
NF-κB in Cell Deaths, Therapeutic Resistance and Nanotherapy of Tumors: Recent Advances

Abstract
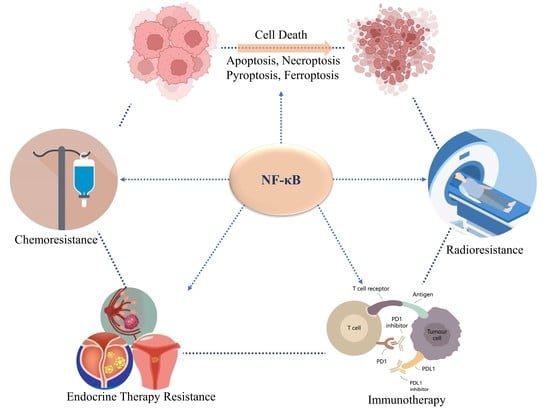
1. Introduction
2. NF-κB Family Members and NF-κB Signaling Pathways
3. NF-κB Function in Tumor Cell Death
3.1. NF-κB Interacts with Apoptosis and Necroptosis
3.2. NF-κB and Pyroptosis
3.3. NF-κB Regulates Ferroptosis
4. Association of NF-κB with Therapeutic Resistance
4.1. NF-κB Induces Chemotherapy Resistance
4.2. NF-κB Promotes Radiotherapy Resistance
4.3. NF-κB and Endocrine Therapy Resistance
4.4. NF-κB Functions in Immunotherapy
5. NF-κB-Based NDS
6. Conclusions and Future Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vasan, N.; Baselga, J.; Hyman, D.M. A View on Drug Resistance in Cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.-X.; Zhou, P.-K. DNA Damage Response Signaling Pathways and Targets for Radiotherapy Sensitization in Cancer. Signal. Transduct. Target. Ther. 2020, 5, 60. [Google Scholar] [CrossRef] [PubMed]
- Vesely, M.D.; Zhang, T.; Chen, L. Resistance Mechanisms to Anti-PD Cancer Immunotherapy. Annu. Rev. Immunol. 2022, 40, 45–74. [Google Scholar] [CrossRef] [PubMed]
- Hanker, A.B.; Sudhan, D.R.; Arteaga, C.L. Overcoming Endocrine Resistance in Breast Cancer. Cancer Cell 2020, 37, 496–513. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, B.A.; El-Deiry, W.S. Targeting Apoptosis in Cancer Therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef]
- Bedoui, S.; Herold, M.J.; Strasser, A. Emerging Connectivity of Programmed Cell Death Pathways and Its Physiological Implications. Nat. Rev. Mol. Cell Biol. 2020, 21, 678–695. [Google Scholar] [CrossRef]
- Peng, F.; Liao, M.; Qin, R.; Zhu, S.; Peng, C.; Fu, L.; Chen, Y.; Han, B. Regulated Cell Death (RCD) in Cancer: Key Pathways and Targeted Therapies. Signal. Transduct. Target. Ther. 2022, 7, 286. [Google Scholar] [CrossRef]
- Gao, W.; Wang, X.; Zhou, Y.; Wang, X.; Yu, Y. Autophagy, Ferroptosis, Pyroptosis, and Necroptosis in Tumor Immunotherapy. Signal. Transduct. Target. Ther. 2022, 7, 196. [Google Scholar] [CrossRef] [PubMed]
- Christgen, S.; Tweedell, R.E.; Kanneganti, T.-D. Programming Inflammatory Cell Death for Therapy. Pharm. Ther. 2022, 232, 108010. [Google Scholar] [CrossRef]
- Tong, X.; Tang, R.; Xiao, M.; Xu, J.; Wang, W.; Zhang, B.; Liu, J.; Yu, X.; Shi, S. Targeting Cell Death Pathways for Cancer Therapy: Recent Developments in Necroptosis, Pyroptosis, Ferroptosis, and Cuproptosis Research. J. Hematol. Oncol. 2022, 15, 174. [Google Scholar] [CrossRef]
- Lei, G.; Zhuang, L.; Gan, B. Targeting Ferroptosis as a Vulnerability in Cancer. Nat. Rev. Cancer 2022, 22, 381–396. [Google Scholar] [CrossRef]
- Gong, Y.; Fan, Z.; Luo, G.; Yang, C.; Huang, Q.; Fan, K.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. The Role of Necroptosis in Cancer Biology and Therapy. Mol. Cancer 2019, 18, 100. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.-K.; Li, C.-Y.; Lin, I.-L.; Syue, W.-J.; Chen, Y.-F.; Cheng, K.-C.; Teng, Y.-N.; Lin, Y.-H.; Yen, C.-H.; Chiu, C.-C. Inflammation-Related Pyroptosis, a Novel Programmed Cell Death Pathway, and Its Crosstalk with Immune Therapy in Cancer Treatment. Theranostics 2021, 11, 8813–8835. [Google Scholar] [CrossRef]
- Tang, R.; Xu, J.; Zhang, B.; Liu, J.; Liang, C.; Hua, J.; Meng, Q.; Yu, X.; Shi, S. Ferroptosis, Necroptosis, and Pyroptosis in Anticancer Immunity. J. Hematol. Oncol. 2020, 13, 110. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-ΚB, Inflammation, Immunity and Cancer: Coming of Age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Zinatizadeh, M.R.; Schock, B.; Chalbatani, G.M.; Zarandi, P.K.; Jalali, S.A.; Miri, S.R. The Nuclear Factor Kappa B (NF-κB) Signaling in Cancer Development and Immune Diseases. Genes. Dis. 2021, 8, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Greten, F.R. NF-κB: Linking Inflammation and Immunity to Cancer Development and Progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-ΚB Pathway for the Therapy of Diseases: Mechanism and Clinical Study. Signal. Transduct. Target. Ther. 2020, 5, 209. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-ΚB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, B.; Schmid, J.A. The Complexity of NF-ΚB Signaling in Inflammation and Cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef]
- Capece, D.; Verzella, D.; Flati, I.; Arboretto, P.; Cornice, J.; Franzoso, G. NF-ΚB: Blending Metabolism, Immunity, and Inflammation. Trends Immunol. 2022, 43, 757–775. [Google Scholar] [CrossRef]
- DiDonato, J.A.; Mercurio, F.; Karin, M. NF-ΚB and the Link between Inflammation and Cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef]
- Xin, Y.; Huang, M.; Guo, W.W.; Huang, Q.; Zhang, L.Z.; Jiang, G. Nano-Based Delivery of RNAi in Cancer Therapy. Mol. Cancer 2017, 16, 134. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zou, M.; Xu, Y.; Lin, P.; Lei, C.; Xia, X. Nano Drug Delivery System for Tumor Immunotherapy: Next-Generation Therapeutics. Front. Oncol. 2022, 12, 864301. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhao, J.; Jiang, H.; Wang, X. Tumor-Targeted Nano-Delivery System of Therapeutic RNA. Mater. Horiz. 2022, 9, 1111–1140. [Google Scholar] [CrossRef] [PubMed]
- Afshari, A.R.; Sanati, M.; Mollazadeh, H.; Kesharwani, P.; Johnston, T.P.; Sahebkar, A. Nanoparticle-Based Drug Delivery Systems in Cancer: A Focus on Inflammatory Pathways. Semin. Cancer Biol. 2022, 86, 860–872. [Google Scholar] [CrossRef] [PubMed]
- Christian, F.; Smith, E.L.; Carmody, R.J. The Regulation of NF-ΚB Subunits by Phosphorylation. Cells 2016, 5, 12. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Signaling to NF-κB. Genes. Dev. 2004, 18, 2195–2224. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NF-κB Family of Transcription Factors and Its Regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Karin, M.; Ben-Neriah, Y. Phosphorylation Meets Ubiquitination: The Control of NF-[Kappa]B Activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef] [PubMed]
- Baud, V.; Karin, M. Is NF-κB a Good Target for Cancer Therapy? Hopes and Pitfalls. Nat. Rev. Drug. Discov. 2009, 8, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFκB System. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef]
- Chen, Z.J.; Parent, L.; Maniatis, T. Site-Specific Phosphorylation of IkappaBalpha by a Novel Ubiquitination-Dependent Protein Kinase Activity. Cell 1996, 84, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Nuclear Factor-Kappa B. Int. J. Biochem. Cell Biol. 1997, 29, 867–870. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-C. Non-Canonical NF-ΚB Signaling Pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Sundaram, C.; Reuter, S.; Aggarwal, B.B. Inhibiting NF-ΚB Activation by Small Molecules as a Therapeutic Strategy. Biochim. Biophys. Acta 2010, 1799, 775–787. [Google Scholar] [CrossRef]
- Chen, J.; Chen, Z.J. Regulation of NF-ΚB by Ubiquitination. Curr. Opin. Immunol. 2013, 25, 4–12. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Shared Principles in NF-κB Signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular Mechanisms of Necroptosis: An Ordered Cellular Explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef]
- Kaczmarek, A.; Vandenabeele, P.; Krysko, D.V. Necroptosis: The Release of Damage-Associated Molecular Patterns and Its Physiological Relevance. Immunity 2013, 38, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Tschopp, J. Induction of TNF Receptor I-Mediated Apoptosis via Two Sequential Signaling Complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef]
- Ju, E.; Park, K.A.; Shen, H.-M.; Hur, G.M. The Resurrection of RIP Kinase 1 as an Early Cell Death Checkpoint Regulator-a Potential Target for Therapy in the Necroptosis Era. Exp. Mol. Med. 2022, 54, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Thapa, R.J.; Basagoudanavar, S.H.; Nogusa, S.; Irrinki, K.; Mallilankaraman, K.; Slifker, M.J.; Beg, A.A.; Madesh, M.; Balachandran, S. NF-κB Protects Cells from Gamma Interferon-Induced RIP1-Dependent Necroptosis. Mol. Cell Biol. 2011, 31, 2934–2946. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, L.; Kim, M.K.; Noonan, A.M.; Sagher, E.; Kohlhammer, H.; Wright, G.; Lyle, L.T.; Steeg, P.S.; Anver, M.; Bowtell, D.D.; et al. A Dual Role for Caspase8 and NF-ΚB Interactions in Regulating Apoptosis and Necroptosis of Ovarian Cancer, with Correlation to Patient Survival. Cell Death Discov. 2015, 1, 15053. [Google Scholar] [CrossRef] [PubMed]
- Bhosale, P.B.; Abusaliya, A.; Kim, H.H.; Ha, S.E.; Park, M.Y.; Jeong, S.H.; Vetrivel, P.; Heo, J.D.; Kim, J.-A.; Won, C.k.; et al. Apigetrin Promotes TNFα-Induced Apoptosis, Necroptosis, G2/M Phase Cell Cycle Arrest, and ROS Generation through Inhibition of NF-ΚB Pathway in Hep3B Liver Cancer Cells. Cells 2022, 11, 2734. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; Li, S.; Wang, J.; Zhao, L.; Yan, Y.; Wu, T.; Zhang, J.; Wang, C. C-Phycocyanin Suppresses the In Vitro Proliferation and Migration of Non-Small-Cell Lung Cancer Cells through Reduction of RIPK1/NF-ΚB Activity. Mar. Drugs 2019, 17, 362. [Google Scholar] [CrossRef] [PubMed]
- Beg, A.A.; Baltimore, D. An Essential Role for NF-κB in Preventing TNF-Alpha-Induced Cell Death. Science 1996, 274, 782–784. [Google Scholar] [CrossRef]
- Fritsch, M.; Günther, S.D.; Schwarzer, R.; Albert, M.-C.; Schorn, F.; Werthenbach, J.P.; Schiffmann, L.M.; Stair, N.; Stocks, H.; Seeger, J.M.; et al. Caspase-8 Is the Molecular Switch for Apoptosis, Necroptosis and Pyroptosis. Nature 2019, 575, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Bist, P.; Leow, S.C.; Phua, Q.H.; Shu, S.; Zhuang, Q.; Loh, W.T.; Nguyen, T.H.; Zhou, J.B.; Hooi, S.C.; Lim, L.H.K. Annexin-1 Interacts with NEMO and RIP1 to Constitutively Activate IKK Complex and NF-ΚB: Implication in Breast Cancer Metastasis. Oncogene 2011, 30, 3174–3185. [Google Scholar] [CrossRef]
- Ea, C.-K.; Deng, L.; Xia, Z.-P.; Pineda, G.; Chen, Z.J. Activation of IKK by TNFalpha Requires Site-Specific Ubiquitination of RIP1 and Polyubiquitin Binding by NEMO. Mol. Cell 2006, 22, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Declercq, W.; Vanden Berghe, T.; Vandenabeele, P. RIP Kinases at the Crossroads of Cell Death and Survival. Cell 2009, 138, 229–232. [Google Scholar] [CrossRef]
- Schneider, A.T.; Gautheron, J.; Feoktistova, M.; Roderburg, C.; Loosen, S.H.; Roy, S.; Benz, F.; Schemmer, P.; Büchler, M.W.; Nachbur, U.; et al. RIPK1 Suppresses a TRAF2-Dependent Pathway to Liver Cancer. Cancer Cell 2017, 31, 94–109. [Google Scholar] [CrossRef]
- Kelliher, M.A.; Grimm, S.; Ishida, Y.; Kuo, F.; Stanger, B.Z.; Leder, P. The Death Domain Kinase RIP Mediates the TNF-Induced NF-κB Signal. Immunity 1998, 8, 297–303. [Google Scholar] [CrossRef]
- Festjens, N.; Vanden Berghe, T.; Cornelis, S.; Vandenabeele, P. RIP1, a Kinase on the Crossroads of a Cell’s Decision to Live or Die. Cell Death Differ. 2007, 14, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Liang, W.; Ma, Z.; Xu, D.; Cao, S.; Lu, X.; Liu, N.; Shan, B.; Qian, L.; Yuan, J. Necroptosis Promotes Cell-Autonomous Activation of Proinflammatory Cytokine Gene Expression. Cell Death Dis. 2018, 9, 500. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-H.; Kim, H.-R.; Park, S.-Y.; Hwang, S.-M.; Hong, S.M.; Park, S.; Kang, H.C.; Morgan, M.J.; Cha, J.-H.; Lee, D.; et al. RIPK3 Activation Induces TRIM28 Derepression in Cancer Cells and Enhances the Anti-Tumor Microenvironment. Mol. Cancer 2021, 20, 107. [Google Scholar] [CrossRef]
- Oliver Metzig, M.; Fuchs, D.; Tagscherer, K.E.; Gröne, H.-J.; Schirmacher, P.; Roth, W. Inhibition of Caspases Primes Colon Cancer Cells for 5-Fluorouracil-Induced TNF-α-Dependent Necroptosis Driven by RIP1 Kinase and NF-ΚB. Oncogene 2016, 35, 3399–3409. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, Pyroptosis and Apoptosis: An Intricate Game of Cell Death. Cell Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef] [PubMed]
- Kesavardhana, S.; Malireddi, R.K.S.; Kanneganti, T.-D. Caspases in Cell Death, Inflammation, and Pyroptosis. Annu. Rev. Immunol. 2020, 38, 567–595. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and Diseases. Signal. Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef]
- Xia, X.; Wang, X.; Cheng, Z.; Qin, W.; Lei, L.; Jiang, J.; Hu, J. The Role of Pyroptosis in Cancer: Pro-Cancer or pro-“host”? Cell Death Dis. 2019, 10, 650. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kang, Y.; Li, Y.; Sun, L.; Zhang, J.; Qian, S.; Luo, K.; Jiang, Y.; Sun, L.; Xu, F. Gasdermin D in Different Subcellular Locations Predicts Diverse Progression, Immune Microenvironment and Prognosis in Colorectal Cancer. J. Inflamm. Res. 2021, 14, 6223–6235. [Google Scholar] [CrossRef]
- Tan, Y.; Sun, R.; Liu, L.; Yang, D.; Xiang, Q.; Li, L.; Tang, J.; Qiu, Z.; Peng, W.; Wang, Y.; et al. Tumor Suppressor DRD2 Facilitates M1 Macrophages and Restricts NF-ΚB Signaling to Trigger Pyroptosis in Breast Cancer. Theranostics 2021, 11, 5214–5231. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.; Guo, J.; Yang, C. Tanshinone II A Enhances Pyroptosis and Represses Cell Proliferation of HeLa Cells by Regulating MiR-145/GSDMD Signaling Pathway. Biosci. Rep. 2020, 40, BSR20200259. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Chen, L.; Dong, Z.; Zhao, Y.; Deng, H.; Wu, J.; Wu, X.; Li, W. Piperlongumine Analogue L50377 Induces Pyroptosis via ROS Mediated NF-ΚB Suppression in Non-Small-Cell Lung Cancer. Chem. -Biol. Interact. 2019, 313, 108820. [Google Scholar] [CrossRef]
- Teng, J.F.; Mei, Q.B.; Zhou, X.G.; Tang, Y.; Xiong, R.; Qiu, W.Q.; Pan, R.; Law, B.Y.; Wong, V.K.; Yu, C.L.; et al. Polyphyllin VI Induces Caspase-1-Mediated Pyroptosis via the Induction of ROS/NF-ΚB/NLRP3/GSDMD Signal Axis in Non-Small Cell Lung Cancer. Cancers 2020, 12, 193. [Google Scholar] [CrossRef]
- Zheng, Z.; Bian, Y.; Zhang, Y.; Ren, G.; Li, G. Metformin Activates AMPK/SIRT1/NF-ΚB Pathway and Induces Mitochondrial Dysfunction to Drive Caspase3/GSDME-Mediated Cancer Cell Pyroptosis. Cell Cycle 2020, 19, 1089–1104. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Gan, L.; Xu, Y.; Luo, D.; Ren, Q.; Wu, S.; Sun, C. Melatonin Alleviates Inflammasome-Induced Pyroptosis through Inhibiting NF-ΚB/GSDMD Signal in Mice Adipose Tissue. J. Pineal Res. 2017, 63, e12414. [Google Scholar] [CrossRef]
- Xu, B.; Jiang, M.; Chu, Y.; Wang, W.; Chen, D.; Li, X.; Zhang, Z.; Zhang, D.; Fan, D.; Nie, Y.; et al. Gasdermin D Plays a Key Role as a Pyroptosis Executor of Non-Alcoholic Steatohepatitis in Humans and Mice. J. Hepatol. 2018, 68, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.; Xu, W.; Bucher, P.; Grimm, M.; Konantz, M.; Horn, H.; Zapukhlyak, M.; Berning, P.; Brändle, M.; Jarboui, M.-A.; et al. Dimethyl Fumarate Induces Ferroptosis and Impairs NF-ΚB/STAT3 Signaling in DLBCL. Blood 2021, 138, 871–884. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Gao, T.; Pang, B.; Su, X.; Guo, C.; Zhang, R.; Pang, Q. RNA Binding Protein NKAP Protects Glioblastoma Cells from Ferroptosis by Promoting SLC7A11 MRNA Splicing in an M6A-Dependent Manner. Cell Death Dis. 2022, 13, 73. [Google Scholar] [CrossRef]
- Kleinschmidt, E.G.; Miller, N.L.G.; Ozmadenci, D.; Tancioni, I.; Osterman, C.D.; Barrie, A.M.; Taylor, K.N.; Ye, A.; Jiang, S.; Connolly, D.C.; et al. Rgnef Promotes Ovarian Tumor Progression and Confers Protection from Oxidative Stress. Oncogene 2019, 38, 6323–6337. [Google Scholar] [CrossRef] [PubMed]
- Yao, F.; Deng, Y.; Zhao, Y.; Mei, Y.; Zhang, Y.; Liu, X.; Martinez, C.; Su, X.; Rosato, R.R.; Teng, H.; et al. A Targetable LIFR−NF-ΚB−LCN2 Axis Controls Liver Tumorigenesis and Vulnerability to Ferroptosis. Nat. Commun. 2021, 12, 7333. [Google Scholar] [CrossRef] [PubMed]
- Jomen, W.; Ohtake, T.; Akita, T.; Suto, D.; Yagi, H.; Osawa, Y.; Kohgo, Y. Iron Chelator Deferasirox Inhibits NF-ΚB Activity in Hepatoma Cells and Changes Sorafenib-Induced Programmed Cell Deaths. Biomed. Pharmacother. 2022, 153, 113363. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, L.; Jiang, S.; Yang, T.; Lan, J.; Lei, Y.; Tan, H.; Pan, K. HMGB1 Mediates Lipopolysaccharide-Induced Inflammation via Interacting with GPX4 in Colon Cancer Cells. Cancer Cell Int. 2020, 20, 205. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.-C.; Wu, B.; Zhang, J.-J.; Zhang, W. Lupeol Triggers Oxidative Stress, Ferroptosis, Apoptosis and Restrains Inflammation in Nasopharyngeal Carcinoma via AMPK/NF-ΚB Pathway. Immunopharmacol. Immunotoxicol. 2022, 44, 621–631. [Google Scholar] [CrossRef]
- Li, S.; He, Y.; Chen, K.; Sun, J.; Zhang, L.; He, Y.; Yu, H.; Li, Q. RSL3 Drives Ferroptosis through NF-ΚB Pathway Activation and GPX4 Depletion in Glioblastoma. Oxid. Med. Cell Longev. 2021, 2021, 2915019. [Google Scholar] [CrossRef] [PubMed]
- Nunes, T.; Hamdan, D.; Leboeuf, C.; El Bouchtaoui, M.; Gapihan, G.; Nguyen, T.T.; Meles, S.; Angeli, E.; Ratajczak, P.; Lu, H.; et al. Targeting Cancer Stem Cells to Overcome Chemoresistance. Int. J. Mol. Sci. 2018, 19, 4036. [Google Scholar] [CrossRef]
- Chen, F.; Qin, X.; Xu, G.; Gou, S.; Jin, X. Reversal of Cisplatin Resistance in Human Gastric Cancer Cells by a Wogonin-Conjugated Pt(IV) Prodrug via Attenuating Casein Kinase 2-Mediated Nuclear Factor-ΚB Pathways. Biochem. Pharm. 2017, 135, 50–68. [Google Scholar] [CrossRef]
- Cao, L.; Zhu, S.; Lu, H.; Soutto, M.; Bhat, N.; Chen, Z.; Peng, D.; Lin, J.; Lu, J.; Li, P.; et al. Helicobacter Pylori-Induced RASAL2 Through Activation of Nuclear Factor-ΚB Promotes Gastric Tumorigenesis via β-Catenin Signaling Axis. Gastroenterology 2022, 162, 1716–1731. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Sun, X.; Feng, Y.; Wang, Y.; Zhang, L.; Wang, Y.; Fang, Z.; Azami, N.L.B.; Sun, M.; Li, Q. Dihydromyricetin Reverses MRP2-Induced Multidrug Resistance by Preventing NF-ΚB-Nrf2 Signaling in Colorectal Cancer Cell. Phytomedicine 2021, 82, 153414. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yin, H.; Zhang, H.; Hu, J.; Lu, H.; Li, C.; Cao, M.; Yan, S.; Cai, L. NF-ΚB-Driven Improvement of EHD1 Contributes to Erlotinib Resistance in EGFR-Mutant Lung Cancers. Cell Death Dis. 2018, 9, 418. [Google Scholar] [CrossRef]
- Jiang, X.; Xu, Y.; Yuan, L.; Zhang, L.; Huang, M.; Ye, Z.; Su, M.; Chen, X.; Zhu, H.; Ye, R.D.; et al. TGFβ2-Mediated Epithelial–Mesenchymal Transition and NF-ΚB Pathway Activation Contribute to Osimertinib Resistance. Acta Pharm. Sin. 2021, 42, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Shostak, K.; Chariot, A. EGFR and NF-ΚB: Partners in Cancer. Trends Mol. Med. 2015, 21, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Liang, C.; Hua, J.; Zhang, B.; Liu, J.; Zhang, Y.; Wei, M.; Yu, X.; Xu, J.; Shi, S. A MiR-146a-5p/TRAF6/NF-κB P65 Axis Regulates Pancreatic Cancer Chemoresistance: Functional Validation and Clinical Significance. Theranostics 2020, 10, 3967–3979. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Fu, Z.; Cao, J.; Liu, Y.; Wu, J.; Li, Q.; Chen, Y. MicroRNA-132 and MicroRNA-212 Mediate Doxorubicin Resistance by down-Regulating the PTEN-AKT/NF-ΚB Signaling Pathway in Breast Cancer. Biomed. Pharm. 2018, 102, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Han, X.; Lu, C.; Yang, T.; Qiao, P.; Sun, Y.; Yu, Z. Ubiquitination of NF-ΚB P65 by FBXW2 Suppresses Breast Cancer Stemness, Tumorigenesis, and Paclitaxel Resistance. Cell Death Differ. 2022, 29, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Drain, A.P.; Zahir, N.; Northey, J.J.; Zhang, H.; Huang, P.-J.; Maller, O.; Lakins, J.N.; Yu, X.; Leight, J.L.; Alston-Mills, B.P.; et al. Matrix Compliance Permits NF-ΚB Activation to Drive Therapy Resistance in Breast Cancer. J. Exp. Med. 2021, 218, e20191360. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Periyasamy, M.; Singh, A.K.; Gemma, C.; Farzan, R.; Allsopp, R.C.; Shaw, J.A.; Charmsaz, S.; Young, L.S.; Cunnea, P.; Coombes, R.C.; et al. Induction of APOBEC3B Expression by Chemotherapy Drugs Is Mediated by DNA-PK-Directed Activation of NF-ΚB. Oncogene 2021, 40, 1077–1090. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Wu, K.; Long, Z.; Zhou, X.; Zhong, C.; Wang, S.; Lai, H.; Guo, Y.; Lv, D.; Lu, J.; et al. Gut Dysbiosis Promotes Prostate Cancer Progression and Docetaxel Resistance via Activating NF-ΚB-IL6-STAT3 Axis. Microbiome 2022, 10, 94. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-L.; Wang, C.-C.; Lin, Y.-J.; Wu, C.-P.; Hsieh, C.-H. Cycling Hypoxia Induces Chemoresistance through the Activation of Reactive Oxygen Species-Mediated B-Cell Lymphoma Extra-Long Pathway in Glioblastoma Multiforme. J. Transl. Med. 2015, 13, 389. [Google Scholar] [CrossRef] [PubMed]
- Raghava Kurup, R.; Oakes, E.K.; Vadlamani, P.; Nwosu, O.; Danthi, P.; Hundley, H.A. ADAR3 Activates NF-ΚB Signaling and Promotes Glioblastoma Cell Resistance to Temozolomide. Sci. Rep. 2022, 12, 13362. [Google Scholar] [CrossRef]
- Yuan, J.; Zhang, Q.; Wu, S.; Yan, S.; Zhao, R.; Sun, Y.; Tian, X.; Zhou, K. MiRNA-223-3p Modulates Ibrutinib Resistance through Regulation of the CHUK/Nf-Κb Signaling Pathway in Mantle Cell Lymphoma. Exp. Hematol. 2021, 103, 52–59.e2. [Google Scholar] [CrossRef] [PubMed]
- Mark, C.; Warrick, J.; Callander, N.S.; Hematti, P.; Miyamoto, S. A Hyaluronan and Proteoglycan Link Protein 1 Matrikine: Role of Matrix Metalloproteinase 2 in Multiple Myeloma NF-ΚB Activation and Drug Resistance. Mol. Cancer Res. 2022, 20, 1456–1466. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-L.; Tang, C.; Zhang, M.-Y.; Huang, W.-L.; Xu, Y.; Sun, H.-Y.; Yang, F.; Song, L.-L.; Wang, H.; Mu, L.-L.; et al. Blocking ATM-Dependent NF-ΚB Pathway Overcomes Niche Protection and Improves Chemotherapy Response in Acute Lymphoblastic Leukemia. Leukemia 2019, 33, 2365–2378. [Google Scholar] [CrossRef]
- Yi, H.; Liang, L.; Wang, H.; Luo, S.; Hu, L.; Wang, Y.; Shen, X.; Xiao, L.; Zhang, Y.; Peng, H.; et al. Albendazole Inhibits NF-ΚB Signaling Pathway to Overcome Tumor Stemness and Bortezomib Resistance in Multiple Myeloma. Cancer Lett. 2021, 520, 307–320. [Google Scholar] [CrossRef]
- Peng, C.; Ouyang, Y.; Lu, N.; Li, N. The NF-ΚB Signaling Pathway, the Microbiota, and Gastrointestinal Tumorigenesis: Recent Advances. Front. Immunol. 2020, 11, 1387. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, Y.; Gerhard, M.; Gao, J.-J.; Mejias-Luque, R.; Zhang, L.; Vieth, M.; Ma, J.-L.; Bajbouj, M.; Suchanek, S.; et al. Effect of Helicobacter Pylori on Gastrointestinal Microbiota: A Population-Based Study in Linqu, a High-Risk Area of Gastric Cancer. Gut 2020, 69, 1598–1607. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, X.; Mi, Z.; Jiang, X.; Sun, L.; Zheng, B.; Wang, J.; Meng, M.; Zhang, L.; Wang, Z.; et al. STAT3/MiR-135b/NF-ΚB Axis Confers Aggressiveness and Unfavorable Prognosis in Non-Small-Cell Lung Cancer. Cell Death Dis. 2021, 12, 493. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Zhu, S.; Li, X.; Li, Z.; Huang, H.; Gao, W.; Liu, Y.; Zhu, H.; Yu, X. The NF-ΚB/MiR-488/ERBB2 Axis Modulates Pancreatic Cancer Cell Malignancy and Tumor Growth through Cell Cycle Signaling. Cancer Biol. Ther. 2022, 23, 294–309. [Google Scholar] [CrossRef]
- Kara, G.; Calin, G.A.; Ozpolat, B. RNAi-Based Therapeutics and Tumor Targeted Delivery in Cancer. Adv. Drug. Deliv. Rev. 2022, 182, 114113. [Google Scholar] [CrossRef]
- Wang, B.; Mao, J.-H.; Wang, B.-Y.; Wang, L.-X.; Wen, H.-Y.; Xu, L.-J.; Fu, J.-X.; Yang, H. Exosomal MiR-1910-3p Promotes Proliferation, Metastasis, and Autophagy of Breast Cancer Cells by Targeting MTMR3 and Activating the NF-ΚB Signaling Pathway. Cancer Lett. 2020, 489, 87–99. [Google Scholar] [CrossRef]
- Xia, Y.; Shen, S.; Verma, I.M. NF-ΚB, an Active Player in Human Cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.Y.; Wu, C.-Y.; Yu, J. The Role of Gut Microbiota in Cancer Treatment: Friend or Foe? Gut 2020, 69, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Ting, N.L.-N.; Lau, H.C.-H.; Yu, J. Cancer Pharmacomicrobiomics: Targeting Microbiota to Optimise Cancer Therapy Outcomes. Gut 2022, 71, 1412–1425. [Google Scholar] [CrossRef] [PubMed]
- Hou, K.; Wu, Z.-X.; Chen, X.-Y.; Wang, J.-Q.; Zhang, D.; Xiao, C.; Zhu, D.; Koya, J.B.; Wei, L.; Li, J.; et al. Microbiota in Health and Diseases. Signal. Transduct. Target. Ther. 2022, 7, 135. [Google Scholar] [CrossRef]
- Tang, G.; Luo, L.; Zhang, J.; Zhai, D.; Huang, D.; Yin, J.; Zhou, Q.; Zhang, Q.; Zheng, G. LncRNA LINC01057 Promotes Mesenchymal Differentiation by Activating NF-ΚB Signaling in Glioblastoma. Cancer Lett. 2021, 498, 152–164. [Google Scholar] [CrossRef]
- Silberstein, J.; Tuchman, S.; Grant, S.J. What Is Multiple Myeloma? JAMA 2022, 327, 497. [Google Scholar] [CrossRef]
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef]
- Sánchez-Aguilera, A.; Méndez-Ferrer, S. The Hematopoietic Stem-Cell Niche in Health and Leukemia. Cell Mol. Life Sci. 2017, 74, 579–590. [Google Scholar] [CrossRef]
- Boulais, P.E.; Frenette, P.S. Making Sense of Hematopoietic Stem Cell Niches. Blood 2015, 125, 2621–2629. [Google Scholar] [CrossRef]
- Lau, T.-Y.; Kwan, H.-Y. Fucoxanthin Is a Potential Therapeutic Agent for the Treatment of Breast Cancer. Mar. Drugs 2022, 20, 370. [Google Scholar] [CrossRef]
- Narayanan, S.; Cai, C.-Y.; Assaraf, Y.G.; Guo, H.-Q.; Cui, Q.; Wei, L.; Huang, J.-J.; Ashby, C.R.; Chen, Z.-S. Targeting the Ubiquitin-Proteasome Pathway to Overcome Anti-Cancer Drug Resistance. Drug. Resist. Updat. 2020, 48, 100663. [Google Scholar] [CrossRef]
- Liu, X.; Hu, Z.; Qu, J.; Li, J.; Gong, K.; Wang, L.; Jiang, J.; Li, X.; He, R.; Duan, L.; et al. AKR1B10 Confers Resistance to Radiotherapy via FFA/TLR4/NF-ΚB Axis in Nasopharyngeal Carcinoma. Int. J. Biol. Sci. 2021, 17, 756–767. [Google Scholar] [CrossRef]
- Schoetz, U.; Klein, D.; Hess, J.; Shnayien, S.; Spoerl, S.; Orth, M.; Mutlu, S.; Hennel, R.; Sieber, A.; Ganswindt, U.; et al. Early Senescence and Production of Senescence-Associated Cytokines Are Major Determinants of Radioresistance in Head-and-Neck Squamous Cell Carcinoma. Cell Death Dis. 2021, 12, 1162. [Google Scholar] [CrossRef]
- Dai, D.; Zhou, H.; Yin, L.; Ye, F.; Yuan, X.; You, T.; Zhao, X.; Long, W.; Wang, D.; He, X.; et al. PELI1 Promotes Radiotherapy Sensitivity by Inhibiting Noncanonical NF-ΚB in Esophageal Squamous Cancer. Mol. Oncol. 2022, 16, 1384–1401. [Google Scholar] [CrossRef]
- Sulman, E.P.; Ismaila, N.; Armstrong, T.S.; Tsien, C.; Batchelor, T.T.; Cloughesy, T.; Galanis, E.; Gilbert, M.; Gondi, V.; Lovely, M.; et al. Radiation Therapy for Glioblastoma: American Society of Clinical Oncology Clinical Practice Guideline Endorsement of the American Society for Radiation Oncology Guideline. J. Clin. Oncol. 2017, 35, 361–369. [Google Scholar] [CrossRef]
- Gu, J.; Mu, N.; Jia, B.; Guo, Q.; Pan, L.; Zhu, M.; Zhang, W.; Zhang, K.; Li, W.; Li, M.; et al. Targeting Radiation-Tolerant Persister Cells as a Strategy for Inhibiting Radioresistance and Recurrence in Glioblastoma. Neuro-Oncol. 2022, 24, 1056–1070. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, S.; Li, H.-L.; Luo, H.; Wu, X.; Lu, J.; Wang, H.-W.; Chen, Y.; Chen, D.; Wu, W.-T.; et al. FOSL1 Promotes Proneural-to-Mesenchymal Transition of Glioblastoma Stem Cells via UBC9/CYLD/NF-ΚB Axis. Mol. Ther. 2022, 30, 2568–2583. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Xu, J.; Fan, Y.; Zhang, Z.; Wang, H.; Qian, M.; Zhang, P.; Deng, L.; Shen, J.; Xue, H.; et al. ARPC1B Promotes Mesenchymal Phenotype Maintenance and Radiotherapy Resistance by Blocking TRIM21-Mediated Degradation of IFI16 and HuR in Glioma Stem Cells. J. Exp. Clin. Cancer Res. 2022, 41, 323. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Guo, Q.; Luo, Z.; Wang, Y.; Weng, J.; Chen, Y.; Liang, W.; Li, Y.; Zhang, Y.; Chen, K.; et al. TXN Inhibitor Impedes Radioresistance of Colorectal Cancer Cells with Decreased ALDH1L2 Expression via TXN/NF-ΚB Signaling Pathway. Br. J. Cancer 2022, 127, 637–648. [Google Scholar] [CrossRef]
- Khongthong, P.; Roseweir, A.K.; Edwards, J. The NF-κB Pathway and Endocrine Therapy Resistance in Breast Cancer. Endocr. Relat. Cancer 2019, 26, R369–R380. [Google Scholar] [CrossRef]
- Wang, X.; Fang, Y.; Sun, W.; Xu, Z.; Zhang, Y.; Wei, X.; Ding, X.; Xu, Y. Endocrinotherapy Resistance of Prostate and Breast Cancer: Importance of the NF-κB Pathway (Review). Int. J. Oncol. 2020, 56, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wang, T.; Wang, H.; Zhang, L.; Xu, F.; Fang, R.; Li, L.; Cai, X.; Wu, Y.; Zhang, W.; et al. Oncoprotein HBXIP Enhances HOXB13 Acetylation and Co-Activates HOXB13 to Confer Tamoxifen Resistance in Breast Cancer. J. Hematol. Oncol. 2018, 11, 26. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, J.; Li, J.; Zhou, X.; Yin, L.; Wang, Y.; Gu, Y.; Niu, X.; Yang, Y.; Ji, H.; et al. HMGB1 Is a Key Factor for Tamoxifen Resistance and Has the Potential to Predict the Efficacy of CDK4/6 Inhibitors in Breast Cancer. Cancer Sci. 2021, 112, 1603–1613. [Google Scholar] [CrossRef]
- Sikora, M.J.; Jacobsen, B.M.; Levine, K.; Chen, J.; Davidson, N.E.; Lee, A.V.; Alexander, C.M.; Oesterreich, S. WNT4 Mediates Estrogen Receptor Signaling and Endocrine Resistance in Invasive Lobular Carcinoma Cell Lines. Breast Cancer Res. 2016, 18, 92. [Google Scholar] [CrossRef]
- Azuma, K.; Ikeda, K.; Suzuki, T.; Aogi, K.; Horie-Inoue, K.; Inoue, S. TRIM47 Activates NF-ΚB Signaling via PKC-ε/PKD3 Stabilization and Contributes to Endocrine Therapy Resistance in Breast Cancer. Proc. Natl. Acad. Sci. 2021, 118, e2100784118. [Google Scholar] [CrossRef]
- Fu, X.; De Angelis, C.; Schiff, R. Interferon Signaling in Estrogen Receptor–Positive Breast Cancer: A Revitalized Topic. Endocrinology 2022, 163, bqab235. [Google Scholar] [CrossRef] [PubMed]
- Smart, E.; Semina, S.E.; Frasor, J. Update on the Role of NFκB in Promoting Aggressive Phenotypes of Estrogen Receptor-Positive Breast Cancer. Endocrinology 2020, 161, bqaa152. [Google Scholar] [CrossRef] [PubMed]
- Kastrati, I.; Joosten, S.E.P.; Semina, S.E.; Alejo, L.H.; Brovkovych, S.D.; Stender, J.D.; Horlings, H.M.; Kok, M.; Alarid, E.T.; Greene, G.L.; et al. The NF-ΚB Pathway Promotes Tamoxifen Tolerance and Disease Recurrence in Estrogen Receptor-Positive Breast Cancers. Mol. Cancer Res. 2020, 18, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Frasor, J.; El-Shennawy, L.; Stender, J.D.; Kastrati, I. NFκB Affects Estrogen Receptor Expression and Activity in Breast Cancer through Multiple Mechanisms. Mol. Cell Endocrinol. 2015, 418 Pt 3, 235–239. [Google Scholar] [CrossRef]
- Kerr, A.J.; Dodwell, D.; McGale, P.; Holt, F.; Duane, F.; Mannu, G.; Darby, S.C.; Taylor, C.W. Adjuvant and Neoadjuvant Breast Cancer Treatments: A Systematic Review of Their Effects on Mortality. Cancer Treat. Rev. 2022, 105, 102375. [Google Scholar] [CrossRef]
- Roseweir, A.K.; Bennett, L.; Dickson, A.; Cheng, K.; Quintayo, M.-A.; Bayani, J.; McMillan, D.C.; Horgan, P.G.; van de Velde, C.J.H.; Seynaeve, C.; et al. Predictive Biomarkers for Endocrine Therapy: Retrospective Study in Tamoxifen and Exemestane Adjuvant Multinational (TEAM) Trial. J. Natl. Cancer Inst. 2018, 110, 616–627. [Google Scholar] [CrossRef]
- Gnant, M.; Fitzal, F.; Rinnerthaler, G.; Steger, G.G.; Greil-Ressler, S.; Balic, M.; Heck, D.; Jakesz, R.; Thaler, J.; Egle, D.; et al. Duration of Adjuvant Aromatase-Inhibitor Therapy in Postmenopausal Breast Cancer. N. Engl. J. Med. 2021, 385, 395–405. [Google Scholar] [CrossRef]
- Thomas-Jardin, S.E.; Dahl, H.; Nawas, A.F.; Bautista, M.; Delk, N.A. NF-ΚB Signaling Promotes Castration-Resistant Prostate Cancer Initiation and Progression. Pharm. 2020, 211, 107538. [Google Scholar] [CrossRef]
- Jung, A.R.; Kim, G.E.; Kim, M.Y.; Ha, U.-S.; Hong, S.-H.; Lee, J.Y.; Kim, S.W.; Park, Y.H. HMGB1 Promotes Tumor Progression and Invasion through HMGB1/TNFR1/NF-ΚB Axis in Castration-Resistant Prostate Cancer. Am. J. Cancer Res. 2021, 11, 2215–2227. [Google Scholar] [CrossRef]
- Morel, K.L.; Hamid, A.A.; Clohessy, J.G.; Pandell, N.; Ellis, L.; Sweeney, C.J. NF-ΚB Blockade with Oral Administration of Dimethylaminoparthenolide (DMAPT), Delays Prostate Cancer Resistance to Androgen Receptor (AR) Inhibition and Inhibits AR Variants. Mol. Cancer Res. 2021, 19, 1137–1145. [Google Scholar] [CrossRef]
- Ma, X.; Zhao, T.; Yan, H.; Guo, K.; Liu, Z.; Wei, L.; Lu, W.; Qiu, C.; Jiang, J. Fatostatin Reverses Progesterone Resistance by Inhibiting the SREBP1-NF-ΚB Pathway in Endometrial Carcinoma. Cell Death Dis. 2021, 12, 544. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. Immune Checkpoint Targeting in Cancer Therapy: Toward Combination Strategies with Curative Potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef]
- Zhou, Y.; Jin, X.; Yu, H.; Qin, G.; Pan, P.; Zhao, J.; Chen, T.; Liang, X.; Sun, Y.; Wang, B.; et al. HDAC5 Modulates PD-L1 Expression and Cancer Immunity via P65 Deacetylation in Pancreatic Cancer. Theranostics 2022, 12, 2080–2094. [Google Scholar] [CrossRef]
- Antonangeli, F.; Natalini, A.; Garassino, M.C.; Sica, A.; Santoni, A.; Di Rosa, F. Regulation of PD-L1 Expression by NF-ΚB in Cancer. Front. Immunol. 2020, 11, 584626. [Google Scholar] [CrossRef]
- Somani, V.K.; Zhang, D.; Dodhiawala, P.B.; Lander, V.E.; Liu, X.; Kang, L.-I.; Chen, H.-P.; Knolhoff, B.L.; Li, L.; Grierson, P.M.; et al. IRAK4 Signaling Drives Resistance to Checkpoint Immunotherapy in Pancreatic Ductal Adenocarcinoma. Gastroenterology 2022, 162, 2047–2062. [Google Scholar] [CrossRef]
- Cai, H.; Yan, L.; Liu, N.; Xu, M.; Cai, H. IFI16 Promotes Cervical Cancer Progression by Upregulating PD-L1 in Immunomicroenvironment through STING-TBK1-NF-κB Pathway. Biomed. Pharm. 2020, 123, 109790. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhu, S.; Du, Y.; Xu, F.; Sun, W.; Xu, Z.; Wang, X.; Qian, P.; Zhang, Q.; Feng, J.; et al. RelB Upregulates PD-L1 and Exacerbates Prostate Cancer Immune Evasion. J. Exp. Clin. Cancer Res. 2022, 41, 66. [Google Scholar] [CrossRef]
- Rong, D.; Sun, G.; Zheng, Z.; Liu, L.; Chen, X.; Wu, F.; Gu, Y.; Dai, Y.; Zhong, W.; Hao, X.; et al. MGP Promotes CD8+ T Cell Exhaustion by Activating the NF-ΚB Pathway Leading to Liver Metastasis of Colorectal Cancer. Int. J. Biol. Sci. 2022, 18, 2345–2361. [Google Scholar] [CrossRef]
- Jin, X.; Ding, D.; Yan, Y.; Li, H.; Wang, B.; Ma, L.; Ye, Z.; Ma, T.; Wu, Q.; Rodrigues, D.N.; et al. Phosphorylated RB Promotes Cancer Immunity by Inhibiting NF-ΚB Activation and PD-L1 Expression. Mol. Cell 2019, 73, 22–35.e6. [Google Scholar] [CrossRef]
- Yu, Z.; Li, Y.; Li, Y.; Zhang, J.; Li, M.; Ji, L.; Tang, Y.; Zheng, Y.; Sheng, J.; Han, Q.; et al. Bufalin Stimulates Antitumor Immune Response by Driving Tumor-Infiltrating Macrophage toward M1 Phenotype in Hepatocellular Carcinoma. J. Immunother. Cancer 2022, 10, e004297. [Google Scholar] [CrossRef] [PubMed]
- Grinberg-Bleyer, Y.; Oh, H.; Desrichard, A.; Bhatt, D.M.; Caron, R.; Chan, T.A.; Schmid, R.M.; Klein, U.; Hayden, M.S.; Ghosh, S. NF-ΚB c-Rel Is Crucial for the Regulatory T Cell Immune Checkpoint in Cancer. Cell 2017, 170, 1096–1108.e13. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Zhao, Q.; Gao, Z.; Lao, X.-M.; Lin, W.-M.; Chen, D.-P.; Mu, M.; Huang, C.-X.; Liu, Z.-Y.; Li, B.; et al. The Local Immune Landscape Determines Tumor PD-L1 Heterogeneity and Sensitivity to Therapy. J. Clin. Investig. 2019, 129, 3347–3360. [Google Scholar] [CrossRef]
- Kumari, M.; Purohit, M.P.; Patnaik, S.; Shukla, Y.; Kumar, P.; Gupta, K.C. Curcumin Loaded Selenium Nanoparticles Synergize the Anticancer Potential of Doxorubicin Contained in Self-Assembled, Cell Receptor Targeted Nanoparticles. Eur. J. Pharm. Biopharm. 2018, 130, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.; Choi, Y.J.; Jeong, M.S.; Park, Y.I.; Motoyama, K.; Kim, M.W.; Kwon, S.-H.; Choi, J.H. Hyaluronic Acid-Decorated Glycol Chitosan Nanoparticles for PH-Sensitive Controlled Release of Doxorubicin and Celecoxib in Nonsmall Cell Lung Cancer. Bioconjugate Chem. 2020, 31, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Saneja, A.; Nayak, D.; Srinivas, M.; Kumar, A.; Khare, V.; Katoch, A.; Goswami, A.; Vishwakarma, R.A.; Sawant, S.D.; Gupta, P.N. Development and Mechanistic Insight into Enhanced Cytotoxic Potential of Hyaluronic Acid Conjugated Nanoparticles in CD44 Overexpressing Cancer Cells. Eur. J. Pharm. Sci. 2017, 97, 79–91. [Google Scholar] [CrossRef]
- Sun, T.; Gao, J.; Han, D.; Shi, H.; Liu, X. Fabrication and Characterization of Solid Lipid Nano-Formulation of Astraxanthin against DMBA-Induced Breast Cancer via Nrf-2-Keap1 and NF-κB and MTOR/Maf-1/PTEN Pathway. Drug. Deliv. 2019, 26, 975–988. [Google Scholar] [CrossRef]
- Kim, K.M.; Jung, J. Upregulation of G Protein-Coupled Estrogen Receptor by Chrysin-Nanoparticles Inhibits Tumor Proliferation and Metastasis in Triple Negative Breast Cancer Xenograft Model. Front. Endocrinol. 2020, 11, 560605. [Google Scholar] [CrossRef]
- Kannappan, V.; Liu, Y.; Wang, Z.; Azar, K.; Kurusamy, S.; Kilari, R.S.; Armesilla, A.L.; Morris, M.R.; Najlah, M.; Liu, P.; et al. PLGA-Nano-Encapsulated Disulfiram Inhibits Hypoxia-Induced NF-ΚB, Cancer Stem Cells, and Targets Glioblastoma In Vitro and In Vivo. Mol. Cancer Ther. 2022, 21, 1273–1284. [Google Scholar] [CrossRef]
- Rauch, D.A.; Harding, J.C.; Ratner, L.; Wickline, S.A.; Pan, H. Targeting NF-ΚB with Nanotherapy in a Mouse Model of Adult T-Cell Leukemia/Lymphoma. Nanomaterials 2021, 11, 1582. [Google Scholar] [CrossRef]
- Ibaraki, H.; Kanazawa, T.; Owada, M.; Iwaya, K.; Takashima, Y.; Seta, Y. Anti-Metastatic Effects on Melanoma via Intravenous Administration of Anti-NF-ΚB SiRNA Complexed with Functional Peptide-Modified Nano-Micelles. Pharmaceutics 2020, 12, 64. [Google Scholar] [CrossRef]
- Gao, X.; Wu, Y.; Qiao, L.; Feng, X. SENP2 Suppresses NF-ΚB Activation and Sensitizes Breast Cancer Cells to Doxorubicin. Eur. J. Pharm. 2019, 854, 179–186. [Google Scholar] [CrossRef]
- Kumari, M.; Ray, L.; Purohit, M.P.; Patnaik, S.; Pant, A.B.; Shukla, Y.; Kumar, P.; Gupta, K.C. Curcumin Loading Potentiates the Chemotherapeutic Efficacy of Selenium Nanoparticles in HCT116 Cells and Ehrlich’s Ascites Carcinoma Bearing Mice. Eur. J. Pharm. Biopharm. 2017, 117, 346–362. [Google Scholar] [CrossRef]
- Lim, H.-K.; Kim, K.M.; Jeong, S.-Y.; Choi, E.K.; Jung, J. Chrysin Increases the Therapeutic Efficacy of Docetaxel and Mitigates Docetaxel-Induced Edema. Integr. Cancer Ther. 2017, 16, 496–504. [Google Scholar] [CrossRef]
- Chen, D.; Cui, Q.C.; Yang, H.; Dou, Q.P. Disulfiram, a Clinically Used Anti-Alcoholism Drug and Copper-Binding Agent, Induces Apoptotic Cell Death in Breast Cancer Cultures and Xenografts via Inhibition of the Proteasome Activity. Cancer Res. 2006, 66, 10425–10433. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agents | The Function of NF-κB (PRO, Promotion; INH, Inhibition) | Regulation Mechanism | Specific Effects on Various Tumor Types | Reference |
|---|---|---|---|---|
| Polyphyllin VI | PRO | activates the NF-κB/NLRP3/caspase 1/GSDMD pathway, and promotes pyroptosis | Anti-NSCLC | [68] |
| Metformin | PRO | activates the AMPK/SIRT1/NF-κB/Bax-cytC/caspase 3/GSDME pathway, and promotes pyroptosis | Anti-HCC, breast cancer, and CRC | [69] |
| Lupeol | PRO | activates the AMPK/NF-κB pathway, decreases GPX4, and triggers ferroptosis | Anti-NPC | [78] |
| RSL3 | PRO | activates NF-κB, decreases GPX4, and induces ferroptosis | Anti-GBM | [79] |
| H. pylori | PRO | activates the NF-κB/RASAL2/AKT/β-catenin pathway | Promotes gastric tumorigenesis, and chemoresistance of platinum and fluorouracil | [82] |
| Osimertinib | PRO | activates the TGFβ2/NF-κB/SMAD2/EMT pathway | Promotes osimertinib resistance in NSCLC | [85] |
| Etoposide and Cisplatin | PRO | activates the DNA-PKcs/ATM/NF-κB/APOBEC3B pathway | Promotes breast cancer progression and therapy resistance | [92] |
| LPS | PRO | activates the NF-κB/IL-6/STAT3 pathway | Promotes prostate cancer progression and docetaxel resistance | [93] |
| Ara-C, DNR, and 6-MP | PRO | activates the ATM/TRAF6/NF-κB/GDF15, CCL3, CCL4 pathway | Promotes drug resistance of ALL | [98] |
| Tanshinone II | INH | activates caspase 3/GSDMD axis and promotes pyroptosis | Anti-cervical cancer | [66] |
| Piperlongumine analogue L50377 | INH | induces ROS-mediated NF-κB inhibition and promotes pyroptosis | Anti-NSCLC | [67] |
| DMF | INH | induces lipid peroxidation and ferroptosis | Anti-DLBCL | [72] |
| DFX | INH | inhibits ferroptosis, DFX + SOR promotes apoptosis | Anti-HCC | [76] |
| CDK4/6 kinase suppressors (Palbociclib, Ribociclib, and Abemaciclib) | INH | inhibits the HMGB1/TLR-4/NF-κB pathway | Reverses TAM resistance in breast cancer | [128] |
| DMAPT | INH | oral NF-κB inhibitor | Reverses resistance to ADT in prostate cancer | [140] |
| Fatostatin | INH | inhibits the SREBP1/NF-κB pathway | Reverses progesterone resistance in endometrial cancer | [141] |
| Agents or Nucleic Acid | NPs Encapsulation | Tumor Types | Reference |
|---|---|---|---|
| Curcumin + selenium + DOX | PSHA | CRC | [154] |
| DOX + CXB | HA-GC | NSCLC | [155] |
| Thiotetrazole | PLGA-PEG-HA | pancreatic cancer | [156] |
| Chrysin | PCL-PEG | breast cancer | [158] |
| Astraxanthin | SLN | breast cancer | [157] |
| Disulfiram | PLGA | GBM | [159] |
| siNF-κB | peptide | ATLL | [160] |
| siNF-κB | polymeric micelle | melanoma | [161] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, X.; Sun, L.; Xu, F. NF-κB in Cell Deaths, Therapeutic Resistance and Nanotherapy of Tumors: Recent Advances. Pharmaceuticals 2023, 16, 783. https://doi.org/10.3390/ph16060783
Wu X, Sun L, Xu F. NF-κB in Cell Deaths, Therapeutic Resistance and Nanotherapy of Tumors: Recent Advances. Pharmaceuticals. 2023; 16(6):783. https://doi.org/10.3390/ph16060783
Chicago/Turabian StyleWu, Xuesong, Liang Sun, and Fangying Xu. 2023. "NF-κB in Cell Deaths, Therapeutic Resistance and Nanotherapy of Tumors: Recent Advances" Pharmaceuticals 16, no. 6: 783. https://doi.org/10.3390/ph16060783
APA StyleWu, X., Sun, L., & Xu, F. (2023). NF-κB in Cell Deaths, Therapeutic Resistance and Nanotherapy of Tumors: Recent Advances. Pharmaceuticals, 16(6), 783. https://doi.org/10.3390/ph16060783