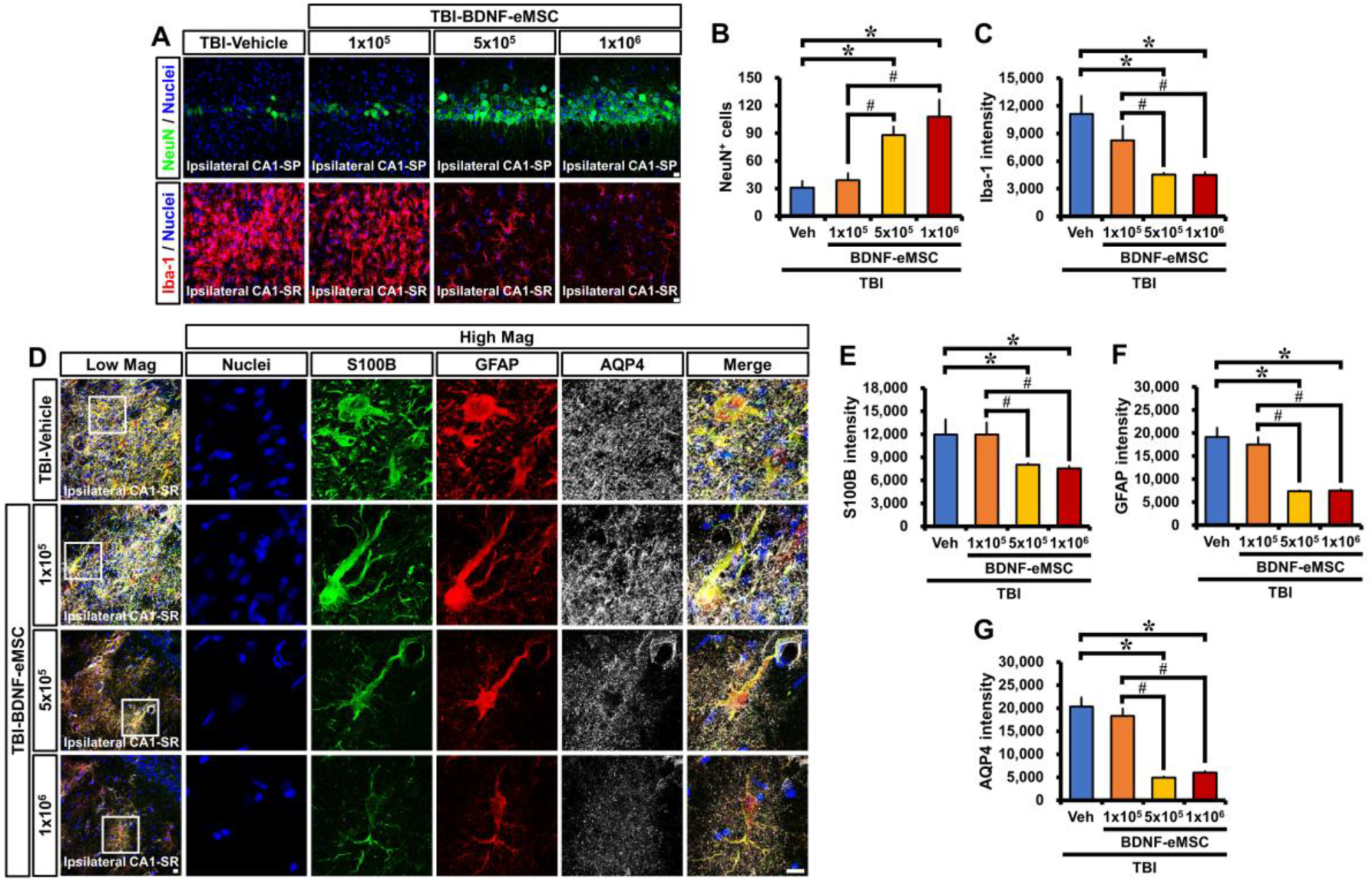
2.3. BDNF-eMSC Administration Reduced Neuronal Death and Glial Activation after TBI
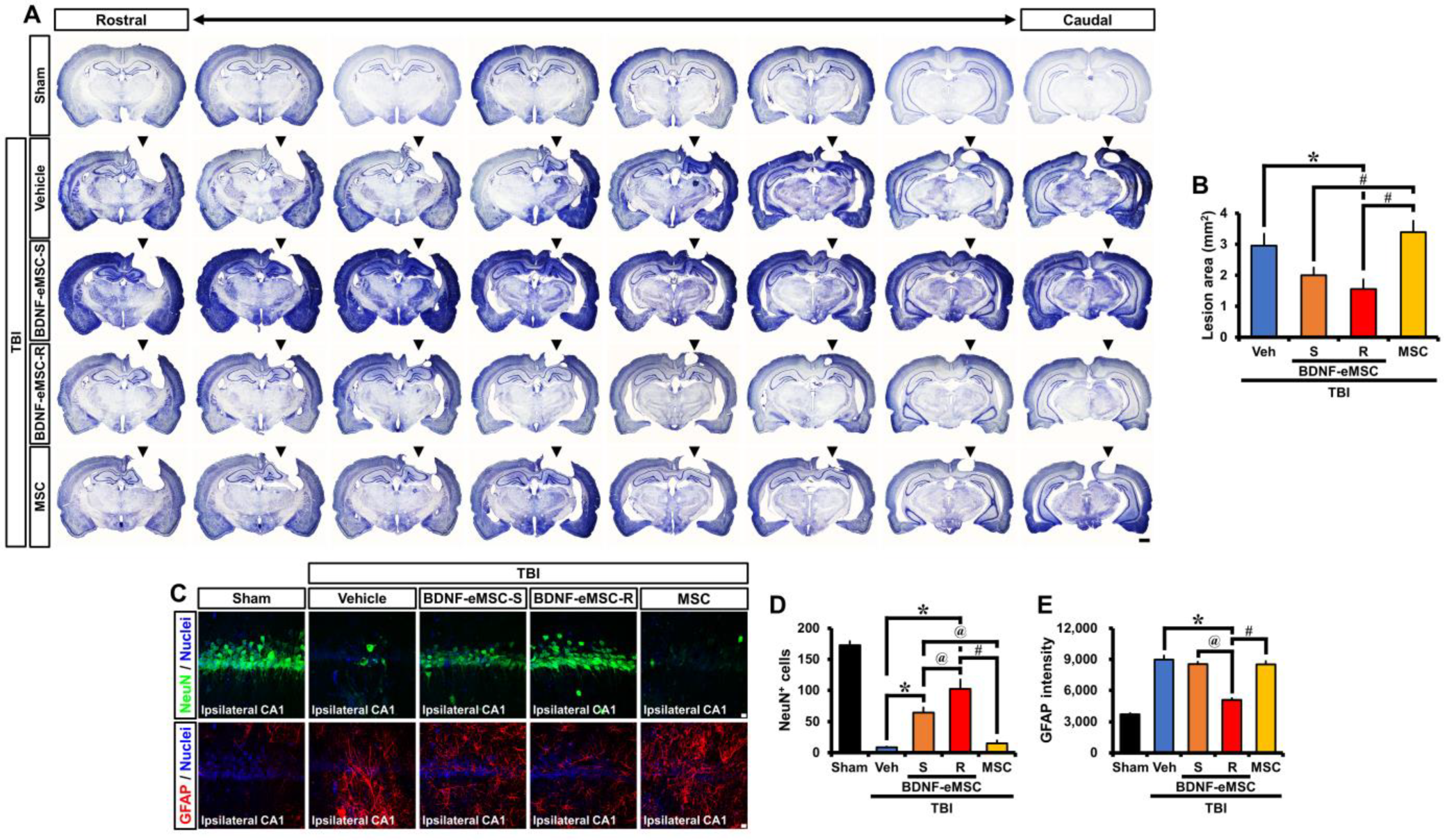
2.4. Long-Term Neuroprotective Effects of Repeated BDNF-eMSC Administration against TBI
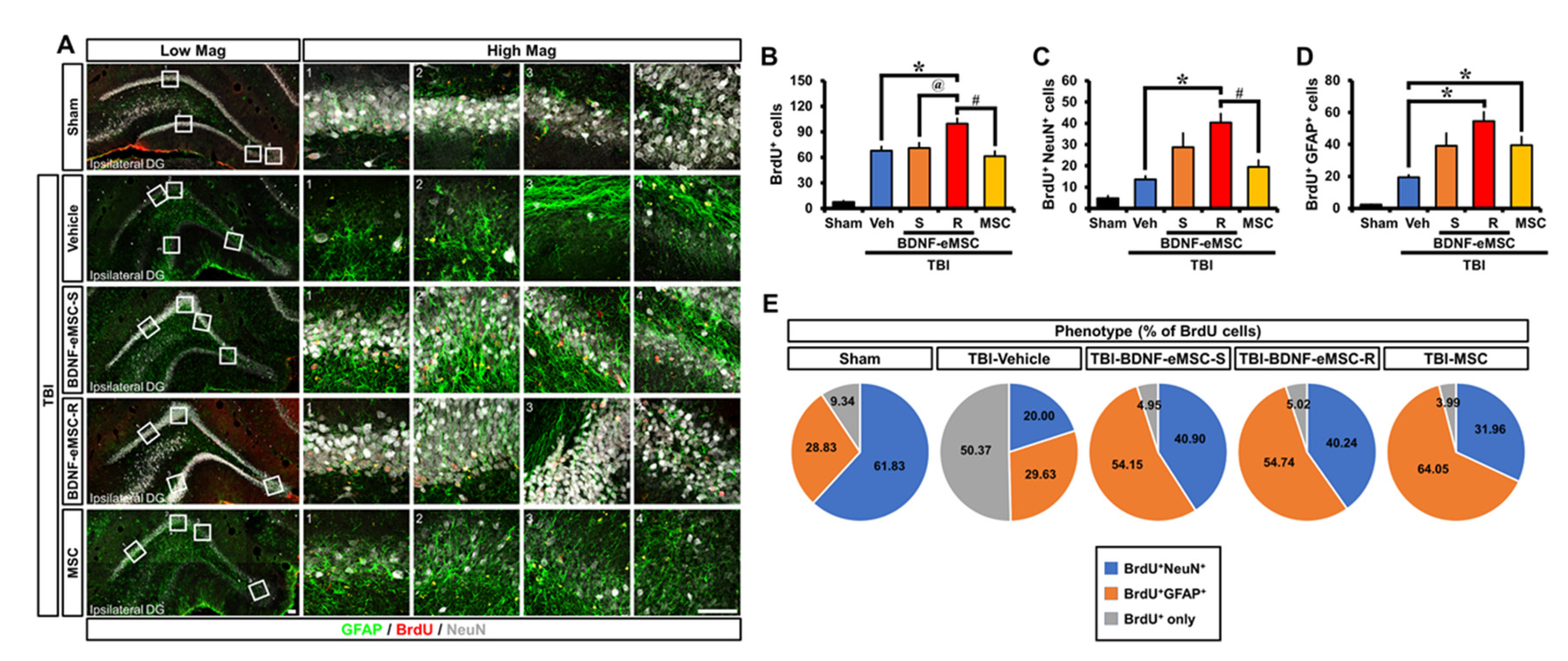
2.5. Repeated BDNF-eMSC Administration Increased Adult Hippocampal Neurogenesis and Gliogenesis after TBI
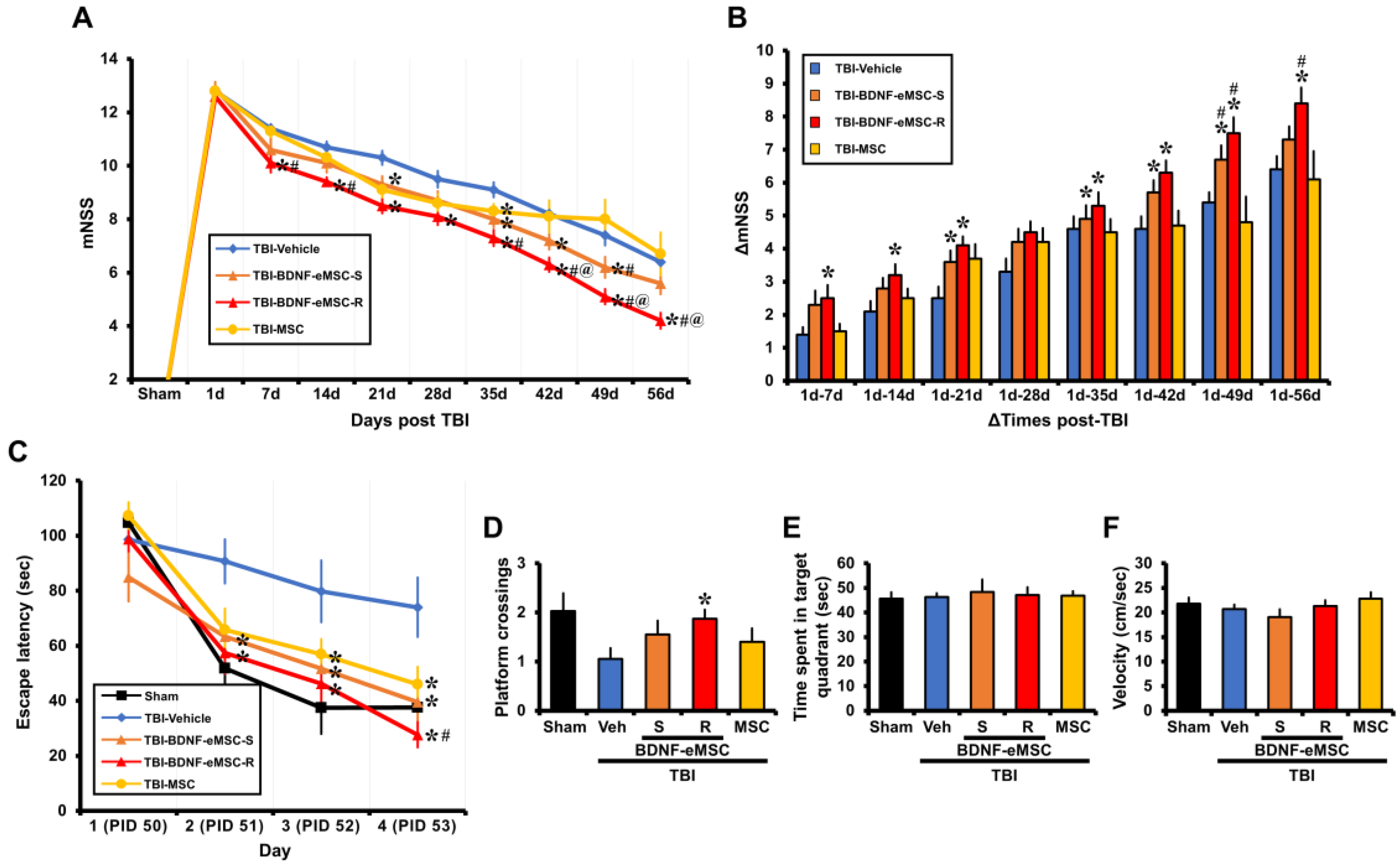
2.6. Repeated BDNF-eMSC Administration Improved Neurological and Cognitive Function after TBI
3. Discussion
4. Materials and Methods
4.1. Cell Preparation
4.2. Analysis of Cell Morphology
4.3. In Vitro Transformation Assay
4.4. Enzyme-Linked Immunosorbent Assay (ELISA)
4.5. Immunoblot Analysis
4.6. In Vitro Scratch Assay
4.7. Proteomic Analysis and Data Processing
4.8. Differential Expression Analysis
4.9. Selection of Signature Proteins
4.10. Gene Ontology Analysis
4.11. Experimental Animals and Ethics Statement
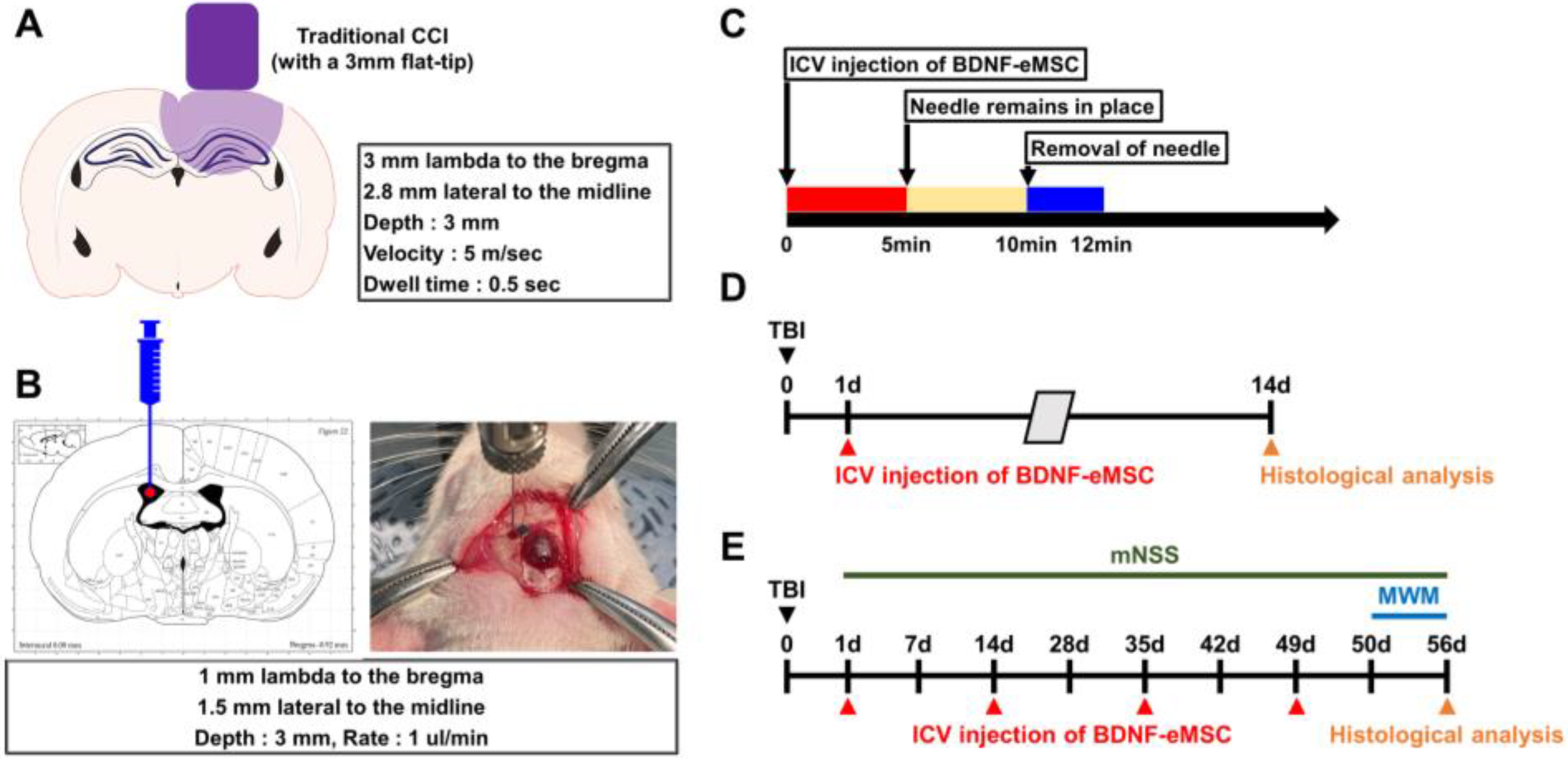
4.12. In Vivo Model for TBI
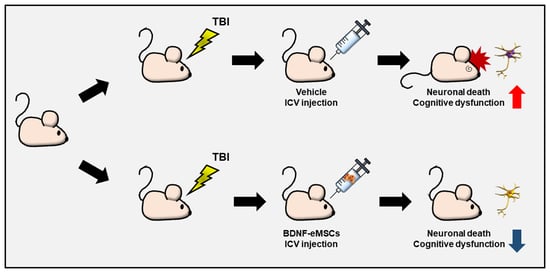
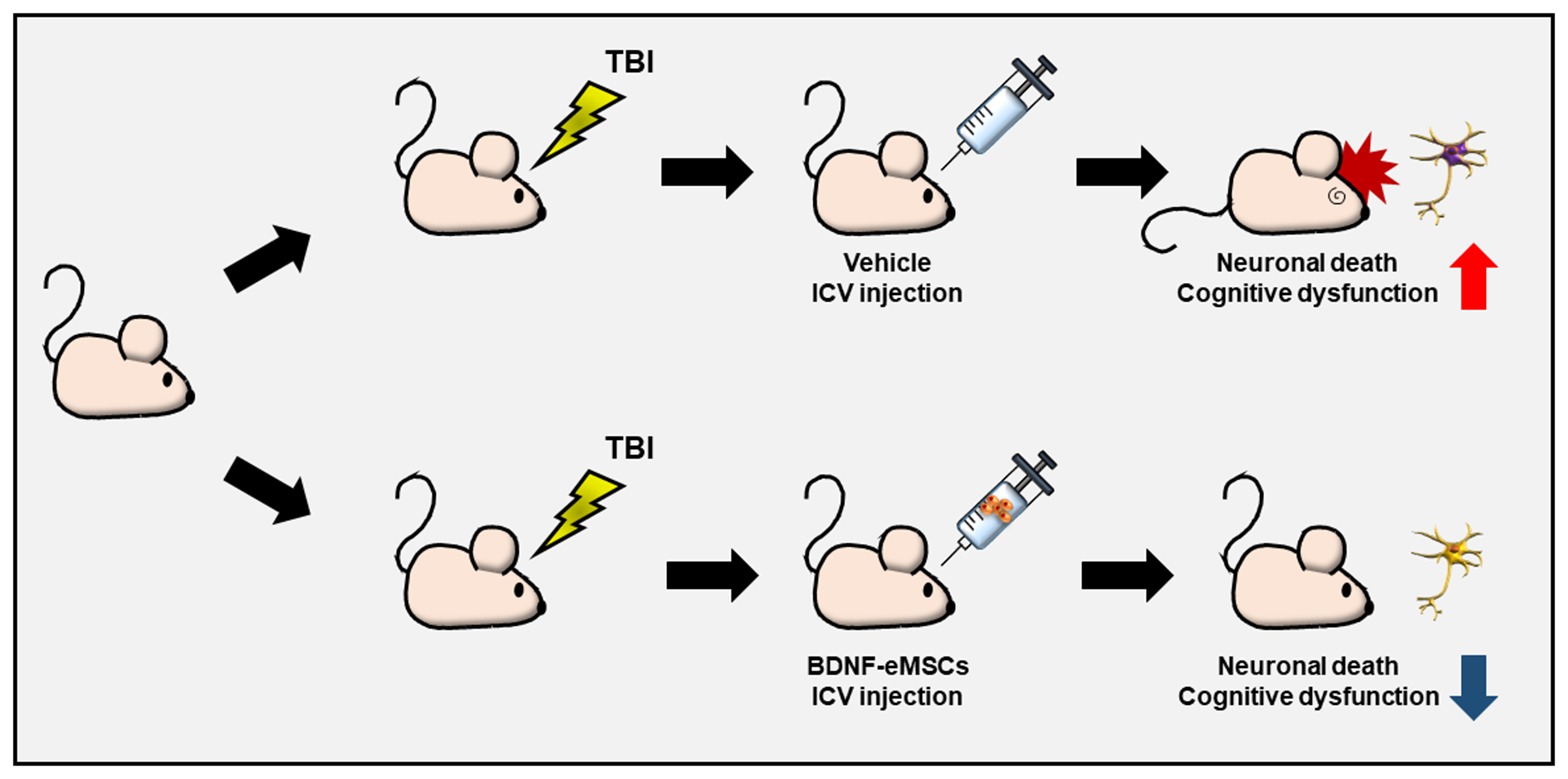
4.13. BDNF-eMSC Administration and Experimental Design
4.14. Tissue Preparation
4.15. Immunofluorescence Analysis
4.16. Measurement of the Lesion Area
4.17. Assessment of Neurological Deficits
4.18. Morris Water Maze Test
4.19. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ghajar, J. Traumatic brain injury. Lancet 2000, 356, 923–929. [Google Scholar] [CrossRef] [PubMed]
- DALYs, G.B.D.; Collaborators, H.; Murray, C.J.; Barber, R.M.; Foreman, K.J.; Abbasoglu Ozgoren, A.; Abd-Allah, F.; Abera, S.F.; Aboyans, V.; Abraham, J.P.; et al. Global, regional, and national disability-adjusted life years (DALYs) for 306 diseases and injuries and healthy life expectancy (HALE) for 188 countries, 1990–2013: Quantifying the epidemiological transition. Lancet 2015, 386, 2145–2191. [Google Scholar] [CrossRef]
- Menon, D.K. Unique challenges in clinical trials in traumatic brain injury. Crit. Care Med. 2009, 37, S129–S135. [Google Scholar] [CrossRef] [PubMed]
- Duncan, T.; Valenzuela, M. Alzheimer’s disease, dementia, and stem cell therapy. Stem Cell Res. Ther. 2017, 8, 111. [Google Scholar] [CrossRef] [PubMed]
- Ali, F.; Stott, S.R.; Barker, R.A. Stem cells and the treatment of Parkinson’s disease. Exp. Neurol. 2014, 260, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.W.; Noh, M.Y.; Kwon, M.S.; Kim, H.Y.; Oh, S.I.; Park, J.; Kim, H.J.; Ki, C.S.; Kim, S.H. Repeated Intrathecal Mesenchymal Stem Cells for Amyotrophic Lateral Sclerosis. Ann. Neurol. 2018, 84, 361–373. [Google Scholar] [CrossRef]
- Xu, X.; Tay, Y.; Sim, B.; Yoon, S.I.; Huang, Y.; Ooi, J.; Utami, K.H.; Ziaei, A.; Ng, B.; Radulescu, C.; et al. Reversal of Phenotypic Abnormalities by CRISPR/Cas9-Mediated Gene Correction in Huntington Disease Patient-Derived Induced Pluripotent Stem Cells. Stem Cell Rep. 2017, 8, 619–633. [Google Scholar] [CrossRef]
- Kim, J.; Shin, K.; Cha, Y.; Ban, Y.H.; Park, S.K.; Jeong, H.S.; Park, D.; Choi, E.K.; Kim, Y.B. Neuroprotective effects of human neural stem cells over-expressing choline acetyltransferase in a middle cerebral artery occlusion model. J. Chem. Neuroanat. 2020, 103, 101730. [Google Scholar] [CrossRef]
- Xu, C.; Fu, F.; Li, X.; Zhang, S. Mesenchymal stem cells maintain the microenvironment of central nervous system by regulating the polarization of macrophages/microglia after traumatic brain injury. Int. J. Neurosci. 2017, 127, 1124–1135. [Google Scholar] [CrossRef]
- Cox, C.S., Jr. Cellular therapy for traumatic neurological injury. Pediatr. Res. 2018, 83, 325–332. [Google Scholar] [CrossRef]
- Zhou, Y.; Shao, A.; Xu, W.; Wu, H.; Deng, Y. Advance of Stem Cell Treatment for Traumatic Brain Injury. Front. Cell Neurosci. 2019, 13, 301. [Google Scholar] [CrossRef] [PubMed]
- Lo Furno, D.; Mannino, G.; Giuffrida, R. Functional role of mesenchymal stem cells in the treatment of chronic neurodegenerative diseases. J. Cell Physiol. 2018, 233, 3982–3999. [Google Scholar] [CrossRef] [PubMed]
- Yu-Taeger, L.; Stricker-Shaver, J.; Arnold, K.; Bambynek-Dziuk, P.; Novati, A.; Singer, E.; Lourhmati, A.; Fabian, C.; Magg, J.; Riess, O.; et al. Intranasal Administration of Mesenchymal Stem Cells Ameliorates the Abnormal Dopamine Transmission System and Inflammatory Reaction in the R6/2 Mouse Model of Huntington Disease. Cells 2019, 8, 595. [Google Scholar] [CrossRef] [PubMed]
- Acheson, A.; Conover, J.C.; Fandl, J.P.; DeChiara, T.M.; Russell, M.; Thadani, A.; Squinto, S.P.; Yancopoulos, G.D.; Lindsay, R.M. A BDNF autocrine loop in adult sensory neurons prevents cell death. Nature 1995, 374, 450–453. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in neuronal development and function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef]
- Binder, D.K.; Scharfman, H.E. Brain-derived neurotrophic factor. Growth Factors 2004, 22, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.S.; Shen, L.L.; Zhu, C.; Bu, X.L.; Liu, Y.H.; Liu, C.H.; Yao, X.Q.; Zhang, L.L.; Zhou, H.D.; Walker, D.G.; et al. Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer’s disease. Transl. Psychiatry 2016, 6, e907. [Google Scholar] [CrossRef]
- Hernandez-Chan, N.G.; Bannon, M.J.; Orozco-Barrios, C.E.; Escobedo, L.; Zamudio, S.; De la Cruz, F.; Gongora-Alfaro, J.L.; Armendariz-Borunda, J.; Reyes-Corona, D.; Espadas-Alvarez, A.J.; et al. Neurotensin-polyplex-mediated brain-derived neurotrophic factor gene delivery into nigral dopamine neurons prevents nigrostriatal degeneration in a rat model of early Parkinson’s disease. J. Biomed. Sci. 2015, 22, 59. [Google Scholar] [CrossRef]
- Giampa, C.; Montagna, E.; Dato, C.; Melone, M.A.; Bernardi, G.; Fusco, F.R. Systemic delivery of recombinant brain derived neurotrophic factor (BDNF) in the R6/2 mouse model of Huntington’s disease. PLoS ONE 2013, 8, e64037. [Google Scholar] [CrossRef]
- Schabitz, W.R.; Steigleder, T.; Cooper-Kuhn, C.M.; Schwab, S.; Sommer, C.; Schneider, A.; Kuhn, H.G. Intravenous brain-derived neurotrophic factor enhances poststroke sensorimotor recovery and stimulates neurogenesis. Stroke 2007, 38, 2165–2172. [Google Scholar] [CrossRef]
- Han, B.H.; Holtzman, D.M. BDNF protects the neonatal brain from hypoxic-ischemic injury in vivo via the ERK pathway. J. Neurosci. 2000, 20, 5775–5781. [Google Scholar] [CrossRef] [PubMed]
- Jeong, C.H.; Kim, S.M.; Lim, J.Y.; Ryu, C.H.; Jun, J.A.; Jeun, S.S. Mesenchymal stem cells expressing brain-derived neurotrophic factor enhance endogenous neurogenesis in an ischemic stroke model. Biomed. Res. Int. 2014, 2014, 129145. [Google Scholar] [CrossRef] [PubMed]
- Pollock, K.; Dahlenburg, H.; Nelson, H.; Fink, K.D.; Cary, W.; Hendrix, K.; Annett, G.; Torrest, A.; Deng, P.; Gutierrez, J.; et al. Human Mesenchymal Stem Cells Genetically Engineered to Overexpress Brain-derived Neurotrophic Factor Improve Outcomes in Huntington’s Disease Mouse Models. Mol. Ther. 2016, 24, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Price, R.D.; Milne, S.A.; Sharkey, J.; Matsuoka, N. Advances in small molecules promoting neurotrophic function. Pharmacol. Ther. 2007, 115, 292–306. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.Y.; Sung, D.K.; Chang, Y.S.; Sung, S.I.; Kim, Y.E.; Kim, H.J.; Lee, S.M.; Park, W.S. BDNF-Overexpressing Engineered Mesenchymal Stem Cells Enhances Their Therapeutic Efficacy against Severe Neonatal Hypoxic Ischemic Brain Injury. Int. J. Mol. Sci. 2021, 22, 11395. [Google Scholar] [CrossRef]
- Chen, A.I.; Nguyen, C.N.; Copenhagen, D.R.; Badurek, S.; Minichiello, L.; Ranscht, B.; Reichardt, L.F. TrkB (tropomyosin-related kinase B) controls the assembly and maintenance of GABAergic synapses in the cerebellar cortex. J. Neurosci. 2011, 31, 2769–2780. [Google Scholar] [CrossRef]
- Loane, D.J.; Kumar, A.; Stoica, B.A.; Cabatbat, R.; Faden, A.I. Progressive neurodegeneration after experimental brain trauma: Association with chronic microglial activation. J. Neuropathol. Exp. Neurol. 2014, 73, 14–29. [Google Scholar] [CrossRef]
- Block, M.L.; Hong, J.S. Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mechanism. Prog. Neurobiol. 2005, 76, 77–98. [Google Scholar] [CrossRef]
- Marmarou, A. Pathophysiology of traumatic brain edema: Current concepts. Acta Neurochir. Suppl. 2003, 86, 7–10. [Google Scholar]
- Donkin, J.J.; Vink, R. Mechanisms of cerebral edema in traumatic brain injury: Therapeutic developments. Curr. Opin. Neurol. 2010, 23, 293–299. [Google Scholar] [CrossRef]
- Yang, K.; Perez-Polo, J.R.; Mu, X.S.; Yan, H.Q.; Xue, J.J.; Iwamoto, Y.; Liu, S.J.; Dixon, C.E.; Hayes, R.L. Increased expression of brain-derived neurotrophic factor but not neurotrophin-3 mRNA in rat brain after cortical impact injury. J. Neurosci. Res. 1996, 44, 157–164. [Google Scholar] [CrossRef]
- Oyesiku, N.M.; Evans, C.O.; Houston, S.; Darrell, R.S.; Smith, J.S.; Fulop, Z.L.; Dixon, C.E.; Stein, D.G. Regional changes in the expression of neurotrophic factors and their receptors following acute traumatic brain injury in the adult rat brain. Brain Res. 1999, 833, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Merlio, J.P.; Ernfors, P.; Kokaia, Z.; Middlemas, D.S.; Bengzon, J.; Kokaia, M.; Smith, M.L.; Siesjo, B.K.; Hunter, T.; Lindvall, O.; et al. Increased production of the TrkB protein tyrosine kinase receptor after brain insults. Neuron 1993, 10, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Scheff, S.W. Endogenous neuroprotection factors and traumatic brain injury: Mechanisms of action and implications for therapy. J. Neurotrauma 1994, 11, 3–33. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Pan, S.; Sun, Z.; Dan, Q.; Liu, J. Brain-derived neurotrophic factor-modified umbilical cord mesenchymal stem cell transplantation improves neurological deficits in rats with traumatic brain injury. Int. J. Neurosci. 2014, 124, 524–531. [Google Scholar] [CrossRef]
- Barone, F.C.; Feuerstein, G.Z. Inflammatory mediators and stroke: New opportunities for novel therapeutics. J. Cereb. Blood Flow Metab. 1999, 19, 819–834. [Google Scholar] [CrossRef]
- Chao, C.C.; Hu, S.; Molitor, T.W.; Shaskan, E.G.; Peterson, P.K. Activated microglia mediate neuronal cell injury via a nitric oxide mechanism. J. Immunol. 1992, 149, 2736–2741. [Google Scholar] [CrossRef]
- Hanisch, U.K. Microglia as a source and target of cytokines. Glia 2002, 40, 140–155. [Google Scholar] [CrossRef]
- Park, H.; Poo, M.M. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 2013, 14, 7–23. [Google Scholar] [CrossRef]
- Leal, G.; Comprido, D.; Duarte, C.B. BDNF-induced local protein synthesis and synaptic plasticity. Neuropharmacology 2014, 76 (Pt C), 639–656. [Google Scholar] [CrossRef]
- Leal, G.; Afonso, P.M.; Salazar, I.L.; Duarte, C.B. Regulation of hippocampal synaptic plasticity by BDNF. Brain Res. 2015, 1621, 82–101. [Google Scholar] [CrossRef] [PubMed]
- Aimone, J.B.; Wiles, J.; Gage, F.H. Potential role for adult neurogenesis in the encoding of time in new memories. Nat. Neurosci. 2006, 9, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Saxe, M.D.; Gallina, I.S.; Gage, F.H. Adult-born hippocampal dentate granule cells undergoing maturation modulate learning and memory in the brain. J. Neurosci. 2009, 29, 13532–13542. [Google Scholar] [CrossRef] [PubMed]
- Scharfman, H.; Goodman, J.; Macleod, A.; Phani, S.; Antonelli, C.; Croll, S. Increased neurogenesis and the ectopic granule cells after intrahippocampal BDNF infusion in adult rats. Exp. Neurol. 2005, 192, 348–356. [Google Scholar] [CrossRef]
- Chan, J.P.; Cordeira, J.; Calderon, G.A.; Iyer, L.K.; Rios, M. Depletion of central BDNF in mice impedes terminal differentiation of new granule neurons in the adult hippocampus. Mol Cell Neurosci. 2008, 39, 372–383. [Google Scholar] [CrossRef]
- Gao, X.; Chen, J. Conditional knockout of brain-derived neurotrophic factor in the hippocampus increases death of adult-born immature neurons following traumatic brain injury. J. Neurotrauma 2009, 26, 1325–1335. [Google Scholar] [CrossRef]
- Kempermann, G.; Kuhn, H.G.; Gage, F.H. Genetic influence on neurogenesis in the dentate gyrus of adult mice. Proc. Natl. Acad. Sci. USA 1997, 94, 10409–10414. [Google Scholar] [CrossRef]
- Biebl, M.; Cooper, C.M.; Winkler, J.; Kuhn, H.G. Analysis of neurogenesis and programmed cell death reveals a self-renewing capacity in the adult rat brain. Neurosci. Lett. 2000, 291, 17–20. [Google Scholar] [CrossRef]
- Somayaji, M.R.; Przekwas, A.J.; Gupta, R.K. Combination Therapy for Multi-Target Manipulation of Secondary Brain Injury Mechanisms. Curr. Neuropharmacol. 2018, 16, 484–504. [Google Scholar] [CrossRef]
- Park, S.E.; Lee, N.K.; Na, D.L.; Chang, J.W. Optimal mesenchymal stem cell delivery routes to enhance neurogenesis for the treatment of Alzheimer’s disease: Optimal MSCs delivery routes for the treatment of AD. Histol. Histopathol. 2018, 33, 533–541. [Google Scholar] [CrossRef]
- Falo, M.C.; Reeves, T.M.; Phillips, L.L. Agrin expression during synaptogenesis induced by traumatic brain injury. J. Neurotrauma 2008, 25, 769–783. [Google Scholar] [CrossRef] [PubMed]
- Mondal, K.; Takahashi, H.; Cole, J., 2nd; Del Mar, N.A.; Li, C.; Stephenson, D.J.; Allegood, J.; Cowart, L.A.; Chalfant, C.E.; Reiner, A.; et al. Systemic Elevation of n-3 Polyunsaturated Fatty Acids (n-3-PUFA) Is Associated with Protection against Visual, Motor, and Emotional Deficits in Mice following Closed-Head Mild Traumatic Brain Injury. Mol. Neurobiol. 2021, 58, 5564–5580. [Google Scholar] [CrossRef] [PubMed]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef]
- Eng, J.K.; Jahan, T.A.; Hoopmann, M.R. Comet: An open-source MS/MS sequence database search tool. Proteomics 2013, 13, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Chae, S.; Ahn, B.Y.; Byun, K.; Cho, Y.M.; Yu, M.H.; Lee, B.; Hwang, D.; Park, K.S. A systems approach for decoding mitochondrial retrograde signaling pathways. Sci. Signal 2013, 6, rs4. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.; Rust, A.G.; Ramsey, S.; Smith, J.J.; Leslie, D.M.; Weston, A.D.; de Atauri, P.; Aitchison, J.D.; Hood, L.; Siegel, A.F.; et al. A data integration methodology for systems biology. Proc. Natl. Acad. Sci. USA 2005, 102, 17296–17301. [Google Scholar] [CrossRef]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 2012, 8, e1000412. [Google Scholar] [CrossRef]
- Choi, B.Y.; Jang, B.G.; Kim, J.H.; Lee, B.E.; Sohn, M.; Song, H.K.; Suh, S.W. Prevention of traumatic brain injury-induced neuronal death by inhibition of NADPH oxidase activation. Brain Res. 2012, 1481, 49–58. [Google Scholar] [CrossRef]
- Choi, B.Y.; Lee, S.H.; Choi, H.C.; Lee, S.K.; Yoon, H.S.; Park, J.B.; Chung, W.S.; Suh, S.W. Alcohol dependence treating agent, acamprosate, prevents traumatic brain injury-induced neuron death through vesicular zinc depletion. Transl. Res. 2019, 207, 1–18. [Google Scholar] [CrossRef]
- Xing, Z.; Xia, Z.; Peng, W.; Li, J.; Zhang, C.; Fu, C.; Tang, T.; Luo, J.; Zou, Y.; Fan, R.; et al. Xuefu Zhuyu decoction, a traditional Chinese medicine, provides neuroprotection in a rat model of traumatic brain injury via an anti-inflammatory pathway. Sci. Rep. 2016, 6, 20040. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Sanberg, P.R.; Li, Y.; Wang, L.; Lu, M.; Willing, A.E.; Sanchez-Ramos, J.; Chopp, M. Intravenous administration of human umbilical cord blood reduces behavioral deficits after stroke in rats. Stroke 2001, 32, 2682–2688. [Google Scholar] [CrossRef] [PubMed]
- Brayer, S.W.; Ketcham, S.; Zou, H.; Hurwitz, M.; Henderson, C.; Fuletra, J.; Kumar, K.; Skidmore, E.; Thiels, E.; Wagner, A.K. Developing a clinically relevant model of cognitive training after experimental traumatic brain injury. Neurorehabil. Neural Repair 2015, 29, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Mennenga, S.E.; Gerson, J.E.; Dunckley, T.; Bimonte-Nelson, H.A. Harmine treatment enhances short-term memory in old rats: Dissociation of cognition and the ability to perform the procedural requirements of maze testing. Physiol. Behav. 2015, 138, 260–265. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, B.Y.; Hong, D.K.; Kang, B.S.; Lee, S.H.; Choi, S.; Kim, H.-J.; Lee, S.M.; Suh, S.W. Engineered Mesenchymal Stem Cells Over-Expressing BDNF Protect the Brain from Traumatic Brain Injury-Induced Neuronal Death, Neurological Deficits, and Cognitive Impairments. Pharmaceuticals 2023, 16, 436. https://doi.org/10.3390/ph16030436
Choi BY, Hong DK, Kang BS, Lee SH, Choi S, Kim H-J, Lee SM, Suh SW. Engineered Mesenchymal Stem Cells Over-Expressing BDNF Protect the Brain from Traumatic Brain Injury-Induced Neuronal Death, Neurological Deficits, and Cognitive Impairments. Pharmaceuticals. 2023; 16(3):436. https://doi.org/10.3390/ph16030436
Chicago/Turabian StyleChoi, Bo Young, Dae Ki Hong, Beom Seok Kang, Si Hyun Lee, Seunghyuk Choi, Hyo-Jin Kim, Soon Min Lee, and Sang Won Suh. 2023. "Engineered Mesenchymal Stem Cells Over-Expressing BDNF Protect the Brain from Traumatic Brain Injury-Induced Neuronal Death, Neurological Deficits, and Cognitive Impairments" Pharmaceuticals 16, no. 3: 436. https://doi.org/10.3390/ph16030436
APA StyleChoi, B. Y., Hong, D. K., Kang, B. S., Lee, S. H., Choi, S., Kim, H.-J., Lee, S. M., & Suh, S. W. (2023). Engineered Mesenchymal Stem Cells Over-Expressing BDNF Protect the Brain from Traumatic Brain Injury-Induced Neuronal Death, Neurological Deficits, and Cognitive Impairments. Pharmaceuticals, 16(3), 436. https://doi.org/10.3390/ph16030436