Abstract
Neuroprotective drugs to protect the brain against cerebral ischemia and reperfusion (I/R) injury are urgently needed. Mammalian cell-produced recombinant human erythropoietin (rhuEPOM) has been demonstrated to have excellent neuroprotective functions in preclinical studies, but its neuroprotective properties could not be consistently translated in clinical trials. The clinical failure of rhuEPOM was thought to be mainly due to its erythropoietic activity-associated side effects. To exploit its tissue-protective property, various EPO derivatives with tissue-protective function only have been developed. Among them, asialo-rhuEPO, lacking terminal sialic acid residues, was shown to be neuroprotective but non-erythropoietic. Asialo-rhuEPO can be prepared by enzymatic removal of sialic acid residues from rhuEPOM (asialo-rhuEPOE) or by expressing human EPO gene in glycoengineered transgenic plants (asialo-rhuEPOP). Both types of asialo-rhuEPO, like rhuEPOM, displayed excellent neuroprotective effects by regulating multiple cellular pathways in cerebral I/R animal models. In this review, we describe the structure and properties of EPO and asialo-rhuEPO, summarize the progress on neuroprotective studies of asialo-rhuEPO and rhuEPOM, discuss potential reasons for the clinical failure of rhuEPOM with acute ischemic stroke patients, and advocate future studies needed to develop asialo-rhuEPO as a multimodal neuroprotectant for ischemic stroke treatment.
1. Introduction
Stroke remains a major cause of mortality and long-term disability globally. Although the age-standardized rates of incidence and death from strokes have decreased since 1990, the annual number of deaths due to stroke increased substantially, with over 12 million incidents and more than 100 million prevalent cases worldwide [1]. Consequently, stroke and post-stroke care continue to present major social and economic challenges for society. Strokes are mainly classified as either ischemic or hemorrhagic, with the former accounting for approximately 87% of cases [1,2]. Following an ischemic stroke (IS) caused by the blockage of an artery in the brain, a lack of cerebral blood supply immediately causes severe oxygen and glucose deprivation in the blocked region, leading to suppression of ATP production, reduction of pH, impairment of mitochondrial dynamics, inhibition of glycosylation capacity, and increased production of reactive oxygen species (ROS) [3,4,5,6,7]. These pathophysiological changes result in neuronal cell injury and death [2,3]. Timely reperfusion by thrombolysis or mechanical thrombectomy to re-establish blood flow is an effective therapeutic strategy to salvage damaged nerve cells and improve clinical outcomes by reducing cerebral ischemic damage and preserving brain functions [2,3,6]. However, reperfusion paradoxically exacerbates brain tissue injury [3,8,9]. Therefore, adjunct neuroprotection alongside reperfusion is crucial and may bring better outcomes.
Cerebral ischemia and reperfusion (I/R) injury is a detrimental process, since both I/R processes can cause various pathophysiological changes. The damaging effects induced by ischemia, reperfusion, or their combined action are not separable [3,8,10]. The mechanisms underlying cerebral I/R injury are complex and include metabolism dysfunction, mitochondrial dysregulation, oxidative stress, disruption of blood–brain barrier (BBB), leukocyte infiltration, brain inflammation, and various types of cell death, such as necrosis, apoptosis, autophagy, necroptosis, and pyroptosis [3,9,11,12]. While the detailed mechanisms are not fully understood, these complex pathophysiological changes ultimately lead to infarction and subsequent cognitive impairments [3,10,11]. Nevertheless, the discovery of neuroprotective therapeutics and further understanding the complexity of the I/R-induced injury cascade are crucial for developing improved treatments.
Based on the current understanding of the I/R injury, various pharmacological and mechanical interventions have been explored to protect the brain from its detrimental effects with some efficacy. These interventions include inhibiting apoptosis, promoting angiogenesis, suppressing the immune system and inflammation, reducing ROS production and stress response, and regulating metabolic processes [2,3,7,10,13]. Unfortunately, most of these strategies failed in clinical trials, leaving no safe and effective therapeutic treatment to ameliorate the repercussions of cerebral I/R injury [2,10,13]. Moreover, therapies targeting a single specific mechanism have been found to be insufficient, as I/R injury involves multiple pathophysiological pathways [14,15]. Thus, an agent targeting an array of key cellular pathways has been proposed in order to have better long-term benefits [14,15,16].
Erythropoietin (EPO) is a glycoprotein hormone primarily known for regulating red blood cell (RBC) production (Table 1) [17]. In addition to its hematopoietic activity, EPO and its derivatives have been demonstrated to display remarkable anti-apoptotic and broad tissue-protective effects against damage triggered by I/R injury, or cytotoxic agents in the brain, the heart, the kidneys, and the liver [18,19,20]. EPO and the EPO receptor (EPOR) are functionally expressed in various non-hematopoietic organs [18,21,22], including neurons, glial cells, and endothelial cells in the central nervous system (CNS) [23,24]. Recombinant human EPO produced in mammalian cells (rhuEPOM) has been demonstrated to display remarkable neuroprotection in animal models of ischemic stroke [25,26,27]. Most importantly, numerous studies have also revealed that its tissue-protective properties are mediated through pleiotropic effects, including anti-oxidative, -apoptotic, -inflammatory, and -excitotoxic effects, as well as angiogenic and neurogenic effects [27,28,29,30].

Table 1.
General information on EPO, rhuEPOM, asialo-rhuEPOE, and asialo-rhuEPOP.
Given the broad tissue-protective and pleiotropic effects, rhuEPOM entered into clinical trials with acute IS patients two decades ago [31], which was followed by several other trials [32,33,34]. Unfortunately, its tissue-protective effects to reduce the infarct size from cerebral I/R injury could not be consistently observed in clinical studies [32]. Its hematopoietic activity-associated side effects (HAASEs), such as hypertension and thrombosis, were believed to mask tissue-protective effects. As a result, scientists developed low- or non-erythropoietic tissue-protective derivatives, such as EPOL (EPO derived from genetically modified goat milk with low hematopoietic activity), Neuro-EPO (EPO produced from mammalian cell with low sialic acid content), CEPO (carbamoylated EPO), asialo-rhuEPO (EPO lacking terminal sialic acid) (Figure 1), and MEPO (mutant EPO made by replacing a single amino acid within the erythropoietic motif) and other EPO variants, conceptually devoid of side effects for brain protection as reviewed by Ma et al. [6]. Among them, asialo-rhuEPO was well-documented to be nonerythropoietic but neuroprotective [35,36,37,38,39,40,41]. It has also been demonstrated to have protective effects on other organs (e.g., heart) from I/R injury [42,43,44].
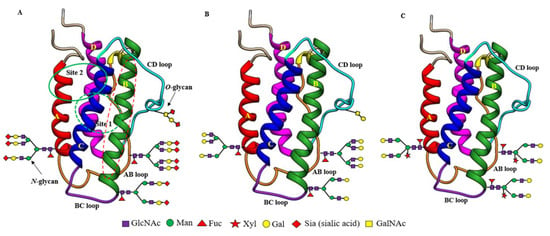
Figure 1.
Structural differences between recombinant sialylated rhuEPOM (A), enzymatically prepared asialo-rhuEPOE (B) and plant-produced asialo-rhuEPOP (C). Their protein structures are the same with two separate hematopoietic receptor (EPOR)2 binding sites (site 1 marked by the green dotted circle at the back side of EPO; site 2 marked by a solid green circle at the front side of EPO). Both binding sites are present distal to the carbohydrate chains. Helix B marked by the red dotted oval is proposed to be involved in EPO-mediated protective functions. All three types of EPO differ with respect to the structure of their N-glycan chains. RhuEPOM bears sialic acid residues (red diamond) as terminal sugar on bi-, tri- and tetrantennary N-glycans while asialo-rhuEPOE lacks sialic acid residues and contains β1,4-galactose residues (yellow circles) as terminal sugars. Asialo-rhuEPOP lacks terminal sialic acid residues like asialo-rhuEPOE. In addition, asialo-rhuEPOP bears biantennary N-glycan chains with plant-specific xylose and fucose residues and lacks O-glycan chain.
This review summarizes recent progress on neuroprotection studies performed with enzymatically prepared asialo-rhuEPO (asialo-rhuEPOE) and plant-produced asialo-rhuEPO (asialo-rhuEPOP) as well as rhuEPOM (Table 1) on the cerebral I/R injury. It also discusses potential factors responsible for rhuEPOM’s clinical failures and the crucial studies needed on asialo-rhuEPO to warrant its success in future clinical trials.
2. EPO and RhuEPOM
2.1. EPO Structures and Properties
As mentioned earlier, EPO (Table 1) is a glycoprotein secreted primarily by the kidneys in adults [45], with small amounts secreted by the liver [46] and the brain [47]. EPO protein encoded by the EPO gene is 193 amino acids long with a 27 amino acid long N-terminal signal peptide. Shortly before secretion, the signal peptide is cleaved, leaving behind matured EPO with only 166 amino acids. However, the physiologically active form circulating in the plasma is 165 amino acids long because of the removal of C-terminal Arg166 by proteolysis [48]. The active mature EPO contains two intramolecular disulfide bonds (Cys7-Cys161, Cys29-Cys33) and bears three N-glycan chains attached to Asn at positions 24, 38, and 83 and an O-glycan chain at Ser 126 [49]. Therefore, EPO is a heavily glycosylated protein with ~40% carbohydrate content and a molecular weight of ~30,400 Daltons [50]. The three-dimensional structure determined by NMR [51] and X-ray crystallography [52] showed that EPO is an elongated molecule consisting of a left-handed, four-helix bundle (Figure 1A), typically present in the hematopoietic growth factor family members. The four long helices (A, B, C, and D) are arranged in an up-up-down-down direction, linked by two long cross-over loops (AB and CD) and one short loop (BC) (Figure 1A). Two distinct patches of amino acids on the protein surface were shown to form two spatially separate binding sites for its homodimeric hematopoietic receptor (EPOR)2: a high-affinity binding site (Site 1, dotted green circle at the back side of EPO), comprising helix D and the AB loop, and a low-affinity binding site (Site 2, solid green circle at the front side of EPO) in the A and C helical bundle (Figure 1A). EPO thus forms a 2:1 homodimeric (EPOR)2:EPO complex.
The carbohydrate (N-glycan) chains in EPO are clustered at one end of the molecule, distal from the receptor-binding sites (Figure 1A). Although the carbohydrate chains constitute approximately 40% of the mass of EPO, they are thought to cover much of the surface of the molecule, but not to be involved in (EPOR)2 binding even though they have been shown to influence EPO’s in vivo hematopoietic activity [53]. The smaller O-glycan chain at Ser 126 has not been found to have any important roles in both in vitro and in vivo hematopoietic activity of EPO yet [53,54] while the N-glycan chains are proven to be indispensable for its secretion, proper folding, stability, and in vivo hematopoietic activity [53,55,56]. Each N-glycan chain in EPO is branched containing 2 to 4 arms (or antennas) capped with negatively charged sugar residue, sialic acid (N-acetylneuraminic acid, Neu5Ac) (Figure 1 and Figure 2), which imparts a net negative charge on the protein molecule, giving EPO an acidic pI in the range of 3.92 to 5.11 (Table 1) [57]. Theoretically, EPO can have around 14 sialic acid residues, with 12 present at 3 N-glycan chains and 2 presenting at 1 O-glycan chain.
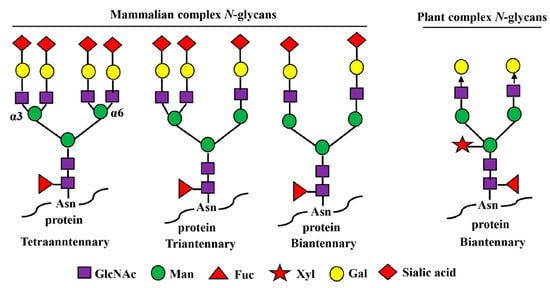
Figure 2.
Mammalian- and plant-produced complex-type N-glycans. Complex N-glycans refer to those in which both the α3- and α6-linked mannose residues are substituted with GlcNAc moieties. In mammals, the N-glycan chains can be bi-, tri-, and tetraantennary, and the GlcNAc residues in each glycan chain are further extended with β1,4-galactose residues and terminal sialic acid residues. The sialic acid in humans is N-acetyl-neuraminic acid (Neu5Ac) while other mammals have both Neu5Ac and N-glycolylneuraminic acids (GlcNGc). In plants, their complex N-glycan chains are biantennary, but do not bear β1,4-galactose and sialic acid residues. The arrow depicts the locations of further expansion of plant complex N-glycans with β1,4-galactose residues by overexpression of human β1,4-galactosyltransferase gene (GalT).
Currently, transfected Chinese hamster ovary cells (CHOs) are the major source for large-scale manufacturing of rhuEPOM (Table 1) [50]. Both plasma EPO and rhuEPOM have been reported to exist in numerous glycoforms due to the heterogeneity of their N- and O-glycan chains and varying sialic acid content [58]. The former was reported to bear only mono-, di-, and triantennary N-glycans but lack tetraantennary N-glycan chains while the latter was shown to contain predominantly tetrantennary sialylated N-glycans [58]. The degree of branching has been shown to influence its in vivo hematopoietic activity, and rhuEPOM bearing tetraantennary N-glycans displays higher activity than that with biantennary N-glycans [59]. The terminal sialic acid residues on glycan chains were reported to play a critical role in the in vivo hematopoietic activity of EPO by prolonging its serum half-life (~4–8 h) [60,61]. Removal of sialic acid residues was shown to result in an almost complete loss of its in vivo hematopoietic activity, while its in vitro activity remains unaffected [62,63]. The near complete loss of in vivo hematopoietic activity could be due to the rapid clearance of EPO from the circulation system (short half-life, ~2–3 min) [35,64] likely through asialoglycoprotein receptors present on the liver [62,63].
Concerning the influence of carbohydrate chains on the tissue-protective activity of EPO and its derivatives, the neuroprotection studies from both Yamashita et al. [39] and our own [41] suggest that they do not have any influence on the short-term protective effects because both rhuEPOM and asialo-rhuEPOP displayed similar protection in in vivo model of transient I/R injury. However, there is no report of their impact on long-term outcomes of EPO-mediated neuroprotection. Concerning EPO protein regions involved in binding to receptors to transduce tissue-protective signals, both binding sites 1 and 2 that are essential for erythropoiesis were shown to be not required for tissue-protective function, because chemical and mutational modifications of amino acid residues within these sites did not abolish the protective function of EPO and its derivatives [65,66]. Brines et al. [67] proposed that the region (Gln58-Ser82) comprising the B helix in EPO (see Figure 1A,B helix colored green with dotted red oval) is sufficient for its tissue-protective function. Thus, the regions of EPO responsible for its hematopoietic and tissue-protective functions are likely different.
2.2. Neuroprotective Function of RhuEPOM in Preclinical Studies
EPO was originally thought to function as the regulator of erythropoiesis only. However, in 1987 it was recognized that rhuEPOM possesses cognition-enhancing properties when given to anemia patients suffering from kidney failure or under chemotherapy [68]. Research performed in the following decade showed that EPO and EPOR are in fact widely distributed in the mammalian brain [69,70], and are up-regulated after ischemic infarction or hypoxic damage [71]. Targeted deletion of EPO and EPOR in mice showed, in addition to defective erythropoiesis, increased apoptosis in the mouse brain, suggesting that EPO signaling is required for brain development [72,73]. The above observations led to the hypothesis that both endogenously induced EPO and exogenously added rhuEPOM could provide neuroprotection.
In a series of elegant experiments, the Sasaki group [25,74] showed that exogenous rhuEPOM infused directly into the brain resulted in strong protection against ischemic injury, whereas soluble EPOR infused into the brain worsened the injury by neutralization of endogenous EPO. In subsequent studies, intracerebral injection of rhuEPOM before permanent occlusion of the middle cerebral artery (MCA) in mice revealed a 50% reduction of infarct volume [75], whereas Brines et al. [18] using a rodent model of cerebral ischemia where MCA was transiently occluded, showed that systemic administration of rhuEPOM before or up to 6 h after ischemia reduced injury by 50–75%. Intraperitoneal administration of rhuEPOM (5000 IU/kg bw) in rats was shown to reduce the ischemic area in the cerebrum, cerebellum, and brain stem [76], while Villa et al. [77] reported similar doses of exogenous rhuEPOM reducing astrocyte activation and the recruitment of leukocytes and microglia into an infarction produced by MCA occlusion (MCAO) in rats and decreasing the production of inflammatory cytokines. Gunnarson et al. [78] reported that EPO decreased the susceptibility of the brain to edema via regulation of brain-water homeostasis. We have also observed that intravenous (IV) administration of rhuEPOM (44 µg/kg bw; EPO: ~8 ng/IU) in mice at the restoration of blood flow following MCAO reduced the infarct volume and brain edema by 50% [41]. In other animal studies involving observation of functional outcomes in addition to infarct volume, rhuEPOM was shown to improve learning ability [74], navigation disability [25], sensory neglect, and sensorimotor functions [79,80]. RhuEPOM was also reported to be neuroprotective in animal models of hemorrhagic stroke [81] and traumatic brain injury (TBI) [82,83]. All of the above-discussed studies, together with many others not mentioned in this article, demonstrated that EPO is neuroprotective and could possibly be used to treat brain injury.
2.3. Neuroprotection Mechanisms of EPO
Following the discovery of its excellent neuroprotective effects, the mechanisms of EPO-mediated neuroprotection were also partly elucidated. Numerous studies have shown that it protects the CNS by limiting the production of ROS and glutamate, reducing BBB disruption, modulating inflammation, balancing the mitochondrial fission–fusion dynamics, attenuating apoptosis, stimulating angiogenesis, and inducing neurogenesis. RhuEPOM was shown to block the generation of ROS [84] and preserve the BBB integrity [85,86] and the cellular integrity in neurons [87] and inflammatory cells of the nervous system [88]. It has been reported to protect neurons from glutamate toxicity either by repressing Ca2+ influx [89] or through the suppression of Ca2+/calmodulin-dependent neuronal nitric oxide synthase (nNOS) [25]. The neuroprotective effects of EPO are now believed mainly due to its anti-apoptotic properties, akin to that observed in erythropoiesis. In the case of neurons, its protective effects were shown to occur through the prevention of mitochondrial depolarization via control of BAD, BAX, and PUMA, and the balance of mitochondrial dynamics through restoration of I/R injury-dysregulated mitochondrial fission and fusion proteins, leading to prevention of cytochrome c (cyt c) release and activation of caspases [41,87,90,91,92]. Moreover, rhuEPOM treatment was found to upregulate the anti-apoptotic proteins X-linked inhibitor of apoptosis (XIAP) and inhibitor of apoptosis proteins (c-IAP2) [93]. RhuEPOM was also reported to be anti-inflammatory, and has been shown to reduce the infarct volume in an animal model of cerebral ischemia by decreasing the production of pro-inflammatory molecules like TNF, IL-6, and chemokine MCP-1 [77]. The anti-inflammatory effect of EPO is reported to be due to the inhibition of apoptosis, as well as the reduction of BBB disruption to regulate immune-cell trafficking across the BBB, and attenuation of microglia activation [6,86,94].
Besides direct effects on neurons, EPO-mediated neuroprotection is believed to be due to improvement in cerebral blood flow (CBF) and brain perfusion by promoting new vessel growth. Beleslin-Cokic et al. [95] showed that EPO can stimulate endothelial nitric oxide synthase (eNOS) and nitric oxide (NO) production, which could improve CBF and contribute to neuroprotection. Furthermore, Cruz-Navarro et al. [96], using wild-type and eNOS-deficient mice, reported that EPO-mediated improvement in CBF in a traumatic brain injury model is eNOS-dependent. The angiogenic effect of rhuEPOM was also confirmed in mice genetically engineered to lack EPO and EPOR, whose embryos displayed severe defects in angiogenesis [97], while rhuEPOM treatment could promote the angiogenesis through the activation of AMPK-KLF2 signaling pathways [98]. EPO also exhibits neurotrophic function. It was shown to promote the survival and differentiation of dopaminergic precursor neurons [99]. In addition, endogenously produced EPO under hypoxia was reported to act directly on neuronal stem cells in the forebrain, suggesting a direct role in neurogenesis following hypoxia [100]. In sum, these studies revealed that EPO-mediated neuroprotection involves multiple mechanisms.
Concerning the signaling pathway involved in EPO-mediated neuroprotection, many studies have shown that multiple signaling pathways, which differ in importance depending on the cell type, type of injury, and EPO administration with respect to the injury, are activated by EPO. Like erythropoiesis, neuroprotective signals are reported to be initiated by phosphorylation of JAK2, followed by activation of downstream STAT5, PI3K/AKT, and MAPK/ERK1/2 pathways [71,101,102,103,104] (Figure 3). Siren et al. [71] reported that in hippocampal neurons, rhuEPOM restored the levels of phosphorylated STAT5, AKT, and ERK1/2 reduced by hypoxia, suggesting that these three pathways are important for EPO-mediated neuroprotection. In another study using EPOR mutants in which a component of the downstream signaling pathway was either selectively retained or inhibited [103], it was revealed that STAT5 and PI3K/AKT pathways in the SH-SY5Y cell line are critical because their impairments completely abolished the anti-apoptotic effects of rhuEPOM, whereas the MAPK pathway is less important. In addition, the authors suggested that both AKT and STAT5 also contribute to the activation of the NF-κB pathway [103]. Digicaylioglu and Lipton [93] also reported that JAK2 phosphorylated NF-κB’s inhibitory subunit, IκB, releasing the transcription factor NF-κB, and allowing its translocation to the nucleus to activate the expression of neuroprotective genes. In an in vivo study, Kilic et al. [102] also demonstrated that dual activation of ERK-1/2 and Akt is crucial for EPO’s neuroprotective activity. STAT5 activation was shown to inhibit apoptosis by inducing the synthesis of the protein Bcl-xL [105], whereas activation of ERK1/2 and AKT was reported to induce Bcl-xL, inactivate pro-apoptotic BCL2-associated agonist of cell death (BAD), and glycogen synthase kinase 3β (GSK3B), resulting in inhibition of apoptosis [88,102]. In addition to mediating anti-apoptotic effects, STAT5, PI3K/AKT, and MAPK pathways are also thought to be responsible for the antioxidant, anti-inflammation, angiogenesis, and neurogenic effects of EPO. Besides these three pathways, EPO was reported to display its neuroprotective effects through stimulation of eNOS [95] and activation of voltage-gated Ca2+ channels, resulting in NO production [95,106], as well as via regulation of the Wnt1 signaling pathway to prevent microglial early- and late-stage apoptotic injury [88].
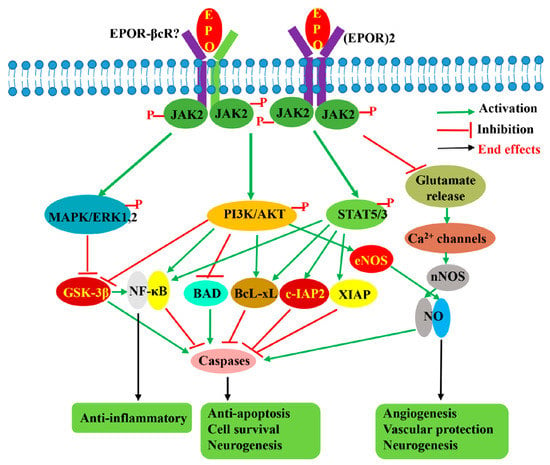
Figure 3.
EPO-induced signaling pathways involved in its neuroprotective effects. EPO by binding to either homodimeric (EPOR)2 receptor or another possible heterodimeric receptor EPOR-βcR activates JAK2 by phosphorylation, followed by activation of downstream STAT5, PI3K/AKT, and MAPK signaling pathways, as well as regulation of voltage-gated Ca2+ channel. The activation of these three signaling pathways results in the activation of anti-apoptotic and anti-inflammatory pathways benefiting cell survival while promoting angiogenesis and neurogenesis. The suppression of glutamate release benefits cell survival and inhibits apoptosis through the regulation of voltage-gated calcium ion channels to lower glutamate excitotoxicity.
2.4. Clinical Trials of RhuEPOM in Acute IS Treatment
After preclinical studies demonstrated its broad tissue-protective properties and pleiotropic effects, rhuEPOM entered clinical trials two decades ago to investigate its neuroprotection against I/R injury [31]. Although several clinical trials were conducted in different locations, unlike preclinical studies, its neuroprotective effects could not be consistently reproduced in clinical studies [31,32,33,34] (Table 2). In the earliest pilot study with 40 acute IS patients, daily IV administration of 33,000 IU rhuEPOM for the first three days, with the first injection within eight hours of the onset of stroke symptoms, showed a significant reduction in infarct size and neurological deficit, recovery of neurocognitive function, and amelioration of stroke-related disability at 30 days [31]. The observed positive effects of rhuEPOM were supported by the other two subsequent small clinical trials [33,34]. In a randomized trial involving 37 patients in the rhuEPOM group and 43 in the control group, a high dose of 56,000 IU (initial IV administration of 16,000 IU, followed by 8000 IU every 12 h over 60 h) was found to be effective in improving neurological function at 14 and 28 days, respectively [33]. Even a low-dose trial with 71 acute IS patients showed improvement in long-term (five years) neurological outcomes and a lower 90-day recurrent stroke rate with two consecutive subcutaneous (SC) administrations of 5000 IU rhuEPOM each at 48 and 72 h after onset of ischemic stroke symptoms [34]. The same group also demonstrated that the improvement in recurrent stroke rate with a low dose of rhuEPOM was through the enhancement of circulating endothelial progenitor cell levels [107]. Unfortunately, a large trial involving 522 acute IS patients with a cumulative dose of 120,000 IU (three doses with 40,000 IU each IV injection at 6, 24, and 48 h after onset of stroke symptoms) did not show any beneficial effects and even displayed harmful outcomes with increased mortality, particularly when combined with rtPA treatment [32].
Table 2.
Clinical trials of rhuEPOM treatment for acute ischemic stroke patients.
2.5. Possible Factors Responsible for Clinical Failure of RhuEPOM in Ischemic Stroke Treatment
After the clinical trials of rhuEPOM against cerebral I/R injury proved unsuccessful, concerns were raised regarding its capacity to cross the BBB due to its large molecular weight of approximately 30.4 kDa [50]. However, several research groups [18,108,109,110] have reported that rhuEPOM can cross the BBB either through EPOR-mediated or non-receptor-mediated transport, indicating that BBB permeability may not be an issue. During cerebral I/R injury, the BBB’s integrity is interrupted, making it easier for injected rhuEPOM to cross the barrier [111]. In addition, rhuEPOM therapy for cardiac and renal protection also showed no clear efficacy in clinical studies, even though animal data had shown outstanding protective effects against I/R injury in the heart [19,112,113] and kidneys [22]. Several systematic reviews and meta-analyses concluded that the observed cardioprotective effects of rhuEPOM to reduce MI size in animal models of myocardial I/R injury could not be reliably translated in clinical settings [114,115,116,117]. Taken together, these findings suggest that the impermeability of the BBB to rhuEPOM is unlikely to be the major reason for its failure in stroke clinical trials, and thus other factors must be responsible.
Besides the issue of BBB permeability, several other factors were also considered responsible for the clinical failures of rhuEPOM in stroke treatment. These include the dosage, the age of the patients, and most critically, its HAASEs, such as hypertension and thrombosis [26,30,117,118,119]. Regarding HAASEs with rhuEPOM administration, studies in transgenic mice have demonstrated that high levels of EPO induced by overexpressing the EPO gene can increase hematocrit, leading to vasoconstriction and cardiac dysfunction [120,121]. Similarly, in humans, rhuEPOM treatment for anemia patients with a dose of 100 IU/kg bw has been found to have pro-thrombotic or platelet-activating effects and cause hypertension in 20–30% of renal patients [122,123]. Since rhuEPOM doses for tissue-protection purposes are at least five times higher than those required for anemia treatment [29], these higher doses can stimulate mass production of RBCs and increase the risk of thrombosis. Therefore, the HAASEs associated with rhuEPOM could be a major issue when used for tissue protection.
The HAASEs of rhuEPOM were considered in previous clinical trials. In order to minimize side effects, lower doses of rhuEPOM were used compared to preclinical studies. However, these lower doses likely pose two new problems: insufficient dose and a narrow therapeutic window, hindering the display of its tissue-protective effects. Preclinical studies on neuroprotection have used doses of rhuEPOM ranging from ~1000 to ~5950 IU/kg bw, with 5000 IU/kg being the most common [6,124]. In contrast, clinical trials have used rhuEPOM doses of ~150 to ~1850 IU/kg bw (assuming an average patient weight of 65 kg) [31,32,33,34]. The clinical doses are several-fold lower than those used in preclinical studies. In cardioprotection clinical trials, lower doses of rhuEPOM [114,117] were also used compared to preclinical studies [119,125,126]. Low doses of rhuEPOM used in clinical trials in general showed no consistent benefits against I/R injury in neuroprotection, cardioprotection, and renal protection, raising concerns about the dosage applied [119,124,127]. Moreover, it is still not clear whether rhuEPOM uses the classical (EPOR)2 or the alternative tissue-protective receptors, such as EPOR-beta common receptor (βcR) and ephrin B4 (EphB4), to display its tissue-protection function. The alternative tissue-protective receptors require much higher concentrations of rhuEPOM than for erythropoiesis [29,127], because both EPOR-βcR [128] and EphB4 [129] were found to have very low affinity and may require a high concentration of rhuEPOM to transduce a tissue-protective signal. Additionally, the administration times of rhuEPOM in clinical studies are generally much later than those in preclinical studies, which may also be a responsible factor in the clinical failure of the drug. Currently, EPO’s therapeutic window for brain protection is not well-established, and may depend on the type of brain injury and the dosage and route of administration. Extending the therapeutic time window could allow rhuEPOM to display its protective effects. In one animal study, high doses of rhuEPOM for cardioprotection could extend the therapeutic time window [125].
The above discussion clearly indicates that in order to bring about stable and beneficial effects, as well as to extend the treatment time window, high doses of rhuEPOM should be used in clinical trials. Unfortunately, high doses of rhuEPOM have been found to correlate with severe HAASEs [30,119,124]. High doses may cause untoward complications, e.g., polycythemia and thrombotic secondary stroke [130]. Some studies have reported increased rates of adverse cardiovascular events with high doses of rhuEPOM [119,131]. Therefore, its HAASEs limited the application of high rhuEPOM dosage in clinical trials. A promising approach to increase rhuEPOM doses and prolong the therapeutic window could be to use nonerythropoietic EPO derivatives, such as asialo-rhuEPO to avoid the HAASEs.
3. Asialo-rhuEPO
3.1. Asialo-rhuEPO Structures and Properties
EPO lacking terminal sialic acid residues is known as asialo-rhuEPO, which contains neutral galactose residues as new terminal sugars instead of acidic sialic acid residues (Table 1; Figure 1B). Removal of sialic acid residues does not impact protein folding [56], but has shown to dramatically alter protein charge, binding capacity to the homodimeric (EPOR)2, in vitro and in vivo hematopoietic activities [60,61], and circulatory half-life [17,35]. In contrast to sialylated EPO with acidic pI, asialo-rhuEPO is a basic protein with a pI of 8.5 and a short circulatory half-life of ~2.5 min [35]. It has been reported to bind to the EPOR four times faster than its sialylated form in vitro [132]. Most importantly, asialo-rhuEPO was demonstrated to lack in vivo hematopoietic activity (non-erythropoietic) even at very high doses [35], and could cross the BBB to display excellent neuroprotective effects [35,36,38,39,41,133].
3.2. Methods of Asialo-rhuEPO Production
Currently, there are two methods used to produce asialo-rhuEPO (Table 1). It is commonly prepared by enzymatic removal of sialic acid residues (desialylation) from rhuEPOM [35,56]. We designate this enzymatically prepared one as asialo-rhuEPOE. Recently, plants have been successfully glycoengineered to produce asialo-rhuEPO [134,135,136,137], which is designated as asialo-rhuEPOP in this review.
3.2.1. Enzymatic Method
Asialo-rhuEPO in small quantities for basic research was produced by the enzymatic method. In this method, asialo-rhuEPOE was prepared by desialylation of rhuEPOM with commercially available enzymes called neuraminidases (also known as sialidases) [35,56,132]. Neuraminidases catalyze the hydrolysis of α2,3-, α2-6-, α2-8-, and α2-9-linked Neu5Ac (a type of sialic acid typically present on mammalian glycoproteins) from glycoproteins. Although this is a simple and straightforward method to obtain asialo-rhuEPO without the loss of biological activity, it is not an economically viable method for large-scale production because of the high cost (~4000 USD/mg protein) of rhuEPOM [138]. In addition, neuraminidases for the above purpose are unavailable in bulk quantities for large-scale desialylation of rhuEPOM. A mammalian cell-based expression system is also not available to directly express asialo-rhuEPO. Hence, the neuroprotective properties of asialo-rhuEPOE could not be translated into clinical practice. Alternative methods to produce asialo-rhuEPO inexpensively were therefore sought to realize its full therapeutic potential.
3.2.2. Plant-Based Expression Method
Plants have been used as an inexpensive expression system to produce asialo-rhuEPO because they lack sialylation capacity (as they lack the entire enzymatic machinery necessary for the synthesis and transfer of sialic acid residues to glycoproteins) but have the ability to synthesize similar complex biantennary N-glycans like mammalian cells (Figure 2) [139,140,141]. Moreover, transgenic plants expressing wild-type or chimeric human GalT can sufficiently add galactose residues on the N-glycans of produced glycoproteins (Figure 2) [142,143]. The other advantages of using a plant-based expression system are low production cost, lack of human pathogen contamination, and ease of scaling up in production [140,144]. We produced asialo-rhuEPO in tobacco plants by stably co-expressing human EPO and β1,4-galactosyltransferase (GalT) genes [135,136,137], while Parson and co-workers produced it in moss [134].
3.3. Unique Properties of Asialo-rhuEPOP
3.3.1. Asialo-rhuEPOP Carries Plant-Specific Biantennary N-Glycans
Asialo-rhuEPOP accumulates as 28–30 kD bands representing different glycoforms in transgenic tobacco plants [136]. It is 162 amino acids long because of the proteolytic removal of the extreme C-terminal region Thr163-Arg166 [145]. This lost region is not important for the biological activity of EPO [146]. Asialo-rhuEPOP is a basic protein with a theoretical pI of 8.75. All three N-glycosylation sites in asialo-rhuEPOP are occupied with N-glycan chains each bearing terminal mammalian-type β1,4-galactose residues [136,137]. The proportion of β1,4-galactose residues on asialo-rhuEPOP was high (72%) when chimeric GalT was co-expressed with EPO [137], whereas it was low (8%) in the case of co-expressing wild-type GalT [136]. The N-glycan chains in asialo-rhuEPOP are slightly different from that in asialo-rhuEPOE. Asialo-rhuEPOP carries biantennary N-glycans containing plant-specific β1,2-xylose and α1,3-fucose residues, while those N-glycans in asialo-rhuEPOE lack β1,2-xylose but contain a core α1,6-fucose instead of α1,3-fucose (see Figure 2). The presence of biantennary N-glycans with plant-specific β1,2-xylose and α1,3-fucose residues on asialo-rhuEPOP showed no impact on in vitro (EPOR)2 binding, since asialo-rhuEPOP displayed similar affinity for the (EPOR)2 as rhuEPOM [135]. Furthermore, it provided a similar level of protection to the brain as rhuEPOM after I/R injury [41], suggesting that the biantennary N-glycans bearing plant-specific sugars have no impact on its in vivo neuroprotective activity.
3.3.2. Asialo-rhuEPOP Is Non-Erythropoietic and Non-Immunogenic
RhuEPOM doses used for neuroprotection are typically higher (~40 µg/kg bw in a rodent stroke model and 4–36 µg/kg bw in clinical trials) than that used to improve hemoglobin (Hb) levels in anemia patients (~0.8 µg/kg bw) [29]. Therefore, rhuEPOM or any of its derivatives at high doses can increase RBC levels and pose a thrombosis risk if they possess hematopoietic activity. We confirmed a lack of erythropoietic activity in asialo-rhuEPOP by repeated IV injection (twice a week for 5 weeks) in female BALB/c mice at a dose of 44 µg/kg bw (neuroprotective dose), which is 55 times higher than the dose of rhuEPOM typically used to stimulate RBC production. At this high dose, asialo-rhuEPOP showed no increase in Hb concentrations, while rhuEPOM significantly increased Hb concentrations as early as a week after two injections [41]. Consistent with this observation, at the end of the 5-week period, the RBC count in mice receiving asialo-rhuEPOP was 7.8 × 106/mm3, similar to the saline group (7.5 × 106/mm3), while that in mice administered rhuEPOM was 11 × 106/mm3, corresponding to a ~47% increase [41]. These results confirmed that asialo-rhuEPOP is also non-erythropoietic, like asialo-rhuEPOE.
Concerning the plant-specific sugars on asialo-rhuEPOP, there is an ongoing debate whether plant-specific sugars on therapeutic glycoproteins are immunogenic [147,148,149,150]. The immunogenicity of plant-specific sugars on asialo-rhuEPOP was investigated by immunizing BALB/c mice with 44 µg/kg bw (neuroprotective dose) and 88 µg/kg bw protein, along with the same doses of rhuEPOM as a negative control and horse radish peroxidase as a positive control. We detected no antibodies against plant-specific sugars in the sera of mice immunized with asialo-rhuEPOP, indicating that it is non-immunogenic even at high doses [41]. These results suggested that asialo-rhuEPOP is safe for use in clinical practice.
3.4. Neuroprotective Effects of Asialo-rhuEPO
Asialo-rhuEPO is a nonerythropoietic EPO derivative that has been proven to have neuroprotective functions in various studies (Table 3) [35,39,41,133]. Erbayraktar et al. [35] demonstrated that asialo-rhuEPOE is non-erythropoietic and neuroprotective in animal models of cerebral ischemia, spinal cord compression, and sciatic nerve crush. It has also been shown to attenuate neuronal cell death in the hippocampal CA1 region after transient forebrain ischemia [39]. In addition, asialo-rhuEPOE was reported to improve motor behavior and reduce motoneuron loss in the cervical spinal cord of wobbler mice, an animal model of amyotrophic lateral sclerosis, without affecting hematocrit values [133]. Despite its non-erythropoietic nature and excellent neuroprotective effects, no clinical trials have been conducted for ischemic stroke treatment or for other organ injury treatment, likely due to its limited availability and high cost. To circumvent this problem, we produced asialo-rhuEPOP in tobacco plants. In the following sections, we describe the neuroprotective properties of asialo-rhuEPOP and discuss its potential as a multimodal drug for ischemic stroke treatment.
Table 3.
In vitro and in vivo neuroprotective effects of asialo-rhuEPO.
3.4.1. In Vitro and In Vivo Neuroprotective Effects of Asialo-rhuEPOP
We evaluated the in vitro neuroprotective effect of asialo-rhuEPOP by studying its ability to protect neuronal-like cells (N2A) against staurosporine (STS)-induced apoptosis. Our results showed that simultaneous treatment of N2A cells with 1 µM STS and 20 IU/mL asialo-rhuEPOP or rhuEPOM (a positive control) resulted in lower cytotoxicity (47% and 66%, respectively) while treatment with 1 µM STS alone caused 84% cytotoxicity [136]. These results suggest that asialo-rhuEPOP is not only neuroprotective but also more effective (~2-fold) than rhuEPOM in vitro.
The in vivo neuroprotective effect of asialo-rhuEPOP was evaluated using a mouse model of I/R injury and compared with rhuEPOM. We used a dose of 44 µg/kg bw because asialo-rhuEPOE has been found to be neuroprotective at this dose in a mouse model of I/R injury [35]. Following IV administration of asialo-rhuEPOP (rhuEPOM as well) immediately at the restoration of blood flow after 1 h occlusion, we observed a significant decrease in neurological deficits (from 3.1 in I/R–saline group to 1.8–1.9 in asialo-rhuEPOP and rhuEPOM groups), cerebral infarction volume (from 33.0% in I/R–saline group to 15.0% in asialo-rhuEPOP and rhuEPOM groups), and edema volume (from ~30% in I/R–saline group to 14% in asialo-rhuEPOP and rhuEPOM groups) [41]. Consistent with these observations, HE and Nissl staining of brain sections showed lesser cellular damage and higher neuron density in the asialo-rhuEPOP-treated group than in the I/R–saline group. Immunostaining for NeuN, a marker commonly used to assess the functional state of neurons, also revealed a higher number of NeuN-positive cells in the asialo-rhuEPOP-treated group than that in the I/R–saline group [41]. Our results demonstrated that asialo-rhuEPOP is neuroprotective and equipotent to rhuEPOM in reducing brain damage induced by I/R injury.
3.4.2. Neuroprotective Mechanism of Asialo-rhuEPOP
Asialo-rhuEPOE has been shown to exert excellent neuroprotection against cerebral I/R injury [35,36,38,39,40,133], but its neuroprotective mechanism has not been dissected yet. Regarding asialo-rhuEPOP, our studies suggested that it protects the brain from I/R injury by restoring mitochondria fusion–fission-related proteins, preventing I/R injury-induced mitophagy and autophagy markers, and inhibiting apoptosis [41]. Mitochondria are both a source and target of I/R injury and cell death, and their dysfunction occurring via fission and fusion imbalance is considered one of the hallmarks of I/R-induced neuronal cell death [151,152]. Fission regulates the amounts of mitochondria and removes damaged mitochondria, whereas fusion maintains normal mitochondrial activity by complementing damaged mitochondrial contents with the components of healthy mitochondria [152,153]. I/R injury and other cerebral insults promote fission, leading to disturbed mitochondrial dynamics and compromised mitochondrial functions, thereby promoting the release of pro-apoptotic factors, such as cyt c [151,152,154]. Our western blotting and immunofluorescence studies showed that the induced fission-related proteins dynamin-related protein 1 (p-Drp1) and Drp1 receptor fission 1 protein (Fis1) in the brain of the I/R-saline group were significantly restored in both asialo-rhuEPOP- and rhuEPOM-treated groups [41]. Similarly, the fusion-related proteins mitofusin 1 and 2 (Mfn1 and Mfn2) and optic atrophy protein 1 (OPA1), whose levels were reduced in the I/R-saline group, were significantly restored in asialo-rhuEPOP and rhuEPOM groups [41]. These results suggested that, like rhuEPOM, asialo-rhuEPOP can maintain mitochondrial fission–fusion balance under I/R conditions.
Our results also showed that asialo-rhuEPOP treatment and rhuEPOM as well can regulate mitophagy-related markers. Mitochondrial fission is followed by mitophagy to remove damaged organelles [155,156]. In mammalian cells, the PINK1 (PTEN-induced putative kinase protein 1) and parkin (an E3 ubiquitin ligase PARK2) cooperatively sense cellular stress and mediate the removal of damaged mitochondria [155,156]. Our Western blotting results showed that asialo-rhuEPOP (also rhuEPOM) treatment was able to restore both PINK1 and PARK2 levels that were elevated by I/R injury [41], consistent with lower mitochondrial fission anticipated from the restoration of fission-related proteins in EPO-treated groups. In addition to mitophagy, our Western blotting and immunofluorescence results of general autophagy markers (LC3B, p62, and Beclin1) revealed that their increased levels in the I/R–saline group were reinstated back in asialo-rhuEPOP and rhuEPOM groups to the levels similar to the sham group [41]. Furthermore, investigation of apoptotic markers and TUNEL staining revealed that asialo-rhuEPOP-treatment (also rhuEPOM) significantly attenuated I/R-induced Bax/Bcl2 ratio, cyt c release, and caspase 3 caspase cleavage, and reduced TUNEL-positive cells by 50% compared to the I/R–saline control, indicating that both asialo-rhuEPOP and rhuEPOM have similar suppressive effects on I/R injury-induced apoptosis [41]. Together, these results suggested that asialo-rhuEPOP- and rhuEPOM-treatment can restore I/R injury-affected mitochondrial fission–fusion-related proteins and mitophagy- and autophagy-related markers, leading to anti-apoptotic effects and cell survival.
Regarding signaling pathways responsible for displaying the neuroprotective effect of asialo-rhuEPOP, our Western blotting results showed a significant increase in phosphorylation of STAT5, PI3K, AKT, and ERK1/2 in asialo-rhuEPOP- and rhuEPOM-treated groups compared with the I/R–saline group [41]. These results suggest that, like rhuEPOM, the neuroprotective effects of asialo-rhuEPOP are mediated through the activation of STAT5, PI3K/AKT, and MAPK/ERK1/2 signaling pathways as reported previously by others for rhuEPOM [71,101,102,103,104]. These findings also indicate that asialo-rhuEPOP, like rhuEPOM, exhibits pleiotropic effects through these pathways. However, at this time, it remains unknown what the relative contribution of each of these asialo-rhuEPOP-induced signaling pathways is to the regulation of mitochondrial fission–fusion, mitophagy, autophagy, and apoptosis to display neuroprotective effects. In addition, the receptor responsible for transducing asialo-rhuEPOP tissue-protective signal remains to be identified.
4. Future Directions
Since the preclinical results of rhuEPOM to protect against I/R injuries in the brain and other organs could not be consistently observed in clinical trials, more precautions should be taken to perform a clinical trial for ischemic stroke treatment with asialo-rhuEPO, regardless of whether asialo-rhuEPOE or asialo-rhuEPOP is used. Several preclinical experiments may still be required to obtain more information in order to determine whether it is worth carrying out an ischemic stroke clinical study with asialo-rhuEPO.
Firstly, the neuroprotective effects of asialo-rhuEPO should be investigated in an aged-stroke mouse model, as age is the most important risk factor for cerebral ischemia [157,158,159,160]. Most studies on cerebral I/R injury and its therapies were performed in young and healthy animals [10,160]. These young and healthy animals used in preclinical studies are dramatically different from patients recruited into clinical trials who are usually of advanced age with different health conditions [160,161,162]. Furthermore, elderly patients have a higher burden of surgical risk factors with reduced functional capacity and increased comorbidities compared to younger patients [161,163]. The aged brain often displays a compromised ability to resolve stroke-mediated inflammation and shows poor functional recovery compared to the young brain [158,160]. Ischemic stroke experiments with aged rats have also shown significant differences in neuroinflammation, accelerated apoptosis, and precipitous infarct development compared to young and healthy animals after an ischemic insult [164]. Hence, preclinical studies with young animal models might not represent aged acute IS patients well [162,165]. It is essential to study neuroprotective effects of the asialo-rhuEPO in aged animal models, which more closely mimics the clinical situation.
Secondly, it is necessary to investigate the dosage of asialo-rhuEPO to determine an optimal dose and subsequent therapeutic time window. High doses and repeated administration of both asialo-rhuEPOE and asialo-rhuEPOP revealed no increase in either RBC counts or Hb levels [35,41]. These results are good indicators that asialo-rhuEPO is indeed non-erythropoietic, leading us to believe that it should be free of HAASEs. The asialo-rhuEPO dose commonly used in animal experiments is ~5000 IU/kg bw [35,41], which was based on the previously optimal dose found in studies on rhuEPOM [18,124]. Further studies with different doses of asialo-rhuEPO in stroke animal models could allow us to determine whether its dose can be increased further to bring better or stable neuroprotective effects. A previous study also revealed that the best administration time is up to 3 h after ischemia with a leaky BBB [166]. It will be also interesting to know under the optimal dose whether the administration time window of asialo-rhuEPO can be extended or not.
Thirdly, it is also important to know whether asialo-rhuEPO without hematopoietic activity will bring out better long-term neuroprotective functions to minimize I/R-induced brain injury than rhuEPOM with hematopoietic activity. Currently, most neuroprotective effects of asialo-rhuEPO treatment were observed in short-term studies with the doses optimized from rhuEPOM studies. The short-term neuroprotective effects of asialo-rhuEPO were found to be similar to those observed in rhuEPOM treatment [35,41]. Based on their different hematopoietic properties, asialo-rhuEPOP free of HAASEs might have better long-term neuroprotective functions than rhuEPOM when they are used to treat I/R-induced brain injury. Therefore, investigating its long-term rather than short-term effects, especially in aged mice, would be more meaningful before moving asialo-rhuEPO to clinical tests.
Lastly, the tissue-protective receptor for EPO remains elusive [6,167]. RhuEPOM and asialo-rhuEPO with different terminal sugars and protein charges may use distinct receptors and certain unique mechanisms to attenuate I/R injury. To understand whether two different activities (erythropoiesis and tissue protection) of EPO use different receptors and to identify any alternative tissue-protective EPO receptor have basic and practical significance, which will lay a solid foundation to exploit EPO derivatives like asialo-rhuEPO as potential tissue-protective drugs. Hence, further investigation of whether the erythropoietic and tissue-protective activities of EPO are mediated through the same receptor or different receptor(s) is crucial for future applications. Asialo-rhuEPO displays only a cytoprotective function, and could be an ideal EPO derivative to be used to identify and study EPO tissue-protective receptor(s).
Author Contributions
The literature search and the manuscript drafted, F.S.K. and J.X.; review and edition, C.-Y.H., P.A.L. and D.C.S.; funding acquisition, J.X. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by National Institute of General Medical Sciences grant SC1GM111178 to Jiahua Xie.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing not applicable.
Conflicts of Interest
J.X., F.K. and C.-Y.H. are inventors of filed patent “Methods for the production of cytoprotective asialo-erythropoietin in plants and its purification from plant tissues” (PCT NUMBER: US2013031382, pending).
References
- Feigin, V.L.; Norrving, B.; Mensah, G.A. Global Burden of Stroke. Circ. Res. 2017, 120, 439–448. [Google Scholar] [CrossRef]
- Barthels, D.; Das, H. Current advances in ischemic stroke research and therapies. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2018, 1866, 165260. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—From mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Robbins, N.M.; Swanson, R.A. Opposing Effects of Glucose on Stroke and Reperfusion Injury. Stroke 2014, 45, 1881–1886. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Li, X. Targeting O-GlcNAcylation in ischemic stroke. Neural Regen. Res. 2022, 17, 2427. [Google Scholar] [CrossRef]
- Ma, Y.; Zhou, Z.; Yang, G.-Y.; Ding, J.; Wang, X. The Effect of Erythropoietin and Its Derivatives on Ischemic Stroke Therapy: A Comprehensive Review. Front. Pharmacol. 2022, 13, 743926. [Google Scholar] [CrossRef]
- Xie, J.; Kittur, F.S.; Li, P.A.; Hung, C.-Y. Rethinking the necessity of low glucose intervention for cerebral ischemia/reperfusion injury. Neural Regen. Res. 2022, 17, 1397. [Google Scholar] [CrossRef]
- Pan, J.; Konstas, A.-A.; Bateman, B.; Ortolano, G.A.; Pile-Spellman, J. Reperfusion injury following cerebral ischemia: Pathophysiology, MR imaging, and potential therapies. Neuroradiology 2006, 49, 93–102. [Google Scholar] [CrossRef]
- Mallet, R.T.; Ryou, M.-G. Erythropoietin: Endogenous Protection of Ischemic Brain. Vitam. Horm. 2017, 105, 197–232. [Google Scholar] [CrossRef]
- Xiong, X.-Y.; Liu, L.; Yang, Q.-W. Refocusing Neuroprotection in Cerebral Reperfusion Era: New Challenges and Strategies. Front. Neurol. 2018, 9, 249. [Google Scholar] [CrossRef]
- Lin, L.; Wang, X.; Yu, Z. Ischemia-reperfusion Injury in the Brain: Mechanisms and Potential Therapeutic Strategies. Biochem. Pharmacol. Open Access 2016, 5, 213. [Google Scholar] [CrossRef]
- Gou, X.; Xu, D.; Li, F.; Hou, K.; Fang, W.; Li, Y. Pyroptosis in stroke-new insights into disease mechanisms and therapeutic strategies. J. Physiol. Biochem. 2021, 77, 511–529. [Google Scholar] [CrossRef] [PubMed]
- Muresanu, D.F.; Strilciuc, S.; Stan, A. Current Drug Treatment of Acute Ischemic Stroke: Challenges and Opportunities. CNS Drugs 2019, 33, 841–847. [Google Scholar] [CrossRef]
- Fisher, M.; Dávalos, A.; Rogalewski, A.; Schneider, A.; Ringelstein, E.B.; Schabitz, W.-R. Toward a Multimodal Neuroprotective Treatment of Stroke. Stroke 2006, 37, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, Z.G.; Chopp, M. The neurovascular unit and combination treatment strategies for stroke. Trends Pharmacol. Sci. 2012, 33, 415–422. [Google Scholar] [CrossRef]
- Jin, X.-C.; Liang, L.-J.; Yang, J.-M. Cocktail treatment, a promising strategy to treat acute cerebral ischemic stroke. Med. Gas Res. 2016, 6, 33–38. [Google Scholar] [CrossRef]
- Jelkmann, W. Erythropoietin: Structure, control of production, and function. Physiol. Rev. 1992, 72, 449–489. [Google Scholar] [CrossRef]
- Brines, M.L.; Ghezzi, P.; Keenan, S.; Agnello, D.; de Lanerolle, N.C.; Cerami, C.; Itri, L.M.; Cerami, A. Erythropoietin crosses the blood–brain barrier to protect against experimental brain injury. Proc. Natl. Acad. Sci. USA 2000, 97, 10526–10531. [Google Scholar] [CrossRef]
- Calvillo, L.; Latini, R.; Kajstura, J.; Leri, A.; Anversa, P.; Ghezzi, P.; Salio, M.; Cerami, A.; Brines, M. Recombinant human erythropoietin protects the myocardium from ischemia-reperfusion injury and promotes beneficial remodeling. Proc. Natl. Acad. Sci. USA 2003, 100, 4802–4806. [Google Scholar] [CrossRef]
- Vesey, D.A. Erythropoietin protects against ischaemic acute renal injury. Nephrol. Dial. Transplant. 2004, 19, 348–355. [Google Scholar] [CrossRef]
- Sharples, E.; Yaqoob, M. Erythropoietin in Experimental Acute Renal Failure. Nephron Exp. Nephrol. 2006, 104, e83–e88. [Google Scholar] [CrossRef] [PubMed]
- Moore, E.; Bellomo, R. Erythropoietin (EPO) in acute kidney injury. Ann. Intensiv. Care 2011, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Marti, H.H.; Gassmann, M.; Wenger, R.H.; Kvietikova, I.; Morganti-Kossmann, M.C.; Kossmann, T.; Trentz, O.; Bauer, C. Detection of erythropoietin in human liquor: Intrinsic erythropoietin production in the brain. Kidney Int. 1997, 51, 416–418. [Google Scholar] [CrossRef] [PubMed]
- Maiese, K.; Li, F.; Chong, Z.Z. Erythropoietin in the brain: Can the promise to protect be fulfilled? Trends Pharmacol. Sci. 2004, 25, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Sakanaka, M.; Wen, T.-C.; Matsuda, S.; Morishita, E.; Nagao, M.; Sasaki, R. In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc. Natl. Acad. Sci. USA 1998, 95, 4635–4640. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.S.; Nandra, K.K.; Thiemermann, C. Bench-to-bedside review: Erythropoietin and its derivatives as therapies in critical care. Crit. Care 2012, 16, 229. [Google Scholar] [CrossRef]
- Souvenir, R.; Doycheva, D.; Zhang, J.H.; Tang, J. Erythropoietin in stroke therapy: Friend or foe. Curr. Med. Chem. 2015, 22, 1205–1213. [Google Scholar] [CrossRef]
- Brines, M.; Cerami, A. Emerging biological roles for erythropoietin in the nervous system. Nat. Rev. Neurosci. 2005, 6, 484–494. [Google Scholar] [CrossRef]
- Brines, M.; Cerami, A. Erythropoietin-mediated tissue protection: Reducing collateral damage from the primary injury response. J. Intern. Med. 2008, 264, 405–432. [Google Scholar] [CrossRef]
- Sølling, C. Organ-Protective and Immunomodulatory Effects of Erythropoietin—An Update on Recent Clinical Trials. Basic Clin. Pharmacol. Toxicol. 2011, 110, 113–121. [Google Scholar] [CrossRef]
- Ehrenreich, H.; Hasselblatt, M.; Dembowski, C.; Cepek, L.; Lewczuk, P.; Stiefel, M.; Rustenbeck, H.-H.; Breiter, N.; Jacob, S.; Knerlich, F.; et al. Erythropoietin Therapy for Acute Stroke Is Both Safe and Beneficial. Mol. Med. 2002, 8, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Ehrenreich, H.; Weissenborn, K.; Prange, H.; Schneider, D.; Weimar, C.; Wartenberg, K.; Schellinger, P.D.; Bohn, M.; Becker, H.; Wegrzyn, M.; et al. Recombinant Human Erythropoietin in the Treatment of Acute Ischemic Stroke. Stroke 2009, 40, e647–e656. [Google Scholar] [CrossRef] [PubMed]
- Asadi, B.; Askari, G.R.; Khorvash, F.; Bagherpur, A.; Mehrabi, F.; Karimi, M.; Ghasemi, M.; Najjaran, A. Neuroprotective Effects of Erythropoietin in Acute Ischemic Stroke. Int. J. Prev. Med. 2013, 4, S306–S312. [Google Scholar] [PubMed]
- Tsai, T.-H.; Lu, C.-H.; Wallace, C.G.; Chang, W.-N.; Chen, S.-F.; Huang, C.-R.; Tsai, N.-W.; Lan, M.-Y.; Sung, P.-H.; Liu, C.-F.; et al. Erythropoietin improves long-term neurological outcome in acute ischemic stroke patients: A randomized, prospective, placebo-controlled clinical trial. Crit. Care 2015, 19, 1–9. [Google Scholar] [CrossRef]
- Erbayraktar, S.; Grasso, G.; Sfacteria, A.; Xie, Q.-W.; Coleman, T.; Kreilgaard, M.; Torup, L.; Sager, T.; Erbayraktar, Z.; Gokmen, N.; et al. Asialoerythropoietin is a nonerythropoietic cytokine with broad neuroprotective activity in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 6741–6746. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhu, C.; Wang, X.; Gerwien, J.G.; Schrattenholz, A.; Sandberg, M.; Leist, M.; Blomgren, K. The nonerythropoietic asialoerythropoietin protects against neonatal hypoxia-ischemia as potently as erythropoietin. J. Neurochem. 2004, 91, 900–910. [Google Scholar] [CrossRef]
- Grasso, G.; Sfacteria, A.; Erbayraktar, S.; Passalacqua, M.; Meli, F.; Gokmen, N.; Yilmaz, O.; La Torre, D.; Buemi, M.; Iacopino, D.G.; et al. Amelioration of spinal cord compressive injury by pharmacological preconditioning with erythropoietin and a nonerythropoietic erythropoietin derivative. J. Neurosurg. Spine 2006, 4, 310–318. [Google Scholar] [CrossRef]
- Price, C.D.; Yang, Z.; Karlnoski, R.; Kumar, D.; Chaparro, R.; Camporesi, E.M. Effect of continuous infusion of asialoerythropoietin on short-term changes in infarct volume, penumbra apoptosis and behaviour following middle cerebral artery occlusion in rats. Clin. Exp. Pharmacol. Physiol. 2010, 37, 185–192. [Google Scholar] [CrossRef]
- Yamashita, T.; Nonoguchi, N.; Ikemoto, T.; Miyatake, S.-I.; Kuroiwa, T. Asialoerythropoietin attenuates neuronal cell death in the hippocampal CA1 region after transient forebrain ischemia in a gerbil model. Neurol. Res. 2010, 32, 957–962. [Google Scholar] [CrossRef]
- Ishii, T.; Asai, T.; Oyama, D.; Fukuta, T.; Yasuda, N.; Shimizu, K.; Minamino, T.; Oku, N. Amelioration of cerebral ischemia–reperfusion injury based on liposomal drug delivery system with asialo-erythropoietin. J. Control. Release 2012, 160, 81–87. [Google Scholar] [CrossRef]
- He, M.; Kittur, F.S.; Hung, C.-Y.; Zhang, J.; Jing, L.; Sane, D.C.; Li, P.A.; Xie, J. A Novel Plant-Produced Asialo-rhuEPO Protects Brain from Ischemic Damage Without Erythropoietic Action. Transl. Stroke Res. 2021, 13, 338–354. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Sawada, T.; Kubota, K. Asialoerythropoietin Has Strong Renoprotective Effects Against Ischemia-Reperfusion Injury in a Murine Model. Transplantation 2007, 84, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Ogino, A.; Takemura, G.; Kawasaki, M.; Tsujimoto, A.; Kanamori, H.; Li, L.; Goto, K.; Maruyama, R.; Kawamura, I.; Takeyama, T.; et al. Erythropoietin Receptor Signaling Mitigates Renal Dysfunction-Associated Heart Failure by Mechanisms Unrelated to Relief of Anemia. J. Am. Coll. Cardiol. 2010, 56, 1949–1958. [Google Scholar] [CrossRef]
- Takeyama, T.; Takemura, G.; Kanamori, H.; Kawaguchi, T.; Ogino, A.; Watanabe, T.; Morishita, K.; Tsujimoto, A.; Goto, K.; Maruyama, R.; et al. Asialoerythropoietin, a Nonerythropoietic Derivative of Erythropoietin, Displays Broad Anti-Heart Failure Activity. Circ. Heart Fail. 2012, 5, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, L.O.; Goldwasser, E.; Fried, W.; Plzak, L. Role of the Kidney in Erythropoiesis. Nature 1957, 179, 633–634. [Google Scholar] [CrossRef]
- Erslev, A.J.; Caro, J.; Kansu, E.; Silver, R. Renal and Extrarenal Erythropoietin Production in Anaemic Rats. Br. J. Haematol. 1980, 45, 65–72. [Google Scholar] [CrossRef]
- Marti, H.H.; Wenger, R.H.; Rivas, L.A.; Straumann, U.; Oigicaylioglu, M.; Henn, V.; Yonekawa, Y.; Bauer, C.; Gassmann, M. Erythropoietin Gene Expression in Human, Monkey and Murine Brain. Eur. J. Neurosci. 1996, 8, 666–676. [Google Scholar] [CrossRef]
- Recny, M.A.; Scoble, H.A.; Kim, Y. Structural characterization of natural human urinary and recombinant DNA-derived erythropoietin. Identification of des-arginine 166 erythropoietin. J. Biol. Chem. 1987, 262, 17156–17163. [Google Scholar] [CrossRef]
- Browne, J.; Cohen, A.; Egrie, J.; Lai, P.; Lin, F.-K.; Strickland, T.; Watson, E.; Stebbing, N. Erythropoietin: Gene Cloning, Protein Structure, and Biological Properties. Cold Spring Harb. Symp. Quant. Biol. 1986, 51, 693–702. [Google Scholar] [CrossRef]
- Jelkmann, W. Physiology and Pharmacology of Erythropoietin. Transfus. Med. Hemother. 2013, 40, 302–309. [Google Scholar] [CrossRef]
- Cheetham, J.C.; Smith, D.M.; Aoki, K.H.; Stevenson, J.L.; Hoeffel, T.J.; Syed, R.S.; Egrie, J.; Harvey, T.S. NMR structure of human erythropoietin and a comparison with its receptor bound conformation. Nat. Struct. Biol. 1998, 5, 861–866. [Google Scholar] [CrossRef]
- Syed, R.S.; Reid, S.W.; Li, C.; Cheetham, J.C.; Aoki, K.H.; Liu, B.; Zhan, H.; Osslund, T.D.; Chirino, A.J.; Zhang, J.; et al. Efficiency of signalling through cytokine receptors depends critically on receptor orientation. Nature 1998, 395, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Wasley, L.; Timony, G.; Murtha, P.; Stoudemire, J.; Dorner, A.; Caro, J.; Krieger, M.; Kaufman, R. The importance of N- and O-linked oligosaccharides for the biosynthesis and in vitro and in vivo biologic activities of erythropoietin. Blood 1991, 77, 2624–2632. [Google Scholar] [CrossRef]
- Thompson, N.K.; Wakarchuk, W. O-glycosylation and its role in therapeutic proteins. Biosci. Rep. 2022, 42, BSR20220094. [Google Scholar] [CrossRef]
- Dubé, S.; Fisher, J.W.; Powell, J.S. Glycosylation at specific sites of erythropoietin is essential for biosynthesis, secretion, and biological function. J. Biol. Chem. 1988, 263, 17516–17521. [Google Scholar] [CrossRef] [PubMed]
- Narhi, L.; Arakawa, T.; Aoki, K.; Elmore, R.; Rohde, M.; Boone, T.; Strickland, T. The effect of carbohydrate on the structure and stability of erythropoietin. J. Biol. Chem. 1991, 266, 23022–23026. [Google Scholar] [CrossRef] [PubMed]
- Lasne, F.; De Ceaurriz, J. Recombinant erythropoietin in urine. Nature 2000, 405, 635. [Google Scholar] [CrossRef] [PubMed]
- Skibeli, V.; Nissen-Lie, G.; Torjesen, P. Sugar profiling proves that human serum erythropoietin differs from recombinant human erythropoietin. Blood 2001, 98, 3626–3634. [Google Scholar] [CrossRef]
- Takeuchi, M.; Inoue, N.; Strickland, T.W.; Kubota, M.; Wada, M.; Shimizu, R.; Hoshi, S.; Kozutsumi, H.; Takasaki, S.; Kobata, A. Relationship between sugar chain structure and biological activity of recombinant human erythropoietin produced in Chinese hamster ovary cells. Proc. Natl. Acad. Sci. USA 1989, 86, 7819–7822. [Google Scholar] [CrossRef]
- Lukowsky, W.A.; Painter, R.H. Studies on the Role of Sialic Acid in the Physical and Biological Properties of Erythropoietin. Can. J. Biochem. 1972, 50, 909–917. [Google Scholar] [CrossRef]
- Goldwasser, E.; Kung, C.K.-H.; Eliason, J. On the Mechanism of Erythropoietin-induced Differentiation. J. Biol. Chem. 1974, 249, 4202–4206. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M.; Sasaki, H.; Lopez, L. Survival of recombinant erythropoietin in the circulation: The role of carbohydrates. Blood 1989, 73, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Spivak, J.; Hogans, B. The in vivo metabolism of recombinant human erythropoietin in the rat. Blood 1989, 73, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Jelkmann, W. The enigma of the metabolic fate of circulating erythropoietin (Epo) in view of the pharmacokinetics of the recombinant drugs rhEpo and NESP. Eur. J. Haematol. 2002, 69, 265–274. [Google Scholar] [CrossRef]
- Leist, M.; Ghezzi, P.; Grasso, G.; Bianchi, R.; Villa, P.; Fratelli, M.; Savino, C.; Bianchi, M.; Nielsen, J.; Gerwien, J.; et al. Derivatives of Erythropoietin That Are Tissue Protective But Not Erythropoietic. Science 2004, 305, 239–242. [Google Scholar] [CrossRef]
- Gan, Y.; Xing, J.; Jing, Z.; Stetler, R.A.; Zhang, F.; Luo, Y.; Ji, X.; Gao, Y.; Cao, G. Mutant Erythropoietin Without Erythropoietic Activity Is Neuroprotective Against Ischemic Brain Injury. Stroke 2012, 43, 3071–3077. [Google Scholar] [CrossRef]
- Brines, M.; Patel, N.S.A.; Villa, P.; Brines, C.; Mennini, T.; De Paola, M.; Erbayraktar, Z.; Erbayraktar, S.; Sepodes, B.; Thiemermann, C.; et al. Nonerythropoietic, tissue-protective peptides derived from the tertiary structure of erythropoietin. Proc. Natl. Acad. Sci. USA 2008, 105, 10925–10930. [Google Scholar] [CrossRef]
- Marsh, J.T.; Brown, W.S.; Wolcott, D.; Carr, C.R.; Harper, R.; Schweitzer, S.V.; Nissenson, A.R. rHuEPO treatment improves brain and cognitive function of anemic dialysis patients. Kidney Int. 1991, 39, 155–163. [Google Scholar] [CrossRef]
- Genc, S.; Koroglu, T.F.; Genc, K. Erythropoietin and the nervous system. Brain Res. 2004, 1000, 19–31. [Google Scholar] [CrossRef]
- Marti, H.H. Erythropoietin and the hypoxic brain. J. Exp. Biol. 2004, 207, 3233–3242. [Google Scholar] [CrossRef]
- Sirén, A.-L.; Knerlich, F.; Poser, W.; Gleiter, C.H.; Brück, W.; Ehrenreich, H. Erythropoietin and erythropoietin receptor in human ischemic/hypoxic brain. Acta Neuropathol. 2001, 101, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Shacka, J.J.; Eells, J.B.; Suarez-Quian, C.; Przygodzki, R.M.; Beleslin-Cokic, B.; Lin, C.-S.; Nikodem, V.M.; Hempstead, B.; Flanders, K.C.; et al. Erythropoietin receptor signalling is required for normal brain development. Development 2002, 129, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Tsai, P.T.; Ohab, J.J.; Kertesz, N.; Groszer, M.; Matter, C.; Gao, J.; Liu, X.; Wu, H.; Carmichael, S.T. A Critical Role of Erythropoietin Receptor in Neurogenesis and Post-Stroke Recovery. J. Neurosci. 2006, 26, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Sadamoto, Y.; Igase, K.; Sakanaka, M.; Sato, K.; Otsuka, H.; Sakaki, S.; Masuda, S.; Sasaki, R. Erythropoietin Prevents Place Navigation Disability and Cortical Infarction in Rats with Permanent Occlusion of the Middle Cerebral Artery. Biochem. Biophys. Res. Commun. 1998, 253, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Bernaudin, M.; Marti, H.H.; Roussel, S.; Divoux, D.; Nouvelot, A.; MacKenzie, E.T.; Petit, E. A Potential Role for Erythropoietin in Focal Permanent Cerebral Ischemia in Mice. J. Cereb. Blood Flow Metab. 1999, 19, 643–651. [Google Scholar] [CrossRef]
- Aluclu, M.U.; Acar, A.; Guzel, A.; Bahceci, S.; Yaldiz, M. Evaluation of erythropoietin effects on cerebral ischemia in rats. Neuro Endocrinol. Lett. 2007, 28, 170–174. [Google Scholar]
- Villa, P.; Bigini, P.; Mennini, T.; Agnello, D.; Laragione, T.; Cagnotto, A.; Viviani, B.; Marinovich, M.; Cerami, A.; Coleman, T.R.; et al. Erythropoietin Selectively Attenuates Cytokine Production and Inflammation in Cerebral Ischemia by Targeting Neuronal Apoptosis. J. Exp. Med. 2003, 198, 971–975. [Google Scholar] [CrossRef]
- Gunnarson, E.; Song, Y.; Kowalewski, J.M.; Brismar, H.; Brines, M.; Cerami, A.; Andersson, U.; Zelenina, M.; Aperia, A. Erythropoietin modulation of astrocyte water permeability as a component of neuroprotection. Proc. Natl. Acad. Sci. USA 2009, 106, 1602–1607. [Google Scholar] [CrossRef]
- Chang, Y.S.; Mu, D.; Wendland, M.; Sheldon, R.A.; Vexler, Z.S.; McQuillen, P.S.; Ferriero, D.M. Erythropoietin Improves Functional and Histological Outcome in Neonatal Stroke. Pediatr. Res. 2005, 58, 106–111. [Google Scholar] [CrossRef]
- Spandou, E.; Papadopoulou, Z.; Soubasi, V.; Karkavelas, G.; Simeonidou, C.; Pazaiti, A.; Guiba-Tziampiri, O. Erythropoietin prevents long-term sensorimotor deficits and brain injury following neonatal hypoxia–ischemia in rats. Brain Res. 2005, 1045, 22–30. [Google Scholar] [CrossRef]
- Grasso, G.; Buemi, M.; Alafaci, C.; Sfacteria, A.; Passalacqua, M.; Sturiale, A.; Calapai, G.; De Vico, G.; Piedimonte, G.; Salpietro, F.M.; et al. Beneficial effects of systemic administration of recombinant human erythropoietin in rabbits subjected to subarachnoid hemorrhage. Proc. Natl. Acad. Sci. USA 2002, 99, 5627–5631. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Mahmood, A.; Qu, C.; Goussev, A.; Schallert, T.; Chopp, M. Erythropoietin Enhances Neurogenesis and Restores Spatial Memory in Rats after Traumatic Brain Injury. J. Neurotrauma 2005, 22, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Lu, D.; Qu, C.; Goussev, A.; Schallert, T.; Mahmood, A.; Chopp, M. Effects of erythropoietin on reducing brain damage and improving functional outcome after traumatic brain injury in mice. J. Neurosurg. 2008, 109, 510–521. [Google Scholar] [CrossRef] [PubMed]
- Dang, J.; Jia, R.; Tu, Y.; Xiao, S.; Ding, G. Erythropoietin prevents reactive oxygen species generation and renal tubular cell apoptosis at high glucose level. Biomed. Pharmacother. 2010, 64, 681–685. [Google Scholar] [CrossRef]
- Wang, R.; Wu, X.; Liang, J.; Qi, Z.; Liu, X.; Min, L.; Ji, X.; Luo, Y.; Zhao, H. Intra-artery infusion of recombinant human erythropoietin reduces blood–brain barrier disruption in rats following cerebral ischemia and reperfusion. Int. J. Neurosci. 2014, 125, 693–702. [Google Scholar] [CrossRef]
- Thériault, P.; Le Béhot, A.; ElAli, A.; Rivest, S. Sub-acute systemic erythropoietin administration reduces ischemic brain injury in an age-dependent manner. Oncotarget 2016, 7, 35552–35561. [Google Scholar] [CrossRef][Green Version]
- Chong, Z.Z.; Kang, J.-Q.; Maiese, K. Erythropoietin fosters both intrinsic and extrinsic neuronal protection through modulation of microglia, Akt1, Bad, and caspase-mediated pathways. Br. J. Pharmacol. 2003, 138, 1107–1118. [Google Scholar] [CrossRef]
- Shang, Y.C.; Chong, Z.Z.; Wang, S.; Maiese, K. Erythropoietin and Wnt1 govern pathways of mTOR, Apaf-1, and XIAP in inflammatory microglia. Curr. Neurovascular Res. 2011, 8, 270–285. [Google Scholar] [CrossRef]
- Morishita, E.; Masuda, S.; Nagao, M.; Yasuda, Y.; Sasaki, R. Erythropoietin receptor is expressed in rat hippocampal and cerebral cortical neurons, and erythropoietin prevents in vitro glutamate-induced neuronal death. Neuroscience 1996, 76, 105–116. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Lin, S.-H.; Kang, J.-Q.; Maiese, K. Erythropoietin prevents early and late neuronal demise through modulation of Akt1 and induction of caspase 1, 3, and 8. J. Neurosci. Res. 2002, 71, 659–669. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Kang, J.-Q.; Maiese, K. Apaf-1, Bcl-xL, Cytochrome c, and Caspase-9 Form the Critical Elements for Cerebral Vascular Protection by Erythropoietin. J. Cereb. Blood Flow Metab. 2003, 23, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Kook, Y.-H.; Ka, M.; Um, M. Neuroprotective cytokines repress PUMA induction in the 1-methyl-4-phenylpyridinium (MPP+) model of Parkinson’s disease. Biochem. Biophys. Res. Commun. 2011, 411, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Digicaylioglu, M.; Lipton, S.A. Erythropoietin-mediated neuroprotection involves cross-talk between Jak2 and NF-κB signalling cascades. Nature 2001, 412, 641–647. [Google Scholar] [CrossRef]
- Bond, W.S.; Rex, T.S. Evidence That Erythropoietin Modulates Neuroinflammation through Differential Action on Neurons, Astrocytes, and Microglia. Front. Immunol. 2014, 5, 523. [Google Scholar] [CrossRef] [PubMed]
- Beleslin-Cokic, B.B.; Cokic, V.P.; Yu, X.; Weksler, B.B.; Schechter, A.N.; Noguchi, C.T. Erythropoietin and hypoxia stimulate erythropoietin receptor and nitric oxide production by endothelial cells. Blood 2004, 104, 2073–2080. [Google Scholar] [CrossRef]
- Navarro, J.C.; Pillai, S.; Ponce, L.L.; Van, M.; Goodman, J.C.; Robertson, C.S. Endothelial Nitric Oxide Synthase Mediates the Cerebrovascular Effects of Erythropoietin in Traumatic Brain Injury. Front. Immunol. 2014, 5, 494. [Google Scholar] [CrossRef]
- Kertesz, N.; Wu, J.; Chen, T.H.-P.; Sucov, H.M.; Wu, H. The role of erythropoietin in regulating angiogenesis. Dev. Biol. 2004, 276, 101–110. [Google Scholar] [CrossRef]
- Chen, G.-H.; Li, X.-L.; Deng, Y.-Q.; Zhou, F.-M.; Zou, W.-Q.; Jiang, W.-X.; Shangguan, S.-Q.; Lu, Z.-N. The Molecular Mechanism of EPO Regulates the Angiogenesis after Cerebral Ischemia through AMPK-KLF2 Signaling Pathway. Crit. Rev. Eukaryot. Gene Expr. 2019, 29, 105–112. [Google Scholar] [CrossRef]
- Studer, L.; Csete, M.; Lee, S.-H.; Kabbani, N.; Walikonis, J.; Wold, B.; McKay, R. Enhanced Proliferation, Survival, and Dopaminergic Differentiation of CNS Precursors in Lowered Oxygen. J. Neurosci. 2000, 20, 7377–7383. [Google Scholar] [CrossRef]
- Shingo, T.; Sorokan, S.T.; Shimazaki, T.; Weiss, S. Erythropoietin regulates the in vitro and in vivo production of neuronal progenitors by mammalian forebrain neural stem cells. J. Neurosci. 2001, 21, 9733–9743. [Google Scholar] [CrossRef]
- Digicaylioglu, M.; Garden, G.; Timberlake, S.; Fletcher, L.; Lipton, S.A. Acute neuroprotective synergy of erythropoietin and insulin-like growth factor I. Proc. Natl. Acad. Sci. USA 2004, 101, 9855–9860. [Google Scholar] [CrossRef] [PubMed]
- Kilic, E.; Kilic, U.; Soliz, J.; Bassetti, C.L.; Gassmann, M.; Hermann, D.M. Brain-derived erythropoietin protects from focal cerebral ischemia by dual activation of ERK-1/-2 and Akt pathways. FASEB J. 2005, 19, 2026–2028. [Google Scholar] [CrossRef]
- Um, M.; Lodish, H.F. Antiapoptotic Effects of Erythropoietin in Differentiated Neuroblastoma SH-SY5Y Cells Require Activation of Both the STAT5 and AKT Signaling Pathways. J. Biol. Chem. 2006, 281, 5648–5656. [Google Scholar] [CrossRef]
- Sergio, C.-M.; Rolando, C.-A. Erythropoietin regulates signaling pathways associated with neuroprotective events. Exp. Brain Res. 2022, 240, 1303–1315. [Google Scholar] [CrossRef] [PubMed]
- Wen, T.-C.; Sadamoto, Y.; Tanaka, J.; Zhu, P.; Nakata, K.; Ma, Y.-J.; Hata, R.; Sakanaka, M. Erythropoietin protects neurons against chemical hypoxia and cerebral ischemic injury by up-regulating Bcl-xL expression. J. Neurosci. Res. 2002, 67, 795–803. [Google Scholar] [CrossRef]
- Yamamoto, M.; Koshimura, K.; Sohmiya, M.; Murakami, Y.; Kato, Y. Effect of erythropoietin on nitric oxide production in the rat hippocampus using in vivo brain microdialysis. Neuroscience 2004, 128, 163–168. [Google Scholar] [CrossRef]
- Yip, H.-K.; Tsai, T.-H.; Lin, H.-S.; Chen, S.-F.; Sun, C.-K.; Leu, S.; Yuen, C.-M.; Tan, T.-Y.; Lan, M.-Y.; Liou, C.-W.; et al. Effect of erythropoietin on level of circulating endothelial progenitor cells and outcome in patients after acute ischemic stroke. Crit. Care 2011, 15, R40. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Jumbe, N.L.; Farrell, C.L.; Niehoff, M.L.; Heatherington, A.C. Passage of erythropoietic agents across the blood–brain barrier: A comparison of human and murine erythropoietin and the analog darbepoetin alfa. Eur. J. Pharmacol. 2004, 505, 93–101. [Google Scholar] [CrossRef]
- Ábrahám, C.S.; Harada, N.; Deli, M.A.; Niwa, M. Transient forebrain ischemia increases the blood-brain barrier permeability for albumin in stroke-prone spontaneously hypertensive rats. Cell. Mol. Neurobiol. 2002, 22, 455–462. [Google Scholar] [CrossRef]
- Xenocostas, A.; Cheung, W.K.; Farrell, F.; Zakszewski, C.; Kelley, M.; Lutynski, A.; Crump, M.; Lipton, J.H.; Kiss, T.L.; Lau, C.Y.; et al. The pharmacokinetics of erythropoietin in the cerebrospinal fluid after intravenous administration of recombinant human erythropoietin. Eur. J. Clin. Pharmacol. 2005, 61, 189–195. [Google Scholar] [CrossRef]
- Ishii, T.; Asai, T.; Urakami, T.; Oku, N. Accumulation of macromolecules in brain parenchyma in acute phase of cerebral infarction/reperfusion. Brain Res. 2010, 1321, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Moon, C.; Krawczyk, M.; Ahn, D.; Ahmet, I.; Paik, D.; Lakatta, E.G.; Talan, M.I. Erythropoietin reduces myocardial infarction and left ventricular functional decline after coronary artery ligation in rats. Proc. Natl. Acad. Sci. USA 2003, 100, 11612–11617. [Google Scholar] [CrossRef] [PubMed]
- Parsa, C.J.; Matsumoto, A.; Kim, J.; Riel, R.U.; Pascal, L.S.; Walton, G.B.; Thompson, R.B.; Petrofski, J.A.; Annex, B.H.; Stamler, J.S.; et al. A novel protective effect of erythropoietin in the infarcted heart. J. Clin. Investig. 2003, 112, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Ning, N.; Niu, X.; Dang, Y.; Dong, X.; Wei, J.; Zhu, C. Erythropoietin treatment in patients with acute myocardial infarction: A meta-analysis of randomized controlled trials. Am. Heart J. 2012, 164, 715–727. [Google Scholar] [CrossRef]
- Wen, Y.; Xu, J.; Ma, X.; Gao, Q. High-Dose Erythropoietin in Acute ST-Segment Elevation Myocardial Infarction: A Meta-Analysis of Randomized Controlled Trials. Am. J. Cardiovasc. Drugs 2013, 13, 435–442. [Google Scholar] [CrossRef]
- Ali-Hassan-Sayegh, S.; Mirhosseini, S.J.; Tahernejad, M.; Mahdavi, P.; Haddad, F.; Shahidzadeh, A.; Lotfaliani, M.R.; Sedaghat-Hamedani, F.; Kayvanpour, E.; Weymann, A.; et al. Administration of erythropoietin in patients with myocardial infarction: Does it make sense? An updated and comprehensive meta-analysis and systematic review. Cardiovasc. Revascularization Med. 2015, 16, 179–189. [Google Scholar] [CrossRef]
- Jean-Baptiste, W.; Ali, A.Y.; Inyang, B.; Koshy, F.S.; George, K.; Poudel, P.; Chalasani, R.; Goonathilake, M.R.; Waqar, S.; George, S.; et al. Are There Any Cardioprotective Effects or Safety Concerns of Erythropoietin in Patients With Myocardial Infarction? A Systematic Review. Cureus 2022, 14, e25671. [Google Scholar] [CrossRef]
- Bennett, C.L.; Silver, S.M.; Djulbegovic, B.; Samaras, A.T.; Blau, C.A.; Gleason, K.J.; Barnato, S.E.; Elverman, K.M.; Courtney, D.M.; McKoy, J.M.; et al. Venous Thromboembolism and Mortality Associated With Recombinant Erythropoietin and Darbepoetin Administration for the Treatment of Cancer-Associated Anemia. JAMA 2008, 299, 914–924. [Google Scholar] [CrossRef]
- Steppich, B.; Groha, P.; Ibrahim, T.; Schunkert, H.; Laugwitz, K.-L.; Hadamitzky, M.; Kastrati, A.; Ott, I. Effect of Erythropoietin in patients with acute myocardial infarction: Five-year results of the REVIVAL-3 trial. BMC Cardiovasc. Disord. 2017, 17, 1–8. [Google Scholar] [CrossRef]
- Ruschitzka, F.T.; Wenger, R.H.; Stallmach, T.; Quaschning, T.; de Wit, C.; Wagner, K.; Labugger, R.; Kelm, M.; Noll, G.; Rülicke, T.; et al. Nitric oxide prevents cardiovascular disease and determines survival in polyglobulic mice overexpressing erythropoietin. Proc. Natl. Acad. Sci. USA 2000, 97, 11609–11613. [Google Scholar] [CrossRef]
- Quaschning, T.; Ruschitzka, F.; Stallmach, T.; Shaw, S.; Morawietz, H.; Goettsch, W.; Hermann, M.; Slowinski, T.; Theuring, F.; Hocher, B.; et al. Erythropoietin-induced excessive erythrocytosis activates the tissue endothelin system in mice. FASEB J. 2002, 17, 259–261. [Google Scholar] [CrossRef] [PubMed]
- Lim, V.S. Recombinant human erythropoietin in predialysis patients. Am. J. Kidney Dis. 1991, 18, 34–37. [Google Scholar] [PubMed]
- Smith, K.J.; Bleyer, A.J.; Little, W.C.; Sane, D.C. The cardiovascular effects of erythropoietin. Cardiovasc. Res. 2003, 59, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.M.R.; Apaiajai, N.; Chattipakorn, N.; Chattipakorn, S.C. The Protective and Reparative Role of Colony-Stimulating Factors in the Brain with Cerebral Ischemia/Reperfusion Injury. Neuroendocrinology 2020, 111, 1029–1065. [Google Scholar] [CrossRef] [PubMed]
- Talan, M.I.; Ahmet, I.; Lakatta, E. Did Clinical Trials in Which Erythropoietin Failed to Reduce Acute Myocardial Infarct Size Miss a Narrow Therapeutic Window? PLoS ONE 2012, 7, e34819. [Google Scholar] [CrossRef]
- Talan, M.I.; Latini, R. Myocardial infarction: Cardioprotection by erythropoietin. Breast Cancer 2013, 982, 265–302. [Google Scholar] [CrossRef]
- Pearl, R.G. Erythropoietin and organ protection: Lessons from negative clinical trials. Crit. Care 2014, 18, 1–3. [Google Scholar] [CrossRef][Green Version]
- Brines, M.; Cerami, A. The Receptor That Tames the Innate Immune Response. Mol. Med. 2011, 18, 486–496. [Google Scholar] [CrossRef]
- Pradeep, S.; Huang, J.; Mora, E.M.; Nick, A.M.; Cho, M.S.; Wu, S.Y.; Noh, K.; Pecot, C.V.; Rupaimoole, R.; Stein, M.A.; et al. Erythropoietin Stimulates Tumor Growth via EphB4. Cancer Cell 2015, 28, 610–622. [Google Scholar] [CrossRef]
- Zhang, F.; Xing, J.; Liou, A.K.-F.; Wang, S.; Gan, Y.; Luo, Y.; Ji, X.; Stetler, R.A.; Chen, J.; Cao, G. Enhanced Delivery of Erythropoietin Across the Blood–Brain Barrier for Neuroprotection Against Ischemic Neuronal Injury. Transl. Stroke Res. 2010, 1, 113–121. [Google Scholar] [CrossRef]
- Najjar, S.S.; Rao, S.V.; Melloni, C.; Raman, S.V.; Povsic, T.J.; Melton, L.; Barsness, G.W.; Prather, K.; Heitner, J.F.; Kilaru, R.; et al. Intravenous Erythropoietin in Patients With ST-Segment Elevation Myocardial Infarction. JAMA 2011, 305, 1863–1872. [Google Scholar] [CrossRef]
- Imai, N.; Higuchi, M.; Kawamura, A.; Tomonoh, K.; Oh-Eda, M.; Fujiwara, M.; Shimonaka, Y.; Ochi, N. Physicochemical and biological characterization of asialoerythropoietin. Suppressive effects of sialic acid in the expression of biological activity of human erythropoietin in vitro. JBIC J. Biol. Inorg. Chem. 1990, 194, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Mennini, T.; De Paola, M.; Bigini, P.; Mastrotto, C.; Fumagalli, E.; Barbera, S.; Mengozzi, M.; Viviani, B.; Corsini, E.; Marinovich, M.; et al. Nonhematopoietic Erythropoietin Derivatives Prevent Motoneuron Degeneration In Vitro and In Vivo. Mol. Med. 2006, 12, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.; Altmann, F.; Arrenberg, C.K.; Koprivova, A.; Beike, A.K.; Stemmer, C.; Gorr, G.; Reski, R.; Decker, E.L. Moss-based production of asialo-erythropoietin devoid of Lewis A and other plant-typical carbohydrate determinants. Plant Biotechnol. J. 2012, 10, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Kittur, F.S.; Hung, C.-Y.; Darlington, D.E.; Sane, D.C.; Xie, J. N-Glycosylation engineering of tobacco plants to produce asialoerythropoietin. Plant Cell Rep. 2012, 31, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Kittur, F.S.; Bah, M.; Archer-Hartmann, S.; Hung, C.-Y.; Azadi, P.; Ishihara, M.; Sane, D.C.; Xie, J. Cytoprotective Effect of Recombinant Human Erythropoietin Produced in Transgenic Tobacco Plants. PLoS ONE 2013, 8, e76468. [Google Scholar] [CrossRef]
- Kittur, F.S.; Hung, C.-Y.; Zhu, C.; Shajahan, A.; Azadi, P.; Thomas, M.D.; Pearce, J.L.; Gruber, C.; Kallolimath, S.; Xie, J. Glycoengineering tobacco plants to stably express recombinant human erythropoietin with different N-glycan profiles. Int. J. Biol. Macromol. 2020, 157, 158–169. [Google Scholar] [CrossRef]
- Weise, A.; Altmann, F.; Rodriguez-Franco, M.; Sjoberg, E.R.; Bäumer, W.; Launhardt, H.; Kietzmann, M.; Gorr, G. High-level expression of secreted complex glycosylated recombinant human erythropoietin in the Physcomitrella Delta-fuc-t Delta-xyl-t mutant. Plant Biotechnol. J. 2007, 5, 389–401. [Google Scholar] [CrossRef]
- Wee, E.G.-T.; Sherrier, D.J.; Prime, T.A.; DuPree, P. Targeting of Active Sialyltransferase to the Plant Golgi Apparatus. Plant Cell 1998, 10, 1759–1768. [Google Scholar] [CrossRef]
- Ma, J.K.-C.; Drake, P.M.W.; Christou, P. The production of recombinant pharmaceutical proteins in plants. Nat. Rev. Genet. 2003, 4, 794–805. [Google Scholar] [CrossRef]
- Gomord, V.; Faye, L. Posttranslational modification of therapeutic proteins in plants. Curr. Opin. Plant Biol. 2004, 7, 171–181. [Google Scholar] [CrossRef]
- Palacpac, N.Q.; Yoshida, S.; Sakai, H.; Kimura, Y.; Fujiyama, K.; Yoshida, T.; Seki, T. Stable expression of human β1,4-galactosyltransferase in plant cells modifies N-linked glycosylation patterns. Proc. Natl. Acad. Sci. USA 1999, 96, 4692–4697. [Google Scholar] [CrossRef] [PubMed]
- Bakker, H.; Rouwendal, G.J.A.; Karnoup, A.S.; Florack, D.E.A.; Stoopen, G.M.; Helsper, J.P.F.G.; van Ree, R.; van Die, I.; Bosch, D. An antibody produced in tobacco expressing a hybrid β-1,4-galactosyltransferase is essentially devoid of plant carbohydrate epitopes. Proc. Natl. Acad. Sci. USA 2006, 103, 7577–7582. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.; Streatfield, S.J.; Wycoff, K. Medical molecular farming: Production of antibodies, biopharmaceuticals and edible vaccines in plants. Trends Plant Sci. 2001, 6, 219–226. [Google Scholar] [CrossRef]
- Kittur, F.S.; Lalgondar, M.; Hung, C.-Y.; Sane, D.C.; Xie, J. C-Terminally fused affinity Strep-tag II is removed by proteolysis from recombinant human erythropoietin expressed in transgenic tobacco plants. Plant Cell Rep. 2014, 34, 507–516. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wen, D.; Boissel, J.; Showers, M.; Ruch, B.; Bunn, H. Erythropoietin structure-function relationships. Identification of functionally important domains. J. Biol. Chem. 1994, 269, 22839–22846. [Google Scholar] [CrossRef]
- Chargelegue, D.; Vine, N.D.; Van Dolleweerd, C.J.; Drake, P.M.; Ma, J.K.-C. A murine monoclonal antibody produced in transgenic plants with plant-specific glycans is not immunogenic in mice. Transgenic Res. 2000, 9, 187–194. [Google Scholar] [CrossRef]
- Jin, C.; Altmann, F.; Strasser, R.; Mach, L.; Schähs, M.; Kunert, R.; Rademacher, T.; Glössl, J.; Steinkellner, H. A plant-derived human monoclonal antibody induces an anti-carbohydrate immune response in rabbits. Glycobiology 2007, 18, 235–241. [Google Scholar] [CrossRef]
- Ward, B.J.; Landry, N.; Trépanier, S.; Mercier, G.; Dargis, M.; Couture, M.; D’aoust, M.-A.; Vézina, L.-P. Human antibody response to N-glycans present on plant-made influenza virus-like particle (VLP) vaccines. Vaccine 2014, 32, 6098–6106. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, D.; Shi, J.; Yang, D. Expression of α-1,6-fucosyltransferase (FUT8) in rice grain and immunogenicity evaluation of plant-specific glycans. J. Biotechnol. 2017, 242, 111–121. [Google Scholar] [CrossRef]
- Cheung, E.C.C.; McBride, H.M.; Slack, R.S. Mitochondrial dynamics in the regulation of neuronal cell death. Apoptosis 2007, 12, 979–992. [Google Scholar] [CrossRef] [PubMed]
- Balog, J.; Mehta, S.L.; Vemuganti, R. Mitochondrial fission and fusion in secondary brain damage after CNS insults. J. Cereb. Blood Flow Metab. 2016, 36, 2022–2033. [Google Scholar] [CrossRef] [PubMed]
- Otera, H.; Mihara, K. Mitochondrial Dynamics: Functional Link with Apoptosis. Int. J. Cell Biol. 2012, 2012, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Bukowski, M.J.; Wider, J.M.; Reynolds, C.A.; Calo, L.; Lepore, B.; Tousignant, R.; Jones, M.; Przyklenk, K.; Sanderson, T.H. Mitochondrial dynamics following global cerebral ischemia. Mol. Cell. Neurosci. 2016, 76, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Burman, J.L.; Pickles, S.; Wang, C.; Sekine, S.; Vargas, J.N.S.; Zhang, Z.; Youle, A.M.; Nezich, C.L.; Wu, X.; Hammer, J.A.; et al. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J. Cell Biol. 2017, 216, 3231–3247. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef] [PubMed]
- Markus, T.M.; Tsai, S.-Y.; Bs, M.R.B.; Farrer, R.G.; O’Brien, T.E.; Kindler-Baumann, D.R.; Rausch, M.; Rudin, M.; Wiessner, C.; Mir, A.K.; et al. Recovery and brain reorganization after stroke in adult and aged rats. Ann. Neurol. 2005, 58, 950–953. [Google Scholar] [CrossRef]
- Yang, W.; Paschen, W. Is age a key factor contributing to the disparity between success of neuroprotective strategies in young animals and limited success in elderly stroke patients? Focus on protein homeostasis. J. Cereb. Blood Flow Metab. 2017, 37, 3318–3324. [Google Scholar] [CrossRef]
- Popa-Wagner, A.; Buga, A.-M.; Turner, R.C.; Rosen, C.L.; Toescu, E. Cerebrovascular Disorders: Role of Aging. J. Aging Res. 2012, 2012, 1–4. [Google Scholar] [CrossRef]
- Popa-Wagner, A.; Glavan, D.-G.; Olaru, A.; Olaru, D.-G.; Margaritescu, O.; Tica, O.; Surugiu, R.; Sandu, R.E. Present Status and Future Challenges of New Therapeutic Targets in Preclinical Models of Stroke in Aged Animals with/without Comorbidities. Int. J. Mol. Sci. 2018, 19, 356. [Google Scholar] [CrossRef]
- Chen, R.-L.; Balami, J.S.; Esiri, M.M.; Chen, L.-K.; Buchan, A.M. Ischemic stroke in the elderly: An overview of evidence. Nat. Rev. Neurol. 2010, 6, 256–265. [Google Scholar] [CrossRef]
- Sommer, C.J. Ischemic stroke: Experimental models and reality. Acta Neuropathol. 2017, 133, 245–261. [Google Scholar] [CrossRef]
- Hankey, G. Population Impact of Potentially Modifiable Risk Factors for Stroke. Stroke 2020, 51, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Simon, F.; Floros, N.; Ibing, W.; Schelzig, H.; Knapsis, A. Neurotherapeutic potential of erythropoietin after ischemic injury of the central nervous system. Neural Regen. Res. 2019, 14, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Popa-Wagner, A.; Buga, A.-M.; Doeppner, T.R.; Hermann, D.M. Stem cell therapies in preclinical models of stroke associated with aging. Front. Cell. Neurosci. 2014, 8, 347. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-K.; Yang, M.-T.; Kang, K.-H.; Liou, H.-C.; Lu, D.-H.; Fu, W.-M.; Lin, W.-L. Targeted Delivery of Erythropoietin by Transcranial Focused Ultrasound for Neuroprotection against Ischemia/Reperfusion-Induced Neuronal Injury: A Long-Term and Short-Term Study. PLoS ONE 2014, 9, e90107. [Google Scholar] [CrossRef]
- Ostrowski, D.; Heinrich, R. Alternative Erythropoietin Receptors in the Nervous System. J. Clin. Med. 2018, 7, 24. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).