Abstract
The past few years have shown an ongoing interest in lipoprotein(a) (Lp(a)), a lipid molecule that has been proven to have atherogenic, thrombogenic, and inflammatory properties. Several lines of evidence, indeed, have demonstrated an increased risk of cardiovascular disease as well as calcific aortic valve stenosis in patients with elevated Lp(a) levels. Statins, the mainstay of lipid-lowering therapy, slightly increase Lp(a) levels, while most other lipid-modifying agents do not significantly alter Lp(a) concentrations, except for proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors. The latter have been shown to reduce Lp(a) levels; however, the clinical significance of this effect has not been clearly elucidated. Of note, the pharmaceutical lowering of Lp(a) may be achieved with novel treatments specifically designed for this purpose (i.e., antisense oligonucleotides (ASOs) and small interfering RNAs (siRNAs)). Large clinical trials with cardiovascular outcomes with these agents are ongoing, and their results are eagerly awaited. Furthermore, several non-lipid-modifying drugs of various classes may influence Lp(a) concentrations. We have searched MEDLINE, EMBASE, and CENTRAL databases up to 28 January 2023 and summarized the effects of established and emerging lipid-modifying drugs and other medications on Lp(a) levels. We also discuss the potent clinical implications of these alterations.
1. Introduction
Lipoprotein(a) (Lp(a)) is a plasma lipoprotein that consists of a low-density lipoprotein (LDL)-like particle in which apolipoprotein B100 (apoB100) is linked by a single disulfide bond to a unique plasminogen-like glycoprotein, i.e., apolipoprotein(a) (apo(a)) [1]. Lp(a) not only promotes atherosclerosis but also has inflammatory, oxidative, thrombotic, and antifibrinolytic properties [1].
The concentration of Lp(a) is almost exclusively genetically determined with an autosomal codominant inheritance pattern and strongly determined by a single gene, the LPA gene [1]. Serum Lp(a) levels are inherited in an autosomal codominant manner [1]. Greater than 90% of the variability in Lp(a) levels is determined by a single gene, i.e., the LPA gene [1]. By the age of 2, the LPA gene is fully expressed [2]. Adult Lp(a) levels are usually reached by approximately the age of five but may further increase until adulthood [2]. Elevations in Lp(a) levels, though, may be observed in pregnancy and menopause [3,4]. Ethnicity also affects Lp(a) concentrations, with median Lp(a) increasing sequentially in Chinese, White, South Asian, and Black individuals [5]. Furthermore, sex influences Lp(a) concentrations; specifically, women have ∼5–10% higher levels compared with men in both Blacks and Whites [6]. Overall, Lp(a) levels are stable over time and are not generally affected by diet, physical activity, or other environmental factors [7].
Lp(a) is a well-recognized, independent risk factor for atherosclerotic cardiovascular disease (ASCVD) [8,9]. Evidence from experimental, observational, and genetic studies has demonstrated that increased Lp(a) is associated with increased risk for coronary heart disease (CHD), ischemic stroke, peripheral artery disease, heart failure, calcific aortic valve stenosis (CAVS), mitral valve stenosis, and possibly retinopathy in diabetic patients [1,10,11,12,13,14,15,16,17,18] (Figure 1). On the contrary, elevated Lp(a) concentrations do not appear to be a risk factor for venous thromboembolism [7,8], as previously considered, whereas very low Lp(a) levels have been linked to an increased risk of incident type 2 diabetes mellitus (T2DM) [19].
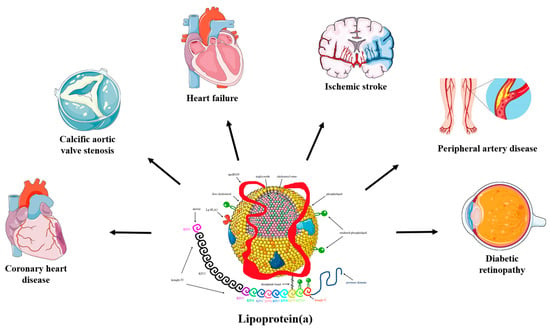
Figure 1.
The pathogenicity of lipoprotein(a). Convincing evidence has emerged from pathophysiological, epidemiological, and genetic studies on the causality of high serum lipoprotein(a) (Lp(a)) levels as a potent risk factor for coronary heart disease (CHD), ischemic stroke, peripheral artery disease, heart failure, calcific aortic valve stenosis (CAVS), mitral valve stenosis, and retinopathy in patients with diabetes. Mendelian randomization and genome-wide association studies support the role of Lp(a) as an independent cardiovascular risk factor.
The thresholds commonly used for high Lp(a) are >30 mg/dL or >50 mg/dL (>75 nmol/L or >125 nmol/L, respectively), as levels above these thresholds have been associated with increased ASCVD risk. Importantly, about 20% of the general population have Lp(a) > 50 mg/dL (>125 nmol/L) [7]. Recent guidelines recommend that Lp(a) should be measured in every adult at least once in a lifetime to identify those with very high Lp(a) levels, i.e., >180 mg/dL (>450 nmol/L), and these individuals are considered to have similar ASCVD risk as individuals with heterozygous familial hypercholesterolemia (FH) [20,21,22]. In addition, Lp(a) measurements should be considered in patients with a personal and/or family history of premature ASCVD or high Lp(a) and also for cardiovascular risk reclassification in those at moderate and high ASCVD risk [20,22]. Screening is also recommended for young adults with ischemic stroke and no other identifiable risk factors [20,22].
Currently, there are no medications specifically approved for Lp(a)-lowering [23,24,25]. However, clinical trials in the advanced stages of development with novel agents targeting Lp(a) are ongoing. Interestingly, some therapeutic agents of various drug classes appear to affect Lp(a) concentrations [23,24,25]. In this narrative review, we discuss the effects of current and future lipid-modifying and non-lipid-modifying medical interventions on Lp(a) levels and their potent clinical implications.
2. Methods
We searched MEDLINE, EMBASE, and CENTRAL databases up to 28 January 2023 using the following terms: lipoprotein(a), cardiovascular risk, hypolipidemic treatment, statins, ezetimibe, proprotein convertase subtilisin/kexin type 9 inhibitors, lipoprotein apheresis, antisense oligonucleotides (ASOs), small interfering RNAs, tamoxifen, thyroid, aspirin, sex hormones, anti-inflammatory, tocilizumab, interleukin-6 inhibitors, and diet. Clinical trials, review articles, and case reports were assessed, whereas the references of these articles were scrutinized for other relevant articles.
3. Lp(a)-Lowering Therapies
3.1. Effects of Lipid-Modifying Interventions on Lp(a) Levels
3.1.1. Lipoprotein Apheresis
The most effective clinically available intervention for Lp(a)-lowering is lipoprotein apheresis (LA) [26]. Lp(a) levels are reduced by 70–80%, acutely, after treatment, but rebound elevations between apheresis sessions, which are typically weekly, biweekly, or less frequently, result in a mean interval Lp(a) reduction of 25–40%, depending on the course and baseline Lp(a) levels [26]. LA is infrequently used worldwide, except in Germany, and long-term studies in patients with high Lp(a) undergoing LA suggest that this therapy may reduce 5-year cardiovascular risk by up to 86% [27]. However, LA remains a semi-invasive, time-consuming, and chronic-expensive therapy with variable adherence [28]. The Hellenic consensus panel on the clinical use of LA proposes that LA treatment still has a role in individuals with hypercholesterolemia, including heterozygous FH for the secondary prevention of ASCVD, if they are under a maximum-tolerated lipid-lowering treatment (LLT) and have Lp(a) levels > 100 mg/dL (>238 nmol/L) [29]. Furthermore, LA may be implemented in individuals with severely elevated Lp(a) (>180 mg/dL (>430 nmol/L)) for the primary prevention of ASCVD if they have LDL cholesterol (LDL-C) > 190 mg/dL under a maximum LLT as well as in individuals with diabetes and Lp(a) > 180 mg/dL (>430 nmol/L) with (a) ASCVD, (b) chronic kidney disease stage 4 or 5, or (c) 24-urine albumin > 300 mg [29].
3.1.2. Statins
The effect of statin treatment on Lp(a) levels remains an area of controversy. Clinical trials of statin therapy have shown mixed results regarding their impact on Lp(a) levels. A large meta-analysis of six trials, including 5256 patients randomized to receive various statins or a placebo, indicated that most statins may increase Lp(a) by an average of 8–24%, although significant heterogeneity in the response of Lp(a) levels to statin administration, as well as between different statins, has been reported [30]. However, another meta-analysis of 39 trials, including 24,448 patients randomized to receive various statins or a placebo, indicated a non-significant 0.1% increase in Lp(a) in the statin groups (vs. the placebo), with no significant differences among the statins or different intensities of statins [31]. Nonetheless, statin therapy has demonstrated a clinical benefit in patients with elevated Lp(a) in both primary and secondary preventions [32,33]. A meta-analysis of seven randomized controlled trials, including 29,069 patients with high Lp(a) levels and a history of cardiovascular events, concluded that those with Lp(a) levels > 50 mg/dL, at the baseline or in treatment with statins, are at a significantly higher risk of ASCVD as compared with those with <30 mg/dL, independent of other conventional risk factors [32]. The data from this meta-analysis suggest that patients with raised concentrations of Lp(a) (>50 mg/dL), representing about 25% of those with previous cardiovascular disease, are at substantial residual risk, despite statin treatment [32]. The mechanisms by which statins influence Lp(a) levels remain unclear [30]. Statins elevate the expression of LPA mRNAs, as well as the synthesis and secretion of the apo(a) protein in HepG2 cells, which may result in an increase in Lp(a) levels [30]. Moreover, statins activate the expression of PCSK9 genes and increase PCSK9 levels, which then enhance Lp(a) production [34,35,36].
3.1.3. Niacin
Niacin decreases Lp(a) in a dose-dependent manner by approximately 30–40% on average [37] but only by 18% in those with the highest Lp(a) levels [38]. The effect of niacin is likely due to a decreased apo(a) production rate [39]. Importantly, studies with cardiovascular outcomes showed no benefit of adding niacin to statins [40,41]. Moreover, niacin use is limited by side effects, such as flushing, gastrointestinal discomfort, and new-onset diabetes [40,41].
In a study conducted a few years ago, the addition of PCSK9 inhibitors to niacin resulted in a 15% additional reduction in Lp(a) compared with that achieved with niacin monotherapy [42].
3.1.4. Ezetimibe
The effect of ezetimibe on Lp(a) is not clear. In a meta-analysis of seven trials of ezetimibe monotherapy, ezetimibe significantly reduced Lp(a) levels by 7.1% [43]. In contrast, in another meta-analysis, ezetimibe, either as monotherapy vs. placebo or in combination with statin vs. statin alone, did not significantly alter Lp(a) concentrations [43]. The mechanisms by which ezetimibe may influence Lp(a) levels have not been clearly elucidated [43]. Ezetimibe likely has anti-inflammatory activity and may affect Lp(a) production, as Lp(a) is an acute-phase reactant [43].
3.1.5. PCSK9 Inhibitors
PCSK9 monoclonal antibodies, i.e., alirocumab and evolocumab, reduce Lp(a) levels by approximately 20–30% [44,45,46]. In the two major PCSK9 inhibitor trials, the FOURIER trial, with evolocumab, and the ODYSSEY OUTCOMES trial, with alirocumab, both PCSK9 inhibitors were associated with an approximately 25% reduction in the risk of major adverse cardiovascular events (MACEs) [47,48]. Secondary analyses of these trials indicated that the main factor determining both the risk for MACEs in the placebo groups as well as the reductions in cardiovascular outcomes in patients treated with PCSK9 inhibitors was Lp(a) concentrations [49]. Indeed, in the ODYSSEY OUTCOMES trial, patients with a recent acute coronary syndrome, LDL-C near 70 mg/dL in their optimized statin treatment, and Lp(a) levels that were at least mildly elevated (≥13.7 mg/dL) derived substantial clinical benefit from the treatment with alirocumab [49]. In contrast, patients with LDL-C near 70 mg/dL and lipoprotein(a) < 13.7 mg/dL had no reduction in MACEs with alirocumab, whereas patients with higher LDL-C levels derived consistent clinical benefit from alirocumab treatment, irrespective of the level of Lp(a) [49]. The exact mechanism by which PCSK9 inhibitors reduce Lp(a) levels remains unclear [50]. Current hypotheses include increased clearance of Lp(a) particles via the LDLR, increased clearance of Lp(a) via other receptors (the LDL receptor-related protein 1, the cluster of differentiation 36 receptor, toll-like receptor 2, scavenger receptor-B1, and plasminogen receptors), as well as a reduction in apo(a) production, secretion, and/or assembly [50].
Inclisiran, a small interfering RNA (siRNA) targeting intracellular PCSK9, has been shown to reduce Lp(a) levels by approximately 20% [51]. The clinical significance of this finding warrants further investigation.
3.1.6. Fibrates
Bezafibrate has been shown to reduce Lp(a) levels by approximately 13–39% [52]. A similarly modest, but not statistically significant, effect was demonstrated for gemfibrozil [53]. Thus, fibrates do not appear to influence Lp(a) concentrations significantly [54].
3.1.7. Lomitapide
Lomitapide, a microsomal triglyceride transfer protein inhibitor, reduces Lp(a) levels by 15–19% [55]. The possible mechanism for Lp(a)-lowering is the decrease in very-low-density lipoprotein (VLDL) and chylomicron synthesis via inhibition of MTP [55]. MTP is located in the endoplasmic reticulum of hepatocytes and enterocytes and is most likely responsible for transferring triglycerides to nascent apoB as it enters the lumen of the endoplasmic lumen [55]. Consequently, MTP inhibition seems to control the number of apoB-containing lipoprotein particles secreted into the bloodstream, including Lp(a) particles [55]. However, its use is restricted to patients with homozygous familial hypercholesterolemia [55] and is not indicated for Lp(a)-lowering [56].
3.1.8. Mipomersen
Mipomersen is an ASO-targeting apoB100 [57]. In clinical trials, mipomersen significantly reduced Lp(a) levels by 21–39% [57,58]. The mechanism for Lp(a)-lowering seems to involve a decrease in the availability of apoB100 for Lp(a) assembly [59]. However, mipomersen has been withdrawn from the market due to side effects, which mainly include an increased risk of hepatic steatosis and hepatic enzyme elevation [57,58,59].
3.1.9. Cholesteryl Transfer Protein (CETP) Inhibitors
CETP mediates the transfer of cholesteryl esters from high-density lipoprotein (HDL) to apoB100-containing particles, including VLDL and LDL, in exchange for triglycerides [60,61,62,63]. Apart from raising HDL cholesterol [60,61,62,63], CETP inhibitors also decrease apoB100, LDL cholesterol, and Lp(a) levels (by approximately 24–36%) [60,61,62,63]. Clinical outcome trials of four CETP inhibitors have been completed, including torcetrapib, dalcetrapib, evacetrapib, and anacetrapib [64]. However, they either had only modest clinical benefits or even clinical futility after prolonged treatment, whereas torcetrapib increased mortality [64]. Thus, CETP inhibitors are not currently approved for clinical use.
We should note, though, that treatment with 5 or 10 mg of obicetrapib, another CETP inhibitor, robustly reduced LDL-C, apoB, non-HDL-C, and Lp(a) (33.8% vs. 56.5%, respectively) and increased HDL cholesterol and apoA1, as an adjunct to high-intensity statins, compared with the placebo in a randomized, double-blind, placebo-controlled, phase 2 trial with a high safety and tolerability profile [65]. Additional phase 3 investigations, including a cardiovascular outcomes trial, are currently underway to further assess the safety and clinical benefits of obicetrapib [65].
3.1.10. Bempedoic Acid
Bempedoic acid inhibits the adenosine triphosphate citrate lyase upstream from the 3-Hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase and decreases cholesterol biosynthesis in the liver. Subsequently, bempedoic acid upregulates LDL receptors and increases the clearance of LDL particles [66]. In a phase 2 trial, bempedoic acid had no significant effect on Lp(a) levels [67]. Similarly, the combination of bempedoic acid with evolocumab was not superior to evolocumab monotherapy in terms of Lp(a) change from the baseline [68]. In a phase 3, randomized, double-blind, placebo-controlled trial (the CLEAR OUTCOMES trial), bempedoic acid (180 mg daily) was associated with a statistically significant and clinically meaningful reduction in the risk of MACEs in 14,014 patients at high risk for ASCVD with documented statin intolerance and elevated LDL cholesterol levels (≥100 mg/dL) [69].
3.1.11. Bile Acid Sequestrants
Bile acid sequestrants (colesevalem, cholestyramine, and colestipol) interrupt the enterohepatic circulation of bile acids [70]. Thus, they increase the conversion of cholesterol into bile acids and upregulate LDL receptors. These drugs have not been shown to affect Lp(a) levels [70].
3.1.12. ASO and siRNA Agents
In the era of RNA-based therapies, ASOs and siRNAs targeting Lp(a) are currently in clinical development [71]. Pelacarsen (formerly IONIS-APO(a)-LRX, AKCEA-APO(a)-LRX, TQJ230) is a second-generation ASO that binds to its target complementary RNA sequence via base pairing, thereby leading to the degradation of the apo(a) mRNA strand and reduced Lp(a) production [72] (Figure 2). Pelacarsen is conjugated to N-acetylgalactosamine (GalNAc), which enables specific targeting to the hepatocytes, thus providing increased drug potency, less systemic toxicity, and less-frequent dosing [72]. In a phase 2 trial, a randomized, double-blind, placebo-controlled, dose-ranging trial involving 286 patients with established ASCVD and screening Lp(a) levels of at least 60 mg/dL (150 nmol/L), pelacarsen effectively reduced Lp(a) levels in a dose-dependent way (i.e., 35% at the dose of 20 mg every 4 weeks, 56% at 40 mg every 4 weeks, 58% at 20 mg every 2 weeks, 72% at 60 mg every 4 weeks, and 80% at 20 mg every week) [73]. The most common adverse effect was injection site reactions [73]. A phase 3 cardiovascular outcome trial (HORIZON trial; ClinicalTrials.gov Identifier: NCT04023552) with pelacarsen is currently ongoing in patients with elevated baseline Lp(a) levels (>70 mg/dL) and established ASCVD (a prior history of myocardial infarction, ischemic stroke, or symptomatic peripheral artery disease). Contrary to phase 2 studies, the dose of 80 mg once monthly subcutaneously is being evaluated.
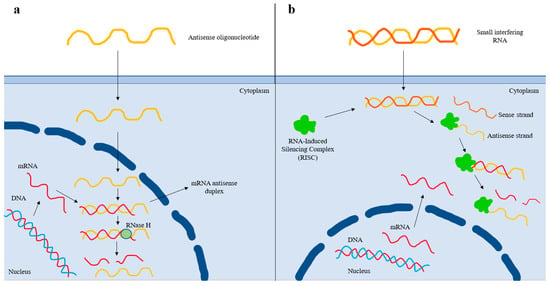
Figure 2.
Mechanism of action of antisense oligonucleotide and small interfering RNA-based therapies. (a) After entering the nucleus, the antisense oligonucleotide (ASO) binds to the complementary sequence of the targeted mRNA. The resulting mRNA antisense duplex is recognized by RNase H, which cleaves the mRNA and, thus, prevents protein translation. (b) After the small interfering RNA (siRNA) enters the cell, it is recognized by the RNA-Induced Silencing Complex (RISC), which removes the sense strand. The resulting complex binds to the complementary mRNA sequence and degrades it, thus preventing protein translation. The antisense strand bound to the RISC forms a recyclable, stable complex, and, as a result, its action against target mRNA strands can be repeated. Thereby, less-frequent dosing of siRNAs is needed compared to that of ASOs.
In contrast to ASOs, siRNAs are double-stranded RNA molecules that dissociate once inside the cell, and the antisense strand is inserted into the RISC (RNA-Induced Silencing Complex) [74]. The antisense strand binds to its homologous target mRNA sequence, leading to its degradation [74]. The antisense strand bound to the RISC forms a recyclable, stable complex, and, as a result, its action against target mRNA strands can be repeated [74]. Thereby less-frequent dosing of siRNAs is needed compared to that of ASOs [74] (Figure 2). Olpasiran (formerly AMG890, ARO-LPA), a GalNAc-conjugated siRNA agent against apo(a), reduced Lp(a) by up to >90% at doses ≥ 9 mg, with its effects persisting for an average of 3 to 6 months with no safety concerns, in a phase 1 clinical trial [75]. Equally encouraging are the results obtained from the phase 2 randomized, double-blind, placebo-controlled, dose-finding trial (a double-blind, randomized, placebo-controlled phase 2 study to evaluate the efficacy, safety, and tolerability of AMG 890 (a GalNAc-conjugated small interfering RNA in subjects with elevated lipoprotein(a); the OCEAN(a)-DOSE trial; NCT04270760) involving patients with established ASCVD and Lp(a) levels of >150 nmol/L. In this trial, 281 patients were randomly assigned to receive one of four doses of olpasiran (10 mg every 12 weeks, 75 mg every 12 weeks, 225 mg every 12 weeks, or 225 mg every 24 weeks) or a matching placebo administered subcutaneously [76]. At 36 weeks, the olpasiran therapy had significantly and substantially reduced the Lp(a) levels in a dose-dependent manner, resulting in placebo-adjusted mean percent changes of −70.5% with the 10 mg dose, −97.4% with the 75 mg dose, −101.1% with the 225 mg dose administered every 12 weeks, and −100.5% with the 225 mg dose administered every 24 weeks [76]. The overall incidence of the adverse events was similar across the trial groups, with the most common olpasiran-related adverse events being injection site reactions, primarily pain [76]. Further research with olpasiran, including cardiovascular outcomes clinical trials, are warranted to assess the role of this agent in clinical practice. Currently, a phase 3 double-blind, randomized, placebo-controlled trial assessing the impact of olpasiran on the risk for CHD death, myocardial infarction, or urgent coronary revascularization in patients with ASCVD and elevated Lp(a) levels (≥200 nmol/L) is ongoing (Olpasiran Trials of Cardiovascular Events and Lipoprotein(a) Reduction (OCEAN(a))—Outcomes Trial; NCT05581303).
Apart from olpasiran, another GalNAc-conjugated siRNA against the apo(a) mRNA, SLN360, reduced circulating Lp(a) by up to 95% in cynomolgus monkeys [77]. A phase 1 trial of SLN360 (NCT04606602) reported a dose-dependent Lp(a) reduction of up to 98% in 32 patients with elevated Lp(a) levels and no known cardiovascular disease [78]. Most patients treated with SLN360 experienced mild to moderate injection site reactions, but further study to determine the safety and efficacy of this siRNA is necessary [78].
Moreover, a phase 1 trial investigating the effect of LY3819469, another GalNAc-conjugated siRNA against the apo(a) mRNA, in patients with elevated Lp(a) levels and increased cardiovascular risk is now ongoing (a study of LY3819469 in healthy participants; NCT04914546).
3.2. Effects of Other Drugs on Lp(a) Levels
3.2.1. Sex Hormone Therapies
Estrogen and its analogs reduce the transcription of the LPA gene [79]. While endogenous sex hormones do not substantially affect Lp(a) levels, postmenopausal hormone replacement therapy can lower Lp(a) levels by approximately 20–25% [80,81]. A systematic review and meta-analysis of 10 clinical trials, which included 2049 postmenopausal women, suggested that anti-estrogen therapy significantly reduced Lp(a) levels by approximately 5.92% [82]. However, adverse effects (e.g., breast cancer, stroke, and thrombosis) may outweigh potential cardiovascular benefits [83,84].
Although previous data suggested that testosterone treatment may lower Lp(a) levels [85], randomized control trials did not confirm this effect [86,87].
Tamoxifen administration in patients with breast cancer has been found to reduce Lp(a) levels by approximately 40% [88,89].
3.2.2. Thyroid Hormone Therapies
Lp(a) levels are decreased in hyperthyroidism and increased in hypothyroidism [90]. Thyromimetic agents (e.g., eprotirome) have been shown to reduce Lp(a) levels [90]. However, in previous programs in animals, these agents were discontinued because of significant adverse effects, such as elevations in liver enzymes and cartilage side effects [90]. Liver-selective thyromimetic agents, such as the thyroid hormone receptor agonist eprotirome and resmetirom (MGL-3196), a liver-directed, orally active thyroid hormone receptor beta agonist, under study for the treatment of nonalcoholic steatohepatitis, reduce Lp(a) levels (by 32.3% and 37.9%, respectively) without extrahepatic adverse events [91,92,93]. In a recent systematic review and meta-analysis, the treatment of overt hyperthyroidism with thyroidectomy, antithyroid drugs, or radioactive iodine increased Lp(a) levels by 20–25%, whereas the treatment of overt and subclinical hypothyroidism with levothyroxine decreased Lp(a) levels by 5–20% [90].
3.2.3. Growth Hormone Replacement Therapy
Growth hormone (GH) replacement therapy has been previously found to increase Lp(a) levels by 25–100% [94]. The increase in the Lp(a) levels induced by GHs may partly contribute to the increased ASCVD risk in patients with acromegaly [94]. In a recent prospective observational study, GH replacement therapy in men with GH deficiency resulted in a significant increase in Lp(a) levels (mean: from 27.4 nmol/L to 34.3 nmol/L) [95]. However, long-term studies with GH replacement therapy in GH deficiency are warranted to address these questions.
3.2.4. Aspirin
The available data regarding the effects of aspirin on Lp(a) concentrations and any potent clinical implications of these alterations are scarce. In a post hoc analysis of the Women’s Health Study, a randomized primary prevention trial, 25,131 healthy White women with Lp(a) > 65 mg/dL received aspirin (100 mg every other day) or a placebo [96]. The treatment with aspirin did not significantly reduce cardiovascular events over a 10-year period (the age-adjusted hazard ratio was 0.75 (the 95% CI: 0.48–1.18, p = 0.22)) [96]. In a subgroup analysis, though, carriers of the rs3798220 variant, which is associated with high circulating Lp(a) levels, with a baseline median Lp(a) ≈ 80 mg/dL, significantly benefitted from aspirin treatment (HR 0.44 and the 95% CI: 0.20–0.94, p = 0.03) [96]. This may imply that the benefit of aspirin treatment depends on the Lp(a) levels, but this warrants validation in further studies [96]. Thus, individuals with high Lp(a) levels might be considered for aspirin therapy if they also have other indications for aspirin therapy (e.g., very high ASCVD risk and low bleeding risk) [7,97].
3.2.5. Anti-Inflammatory Agents
Tocilizumab, a monoclonal antibody targeting interleukin-6, reduced Lp(a) levels by approximately 30–40% when administered in rheumatoid arthritis patients [98]. The LPA gene promoter contains functional interleukin 6-responsive elements; thus, this effect of tocilizumab may be attributed to its inhibitory action in this region of the gene [99].
On the contrary, protease inhibitors and antiretroviral therapy have been associated with increased Lp(a) levels when administered in human immunodeficiency patients with baseline Lp(a) levels > 20–30 mg/dL [100].
3.3. Effects of Dietary Intervention and Physical Activity on Lp(a) Levels
Lifestyle changes, including low-fat diets, have no significant effect on Lp(a) levels [101,102]. Some, but not all, studies reported that isocaloric replacement of dietary saturated fats with carbohydrates or unsaturated fats increased Lp(a) levels by approximately 8–20% [102,103,104]. On the contrary, low-carbohydrate, high-saturated fat diets and diets enriched with walnuts or pecans decreased Lp(a) levels by 15% and 6–15%, respectively [102,105]. Other studies have investigated the effect of dietary supplements (L-carnitine and coenzyme Q10) and specific foods (coffee, tea, and alcoholic beverages, especially red wine) on Lp(a) levels and have shown modest decreases of 10–30% with these interventions [101,106]. Finally, vitamin C, in supplementary (1 g/day) or pharmacologic doses (4.5 g/day), has a neutral effect on plasma Lp(a) levels, as shown in a recent meta-analyses [107,108].
Most studies suggest that physical activity has no or minimal impact on Lp(a) levels, although there are conflicting data, particularly among younger or diabetic populations in whom physical fitness correlated inversely with Lp(a) levels and decreased CVD risk [109,110].
3.4. Effects of Bariatric Surgery on Lp(a) Levels
A prospective observational study of 59 patients with severe obesity (with a median Body Mass Index (BMI) of 48 kg/m2) undergoing metabolic surgery (thirty-one had Roux-en-Y gastric bypass, nineteen had sleeve gastrectomy, and nine had omega loop bypass) demonstrated a significant increase in Lp(a) levels at 12 months following bariatric surgery (10.2 (3.8–31.9) vs. 16.9 (4.9–38.6) mg/dL, p = 0.002) [111]. These results may suggest an increased synthetic liver capacity, leading to improved production and secretion of Lp(a) and, thus, to higher plasma levels [111].
The effects of all the aforementioned agents on Lp(a) concentrations and their possible mechanisms of action are summarized in Table 1 and Table 2, respectively.

Table 1.
Current and emerging lipoprotein(a)-lowering therapies.
Table 2.
Potential mechanisms of action of current and emerging lipoprotein(a) (Lp(a))-lowering therapies.
Several lines of evidence suggest that large absolute reductions in Lp(a) concentrations of ~100 mg/dL are needed to achieve meaningful reductions in ASCVD risk [7,112,113]. Therefore, only those drugs that are specifically designed for the purpose of Lp(a)-lowering could be expected to have beneficial effects on ASCVD outcomes. Ongoing and future studies addressing this question are pending.
4. Conclusions
Lp(a) is a novel risk factor for ASCVD and CAVS, and this risk increases linearly with Lp(a) concentrations. It appears that robust reductions in Lp(a) concentrations should be achieved for meaningful reductions in cardiovascular outcomes. In patients with high Lp(a) levels, more aggressive LDL-C targets, compared with those recommended for individuals with otherwise the same ASCVD risk, are considered. Thus, our initial approach to reduce ASCVD risk in patients with elevated Lp(a) is to further reduce LDL-C. However, several promising agents are in the late stages of development, and the results of phase 3 outcome studies are eagerly awaited. Several other agents, either lipid-modifying or of other drug classes, also alter Lp(a) levels. However, there is no solid evidence that these changes have clinical significance, with the possible exception of PCSK9 inhibitors.
Author Contributions
Conceptualization, A.D.K., G.L. and M.F.; Methodology, A.D.K. and M.F.; Validation, A.D.K., G.L. and M.F.; Formal Analysis, M.F.; Investigation, A.D.K. and M.F.; Resources, A.D.K. and P.S.A.; Data Curation, A.D.K., P.S.A. and M.F.; Writing—Original Draft Preparation, A.D.K. and P.S.A.; Writing—Review & Editing, G.L., M.F. and E.L.; Visualization, G.L. and M.F.; Supervision, G.L., M.F. and E.L.; Project Administration, M.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a research grant from the Hellenic Atherosclerosis Society (Athens, 6 July 2021, Protocol Number 514 Φ/4).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Schmidt, K.; Noureen, A.; Kronenberg, F.; Utermann, G. Thematic Review Series: Lipoprotein (a): Coming of Age at Last: Structure, function, and genetics of lipoprotein (a). J. Lipid Res. 2016, 57, 1339. [Google Scholar] [CrossRef]
- de Boer, L.M.; Hof, M.H.; Wiegman, A.; Stroobants, A.K.; Kastelein, J.J.P.; Hutten, B.A. Lipoprotein(a) levels from childhood to adulthood: Data in nearly 3000 children who visited a pediatric lipid clinic. Atherosclerosis 2022, 349, 227–232. [Google Scholar] [CrossRef]
- Derby, C.A.; Crawford, S.L.; Pasternak, R.C.; Sowers, M.; Sternfeld, B.; Matthews, K.A. Lipid Changes During the Menopause Transition in Relation to Age and Weight: The Study of Women’s Health Across the Nation. Am. J. Epidemiol. 2009, 169, 1352. [Google Scholar] [CrossRef]
- Fanshawe, A.E.; Ibrahim, M. The current status of lipoprotein (a) in pregnancy: A literature review. J. Cardiol. 2013, 61, 99–106. [Google Scholar] [CrossRef]
- Patel, A.P.; Wang, M.; Pirruccello, J.P.; Ellinor, P.T.; Ng, K.; Kathiresan, S.; Khera, A.V. Lipoprotein(a) concentrations and incident atherosclerotic cardiovascular disease: New insights from a large national biobank. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 465. [Google Scholar] [CrossRef]
- Erhart, G.; Lamina, C.; Lehtimäki, T.; Marques-Vidal, P.; Kähönen, M.; Vollenweider, P.; Raitakari, O.T.; Waeber, G.; Thorand, B.; Strauch, K.; et al. Genetic factors explain a major fraction of the 50% lower lipoprotein(a) concentrations in finns. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1230–1241. [Google Scholar] [CrossRef]
- Kronenberg, F.; Mora, S.; Stroes, E.S.G.; Ference, B.A.; Arsenault, B.J.; Berglund, L.; Dweck, M.R.; Koschinsky, M.; Lambert, G.; Mach, F.; et al. Lipoprotein(a) in atherosclerotic cardiovascular disease and aortic stenosis: A European Atherosclerosis Society consensus statement. Eur. Heart J. 2022, 43, 3925–3946. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Langsted, A. Thematic Review Series: Lipoprotein (a): Coming of Age at Last: Lipoprotein (a) as a cause of cardiovascular disease: Insights from epidemiology, genetics, and biology. J. Lipid Res. 2016, 57, 1953. [Google Scholar] [CrossRef]
- Tsimikas, S. A Test in Context: Lipoprotein(a): Diagnosis, Prognosis, Controversies, and Emerging Therapies. J. Am. Coll. Cardiol. 2017, 69, 692–711. [Google Scholar] [CrossRef]
- Kamstrup, P.R.; Nordestgaard, B.G. Elevated Lipoprotein(a) Levels, LPA Risk Genotypes, and Increased Risk of Heart Failure in the General Population. JACC Heart Fail. 2016, 4, 78–87. [Google Scholar] [CrossRef]
- Kamstrup, P.R.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Elevated Lipoprotein(a) and Risk of Aortic Valve Stenosis in the General Population. J. Am. Coll. Cardiol. 2014, 63, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Langsted, A.; Nordestgaard, B.G.; Kamstrup, P.R. Elevated Lipoprotein(a) and Risk of Ischemic Stroke. J. Am. Coll. Cardiol. 2019, 74, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Kamstrup, P.R.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Genetic evidence that lipoprotein(a) associates with atherosclerotic stenosis rather than venous thrombosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1732–1741. [Google Scholar] [CrossRef]
- Langsted, A.; Kamstrup, P.R.; Nordestgaard, B.G. High lipoprotein(a) and high risk of mortality. Eur. Heart J. 2019, 40, 2760–2770. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, G.; Gagliano, C.; Bucolo, C.; Vacante, M.; Salomone, S.; Malaguarnera, M.; Leonardi, D.G.; Motta, M.; Drago, F.; Avitabile, T. Lipoprotein(a) Serum Levels in Diabetic Patients with Retinopathy. BioMed Res. Int. 2013, 2013, 943505. [Google Scholar] [CrossRef]
- Kaltoft, M.; Sigvardsen, P.E.; Afzal, S.; Langsted, A.; Fuchs, A.; Kühl, J.T.; Køber, L.; Kamstrup, P.R.; Kofoed, K.F.; Nordestgaard, B.G. Elevated lipoprotein(a) in mitral and aortic valve calcification and disease: The Copenhagen General Population Study. Atherosclerosis 2021, 349, 166–174. [Google Scholar] [CrossRef]
- Stürzebecher, P.E.; Schorr, J.J.; Klebs, S.H.G.; Laufs, U. Trends and consequences of lipoprotein(a) testing: Cross-sectional and longitudinal health insurance claims database analyses. Atherosclerosis 2023, 367, 24–33. [Google Scholar] [CrossRef]
- Zierfuss, B.; Höbaus, C.; Feldscher, A.; Hannes, A.; Mrak, D.; Koppensteiner, R.; Stangl, H.; Schernthaner, G.H. Lipoprotein (a) and long-term outcome in patients with peripheral artery disease undergoing revascularization. Atherosclerosis 2022, 363, 94–101. [Google Scholar] [CrossRef]
- Gudbjartsson, D.F.; Thorgeirsson, G.; Sulem, P.; Helgadottir, A.; Gylfason, A.; Saemundsdottir, J.; Bjornsson, E.; Norddahl, G.L.; Jonasdottir, A.; Jonasdottir, A.; et al. Lipoprotein(a) Concentration and Risks of Cardiovascular Disease and Diabetes. J. Am. Coll. Cardiol. 2019, 74, 2982–2994. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular riskThe Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Koutsogianni, A.D.; Adamidis, P.S.; Barkas, F.; Liberopoulos, E.; Su, T.C.; Yamashita, S.; Liamis, G.; Rizzo, M. Familial Hypercholesterolemia and Lipoprotein(a): A Gordian Knot in Cardiovascular Prevention. Metabolites 2022, 12, 1065. [Google Scholar] [CrossRef]
- Amiri, M.; Raeisi-Dehkordi, H.; Verkaar, A.J.C.F.; Wu, Y.; van Westing, A.C.; Berk, K.A.; Bramer, W.M.; Aune, D.; Voortman, T. Circulating lipoprotein (a) and all-cause and cause-specific mortality: A systematic review and dose-response meta-analysis. Eur. J. Epidemiol. 2023, 38, 485–499. [Google Scholar] [CrossRef]
- Jawi, M.M.; Frohlich, J.; Chan, S.Y. Lipoprotein(a) the Insurgent: A New Insight into the Structure, Function, Metabolism, Pathogenicity, and Medications Affecting Lipoprotein(a) Molecule. J. Lipids 2020, 2020, 3491764. [Google Scholar] [CrossRef]
- Koutsogianni, A.-D.; Liberopoulos, E.; Tselepis, A.D. Lipoprotein(a): An update on its role in human health and disease. J. Atheroscler. Prev. Treat. 2021, 12, 92–102. [Google Scholar] [CrossRef]
- Koutsogianni, A.D.; Liberopoulos, E.; Tellis, K.; Tselepis, A.D. Oxidized phospholipids and lipoprotein(a): An update. Eur. J. Clin. Investig. 2022, 52, e13710. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, E.; Parhofer, K.G. Lipoprotein apheresis to treat elevated lipoprotein (a). J. Lipid Res. 2016, 57, 1751–1757. [Google Scholar] [CrossRef]
- Roeseler, E.; Julius, U.; Heigl, F.; Spitthoever, R.; Heutling, D.; Breitenberger, P.; Leebmann, J.; Lehmacher, W.; Kamstrup, P.R.; Nordestgaard, B.G.; et al. Lipoprotein apheresis for lipoprotein(a)-Associated cardiovascular disease: Prospective 5 years of follow-up and apolipoprotein(a) characterization. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2019–2027. [Google Scholar] [CrossRef]
- Kayikçioʇlu, M.; Kismali, E.; Can, L.; Payzin, S. Long-term follow-up in patients with homozygous familial hypercholesterolemia; 13-year experience of a university hospital lipid clinic. Turk Kardiyol. Dern. Ars. 2014, 42, 599–611. [Google Scholar] [CrossRef] [PubMed]
- Kolovou, G.; Kolovou, V.; Bilianou, H.; Goumas, G.; Foussas, S.; Grapsa, E.; Garoufi, A.; Karavolias, G.; Mavrogieni, S.; Melidonis, A.; et al. Lipoprotein apheresis: A Hellenic consensus on its clinical use. Hell. J. Cardiol. 2021, 62, 460–462. [Google Scholar] [CrossRef]
- Tsimikas, S.; Gordts, P.L.S.M.; Nora, C.; Yeang, C.; Witztum, J.L. Statin therapy increases lipoprotein(a) levels. Eur. Heart J. 2020, 41, 2275–2284. [Google Scholar] [CrossRef] [PubMed]
- De Boer, L.M.; Oorthuys, A.O.J.; Wiegman, A.; Langendam, M.W.; Kroon, J.; Spijker, R.; Zwinderman, A.H.; Hutten, B.A. Statin therapy and lipoprotein(a) levels: A systematic review and meta-analysis. Eur. J. Prev. Cardiol. 2022, 29, 779–792. [Google Scholar] [CrossRef]
- Willeit, P.; Ridker, P.M.; Nestel, P.J.; Simes, J.; Tonkin, A.M.; Pedersen, T.R.; Schwartz, G.G.; Olsson, A.G.; Colhoun, H.M.; Kronenberg, F.; et al. Baseline and on-statin treatment lipoprotein(a) levels for prediction of cardiovascular events: Individual patient-data meta-analysis of statin outcome trials. Lancet 2018, 392, 1311–1320. [Google Scholar] [CrossRef]
- Khera, A.V.; Everett, B.M.; Caulfield, M.P.; Hantash, F.M.; Wohlgemuth, J.; Ridker, P.M.; Mora, S. Lipoprotein(a) concentrations, rosuvastatin therapy, and residual vascular risk: An analysis from the JUPITER trial (justification for the use of statins in prevention: An intervention trial evaluating rosuvastatin). Circulation 2014, 129, 635–642. [Google Scholar] [CrossRef]
- Villard, E.F.; Thedrez, A.; Blankenstein, J.; Croyal, M.; Tran, T.T.T.; Poirier, B.; Le Bail, J.C.; Illiano, S.; Nobécourt, E.; Krempf, M.; et al. PCSK9 Modulates the Secretion But Not the Cellular Uptake of Lipoprotein(a) Ex Vivo: An Effect Blunted by Alirocumab. JACC Basic Transl. Sci. 2016, 1, 419–427. [Google Scholar] [CrossRef]
- Awan, Z.; Seidah, N.G.; MacFadyen, J.G.; Benjannet, S.; Chasman, D.I.; Ridker, P.M.; Genest, J. Rosuvastatin, Proprotein Convertase Subtilisin/Kexin Type 9 Concentrations, and LDL Cholesterol Response: The JUPITER Trial. Clin. Chem. 2012, 58, 183–189. [Google Scholar] [CrossRef]
- Guo, Y.L.; Liu, J.; Xu, R.X.; Zhu, C.G.; Wu, N.Q.; Jiang, L.X.; Li, J.J. Short-term impact of low-dose atorvastatin on serum proprotein convertase subtilisin/kexin type 9. Clin. Drug Investig. 2013, 33, 877–883. [Google Scholar] [CrossRef]
- Scanu, A.M.; Bamba, R. Niacin and Lipoprotein(a): Facts, Uncertainties, and Clinical Considerations. Am. J. Cardiol. 2008, 101, S44–S47. [Google Scholar] [CrossRef]
- Parish, S.; Hopewell, J.C.; Hill, M.R.; Marcovina, S.; Valdes-Marquez, E.; Haynes, R.; Offer, A.; Pedersen, T.R.; Baigent, C.; Collins, R.; et al. Impact of Apolipoprotein(a) Isoform Size on Lipoprotein(a) Lowering in the HPS2-THRIVE Study. Circ. Genom. Precis. Med. 2018, 11, e001696. [Google Scholar] [CrossRef]
- Chemello, K.; Chan, D.C.; Lambert, G.; Watts, G.F. Recent advances in demystifying the metabolism of lipoprotein(a). Atherosclerosis 2022, 349, 82–91. [Google Scholar] [CrossRef]
- Anderson, T.J.; Boden, W.E.; Desvigne-Nickens, P.; Fleg, J.L.; Kashyap, M.L.; McBride, R.; Probstfield, J.L.; for the AIM-HIGH Investigators. Safety Profile of Extended-Release Niacin in the AIM-HIGH Trial. N. Engl. J. Med. 2014, 371, 288. [Google Scholar] [CrossRef]
- Landray, M.J.; Haynes, R.; Hope-well, J.C.; Parish, S.; Aung, T.; Tomson, J.; Wallendszus, K.; Craig, M.; Jiang, L.; Collins, R.; et al. Effects of extended-release niacin with laropiprant in high-risk patients. N. Engl. J. Med. 2014, 371, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Warden, B.A.; Minnier, J.; Watts, G.F.; Fazio, S.; Shapiro, M.D. Impact of PCSK9 inhibitors on plasma lipoprotein(a) concentrations with or without a background of niacin therapy. J. Clin. Lipidol. 2019, 13, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Sahebkar, A.; Simental-Mendía, L.E.; Pirro, M.; Banach, M.; Watts, G.F.; Sirotri, C.; Al-Rasadi, K.; Atkin, S.L. Impact of ezetimibe on plasma lipoprotein(a) concentrations as monotherapy or in combination with statins: A systematic review and meta-analysis of randomized controlled trials. Sci. Rep. 2018, 8, 17887. [Google Scholar] [CrossRef]
- Gaudet, D.; Kereiakes, D.J.; McKenney, J.M.; Roth, E.M.; Hanotin, C.; Gipe, D.; Du, Y.; Ferrand, A.C.; Ginsberg, H.N.; Stein, E.A. Effect of alirocumab, a monoclonal proprotein convertase subtilisin/kexin 9 antibody, on lipoprotein(a) concentrations (a pooled analysis of 150 mg every two weeks dosing from phase 2 trials). Am. J. Cardiol. 2014, 114, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Raal, F.J.; Giugliano, R.P.; Sabatine, M.S.; Koren, M.J.; Langslet, G.; Bays, H.; Blom, D.; Eriksson, M.; Dent, R.; Wasserman, S.M.; et al. Reduction in Lipoprotein(a) with PCSK9 Monoclonal Antibody Evolocumab (AMG 145): A Pooled Analysis of More Than 1,300 Patients in 4 Phase II Trials. J. Am. Coll. Cardiol. 2014, 63, 1278–1288. [Google Scholar] [CrossRef]
- Stiekema, L.C.A.; Stroes, E.S.G.; Verweij, S.L.; Kassahun, H.; Chen, L.; Wasserman, S.M.; Sabatine, M.S.; Mani, V.; Fayad, Z.A. Persistent arterial wall inflammation in patients with elevated lipoprotein(a) despite strong low-density lipoprotein cholesterol reduction by proprotein convertase subtilisin/kexin type 9 antibody treatment. Eur. Heart J. 2019, 40, 2775. [Google Scholar] [CrossRef]
- Bittner, V.A.; Szarek, M.; Aylward, P.E.; Bhatt, D.L.; Diaz, R.; Edelberg, J.M.; Fras, Z.; Goodman, S.G.; Halvorsen, S.; Hanotin, C.; et al. Effect of Alirocumab on Lipoprotein(a) and Cardiovascular Risk after Acute Coronary Syndrome. J. Am. Coll. Cardiol. 2020, 75, 133–144. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; Fazio, S.; Giugliano, R.P.; Stroes, E.S.G.; Kanevsky, E.; Gouni-Berthold, I.; Im, K.A.; Pineda, A.L.; Wasserman, S.M.; Češka, R.; et al. Lipoprotein(a), PCSK9 Inhibition, and Cardiovascular Risk. Circulation 2019, 139, 1483–1492. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Szarek, M.; Bittner, V.A.; Diaz, R.; Goodman, S.G.; Jukema, J.W.; Landmesser, U.; López-Jaramillo, P.; Manvelian, G.; Pordy, R.; et al. Lipoprotein(a) and Benefit of PCSK9 Inhibition in Patients with Nominally Controlled LDL Cholesterol. J. Am. Coll. Cardiol. 2021, 78, 421–433. [Google Scholar] [CrossRef]
- Liberopoulos, E. Lipoprotein(a) reduction with proprotein convertase subtilisin/kexin type 9 inhibitors: An unsolved mystery. Eur. J. Prev. Cardiol. 2021, 28, 813–815. [Google Scholar] [CrossRef]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.J.; et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef]
- Tenenbaum, A.; Fisman, E.Z. Balanced pan-PPAR activator bezafibrate in combination with statin: Comprehensive lipids control and diabetes prevention? Cardiovasc. Diabetol. 2012, 11, 140. [Google Scholar] [CrossRef]
- Fereshetian, A.G.; Davidson, M.; Haber, H.; Black, D.M. Gemfibrozil Treatment in Patients with Elevated Lipoprotein A. Clin. Drug Investig. 2012, 16, 1–7. [Google Scholar] [CrossRef]
- Von Eckardstein, A. Lipoprotein(a). Eur. Heart J. 2017, 38, 1530–1532. [Google Scholar] [CrossRef]
- Cuchel, M.; Meagher, E.A.; Theron, H.D.T.; Blom, D.J.; Marais, A.D.; Hegele, R.A.; Averna, M.R.; Sirtori, C.R.; Shah, P.K.; Gaudet, D.; et al. Efficacy and Safety of a Microsomal Triglyceride Transfer Protein Inhibitor in Homozygous Familial Hypercholesterolemia. Lancet 2013, 381, 40. [Google Scholar] [CrossRef]
- Vuorio, A.; Watts, G.F.; Kovanen, P.T. Depicting new pharmacological strategies for familial hypercholesterolaemia involving lipoprotein (a). Eur. Heart J. 2017, 38, 3555–3559. [Google Scholar] [CrossRef]
- Santos, R.D.; Raal, F.J.; Catapano, A.L.; Witztum, J.L.; Steinhagen-Thiessen, E.; Tsimikas, S. Mipomersen, an Antisense Oligonucleotide to Apolipoprotein B-100, Reduces Lipoprotein(a) in Various Populations with Hypercholesterolemia: Results of 4 Phase III Trials. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 689–699. [Google Scholar] [CrossRef]
- Fogacci, F.; Ferri, N.; Toth, P.P.; Ruscica, M.; Corsini, A.; Cicero, A.F.G. Efficacy and Safety of Mipomersen: A Systematic Review and Meta-Analysis of Randomized Clinical Trials. Drugs 2019, 79, 751–766. [Google Scholar] [CrossRef]
- Merki, E.; Graham, M.J.; Mullick, A.E.; Miller, E.R.; Crooke, R.M.; Pitas, R.E.; Witztum, J.L.; Tsimikas, S. Antisense Oligonucleotide Directed to Human Apolipoprotein B-100 Reduces Lipoprotein(a) Levels and Oxidized Phospholipids on Human Apolipoprotein B-100 Particles in Lipoprotein(a) Transgenic Mice. Circulation 2008, 118, 743–753. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Ruotolo, G.; Brewer, H.B.; Wang, M.D.; Liu, L.; Willey, M.B.; Deeg, M.A.; Krueger, K.A.; Nissen, S.E. Evacetrapib alone or in combination with statins lowers lipoprotein(a) and total and small LDL particle concentrations in mildly hypercholesterolemic patients. J. Clin. Lipidol. 2016, 10, 519–527.e4. [Google Scholar] [CrossRef]
- Bowman, L.; Chen, F.; Sammons, E.; Hopewell, J.C.; Wallendszus, K.; Stevens, W.; Valdes-Marquez, E.; Wiviott, S.; Cannon, C.P.; Braunwald, E.; et al. Randomized Evaluation of the Effects of Anacetrapib through Lipid-modification (REVEAL)—A large-scale, randomized, placebo-controlled trial of the clinical effects of anacetrapib among people with established vascular disease: Trial design, recruitment, and baseline characteristics. Am. Heart J. 2017, 187, 182. [Google Scholar] [CrossRef] [PubMed]
- Arsenault, B.J.; Petrides, F.; Tabet, F.; Bao, W.; Hovingh, G.K.; Boekholdt, S.M.; Ramin-Mangata, S.; Meilhac, O.; DeMicco, D.; Rye, K.A.; et al. Effect of atorvastatin, cholesterol ester transfer protein inhibition, and diabetes mellitus on circulating proprotein subtilisin kexin type 9 and lipoprotein(a) levels in patients at high cardiovascular risk. J. Clin. Lipidol. 2018, 12, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G.; Ballantyne, C.M.; Barter, P.J.; Kallend, D.; Leiter, L.A.; Leitersdorf, E.; McMurray, J.J.V.; Nicholls, S.J.; Olsson, A.G.; Shah, P.K.; et al. Association of Lipoprotein(a) with Risk of Recurrent Ischemic Events Following Acute Coronary Syndrome: Analysis of the dal-Outcomes Randomized Clinical Trial. JAMA Cardiol. 2018, 3, 164. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.J.; Sniderman, A.D.; Ditmarsch, M.; Dicklin, M.R.; Nicholls, S.J.; Davidson, M.H.; Kastelein, J.J.P. Cholesteryl Ester Transfer Protein Inhibition Reduces Major Adverse Cardiovascular Events by Lowering Apolipoprotein B Levels. Int. J. Mol. Sci. 2022, 23, 9417. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Ditmarsch, M.; Kastelein, J.J.; Rigby, S.P.; Kling, D.; Curcio, D.L.; Alp, N.J.; Davidson, M.H. Lipid lowering effects of the CETP inhibitor obicetrapib in combination with high-intensity statins: A randomized phase 2 trial. Nat. Med. 2022, 28, 1672–1678. [Google Scholar] [CrossRef]
- Bempedoic Acid for LDL-C Lowering: What Do We Know?—American College of Cardiology. Available online: https://www.acc.org/latest-in-cardiology/articles/2020/08/10/08/21/bempedoic-acid-for-ldl-c-lowering (accessed on 4 November 2022).
- Thompson, P.D.; Rubino, J.; Janik, M.J.; Macdougall, D.E.; McBride, S.J.; Margulies, J.R.; Newton, R.S. Use of ETC-1002 to treat hypercholesterolemia in patients with statin intolerance. J. Clin. Lipidol. 2015, 9, 295–304. [Google Scholar] [CrossRef]
- Rubino, J.; MacDougall, D.E.; Sterling, L.R.; Kelly, S.E.; McKenney, J.M.; Lalwani, N.D. Lipid lowering with bempedoic acid added to a proprotein convertase subtilisin/kexin type 9 inhibitor therapy: A randomized, controlled trial. J. Clin. Lipidol. 2021, 15, 593–601. [Google Scholar] [CrossRef]
- CLEAR Outcomes: Bempedoic Acid Reduces Cardiovascular Events. Available online: https://www.practicalcardiology.com/view/clear-outcomes-bempedoic-acid-reduces-cardiovascular-events (accessed on 20 February 2023).
- Eraikhuemen, N.; Lazaridis, D.; Dutton, M.T. Emerging Pharmacotherapy to Reduce Elevated Lipoprotein(a) Plasma Levels. Am. J. Cardiovasc. Drugs 2020, 21, 255–265. [Google Scholar] [CrossRef]
- Greco, M.F.; Sirtori, C.R.; Corsini, A.; Ezhov, M.; Sampietro, T.; Ruscica, M. Lipoprotein(a) Lowering-From Lipoprotein Apheresis to Antisense Oligonucleotide Approach. J. Clin. Med. 2020, 9, 2103. [Google Scholar] [CrossRef]
- Viney, N.J.; van Capelleveen, J.C.; Geary, R.S.; Xia, S.; Tami, J.A.; Yu, R.Z.; Marcovina, S.M.; Hughes, S.G.; Graham, M.J.; Crooke, R.M.; et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): Two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet 2016, 388, 2239–2253. [Google Scholar] [CrossRef]
- Tsimikas, S.; Karwatowska-Prokopczuk, E.; Gouni-Berthold, I.; Tardif, J.-C.; Baum, S.J.; Steinhagen-Thiessen, E.; Shapiro, M.D.; Stroes, E.S.; Moriarty, P.M.; Nordestgaard, B.G.; et al. Lipoprotein(a) Reduction in Persons with Cardiovascular Disease. N. Engl. J. Med. 2020, 382, 244–255. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; López, J.A.G.; Knusel, B.; Gencer, B.; Wang, H.; Wu, Y.; Kassahun, H.; Sabatine, M.S. Study design and rationale for the Olpasiran trials of Cardiovascular Events And lipoproteiN(a) reduction-DOSE finding study (OCEAN(a)-DOSE). Am. Heart J. 2022, 251, 61–69. [Google Scholar] [CrossRef]
- Koren, M.J.; Moriarty, P.M.; Neutel, J.; Baum, S.J.; Hernandez-Illas, M.; Weintraub, H.S.; Hellawell, J.; Varrieur, T.; Sohn, W.; Wang, H.; et al. Abstract 13951: Safety, Tolerability and Efficacy of Single-dose Amg 890, a Novel Sirna Targeting Lp(a), in Healthy Subjects and Subjects with Elevated Lp(a). Circulation 2020, 142, A13951. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; Rosenson, R.S.; Gencer, B.; López, J.A.G.; Lepor, N.E.; Baum, S.J.; Stout, E.; Gaudet, D.; Knusel, B.; Kuder, J.F.; et al. Small Interfering RNA to Reduce Lipoprotein(a) in Cardiovascular Disease. N. Engl. J. Med. 2022, 387, 1855–1864. [Google Scholar] [CrossRef]
- Rider, D.A.; Eisermann, M.; Löffler, K.; Aleku, M.; Swerdlow, D.I.; Dames, S.; Hauptmann, J.; Morrison, E.; Lindholm, M.W.; Schubert, S.; et al. Pre-clinical assessment of SLN360, a novel siRNA targeting LPA, developed to address elevated lipoprotein (a) in cardiovascular disease. Atherosclerosis 2022, 349, 240–247. [Google Scholar] [CrossRef]
- Nissen, S.E.; Wolski, K.; Balog, C.; Swerdlow, D.I.; Scrimgeour, A.C.; Rambaran, C.; Wilson, R.J.; Boyce, M.; Ray, K.K.; Cho, L.; et al. Single Ascending Dose Study of a Short Interfering RNA Targeting Lipoprotein(a) Production in Individuals with Elevated Plasma Lipoprotein(a) Levels. JAMA 2022, 327, 1679–1687. [Google Scholar] [CrossRef]
- Boffelli, D.; Zajchowski, D.A.; Yang, Z.; Lawn, R.M. Estrogen Modulation of Apolipoprotein(a) Expression. J. Biol. Chem. 1999, 274, 15569–15574. [Google Scholar] [CrossRef]
- Anagnostis, P.; Galanis, P.; Chatzistergiou, V.; Stevenson, J.C.; Godsland, I.F.; Lambrinoudaki, I.; Theodorou, M.; Goulis, D.G. The effect of hormone replacement therapy and tibolone on lipoprotein (a) concentrations in postmenopausal women: A systematic review and meta-analysis. Maturitas 2017, 99, 27–36. [Google Scholar] [CrossRef]
- Salpeter, S.R.; Walsh, J.M.E.; Ormiston, T.M.; Greyber, E.; Buckley, N.S.; Salpeter, E.E. Meta-analysis: Effect of hormone-replacement therapy on components of the metabolic syndrome in postmenopausal women. Diabetes Obes. Metab. 2006, 8, 538–554. [Google Scholar] [CrossRef]
- Fogacci, F.; Borghi, C.; Davinelli, S.; Scapagnini, G.; Cicero, A.F.G. Impact of anti-oestrogen therapy on lipoprotein(a) in postmenopausal women: A systematic review and meta-analysis of double-blind placebo-controlled clinical studies. Endocrine 2022, 80, 292–302. [Google Scholar] [CrossRef]
- Hulley, S.; Grady, D.; Bush, T.; Furberg, C.; Herrington, D.; Riggs, B.; Vittinghoff, E. Randomized Trial of Estrogen Plus Progestin for Secondary Prevention of Coronary Heart Disease in Postmenopausal Women. JAMA 1998, 280, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Shlipak, M.G.; Simon, J.A.; Vittinghoff, E.; Lin, F.; Barrett-Connor, E.; Knopp, R.H.; Levy, R.I.; Hulley, S.B. Estrogen and Progestin, Lipoprotein(a), and the Risk of Recurrent Coronary Heart Disease Events after Menopause. JAMA 2000, 283, 1845–1852. [Google Scholar] [CrossRef] [PubMed]
- Marcovina, S.M.; Lippi, G.; Bagatell, C.J.; Bremner, W.J. Testosterone-induced suppression of lipoprotein(a) in normal men; relation to basal lipoprotein(a) level. Atherosclerosis 1996, 122, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Giannoulis, M.G.; Jackson, N.; Shojaee-Moradie, F.; Sonksen, P.H.; Martin, F.C.; Umpleby, A.M. Effects of growth hormone and/or testosterone on very low density lipoprotein apolipoprotein B100 kinetics and plasma lipids in healthy elderly men: A randomised controlled trial. Growth Horm. IGF Res. 2006, 16, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Hartgens, F.; Rietjens, G.; Keizer, H.A.; Kuipers, H.; Wolffenbuttel, B.H.R. Effects of androgenic-anabolic steroids on apolipoproteins and lipoprotein (a). Br. J. Sports Med. 2004, 38, 253–259. [Google Scholar] [CrossRef]
- Shewmon, D.A.; Stock, J.L.; Abusamra, L.C.; Kristan, M.A.; Baker, S.; Heiniluoma, K.M. Tamoxifen decreases lipoprotein(a) in patients with breast cancer. Metab.-Clin. Exp. 1994, 43, 531–532. [Google Scholar] [CrossRef]
- Liberopoulos, E.; Karabina, S.A.; Tselepis, A.; Bairaktari, E.; Nicolaides, C.; Pavlidis, N.; Elisaf, M. Are the Effects of Tamoxifen on the Serum Lipid Profile Modified by Apolipoprotein E Phenotypes? Oncology 2002, 62, 115–120. [Google Scholar] [CrossRef]
- Kotwal, A.; Cortes, T.; Genere, N.; Hamidi, O.; Jasim, S.; Newman, C.B.; Prokop, L.J.; Hassan Murad, M.; Alahdab, F. Treatment of Thyroid Dysfunction and Serum Lipids: A Systematic Review and Meta-analysis. J. Clin. Endocrinol. Metab. 2020, 105, 3683–3694. [Google Scholar] [CrossRef]
- Angelin, B.; Kristensen, J.D.; Eriksson, M.; Carlsson, B.; Klein, I.; Olsson, A.G.; Chester Ridgway, E.; Ladenson, P.W. Reductions in serum levels of LDL cholesterol, apolipoprotein B, triglycerides and lipoprotein(a) in hypercholesterolaemic patients treated with the liver-selective thyroid hormone receptor agonist eprotirome. J. Intern. Med. 2015, 277, 331–342. [Google Scholar] [CrossRef]
- Harrison, S.A.; Bashir, M.R.; Guy, C.D.; Zhou, R.; Moylan, C.A.; Frias, J.P.; Alkhouri, N.; Bansal, M.B.; Baum, S.; Neuschwander-Tetri, B.A.; et al. Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2019, 394, 2012–2024. [Google Scholar] [CrossRef]
- Zhao, M.; Xie, H.; Shan, H.; Zheng, Z.; Li, G.; Li, M.; Hong, L. Development of Thyroid Hormones and Synthetic Thyromimetics in Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 1102. [Google Scholar] [CrossRef]
- Eden, S.; Wiklund, O.; Oscarsson, J.; Rosen, T.; Bengtsson, B.A. Growth hormone treatment of growth hormone-deficient adults results in a marked increase in Lp(a) and HDL cholesterol concentrations. Arterioscler. Thromb. J. Vasc. Biol. 1993, 13, 296–301. [Google Scholar] [CrossRef]
- Glynn, N.; Halsall, D.J.; Boran, G.; Cook, P.; McDermott, J.H.; Smith, D.; Tormey, W.; Thompson, C.J.; O’Gorman, D.; McKenna, M.J.; et al. Growth hormone replacement may influence the biological action of thyroid hormone on liver and bone tissue. Growth Horm. IGF Res. 2021, 57–58, 101393. [Google Scholar] [CrossRef]
- Chasman, D.I.; Shiffman, D.; Zee, R.Y.L.; Louie, J.Z.; Luke, M.M.; Rowland, C.M.; Catanese, J.J.; Buring, J.E.; Devlin, J.J.; Ridker, P.M. Polymorphism in the apolipoprotein(a) gene, plasma lipoprotein(a), cardiovascular disease, and low-dose aspirin therapy. Atherosclerosis 2009, 203, 371–376. [Google Scholar] [CrossRef]
- Lacaze, P.; Bakshi, A.; Riaz, M.; Polekhina, G.; Owen, A.; Bhatia, H.S.; Natarajan, P.; Wolfe, R.; Beilin, L.; Nicholls, S.J.; et al. Aspirin for Primary Prevention of Cardiovascular Events in Relation to Lipoprotein(a) Genotypes. J. Am. Coll. Cardiol. 2022, 80, 1287–1298. [Google Scholar] [CrossRef]
- Schultz, O.; Oberhauser, F.; Saech, J.; Rubbert-Roth, A.; Hahn, M.; Krone, W.; Laudes, M. Effects of Inhibition of Interleukin-6 Signalling on Insulin Sensitivity and Lipoprotein (A) Levels in Human Subjects with Rheumatoid Diseases. PLoS ONE 2010, 5, e14328. [Google Scholar] [CrossRef]
- Wade, D.P.; Clarke, J.G.; Lindahl, G.E.; Liu, A.C.; Zysow, B.R.; Meer, K.; Schwartz, K.; Lawn, R.M. 5′ control regions of the apolipoprotein(a) gene and members of the related plasminogen gene family. Proc. Natl. Acad. Sci. USA 1993, 90, 1369. [Google Scholar] [CrossRef]
- Enkhmaa, B.; Anuurad, E.; Zhang, W.; Li, C.S.; Kaplan, R.; Lazar, J.; Merenstein, D.; Karim, R.; Aouizerat, B.; Cohen, M.; et al. Effect of antiretroviral therapy on allele-associated Lp(a) level in women with HIV in the Women’s Interagency HIV Study. J. Lipid Res. 2018, 59, 1967. [Google Scholar] [CrossRef]
- Santos, H.O.; Kones, R.; Rumana, U.; Earnest, C.P.; Izidoro, L.F.M.; Macedo, R.C.O. Lipoprotein(a): Current Evidence for a Physiologic Role and the Effects of Nutraceutical Strategies. Clin. Ther. 2019, 41, 1780–1797. [Google Scholar] [CrossRef]
- Enkhmaa, B.; Petersen, K.S.; Kris-Etherton, P.M.; Berglund, L. Diet and Lp(a): Does Dietary Change Modify Residual Cardiovascular Risk Conferred by Lp(a)? Nutrients 2020, 12, 2024. [Google Scholar] [CrossRef]
- Clevidence, B.A.; Judd, J.T.; Schaefer, E.J.; Jenner, J.L.; Lichtenstein, A.H.; Muesing, R.A.; Wittes, J.; Sunkin, M.E. Plasma Lipoprotein (a) Levels in Men and Women Consuming Diets Enriched in Saturated, Cis-, or Trans-Monounsaturated Fatty Acids. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1657–1661. [Google Scholar] [CrossRef] [PubMed]
- Silaste, M.L.; Rantala, M.; Alfthan, G.; Aro, A.; Witztum, J.L.; Kesäniemi, Y.A.; Hörkkö, S. Changes in Dietary Fat Intake Alter Plasma Levels of Oxidized, Low-Density Lipoprotein and Lipoprotein(a). Arterioscler. Thromb. Vasc. Biol. 2004, 24, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Ebbeling, C.B.; Knapp, A.; Johnson, A.; Wong, J.M.W.; Greco, K.F.; Ma, C.; Mora, S.; Ludwig, D.S. Effects of a low-carbohydrate diet on insulin-resistant dyslipoproteinemia—Arandomized controlled feeding trial. Am. J. Clin. Nutr. 2022, 115, 154. [Google Scholar] [CrossRef] [PubMed]
- Florentin, M.; Elisaf, M.S.; Rizos, C.V.; Nikolaou, V.; Bilianou, E.; Pitsavos, C.; Liberopoulos, E.N. l-Carnitine/Simvastatin Reduces Lipoprotein (a) Levels Compared with Simvastatin Monotherapy: A Randomized Double-Blind Placebo-Controlled Study. Lipids 2017, 52, 1–9. [Google Scholar] [CrossRef]
- Jenner, J.L.; Jacques, P.F.; Seman, L.J.; Schaefer, E.J. Ascorbic acid supplementation does not lower plasma lipoprotein(a) concentrations. Atherosclerosis 2000, 151, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Bostom, A.G.; Hume, A.L.; Eaton, C.B.; Laurino, J.P.; Yanek, L.R.; Regan, M.S.; McQuade, W.H.; Craig, W.Y.; Perrone, G.; Jacques, P.F. The Effect of High-Dose Ascorbate Supplementation on Plasma Lipoprotein(a) Levels in Patients with Premature Coronary Heart Disease. Pharmacother. J. Hum. Pharmacol. Drug Ther. 1995, 15, 458–464. [Google Scholar]
- Austin, A.; Warty, V.; Janosky, J.; Arslanian, S. The Relationship of Physical Fitness to Lipid and Lipoprotein(a) Levels in Adolescents with IDDm. Diabetes Care 1993, 16, 421–425. [Google Scholar] [CrossRef]
- Theodorou, A.A.; Panayiotou, G.; Volaklis, K.A.; Douda, H.T.; Paschalis, V.; Nikolaidis, M.G.; Smilios, I.; Toubekis, A.; Kyprianou, D.; Papadopoulos, I.; et al. Aerobic, resistance and combined training and detraining on body composition, muscle strength, lipid profile and inflammation in coronary artery disease patients. Res. Sport Med. 2016, 24, 171–184. [Google Scholar] [CrossRef]
- Ho, J.H.; Adam, S.; Liu, Y.; Azmi, S.; Dhage, S.; Syed, A.A.; Ammori, B.J.; Donn, R.; Heald, A.; Gibson, M.J.; et al. Effect of bariatric surgery on plasma levels of oxidised phospholipids, biomarkers of oxidised LDL and lipoprotein(a). J. Clin. Lipidol. 2021, 15, 320–331. [Google Scholar] [CrossRef]
- Durlach, V.; Bonnefont-Rousselot, D.; Boccara, F.; Varret, M.; Di-Filippo Charcosset, M.; Cariou, B.; Valero, R.; Charriere, S.; Farnier, M.; Morange, P.E.; et al. Lipoprotein(a): Pathophysiology, measurement, indication and treatment in cardiovascular disease. A consensus statement from the Nouvelle Société Francophone d’Athérosclérose (NSFA). Arch. Cardiovasc. Dis. 2021, 114, 828–847. [Google Scholar] [CrossRef]
- Burgess, S.; Ference, B.A.; Staley, J.R.; Freitag, D.F.; Mason, A.M.; Nielsen, S.F.; Willeit, P.; Young, R.; Surendran, P.; Karthikeyan, S.; et al. Association of LPA Variants with Risk of Coronary Disease and the Implications for Lipoprotein(a)-Lowering Therapies: A Mendelian Randomization Analysis. JAMA Cardiol. 2018, 3, 619–627. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).