Abstract
Mechanistic target of rapamycin (mTOR) is a protein kinase that regulates cellular growth, development, survival, and metabolism through integration of diverse extracellular and intracellular stimuli. Additionally, mTOR is involved in interplay of signalling pathways that regulate apoptosis and autophagy. In cells, mTOR is assembled into two complexes, mTORC1 and mTORC2. While mTORC1 is regulated by energy consumption, protein intake, mechanical stimuli, and growth factors, mTORC2 is regulated by insulin-like growth factor-1 receptor (IGF-1R), and epidermal growth factor receptor (EGFR). mTOR signalling pathways are considered the hallmark in cancer due to their dysregulation in approximately 70% of cancers. Through downstream regulators, ribosomal protein S6 kinase β-1 (S6K1) and eukaryotic translation initiation factor 4E binding protein 1 (4E-BP1), mTORC1 influences various anabolic and catabolic processes in the cell. In recent years, several mTOR inhibitors have been developed with the aim of treating different cancers. In this review, we will explore the current developments in the mTOR signalling pathway and its importance for being targeted by various inhibitors in anti-cancer therapeutics.
1. Introduction
The mammalian target of rapamycin or mechanistic target of rapamycin (mTOR) is a serine–threonine protein kinase that modulates cellular metabolism, growth, cell proliferation or survival and gene expression phenomena [1]. Structurally, mTOR is a 289-kDa protein that is expressed in almost all the tissues of the body [2]. The phosphorylation of mTOR is triggered at particular residues (S1261, T2446, S2448, S2481) in response to growth factors, mitogens, hormones, and nutrition, whereas a lack of these factors and oxygen can inhibit its activation and enzymatic activity [3]. There are two complexes of mTOR, mTORC1 and mTORC2, which are physically and functionally distinct. While mTORC1 is comprised of mTOR, GβL (positive regulator of mTOR, which increases the kinase activity of mTOR), deptor, and raptor, mTORC2 is comprised of rictor, mTOR, proline rich 5 (PPR5), GβL, deptor and SIN1 (Figure 1). The role of various components of this signalling pathway is summarized briefly in Table 1 [4]. mTOR is linked to many signalling cascades, including phosphoinositide 3-kinase/Protein kinase-B, also known as Akt (PI3K/Akt), LKB1 (Liver kinase B-1/adenosine 5′-monophosphate-activated protein kinase (AMPK), tuberous sclerosis complex subunit 1 TSC1/TSC2/Ras homolog enriched in brain (Rheb), and Vam6/Rag GTPases [5]. It regulates cell proliferation, apoptosis, and autophagy (autophagic cell death) pathways by influencing various key processes like transcription and protein synthesis by integrating diverse signalling stimuli [6].
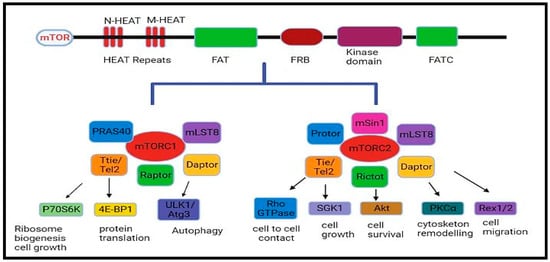
Figure 1.
The mTOR complex, mTORC1, and mTORC2.
mTOR signalling is implicated in various diseases, like cancer, arthritis, insulin resistance, and osteoporosis and in tumour angiogenesis [7]. Several reports support that there is an aberrant activation of the mTOR signalling pathway in cancer [8,9]. Keeping in view the role of this important signalling cascade in crucial stages of cancer development, this review is aimed at highlighting the regulation of mTOR signalling pathway and its involvement in tumourigenesis. Furthermore, the latest findings on mTOR inhibitors in the treatment of various cancers are discussed.

Table 1.
The various components of the mTOR complex, and their functions.
Table 1.
The various components of the mTOR complex, and their functions.
| mTOR Components | Associated with | Mode of Action | Reference |
|---|---|---|---|
| Raptor | mTORC1 | Positive regulator | [10] |
| Rictor | mTORC2 | Positive regulator | [11] |
| Daptor | mTORC1 and mTORC2 | Negative regulator | [12] |
| Ttie/Tel2 | mTORC1 and mTORC2 | Positive regulator | [13] |
| Protor | mTORC2 | Positive regulator | [14] |
| mLST8 | mTORC1 and mTORC2 | Positive regulator | [15] |
| PRAS40 | mTORC1 | Negative regulator | [16] |
| mSin1 | mTORC2 | Positive regulator | [17] |
2. Assembly of mTOR Complex
The mTOR protein is made up of 2549 amino acids and is organized into many domains, like NH2-terminal (N-HEAT), middle HEAT (M-HEAT), FAT domain (also called FKBP12-rapamycin-associated protein, ataxia-telangiectasia and transactivation/transformation), kinase domain with FRB (FKBP12 rapamycin-binding domain site) and a FAT carboxy-terminal domain. [18]. To form a dimer under physiological conditions, the HEAT repeats (12–13) of one mTOR monomer interact with the HEAT repeats (20–23) of another mTOR’s M-HEAT domain as illustrated in Figure 2A,B [19]. Because its C-terminal region shares considerable homology with the catalytic domain of PI3K, mTOR is also considered a member of the phosphatidylinositol-3-kinase superfamily [20]. Earlier, the difference between mTORC1 and mTORC2 was linked to their definite elements and susceptibility to rapamycin that selectively suppresses its activation of the mTORC1/2 complex. Originally it was thought that mTORC1 was rapamycin-sensitive, whereas mTORC2 does not bind to the drug. However, it is now a well-established fact that chronic exposure to rapamycin inhibits both complexes [21]. mTOR, DEP domain-containing mTOR interacting protein (Deptor), Ttie/Tel2 complex, and mammalian lethal with sec-13 protein 8 (mLst8) are the common subunits present in both mTORC1 and mTORC2 [22].
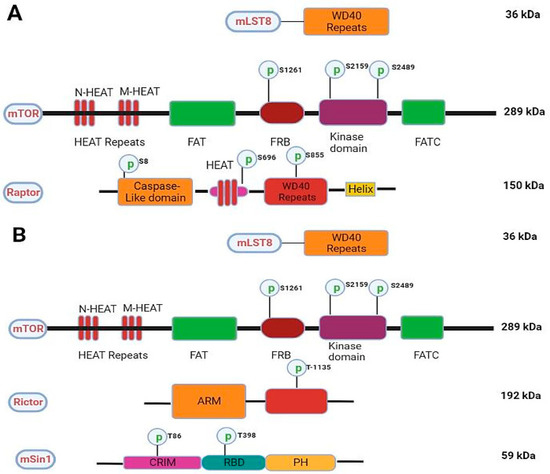
Figure 2.
Schematics of the mTORC1 and mTORC2. (A) showing various domains, phosphorylation sites, and molecular weights in key components of mTORC1. (B) showing various domains, phosphorylation sites, and molecular weights in key components of mTORC2.
3. Regulation of mTORC1
Although mTOR exists in two separate complexes, there exists a cross-talk between mTORC1 and mTORC2. mTORC2 upregulates mTORC1 by activating the IGF-IR-Akt axis [1,23]. mTORC1 inhibits mTORC2 by phosphorylating mSin1 and Rictor on T86/398 and T1135 through its downstream regulator S6K1 via a feedback mechanism [24,25]. The mTORC1 complex is regulated by energy consumption, protein intake, mechanical stimuli, and growth factors (Figure 3). All of these components transmit signals to cells, which are subsequently detected, transformed, and then integrated together, causing cellular functions to alter. Signals are then detected by proteins containing surface receptors and intracellular kinases [26,27]. mTORC1 is mainly regulated by insulin-like growth factor-1 (IGF-1R) and insulin receptor (IR), which are tyrosine kinase receptors whose activation leads to the phosphorylation of insulin receptor substrates [27]. The insulin receptor substrates (IRS) bind and activate Src Homology 2 (SH2) domain-containing phosphatidylinositol-3-kinase, which in turn phosphorylates inositol phospholipids and forms phosphatidylinositol (3,4,5)-triphosphate (PIP3). The PIP3 then interacts with the Pleckstrin homology domain (PH) containing proteins like Akt and 3-phosphoinositide-dependent kinase (PDK1) [28]. The interaction of PIP3 with Akt phosphorylates and activates PDK1. Akt, which is expressed in skeletal muscles, is the main and important upstream regulator of mTORC1. Akt is activated by phosphorylation at serine residue Thr308 by PKD1 [29,30,31]. Active AKT phosphorylates tuberous sclerosis complex-2 (TSC2) leading to its suppression. TSC2 complex is a GTPase-activating protein (GAP) complex, which is comprised of TSC1/2 and TRE2-BUB2-CDC16 domain family member 7 (TBC1D7) [32,33]. Akt activates Ras homolog enriched in the brain, Rheb, a GTP-binding protein, which leads to the activation of mTORC1 [34]. At the lysosomal membrane, GTP-bound Rheb proteins (Rheb-GTP) activate mTORC1 [35]. Very few studies have shown that, apart from insulin and IGF-I modulation, androgens stimulate Akt phosphorylation [36,37,38,39,40,41,42]. Mitogen-activated protein kinase (MEK1/2) phosphorylates mTORC1 by p90 ribosomal S6 kinase and Rs-dependent extracellular signal-regulated kinase (ERK1/2), while the former phosphorylates raptor at S719/722, and later phosphorylates raptor at S696, S863, and S6, thereby up-regulating mTORC1 activity [43,44]. Apart from growth factors, mTORC1 is also regulated by the energy status of the cell. The lack of cellular energy increases the AMP/ATP ratio, which activates (AMPK) AMP-dependent kinase [45]. Activated AMPK phosphorylates two residues of TSC2 viz. Thr1227 and Ser1345, and thus stimulates GAP activity of the complex through inhibition of Rheb, which in turn downregulates or inhibits mTORC1 activity (Figure 3) [46].
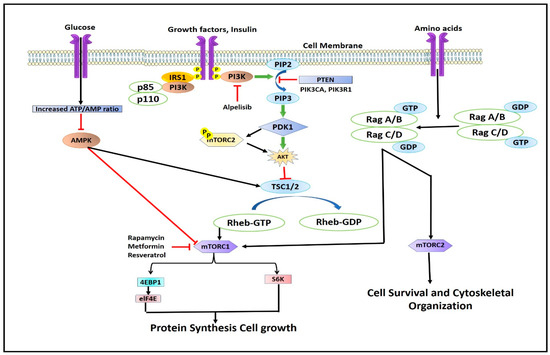
Figure 3.
Illustration describing the regulation of mTOR complex.
4. Downstream Effectors of mTORC1: S6K1 and 4EBP1
mTORC1 phosphorylates its downstream regulator (4EBP1, and S6K1) [47] through the interaction of raptor and TOR signalling (TOS) motif. The TOS motif is a five amino acid sequence located in C-terminus of 4E-BP1 (Phe-Glu-Met-Asp-Asp-I1e) and N-terminus of S6K1 (Phe-Asp-I1e-Leu), which is mandatory for mTORC1 to phosphorylate these proteins in vivo [48,49,50,51]. p70S6K1 is activated by MAPK, PDK1, and SAPK (stress-activated protein kinase). mTORC1 phosphorylates S6K1 at Thr389, which is important for its activation. mTOR inhibitors are known to diminish the Thr389 phosphorylation of the S6K1 [52,53]. Another downstream target of mTORC1 is 4E-BP1, which suppresses protein synthesis by binding to and inhibiting eIF4E (eukaryotic initiation factor 4E) [54,55]. When activated, mTORC1 promotes the dissociation of eIF4E from 4E-BP1, which enables free eIF4E to produce and initiate cap-dependent protein translation. Rapamycin inhibits mTORC1 and promotes dephosphorylation of 4EBP1, which limits denovo protein synthesis [56].
4.1. Eukaryotic Translation Initiation Factor 4E Binding Protein-1
mTOR and other kinases phosphorylate 4EBP1 at numerous serine/threonine sites in reaction to stimuli brought on by mitogens, growth factors, G-protein coupled agonists, and cytokines, which help in the dissociation of eIF4E from 4EBP1. This free eIF4E then binds to large scaffolding proteins, eIF4G, ATP-dependent RNA helicase eIF4A, and eIF4B, facilitating its cap-dependent protein translation [57,58]. The activation of mTOR signalling is often indicated by the phosphorylation of 4E-BP1. 4E-BP1 is a protein that contains seven different phosphorylation sites, namely Thr 37, Thr 46, Thr 70, Ser 65, Ser 83, Ser 101, and Ser, 112 [59]. Numerous experimental findings indicate that mTOR plays a direct role in phosphorylating 4EBP1 and activating eIF4E, which is simulated by various mitogenic signals [60]. The typical mTOR/4E-BP1 cascade may not be the only one that phosphorylates 4E-BP1, as numerous kinases have recently been proven to phosphorylate 4EBP1, either dependently or independently of mTOR [61]. Several signalling mechanisms have been proposed as potential kinases responsible for independent phosphorylation of 4EBP1 independent of mTOR pathway. These include extracellular signal-regulated kinase (ERK), glycogen synthase kinase-3 beta (GSK3β), prim-2 proto-oncogene, serine/threonine kinase (PIM2), ataxia telangiectasia mutated (ATM), cell division cycle protein 2/cyclin-dependent kinase-1 (CDC2/CDK1), p38 mitogen-activated protein kinases (p38MAPK), and leucine-rich repeat kinase 2 (LRRK2) signalling mechanisms. These findings suggest that mTOR inhibitors may not be absolutely effective in inactivating 4EBP1, which is activated alternatively by many other factors as discussed [62].
4.2. Ribosomal Protein S6 Kinase β-1 (S6K1)
The serine/threonine kinase S6K1 is another significant downstream target of mTOR. S6K1 is phosphorylated by activated mTOR on Thr389, which then phosphorylates S6K15, a component of the 40S ribosomal protein [63]. The ribosomal protein S6K Beta 1 (RPS6KB1) gene encodes two isoforms, p70S6K1 and p85S6K1 [64]. This mTOR-S6K1 signalling axis regulates important cellular functions like cell proliferation, metabolism, protein and lipid synthesis, translation, and transcription [64]. Additionally, this axis is responsible for adipocyte metabolism, learning, aging, memory, growth and development, while also controlling insulin sensitivity, glucose homeostasis, and other processes [65]. As a result, S6K1 is thought to be involved in critical functions in regulating cellular physiology [65]. Any disruption in this axis has negative consequences and can lead to the development of serious diseases ranging from metabolic disorders to cancer [64]. As a result, this network has remained a prime focus for different therapies used to treat different pathologies over the years. However, therapeutic interventions targeting mTOR in various cancers, notably renal and breast carcinomas, have resulted in a significant number of relapses due to the presence of feedback mechanisms in the signalling pathway [63]. Overcoming the resistance developed to mTOR inhibitors and other chemotherapeutic agents after chronic exposure to these drugs remains one of the key challenges that the scientific community is facing today.
5. Structure and Regulation of mTORC2
mTORC2 consists of four primary subunits, namely mTOR, RICTOR, mSIN1, and mLST8. Within mTORC1, the essential core subunit is regulatory-associated protein of mTOR (RAPTOR), while mTORC2 possesses distinct core subunits, namely rapamycin-insensitive companion of mTOR (RICTOR) and mammalian stress-activated Map kinase-interacting 1 (mSIN1). While numerous substrates of mTORC1 have been identified, mTORC2 predominantly phosphorylates AGC kinases, such as Akt (also referred to as protein kinase B, PKB), serum- and glucocorticoid-induced kinase 1 (SGK1), and members of the protein kinase C (PKC) family at their hydrophobic motif (HM) and turn motif (TM) [66].
The regulatory mechanisms of mTORC2 activity are lesser known than that of mTORC1. Recent studies suggest that mTORC2 is also regulated by several mechanisms. The platelet-derived growth factor, insulin like growth factor 1 receptor and epidermal growth factor receptor regulates its activity through PI3K signalling pathway, as PI3K helps binding of PIP3 with PH domain of mSin1, which restricts the kinase activity of mTOR, thereby activating mTORC2 and recruits it to the plasma membrane [66,67]. Growth factors stimulate phosphorylation of PI (4,5) P2 to produce phosphatidylinositol 3,4,5-triphosphate at plasma membrane by PI3K, this PIP3 has been proposed to directly activate mTORC2 [68]. Two models, which suggests the activation of mTORC2 are: PIP3 dependent activation, where mTORC2 is localized to plasma membrane, and other is that PIP3 brings conformational change in the mTORC2 leading to increase in its activity. However, emerging evidence suggests that mTORC2 permanently resides at plasma membrane and is constituently active [69].
It is well established that mTORC1 is responsive to both nutrition and growth factors, whereas mTORC2 is primarily controlled by growth factors [70]. Ample evidence suggests that amino acids lead to the activation of mTORC2. A reduction in glutamine catabolites caused by food restriction can activate mTORC2 and cause glutamine–fructose-6-phosphate amidotransferase 1 expression to rise (GFAT1) [71].
While PI3K regulation is the primary focus of most studies on mTORC2, recent research has unveiled the involvement of other signalling pathways in fine tuning mTORC2 activity. These include AMP-activated protein kinase (AMPK), Wnt signalling, and feedback control between mTORC1 and mTORC2.
AMP-activated protein kinase (AMPK) inhibits mTORC1 either by phosphorylating RAPTOR, or increasing the GTPase-activating protein (GAP) activity of TSC complex towards the GTPase Ras homology enriched in brain (RHEB) by phosphorylating TSC2, and hence indirectly activating mTORC2, which is responsible for the adaptation of low energy status of cells [46,72,73]. AMPK phosphorylates both Rictor and mTOR, which is primarily required and enough to increase the kinase activity of mTORC2. However, the molecular mechanism and phosphorylation sites, which regulate mTORC2 activity, are yet to be evaluated fully [74]. There is also a feedback control loops connecting mTORC1 and mTORC2. mTORC1 has a negative effect on mTORC2 through S6K1. S6K1, which is activated by mTORC1, causes inhibitory phosphorylation of IRS1 and reduces the amount of IRS1 protein. Inactivation of mTORC2 occurs as a result of the downregulation of insulin-PI3K signalling [75]. mTORC1 controls mTORC2 through an additional negative feedback loop involving growth factor-bound–receptor protein10 (Grb10) [76].
6. mTORC2 Effectors
mTORC2 is known to phosphorylate AGC kinases family proteins like Protein kinas A (Akt) and Protein Kinase C (PKC). It also phosphorylates glucocorticoid and serum-induced kinase-1 (SGK1) at their hydrophobic motif (HM) and turn motif (TM) [11].
The phosphorylation of HM motif of Akt at Ser473 [77] by mTORC2 is supposed to be a context-dependent event. This has been best illustrated in the study conducted by Estela Jacinto and his colleagues, where they have shown that Akt is incapable of phosphorylating FoxO1/3a, but predominantly phosphorylates other targets like TSC2 and GSK-3 in mSIN1 knockout mouse embryonic fibroblasts (MEFs) [17]. Similar to mTORC1, mTORC2 plays an important role in cell metabolism, regulating processes related to fatty acids, lipids, glucose, amino acid, and nucleotides [1]. It suppresses the activity of xCT (cysteine-glutamate antiporter), which is a solute carrier family 7 member 11 (SLC7A11) by phosphorylating it at xCT Ser26 [78].
7. Role of mTOR Signalling in Cancer at a Glance
Resistance to macrolide antibiotic, rapamycin in the yeast Saccharomyces cerevisiae, led to the discovery of the target of rapamycin (TOR). This eventually led to the discovery of the mammalian target of rapamycin (mTOR) in eukaryotes, which has the biochemical properties as that of the TOR [79]. The mTOR signalling pathway has a greater influence on basic critical cellular activities as its dysregulation disrupts normal physiological functioning in humans, leading to various diseases. It has been linked to various pathological conditions such as neuronal degeneration, obesity, type 2 diabetes mellitus, and cancer [9,80]. Both upstream and downstream effectors of mTORC1 play pivotal roles in the development of human cancers. Genetic mutations and amplifications are the two typical genetic alterations that cause protein molecules to become constitutively active. Hyper-activation of one of these upstream effectors, such as PI3K, Akt, or loss of phosphatase and TENs in homolog deleted on chromosome 10 (PTEN) molecules, triggers mTOR signalling cascade in cancer and plays a role in cellular proliferation, invasion, cytoskeleton rearrangement, metastasis, and cell survival and inhibits initiation of apoptosis and cellular autophagy.
Hanahan and Weinberg described that activation of mTOR signalling is the hallmark of a large number of cancers [81]. According to reports, more than 70% of malignancies have aberrantly overactivated mTOR pathways. It has been amply proven in recent years in cancer patients and animal models that mTOR malfunction promotes carcinogenesis [82,83]. Table 2 outlines how the activation of the mTOR pathway, either by oncogene stimulation or the loss of tumour suppressors can lead to the development of tumour angiogenesis and metastasis in various in-vitro cell lines and in-vivo mouse xenograft models [56].
Table 2.
Tumour-associated genes and cancer.
The loss of PTEN function, receptor tyrosine kinase overexpression, or mutations in Akt and PI3K are not the only factors that can lead to mTOR activation. It can be activated through another mechanism that includes mutation and gene amplification [97]. COSMIC database analysis shows overexpression of mTOR in the skin (8.25%), urinary tract (8.33%), and ovary (9.77%) cancers, and downregulation in central nervous system cancers (13.06%). S2215Y [98] and R2605P are the two distinct point mutations that lead to mTOR activation in cells even under starved conditions as revealed in the COSMIC database. It has also been reported that meningeal cancers (18.63%) have the highest number of mutations in mTOR. The other cancer where mTOR is highly mutated is endometrioid carcinoma (12.63%) [23].
8. Autophagy, Apoptosis, and mTOR Signalling: A Connecting Link
Under normal and pathological circumstances, mTOR is implicated in the suppression of autophagy, which is otherwise triggered by nutrient stress and energy deprivation [99,100]. When autophagy is triggered, lysosomal degradation of cytosolic components occurs. Recent data have shown that mTORC1 phosphorylates UNC-5 like Autophagy Activating Kinase (ULK) 1/2, which results in its deactivation [101]. ULK1/2 phosphorylates ATG13 and FIP200 thereby initiating the autophagic processes [102,103]. mTORC1 also controls autophagy at the transcriptional level by regulating the localization of the transcription factor EB (TFEB), which is responsible for regulating genes involved in autophagy and lysosomes [104,105].
Up-regulation of mTOR1 inhibits glycogen synthase kinase (GSK-3) whose inhibition leads to the suppression of the caspases-3 signalling pathway, which ultimately reduces ROS generation. Decreased ROS generation leads to the inhibition of apoptosis [106,107].
In conclusion, it is inferred that the mTOR signalling pathway may contribute to cancer development by blocking autophagy and apoptotic pathways.
9. mTOR Inhibitors in Cancer
As mTOR plays a crucial role in the development of tumours, mTOR inhibitors have the potential to be effective in various cancer treatments [108]. Rapamycin analogues (rapalogue) have received medical approval for the treatment of various cancers [109]. Additionally, other mTOR inhibitors with various modes of action have been developed. These rapalogues have been subclassified into three generations, some of which are presently undergoing clinical trials in a range of human cancers.
9.1. First-Generation mTOR Inhibitors: Allosteric Inhibitors
Rapamycin and Analogues (Rapalogues)
In a soil sample from Easter Island, a fungus namely Streptomyces hygroscopicus was shown to generate the antifungal metabolite rapamycin (also known as “Rapa Nu” in the native language) (Figure 4A). It is a potent inhibitor of S6K1. Rapamycin specifically binds to the 12-kDa FK506-binding protein (FKBP12) and by doing so, it allosterically inhibits mTORC1. This leads to the inhibition of the intrinsic kinase activity of TOR, including to autophosphorylation, and prevents TOR from reaching its substrates (Figure 4B) [110]. As a result, drugs that exclusively target mTORC1, such as rapamycin, are likely to impede cancer metabolism and are seen as promising anti-cancer therapy agents. However, due to the poor solubility and pharmacokinetic profile of rapamycin have prompted the development of multiple rapamycin analogues (rapalogues). In 2007 and 2009, the Food and Drug Administration (FDA) permitted the use of two water-soluble rapamycin derivatives, temsirolimus and everolimus, respectively, to treat advanced renal malignant carcinoma (RCC).
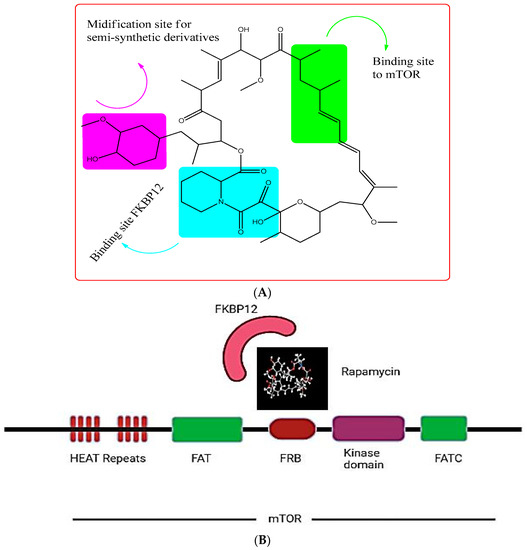
Figure 4.
(A) showing the chemical structure of Rapamycin. (B) showing the rapamycin-mTOR interaction; rapamycin selectively binds and allosterictively inhibits mTORC1 at (FK506-binding protein) FKBP12.
However, the efficacy of rapamycin and rapalogues when used alone seems to be lower than anticipated [111]. Everolimus, an oral mTOR inhibitor, was first developed to suppress the immune system during solid-organ transplantation [112]. It inhibits both B and T cell proliferation and differentiation by blocking the cytokine-driven activation responses of these cells [113,114]. Its potential to act as an antiproliferative agent in various in vitro and in vivo studies led to its approval by FDA for different tumours. Its efficacy in treating these malignancies prompted the scientific world to investigate its impact on cancers having dysregulation of the critical PI3K/Akt/mTOR pathway [115,116]. The drug is known to inhibit HLA-I-stimulated cellular proliferation through the ERK1/2 mTORC1 signalling cascade. Despite the enormous therapeutic potential of mTOR inhibition alone, rapamycin and its derivatives (rapalogues) have limited effectiveness against specific substrates and can activate multiple negative oncogenic feedback loops. This has led to the development of new strategies to overcome these limitations. One approach was to combine rapalogues with other known inhibitors. Table 3 outlines the cellular effects of everolimus.
Table 3.
The impact of everolimus treatment on cell functioning.
Temsirolimus, another rapalogue, has been extensively studied for its potential use in treating blood-related cancers. In clinical settings, it has been administered in combination with other medications. Patients with B-cell non-Hodgkin lymphoma and multiple myeloma who have received other treatments have been evaluated by using a combination of temsirolimus and bortezomib [117,118]. In both studies, participants were given intravenous temsirolimus (25 mg) once a week (on first day of week for 4 weeks), along with intravenous bortezomib (1.6 mg/m2) once a week (on first day of week for 4 weeks), with a treatment duration of 35 days. Around 33% of multiple myeloma patients showed a partial or better response to the treatment. A phase I clinical study has demonstrated that the addition of temsirolimus to rituximab and bendamustine resulted in promising outcomes in terms of both effectiveness and safety profile for patients with mantle cell lymphoma (MCL) and relapsed/refractory cancers [118,119].
9.2. Second-Generation mTOR Inhibitors: mTOR Kinase Inhibitors
In addition to rapalogues, second-generation mTOR inhibitors have generated mTOR to combat cancer in a better way, with one class inhibiting selectively both mTORC1 and mTORC2 without having any effect on other kinases. The other class of these second-generation mTOR inhibitors has shown the capability of inhibiting both mTOR and PI3K (dual inhibitors) [120,121]. As they can inhibit mTOR, PI3K, and Akt, they have the advantage of overcoming feedback loops [122]. A team of researchers who studied two mTOR inhibitors, namely PP242 and PP30, concluded that these inhibitors possess a central pyrazolo [3,4-d]pyrimidine ring with a C4 amino group, distinct heterocyclic substituents, and an N1 isopropyl substituent on C3. Both inhibitors competitively targeted mTORC1 and mTORC2 through ATP binding, exerting more significant effects on cell cycle regulation, cell growth, and proliferation, as well as cap-dependent translation when compared to the conventional inhibitor rapamycin [123]. Table 4 summarizes the second-generation mTOR inhibitors being tested at different phases of clinical trials.
Table 4.
Summary of second-generation mTOR inhibitors.
9.3. Third-Generation mTOR Inhibitor: RapaLink
Since mTOR signalling pathway has an important role in normal cellular function, a complete blockade might have catastrophic effects [132]. Additionally, autophagy gets initiated by the blockade of mTORC1, thus promoting cancer cell survival as seen with the use of AZD8055 [133]. To solve this problem, Rodrick-Outmezguine developed a molecule by linking binding sites rapamycin and INK-128 leading to the generation of a dual mTOR inhibitor called Rapalink [134]. Its property of using the binding sites of both first- and second-generation inhibitors makes it a unique molecule to inhibit and be effective against drug-resistant mTOR mutants that have shown resistance to mTOR kinase inhibitors (TORKi). RapaLink-1 has been found more potent and effective in inhibiting mTOR than Rapamycin and second-generation mTOR inhibitors as evidenced by the treatment of RapaLink-1 in U87MG and LN229 cells. It has also been reported that FKBP12 bound to RapaLink-1 enhances the accumulation of Rapalink-1 molecules inside the cell, and thus makes it a potential drug to inhibit cancer-associated activating mTOR mutant [134,135]. A study conducted by Kazuki and collegues, 2020, reported that Rapalink-1 showed a better response in sunitinib-sensitive and sunitinib-resistant renal cell carcinoma (RCC) than temsirolimus. It has also been stated that rapalink-1 not only inhibits PI3K/AKT/mTOR but has a significant effect on ErbB (erythroblastic leukemia viral oncogene) signalling and ABC transporters [136].
It has been observed that MCF-7 cells treated with Rapalink did not develop resistance to chemotherapeutic drugs. However, significant resistance was observed after three months of treatment with either first- or second-generation mTOR inhibitors [109].
10. Combination Therapies
mTOR is the major signalling pathway involved in the development of resistance to already-existing cancer therapies. In recent years cancer biologists have shifted the focus to developing mTOR-based combination therapies. Positive feedback in PI3K/mTOR signalling pathway has limited the clinical effects of mTOR inhibitors through the activation of AKT by activation of downstream target and nuclear factor kappa B (Nf-κB), which helps in the accumulation of PIP3 and, hence continuous, AKT activation [137]. Built on these assumptions, that simultaneous inhibition of various signalling pathways together will minimize the incidences/chances of resistance, combining mTOR inhibitors with other drugs is now being explored. Numerous clinical trials are being conducted to evaluate the effectiveness of mTOR inhibitors in combination with other targeted therapies or chemotherapeutic drugs. To develop a successful combination therapy, issues related to toxicity and reliable biomarkers are important parameters to be kept in mind while selecting a patient to treat. As a result, a combination therapy of everolimus and exemestance is approved for human epidermal growth factor receptor 2 (HER2)-negative/Estrogen receptor (ER)-positive breast cancer [138]. Another study conducted by Mortzer and colleagues reported that everolimus, when combined with lenvatinib (vascular endothelial growth factor inhibitor), is found to be more effective in metastatic renal cell carcinoma than when used alone [139]. Also phase-II clinical trial of a combination of letrolzole, an aromatase inhibitor, and the rapamycin analogue, everolimus, has shown promising results in oestrogen receptor-positive ovarian cancer, achieving 12-week progressive free survival in 47% of patients [140]. Table 5 summarizes the mTOR-based combination therapies that are being explored in clinical trials. Overall, these studies using mTOR-based combination therapies have suggested that using mTOR inhibitors with other anti-cancer agents to overcome the limitations facing these regimens when using alone.
Since insulin-like growth factor 1 and insulin receptor (IGF-IR) signalling could potentially lead to resistance against mTORC1 inhibitors, a clinical trial was carried out to evaluate the effectiveness of combining cixutumumab, a humanized monoclonal antibody targeting IGF-1R, with temsirolimus. The outcome of the trail was that the combination therapy exhibits better clinical response in patients diagnosed with sarcoma and adrenocortical carcinoma than when temsirolimus was used alone [141].
In another clinical trial, CTRI/2018/05/014178, conducted by Timothy Crook and co-workers in 2020, it was demonstrated that mTOR-based drug combination improved treatment outcomes, like response rate and disease control rate [142].
Despite attempts to use combination therapies, favourable outcomes are not consistently achieved. This was evident in a clinical trial where the combination of everolimus and the epidermal growth factor receptor (EGFR) inhibitor gefitinib showed limited effectiveness in reducing tumour activity in patients diagnosed with metastatic castration-resistant prostate cancer [143]. Similarly in patients with advanced solid tumours, the combination of pimasertib and voxtalisib exhibited inadequate long-term tolerability and restricted effectiveness in reducing tumour activity [144].
The combinations of mTOR inhibitors with other targeted agents or cytotoxics have been shown to delay the emergence of resistance to these agents. They have demonstrated the potential anticancer activity against several types of cancers including hormone positive and HER2- negative breast cancers, whose treatment otherwise is challenge [145].
Figure 5 shows the structures of some of the mTOR inhibitors, while Figure 6 illustrates the mTOR signalling and the effect of its inhibitors.
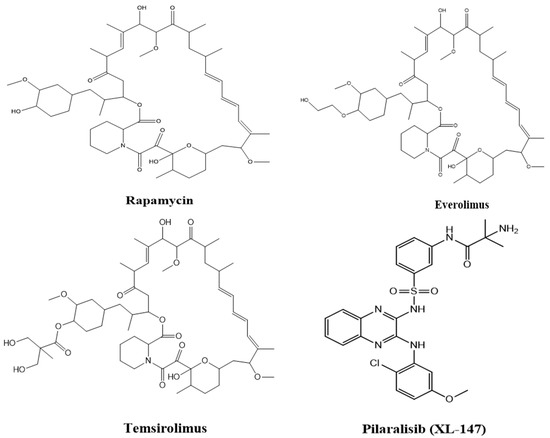
Figure 5.
Structures of various mTOR inhibitors.
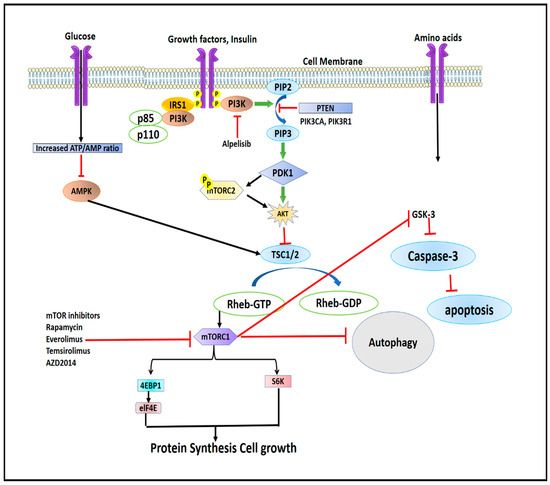
Figure 6.
Illustrating the mTOR signalling and effect of its inhibitors.
Table 5.
Summary of the mTOR-based drug combinations investigated in various cancers.
Table 5.
Summary of the mTOR-based drug combinations investigated in various cancers.
| mTOR Inhibitor | Combined with Drug | Tumour Applied and Type of Study | Outcome | Ref. |
|---|---|---|---|---|
| Everolimus | Trametinib (kinase inhibitor) | Advanced solid tumours/ Phase 1B NCT00955773 | Among 67 patients, 5 patients (7%) achieved partial response (PR) to treatment and 21 (31%) displayed stable disease (SD) | [146] |
| Everolimus | Lenvatinib (multiple receptor kinase inhibitor) | RCC/Phase-II NCT01136733 | Survival rate increased when used in combination | [139] |
| Everolimus | Carboplatin and paclitaxel | LCNEC/ Phase II NCT01317615 | Improvement in overall response rate and tumour regression, combination is effective and well tolerated than using drug alone | [147] |
| Rapamycin | Entinostat (benzamide histone deacetylase inhibitor) | General cancers/ In vitro | This led to the halting of the cell cycle and the start of programmed cell death (apoptosis), it promotes MYC degradation | [148] |
| Rapamycin | AR inhibitor enzalutamide | HCC/ In vitro and In vivo | Enalutamide and rapamycin together yielded stronger anti-HCC activity than each drug alone in vitro and in vivo. Also, combination exhibited more potent antitumour activity in the xenograft tumour model than cultured cancer cells, causing elevated apoptotic cell death and tumour regression | [149] |
| Rapamycin | STX-0119 | Glioblastoma/ In vitro | Combining of two drugs had significant growth inhibitory effect against the TMZ-R U87 cell line. IC50 decreased to 11.3μM (drug combination) from 78 μM (STX-0119) and 30.5 μM (rapamycin) | [150] |
AR: androgen receptor, HCC: hepatic cell carcinoma, LCNEC: large-cell neuroendocrine carcinoma, RCC: renal cell carcinoma.
11. Adverse Events Related to the Use of mTOR Inhibitors
The side effects of modern targeted therapies differ from those observed with traditional chemotherapy. Mammalian target of rapamycin (mTOR) inhibitors have attained significant attention in the field of oncology and exhibit a wide range of toxic effects like headache, mucositis, rashes, and metabolic toxicities including hyperglycemia, hyperlipidemia, and hypophosphatemia. These adverse effects can be mild to severe, which require medical attention [151].
Skin toxicity, usually on face and neck, occurs with the use of mTOR inhibitors. The reported rate of skin rashes with the use of everolimus is 25–50%, temsirolimus 47–76%, and ridaforolimus 48–66% [151,152,153,154].
Development of adverse reactions or events (AR/AE) with the use of mTOR inhibitors requires attention, and it is important to understand their underlying mechanisms for managing such AEs [154]. Pneumonitis [155], metabolic adverse events like hyperglycemia (13–50%) and hyperlipidemia (12%) with the use of everolimus and 11% with the use of temsirolimus [156]. The incidence of mTOR inhibitors associated stomatitis (mIAS) has been also reported in some studies [157]. The use of mTOR inhibitors has been associated with haematological adverse events such as thrombocytopenia and leucopenia/neutropenia and, hence, their use requires routine complete blood counts [158].
Fatigue, weakness, changes in taste perception, and diarrhoea are among the frequently observed adverse effects in clinical trials involving mTOR inhibitors [159].
The novel mTOR inhibitors also have been associated with bone marrow suppression. In a phase III clinical trial of temsirolimus, 45% of patients reported anaemia, and 20% of them had severe anaemia (grade 3–4) [152]. Similarly, in another phase III clinical trial with everolimus, it was revealed that 91% patients had anaemia, with 9% grade three, and 1% with grade four [160].
A meta-analysis conducted by Jian Xu and Deying Tian in 2014 included a total of 5436 patients with various solid tumours from 26 clinical trials. The study findings indicated the following rates of hematologic toxicities associated with mTOR inhibitors: anaemia in 38.8%, with 7.5% of patients experiencing severe anaemia; leucopenia in 19.6%, with 1.8% of patients experiencing severe leucopenia; and neutropenia in 14.9%, with 5.6% of patients experiencing severe neutropenia [161]. Table 6 summarizes the AE/AR associated with the use of mTOR inhibitors in various cancers.
Table 6.
Summary of AR/AE with the use of some of the mTOR inhibitors.
12. mTOR Inhibitors in Combination with Chimeric Antigen Receptor Treatment (CAR-T) Therapy and Immune Check Point Inhibitor (ICI) Therapy
Managing advanced and metastatic cancers poses a formidable challenge, which is why combination therapies are employed to effectively address the underlying cause of the issue. Numerous combination therapies have been investigated so far, and among the recently explored approaches in the treatment of various solid tumours is the combination of CAR-T, ICI, and mTOR-based therapies. A study conducted by Zhigang Nain and co-workers reported that by utilizing rapamycin as a pretreatment to reduce mTORC1 activity, the ability of CAR-T cells to penetrate the bone marrow was enhanced, resulting in an increased elimination of acute myeloid leukaemia (AML) cells in the bone marrow of leukemia xenograft mouse models [162].
Similarly, the combination of mTOR inhibitors with immunotherapies like ICI has been the subject of numerous preclinical studies, which have underscored the potential anticancer advantages of such combinations. In their study, Moore and coworkers revealed that combining rapamycin and anti-PD-L1, a monoclonal antibody (mAb), resulted in increase in survival rate of mice with murine oral cancer cell line immunogenic (MOC1) tumour when compared to the monotherapy [163].
In a similar manner to rapamycin, the targeting of mTOR using kinase inhibitors enhances the anti-cancer effects of checkpoint inhibitors. In fact, the combination of the mTORC1/mTORC2 inhibitor vistusertib with anti-CTLA-4, anti-PD-L1, and anti-PD-1, demonstrated a significant decrease in the growth of MC38 or CT-26 (murine colorectal cancer cell lines) tumours compared to individual therapies [164].
Collectively, these studies provide ample evidence that combining mTOR inhibitors with immune checkpoint modulators and CAR-T therapy have advantages over monotherapy in the context of cancer treatment.
13. Clinical Application of mTOR Inhibitors against Various Cancers
Due to the observed anti-cancer effectiveness of mTOR inhibitors in preclinical studies, either as standalone treatments or in combination with chemotherapy, radiotherapy, and targeted therapy, numerous clinical trials have been completed or are currently underway to assess the efficacy of mTOR inhibitors in the treatment of various types of human cancers. Multiple mTOR inhibitors have received approval for the treatment of human cancers. Many more studies/clinical trials are currently underway to assess the efficacy of mTOR inhibitors [165]. The published data support that use of mTOR inhibitors against oesophageal cancer (everolimus, NCT00985192 by translational oncology research international, USA in 2009) [165], gastric cancer (everolimus, NCT00519324 by Novartis pharmaceuticals in 2009) [166], everolimus plus cisplatin, NCT00632268 by National Taiwan University Hospital in 2008 [167], hepatocellular carcinoma, pancreatic cancer, and colorectal cancer, where a number of mTOR inhibitors have been tested and found safe and effective against the disease.
Based on the clinical data that have been published, the efficacy of using mTOR inhibitors alone against gastrointestinal cancers is restricted and might be due to the feedback regulation mechanisms, which impede the therapeutic impact of mTOR inhibitors. Consequently, the most promising approach for utilizing mTOR inhibitors in the future would involve combining them with other types of drugs such as radiation therapy, antibody drugs, and other cytotoxic drugs.
14. Conclusions
mTOR is the critical pathway in modulating cellular proliferation, apoptosis, and autophagy of cells by influencing transcription and protein synthesis through the integration of diverse extracellular and intracellular signal stimuli. mTORC1 and mTORC2 are two separate complexes, which are cross-talked with each other. Understanding the role of mTOR downstream regulators, 4E-BP1 and S6K1 is important in treating cancer at later stages because of their active role in cellular proliferation, protein synthesis, tumour angiogenesis, and metastasis. Since major developments in understanding this crucial signalling pathway are primarily focused on the use of rapamycin or rapalogues, which primarily inhibit mTORC1 activity, novel dual PI3K/mTOR inhibitors, and selective mTORC1/mTORC2 inhibitors are being evaluated at clinical levels to overcome the challenges like resistance and feedback effects of already known mTOR inhibitors. Also, several studies provide ample evidence that combining mTOR inhibitors with immune checkpoint modulators and CAR-T therapy have advantage over monotherapy in the context of cancer treatment. However, the toxicity and adverse effects of these mTOR inhibitors make it difficult to treat cancer, which has significant negative effects on the patients. As a result, it is important to conduct more clinical trials to understand the underlying mechanisms associated with the use of mTOR inhibitors so that they can be used to counteract these toxicities for their safe and effective treatments.
Author Contributions
S.A.M., L.H. and T.A.; performed a literature survey and drafted the content. S.A.M. and L.H.; wrote the manuscript. G.N.B., A.D. and S.A.M.; conceived the idea. S.A.M. and L.H.; revised and edited the manuscript. S.A.A., S.W. and M.A.A.A.; arranged the funding. G.N.B. and A.D.; supervised the manuscript writing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Deanship of Scientific Research at King Khalid University through the Small Research Groups Project under grant number RGP.1/17/44.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for funding this work through the Small Research Groups Project under grant number (RGP.1/17/44). Author, Suhail Ahmad Mir acknowledges the Department of Science and Technology, Government of India for his INSPIRE, Senior Research Fellowship. Author, Laraibah Hamid acknowledges University Grants Commission, New Delhi, for SJSGC junior Research Fellowship.
Conflicts of Interest
Authors declare no competing interests.
References
- Yin, Y.; Hua, H.; Li, M.; Liu, S.; Kong, Q.; Shao, T.; Wang, J.; Luo, Y.; Wang, Q.; Luo, T.; et al. mTORC2 promotes type I insulin-like growth factor receptor and insulin receptor activation through the tyrosine kinase activity of mTOR. Cell Res. 2016, 26, 46–65. [Google Scholar] [CrossRef]
- Castellanos, M.; Gubern, C.; Kadar, E. mTOR. In Molecules to Medicine with mTOR; Elsevier: Amsterdam, The Netherlands, 2016; pp. 105–122. [Google Scholar] [CrossRef]
- Yoon, M.-S. The Role of Mammalian Target of Rapamycin (mTOR) in Insulin Signaling. Nutrients 2017, 9, 1176. [Google Scholar] [CrossRef]
- Yang, Q.; Guan, K.-L. Expanding mTOR signaling. Cell Res. 2007, 17, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Dowling, R.J.O.; Topisirovic, I.; Fonseca, B.D.; Sonenberg, N. Dissecting the role of mTOR: Lessons from mTOR inhibitors. Biochim. Biophys. Acta Proteins Proteom. 2010, 1804, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, T.P. TOR-dependent control of autophagy: Biting the hand that feeds. Curr. Opin. Cell Biol. 2010, 22, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Li, D.; Du, L.; Zhu, X. piRNAs: Biogenesis and their potential roles in cancer. Cancer Metastasis Rev. 2020, 39, 567–575. [Google Scholar] [CrossRef]
- Mossmann, D.; Park, S.; Hall, M.N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 2018, 18, 744–757. [Google Scholar] [CrossRef]
- Kim, D.-H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR Interacts with Raptor to Form a Nutrient-Sensitive Complex that Signals to the Cell Growth Machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef]
- Oh, W.J.; Jacinto, E. mTOR complex 2 signaling and functions. Cell Cycle 2011, 10, 2305–2316. [Google Scholar] [CrossRef]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR Is an mTOR Inhibitor Frequently Overexpressed in Multiple Myeloma Cells and Required for Their Survival. Cell 2009, 137, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Kaizuka, T.; Hara, T.; Oshiro, N.; Kikkawa, U.; Yonezawa, K.; Takehana, K.; Iemura, S.; Natsume, T.; Mizushima, N. Tti1 and Tel2 Are Critical Factors in Mammalian Target of Rapamycin Complex Assembly. J. Biol. Chem. 2010, 285, 20109–20116. [Google Scholar] [CrossRef] [PubMed]
- Pearce, L.R.; Huang, X.; Boudeau, J.; Pawłowski, R.; Wullschleger, S.; Deak, M.; Ibrahim, A.F.M.; Gourlay, R.; Magnuson, M.A.; Alessi, D.R. Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem. J. 2007, 405, 513–522. [Google Scholar] [CrossRef]
- Kim, D.-H.; Sarbassov, D.D.; Ali, S.M.; Latek, R.R.; Guntur, K.V.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. GβL, a Positive Regulator of the Rapamycin-Sensitive Pathway Required for the Nutrient-Sensitive Interaction between Raptor and mTOR. Mol. Cell 2003, 11, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Harris, T.E.; Roth, R.A.; Lawrence, J.C. PRAS40 Regulates mTORC1 Kinase Activity by Functioning as a Direct Inhibitor of Substrate Binding. J. Biol. Chem. 2007, 282, 20036–20044. [Google Scholar] [CrossRef]
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. SIN1/MIP1 Maintains rictor-mTOR Complex Integrity and Regulates Akt Phosphorylation and Substrate Specificity. Cell 2006, 127, 125–137. [Google Scholar] [CrossRef]
- Yip, C.K.; Murata, K.; Walz, T.; Sabatini, D.M.; Kang, S.A. Structure of the Human mTOR Complex I and Its Implications for Rapamycin Inhibition. Mol. Cell 2010, 38, 768–774. [Google Scholar] [CrossRef]
- Yang, H.; Wang, J.; Liu, M.; Chen, X.; Huang, M.; Tan, D.; Dong, M.-Q.; Wong, C.C.L.; Wang, J.; Xu, Y.; et al. 4.4 Å Resolution Cryo-EM structure of human mTOR Complex 1. Protein Cell 2016, 7, 878–887. [Google Scholar] [CrossRef]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One Drug, Many Effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef]
- Tian, T.; Li, X.; Zhang, J. mTOR Signaling in Cancer and mTOR Inhibitors in Solid Tumor Targeting Therapy. Int. J. Mol. Sci. 2019, 20, 755. [Google Scholar] [CrossRef] [PubMed]
- Popova, N.V.; Jücker, M. The Role of mTOR Signaling as a Therapeutic Target in Cancer. Int. J. Mol. Sci. 2021, 22, 1743. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Asara, J.M.; Manning, B.D. Characterization of Rictor Phosphorylation Sites Reveals Direct Regulation of mTOR Complex 2 by S6K1. Mol. Cell Biol. 2009, 29, 5657–5670. [Google Scholar] [CrossRef]
- Julien, L.-A.; Carriere, A.; Moreau, J.; Roux, P.P. mTORC1-Activated S6K1 Phosphorylates Rictor on Threonine 1135 and Regulates mTORC2 Signaling. Mol. Cell Biol. 2010, 30, 908–921. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Manning, B.D. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat. Cell Biol. 2013, 15, 555–564. [Google Scholar] [CrossRef]
- Myers, M.G.; Backer, J.M.; Sun, X.J.; Shoelson, S.; Hu, P.; Schlessinger, J.; Yoakim, M.; Schaffhausen, B.; White, M.F. IRS-1 activates phosphatidylinositol 3’-kinase by associating with src homology 2 domains of p85. Proc. Natl. Acad. Sci. USA 1992, 89, 10350–10354. [Google Scholar] [CrossRef]
- BODINE, S.C. mTOR Signaling and the Molecular Adaptation to Resistance Exercise. Med. Sci. Sport. Exerc. 2006, 38, 1950–1957. [Google Scholar] [CrossRef]
- Wu, M.; Falasca, M.; Blough, E.R. Akt/protein kinase B in skeletal muscle physiology and pathology. J. Cell Physiol. 2011, 226, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Park, J.; Cron, P.; Hess, D.; Hemmings, B.A. Identification of a PKB/Akt Hydrophobic Motif Ser-473 Kinase as DNA-dependent Protein Kinase. J. Biol. Chem. 2004, 279, 41189–41196. [Google Scholar] [CrossRef]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.J.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr. Biol. 1997, 7, 261–269. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.-L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Potter, C.J.; Pedraza, L.G.; Xu, T. Akt regulates growth by directly phosphorylating Tsc2. Nat. Cell Biol. 2002, 4, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Tee, A.R.; Manning, B.D.; Roux, P.P.; Cantley, L.C.; Blenis, J. Tuberous Sclerosis Complex Gene Products, Tuberin and Hamartin, Control mTOR Signaling by Acting as a GTPase-Activating Protein Complex toward Rheb. Curr. Biol. 2003, 13, 1259–1268. [Google Scholar] [CrossRef]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb Binds and Regulates the mTOR Kinase. Curr. Biol. 2005, 15, 702–713. [Google Scholar] [CrossRef]
- White, J.P.; Baltgalvis, K.A.; Sato, S.; Wilson, L.B.; Carson, J.A. Effect of nandrolone decanoate administration on recovery from bupivacaine-induced muscle injury. J. Appl. Physiol. 2009, 107, 1420–1430. [Google Scholar] [CrossRef]
- Yin, H.-N.; Chai, J.-K.; Yu, Y.-M.; Shen, C.-A.; Wu, Y.-Q.; Yao, Y.-M.; Liu, H.; Liang, L.-M.; Tompkins, R.G.; Sheng, Z.-Y. Regulation of Signaling Pathways Downstream of IGF-I/Insulin by Androgen in Skeletal Muscle of Glucocorticoid-Treated Rats. J. Trauma Inj. Infect. Crit. Care 2009, 66, 1083–1090. [Google Scholar] [CrossRef]
- Jones, A.; Hwang, D.-J.; Narayanan, R.; Miller, D.D.; Dalton, J.T. Effects of a Novel Selective Androgen Receptor Modulator on Dexamethasone-Induced and Hypogonadism-Induced Muscle Atrophy. Endocrinology 2010, 151, 3706–3719. [Google Scholar] [CrossRef]
- Ibebunjo, C.; Eash, J.K.; Li, C.; Ma, Q.; Glass, D.J. Voluntary running, skeletal muscle gene expression, and signaling inversely regulated by orchidectomy and testosterone replacement. Am. J. Physiol. Metab. 2011, 300, E327–E340. [Google Scholar] [CrossRef]
- White, J.P.; Gao, S.; Puppa, M.J.; Sato, S.; Welle, S.L.; Carson, J.A. Testosterone regulation of Akt/mTORC1/FoxO3a signaling in skeletal muscle. Mol. Cell Endocrinol. 2013, 365, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Hourdé, C.; Jagerschmidt, C.; Clément-Lacroix, P.; Vignaud, A.; Ammann, P.; Butler-Browne, G.S.; Ferry, A. Androgen replacement therapy improves function in male rat muscles independently of hypertrophy and activation of the Akt/mTOR pathway. Acta Physiol. 2009, 195, 471–482. [Google Scholar] [CrossRef]
- Ma, L.; Shen, C.; Chai, J.; Yin, H.; Deng, H.; Feng, R. Extracellular Signal–Regulated Kinase–Mammalian Target of Rapamycin Signaling and Forkhead-Box Transcription Factor 3a Phosphorylation Are Involved in Testosterone’s Effect on Severe Burn Injury in a Rat Model. Shock 2015, 43, 85–91. [Google Scholar] [CrossRef]
- Carriere, A.; Romeo, Y.; Acosta-Jaquez, H.A.; Moreau, J.; Bonneil, E.; Thibault, P.; Fingar, D.C.; Roux, P.P. ERK1/2 Phosphorylate Raptor to Promote Ras-dependent Activation of mTOR Complex 1 (mTORC1). J. Biol. Chem. 2011, 286, 567–577. [Google Scholar] [CrossRef]
- Carrière, A.; Cargnello, M.; Julien, L.-A.; Gao, H.; Bonneil, É.; Thibault, P.; Roux, P.P. Oncogenic MAPK Signaling Stimulates mTORC1 Activity by Promoting RSK-Mediated Raptor Phosphorylation. Curr. Biol. 2008, 18, 1269–1277. [Google Scholar] [CrossRef]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 Integrates Wnt and Energy Signals via a Coordinated Phosphorylation by AMPK and GSK3 to Regulate Cell Growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Danesh Pazhooh, R.; Rahnamay Farnood, P.; Asemi, Z.; Mirsafaei, L.; Yousefi, B.; Mirzaei, H. mTOR pathway and DNA damage response: A therapeutic strategy in cancer therapy. DNA Repair 2021, 104, 103142. [Google Scholar] [CrossRef]
- Lim, H.-K.; Choi, Y.-A.; Park, W.; Lee, T.; Ryu, S.H.; Kim, S.-Y.; Kim, J.-R.; Kim, J.-H.; Baek, S.-H. Phosphatidic Acid Regulates Systemic Inflammatory Responses by Modulating the Akt-Mammalian Target of Rapamycin-p70 S6 Kinase 1 Pathway. J. Biol. Chem. 2003, 278, 45117–45127. [Google Scholar] [CrossRef] [PubMed]
- Nojima, H.; Tokunaga, C.; Eguchi, S.; Oshiro, N.; Hidayat, S.; Yoshino, K.; Hara, K.; Tanaka, N.; Avruch, J.; Yonezawa, K. The Mammalian Target of Rapamycin (mTOR) Partner, Raptor, Binds the mTOR Substrates p70 S6 Kinase and 4E-BP1 through Their TOR Signaling (TOS) Motif. J. Biol. Chem. 2003, 278, 15461–15464. [Google Scholar] [CrossRef] [PubMed]
- Schalm, S.S.; Fingar, D.C.; Sabatini, D.M.; Blenis, J. TOS Motif-Mediated Raptor Binding Regulates 4E-BP1 Multisite Phosphorylation and Function. Curr. Biol. 2003, 13, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Schalm, S.S.; Blenis, J. Identification of a Conserved Motif Required for mTOR Signaling. Curr. Biol. 2002, 12, 632–639. [Google Scholar] [CrossRef]
- Dennis, P.B.; Pullen, N.; Kozma, S.C.; Thomas, G. The principal rapamycin-sensitive p70(s6k) phosphorylation sites, T-229 and T-389, are differentially regulated by rapamycin-insensitive kinase kinases. Mol. Cell Biol. 1996, 16, 6242–6251. [Google Scholar] [CrossRef] [PubMed]
- Faivre, S.; Kroemer, G.; Raymond, E. Current development of mTOR inhibitors as anticancer agents. Nat. Rev. Drug Discov. 2006, 5, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Sonenberg, N.; Gingras, A.-C. The mRNA 5′ cap-binding protein eIF4E and control of cell growth. Curr. Opin. Cell Biol. 1998, 10, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Pause, A.; Belsham, G.J.; Gingras, A.-C.; Donzé, O.; Lin, T.-A.; Lawrence, J.C.; Sonenberg, N. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5’-cap function. Nature 1994, 371, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Jastrzebski, K.; Hannan, K.M.; Tchoubrieva, E.B.; Hannan, R.D.; Pearson, R.B. Coordinate regulation of ribosome biogenesis and function by the ribosomal protein S6 kinase, a key mediator of mTOR function. Growth Factors 2007, 25, 209–226. [Google Scholar] [CrossRef] [PubMed]
- Rosenwald, I.B.; Kaspar, R.; Rousseau, D.; Gehrke, L.; Leboulch, P.; Chen, J.-J.; Schmidt, E.V.; Sonenberg, N.; London, I.M. Eukaryotic Translation Initiation Factor 4E Regulates Expression of Cyclin D1 at Transcriptional and Post-transcriptional Levels. J. Biol. Chem. 1995, 270, 21176–21180. [Google Scholar] [CrossRef]
- Rousseau, D.; Kaspar, R.; Rosenwald, I.; Gehrke, L.; Sonenberg, N. Translation initiation of ornithine decarboxylase and nucleocytoplasmic transport of cyclin D1 mRNA are increased in cells overexpressing eukaryotic initiation factor 4E. Proc. Natl. Acad. Sci. USA 1996, 93, 1065–1070. [Google Scholar] [CrossRef]
- Martineau, Y.; Azar, R.; Bousquet, C.; Pyronnet, S. Anti-oncogenic potential of the eIF4E-binding proteins. Oncogene 2013, 32, 671–677. [Google Scholar] [CrossRef]
- Dilling, M.B.; Germain, G.S.; Dudkin, L.; Jayaraman, A.L.; Zhang, X.; Harwood, F.C.; Houghton, P.J. 4E-binding Proteins, the Suppressors of Eukaryotic Initiation Factor 4E, Are Down-regulated in Cells with Acquired or Intrinsic Resistance to Rapamycin. J. Biol. Chem. 2002, 277, 13907–13917. [Google Scholar] [CrossRef] [PubMed]
- Sikalidis, A.K.; Mazor, K.M.; Kang, M.; Liu, H.; Stipanuk, M.H. Total 4EBP1 Is Elevated in Liver of Rats in Response to Low Sulfur Amino Acid Intake. J. Amino Acids 2013, 2013, 864757. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, X.F.S. mTOR-independent 4E-BP1 phosphorylation is associated with cancer resistance to mTOR kinase inhibitors. Cell Cycle 2012, 11, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Park, I.-H.; Bachmann, R.; Shirazi, H.; Chen, J. Regulation of Ribosomal S6 Kinase 2 by Mammalian Target of Rapamycin. J. Biol. Chem. 2002, 277, 31423–31429. [Google Scholar] [CrossRef] [PubMed]
- Grove, J.R.; Banerjee, P.; Balasubramanyam, A.; Coffer, P.J.; Price, D.J.; Avruch, J.; Woodgett, J.R. Cloning and expression of two human p70 S6 kinase polypeptides differing only at their amino termini. Mol. Cell Biol. 1991, 11, 5541–5550. [Google Scholar] [CrossRef] [PubMed]
- Magnuson, B.; Ekim, B.; Fingar, D.C. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem. J. 2012, 441, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Hall, M.N. Regulation of mTORC2 Signaling. Genes 2020, 11, 1045. [Google Scholar] [CrossRef]
- Gan, X.; Wang, J.; Su, B.; Wu, D. Evidence for Direct Activation of mTORC2 Kinase Activity by Phosphatidylinositol 3,4,5-Trisphosphate. J. Biol. Chem. 2011, 286, 10998–11002. [Google Scholar] [CrossRef]
- Yuan, H.-X.; Guan, K.-L. The SIN1-PH Domain Connects mTORC2 to PI3K. Cancer Discov. 2015, 5, 1127–1129. [Google Scholar] [CrossRef]
- Ebner, M.; Sinkovics, B.; Szczygieł, M.; Ribeiro, D.W.; Yudushkin, I. Localization of mTORC2 activity inside cells. J. Cell Biol. 2017, 216, 343–353. [Google Scholar] [CrossRef]
- Tato, I.; Bartrons, R.; Ventura, F.; Rosa, J.L. Amino Acids Activate Mammalian Target of Rapamycin Complex 2 (mTORC2) via PI3K/Akt Signaling. J. Biol. Chem. 2011, 286, 6128–6142. [Google Scholar] [CrossRef]
- Moloughney, J.G.; Kim, P.K.; Vega-Cotto, N.M.; Wu, C.-C.; Zhang, S.; Adlam, M.; Lynch, T.; Chou, P.-C.; Rabinowitz, J.D.; Werlen, G.; et al. mTORC2 Responds to Glutamine Catabolite Levels to Modulate the Hexosamine Biosynthesis Enzyme GFAT1. Mol. Cell 2016, 63, 811–826. [Google Scholar] [CrossRef]
- González, A.; Hall, M.N.; Lin, S.-C.; Hardie, D.G. AMPK and TOR: The Yin and Yang of Cellular Nutrient Sensing and Growth Control. Cell Metab. 2020, 31, 472–492. [Google Scholar] [CrossRef]
- Gao, M.; Kong, Q.; Hua, H.; Yin, Y.; Wang, J.; Luo, T.; Jiang, Y. AMPK-mediated up-regulation of mTORC2 and MCL-1 compromises the anti-cancer effects of aspirin. Oncotarget 2016, 7, 16349–16361. [Google Scholar] [CrossRef]
- Kazyken, D.; Magnuson, B.; Bodur, C.; Acosta-Jaquez, H.A.; Zhang, D.; Tong, X.; Barnes, T.M.; Steinl, G.K.; Patterson, N.E.; Altheim, C.H.; et al. AMPK directly activates mTORC2 to promote cell survival during acute energetic stress. Sci. Signal. 2019, 12, eaav3249. [Google Scholar] [CrossRef] [PubMed]
- Jhanwar-Uniyal, M.; Wainwright, J.V.; Mohan, A.L.; Tobias, M.E.; Murali, R.; Gandhi, C.D.; Schmidt, M.H. Diverse signaling mechanisms of mTOR complexes: mTORC1 and mTORC2 in forming a formidable relationship. Adv. Biol. Regul. 2019, 72, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.P.; Kang, S.A.; Rameseder, J.; Zhang, Y.; Ottina, K.A.; Lim, D.; Peterson, T.R.; Choi, Y.; Gray, N.S.; Yaffe, M.B.; et al. The mTOR-Regulated Phosphoproteome Reveals a Mechanism of mTORC1-Mediated Inhibition of Growth Factor Signaling. Science 2011, 332, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and Regulation of Akt/PKB by the Rictor-mTOR Complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef]
- Gu, Y.; Albuquerque, C.P.; Braas, D.; Zhang, W.; Villa, G.R.; Bi, J.; Ikegami, S.; Masui, K.; Gini, B.; Yang, H.; et al. mTORC2 Regulates Amino Acid Metabolism in Cancer by Phosphorylation of the Cystine-Glutamate Antiporter xCT. Mol. Cell 2017, 67, 128–138.e7. [Google Scholar] [CrossRef]
- Heitman, J.; Movva, N.R.; Hall, M.N. Targets for Cell Cycle Arrest by the Immunosuppressant Rapamycin in Yeast. Science 1991, 253, 905–909. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Forbes, S.A.; Bindal, N.; Bamford, S.; Cole, C.; Kok, C.Y.; Beare, D.; Jia, M.; Shepherd, R.; Leung, K.; Menzies, A.; et al. COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011, 39, D945–D950. [Google Scholar] [CrossRef]
- Ciuffreda, L.; Di Sanza, C.; Incani, U.C.; Milella, M. The mTOR Pathway: A New Target in Cancer Therapy. Curr. Cancer Drug Targets 2010, 10, 484–495. [Google Scholar] [CrossRef]
- Barlund, M.; Forozan, F.; Kononen, J.; Bubendorf, L.; Chen, Y.; Bittner, M.L.; Torhorst, J.; Haas, P.; Bucher, C.; Sauter, G.; et al. Detecting Activation of Ribosomal Protein S6 Kinase by Complementary DNA and Tissue Microarray Analysis. JNCI J. Natl. Cancer Inst. 2000, 92, 1252–1259. [Google Scholar] [CrossRef] [PubMed]
- Pópulo, H.; Soares, P.; Faustino, A.; Rocha, A.S.; Silva, P.; Azevedo, F.; Lopes, J.M. mTOR pathway activation in cutaneous melanoma is associated with poorer prognosis characteristics. Pigment. Cell Melanoma Res. 2011, 24, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Hiramatsu, M.; Ninomiya, H.; Inamura, K.; Nomura, K.; Takeuchi, K.; Satoh, Y.; Okumura, S.; Nakagawa, K.; Yamori, T.; Matsuura, M.; et al. Activation status of receptor tyrosine kinase downstream pathways in primary lung adenocarcinoma with reference of KRAS and EGFR mutations. Lung Cancer 2010, 70, 94–102. [Google Scholar] [CrossRef]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-Kinase–AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a Putative Protein Tyrosine Phosphatase Gene Mutated in Human Brain, Breast, and Prostate Cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef]
- Shaw, R.J.; Cantley, L.C. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef]
- Lu, Z.H.; Shvartsman, M.B.; Lee, A.Y.; Shao, J.M.; Murray, M.M.; Kladney, R.D.; Fan, D.; Krajewski, S.; Chiang, G.G.; Mills, G.B.; et al. Mammalian Target of Rapamycin Activator RHEB Is Frequently Overexpressed in Human Carcinomas and Is Critical and Sufficient for Skin Epithelial Carcinogenesis. Cancer Res. 2010, 70, 3287–3298. [Google Scholar] [CrossRef]
- Armengol, G.; Rojo, F.; Castellví, J.; Iglesias, C.; Cuatrecasas, M.; Pons, B.; Baselga, J.; Ramón y Cajal, S. 4E-Binding Protein 1: A Key Molecular “Funnel Factor” in Human Cancer with Clinical Implications. Cancer Res. 2007, 67, 7551–7555. [Google Scholar] [CrossRef] [PubMed]
- No, J.H.; Jeon, Y.-T.; Park, I.-A.; Kim, Y.-B.; Kim, J.W.; Park, N.-H.; Kang, S.-B.; Han, J.Y.; Lim, J.M.; Song, Y.-S. Activation of mTOR signaling pathway associated with adverse prognostic factors of epithelial ovarian cancer. Gynecol. Oncol. 2011, 121, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, D.J. Tuberous Sclerosis: From Tubers to mTOR. Ann. Hum. Genet. 2003, 67, 87–96. [Google Scholar] [CrossRef]
- Tamguney, T.; Stokoe, D. New insights into PTEN. J. Cell Sci. 2007, 120, 4071–4079. [Google Scholar] [CrossRef] [PubMed]
- Tee, A.R.; Blenis, J. mTOR, translational control and human disease. Semin. Cell Dev. Biol. 2005, 16, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Corradetti, M.N.; Guan, K.-L. Dysregulation of the TSC-mTOR pathway in human disease. Nat. Genet. 2005, 37, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Murugan, A.K. mTOR: Role in cancer, metastasis and drug resistance. Semin. Cancer Biol. 2019, 59, 92–111. [Google Scholar] [CrossRef]
- Sato, T.; Nakashima, A.; Guo, L.; Coffman, K.; Tamanoi, F. Single amino-acid changes that confer constitutive activation of mTOR are discovered in human cancer. Oncogene 2010, 29, 2746–2752. [Google Scholar] [CrossRef]
- Miceli, C.; Leri, M.; Stefani, M.; Bucciantini, M. Autophagy-related proteins: Potential diagnostic and prognostic biomarkers of aging-related diseases. Ageing Res. Rev. 2023, 89, 101967. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.U.; Pathania, A.S.; Wani, A.; Fatima, K.; Mintoo, M.J.; Hamza, B.; Paddar, M.A.; Bhumika, W.; Anand, L.K.; Maqbool, M.S.; et al. Activation of lysosomal mediated cell death in the course of autophagy by mTORC1 inhibitor. Sci. Rep. 2022, 12, 5052. [Google Scholar] [CrossRef]
- Zou, L.; Liao, M.; Zhen, Y.; Zhu, S.; Chen, X.; Zhang, J.; Hao, Y.; Liu, B. Autophagy and beyond: Unraveling the complexity of UNC-51-like kinase 1 (ULK1) from biological functions to therapeutic implications. Acta Pharm. Sin. B 2022, 12, 3743–3782. [Google Scholar] [CrossRef]
- Ganley, I.G.; Lam, D.H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1·ATG13·FIP200 Complex Mediates mTOR Signaling and Is Essential for Autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Jun, C.B.; Ro, S.-H.; Kim, Y.-M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.-H. ULK-Atg13-FIP200 Complexes Mediate mTOR Signaling to the Autophagy Machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Klionsky, D.J. Transcriptional regulation of autophagy and its implications in human disease. Cell Death Differ. 2023, 30, 1416–1429. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ai, J.; Jin, X.; Liu, L.; Lin, T.; Xu, H.; Wei, Q.; Yang, L. IL-8 protects prostate cancer cells from GSK-3β-induced oxidative stress by activating the mTOR signaling pathway. Prostate 2019, 79, 1180–1190. [Google Scholar] [CrossRef]
- Pahlavani, H.A. Exercise-induced signaling pathways to counteracting cardiac apoptotic processes. Front. Cell Dev. Biol. 2022, 10, 950927. [Google Scholar] [CrossRef]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef]
- Ali, E.S.; Mitra, K.; Akter, S.; Ramproshad, S.; Mondal, B.; Khan, I.N.; Islam, M.T.; Sharifi-Rad, J.; Calina, D.; Cho, W.C. Recent advances and limitations of mTOR inhibitors in the treatment of cancer. Cancer Cell Int. 2022, 22, 284. [Google Scholar] [CrossRef]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR Signaling in Growth and Metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef]
- Guertin, D.A.; Sabatini, D.M. The Pharmacology of mTOR Inhibition. Sci. Signal. 2009, 2, pe24. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; Oudard, S.; Hutson, T.E.; Porta, C.; Bracarda, S.; Grünwald, V.; Thompson, J.A.; Figlin, R.A.; Hollaender, N.; et al. Phase 3 trial of everolimus for metastatic renal cell carcinoma. Cancer 2010, 116, 4256–4265. [Google Scholar] [CrossRef] [PubMed]
- Hardinger, K.L.; Koch, M.J.; Brennan, D.C. Current and Future Immunosuppressive Strategies in Renal Transplantation. Pharmacotherapy 2004, 24, 1159–1176. [Google Scholar] [CrossRef] [PubMed]
- Mártinez, J.M.A.; Pulido, L.B.; Bellido, C.B.; Usero, D.D.; Aguilar, L.T.; Moreno, J.L.G.; Artacho, G.S.; Díez-Canedo, J.S.; Gómez, L.M.M.; Bravo, M.Á.G. Rescue Immunosuppression with Mammalian Target of Rapamycin Inhibitor Drugs in Liver Transplantation. Transplant. Proc. 2010, 42, 641–643. [Google Scholar] [CrossRef]
- Zhu, A.X.; Abrams, T.A.; Miksad, R.; Blaszkowsky, L.S.; Meyerhardt, J.A.; Zheng, H.; Muzikansky, A.; Clark, J.W.; Kwak, E.L.; Schrag, D.; et al. Phase 1/2 study of everolimus in advanced hepatocellular carcinoma. Cancer 2011, 117, 5094–5102. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Kudo, M.; Assenat, E.; Cattan, S.; Kang, Y.-K.; Lim, H.Y.; Poon, R.T.P.; Blanc, J.-F.; Vogel, A.; Chen, C.-L.; et al. Effect of Everolimus on Survival in Advanced Hepatocellular Carcinoma After Failure of Sorafenib. JAMA 2014, 312, 57. [Google Scholar] [CrossRef]
- Ghobrial, I.M.; Weller, E.; Vij, R.; Munshi, N.C.; Banwait, R.; Bagshaw, M.; Schlossman, R.; Leduc, R.; Chuma, S.; Kunsman, J.; et al. Weekly bortezomib in combination with temsirolimus in relapsed or relapsed and refractory multiple myeloma: A multicentre, phase 1/2, open-label, dose-escalation study. Lancet Oncol. 2011, 12, 263–272. [Google Scholar] [CrossRef]
- Fenske, T.S.; Shah, N.M.; Kim, K.M.; Saha, S.; Zhang, C.; Baim, A.E.; Farnen, J.P.; Onitilo, A.A.; Blank, J.H.; Ahuja, H.; et al. A phase 2 study of weekly temsirolimus and bortezomib for relapsed or refractory B-cell non-Hodgkin lymphoma: A Wisconsin Oncology Network study. Cancer 2015, 121, 3465–3471. [Google Scholar] [CrossRef]
- Hess, G.; Keller, U.; Scholz, C.W.; Witzens-Harig, M.; Atta, J.; Buske, C.; Kirschey, S.; Ruckes, C.; Medler, C.; van Oordt, C.; et al. Safety and efficacy of Temsirolimus in combination with Bendamustine and Rituximab in relapsed mantle cell and follicular lymphoma. Leukemia 2015, 29, 1695–1701. [Google Scholar] [CrossRef]
- Mi, W.; Ye, Q.; Liu, S.; She, Q.-B. AKT inhibition overcomes rapamycin resistance by enhancing the repressive function of PRAS40 on mTORC1/4E-BP1 axis. Oncotarget 2015, 6, 13962–13977. [Google Scholar] [CrossRef]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An ATP-competitive Mammalian Target of Rapamycin Inhibitor Reveals Rapamycin-resistant Functions of mTORC1. J. Biol. Chem. 2009, 284, 8023–8032. [Google Scholar] [CrossRef]
- Dienstmann, R.; Rodon, J.; Serra, V.; Tabernero, J. Picking the Point of Inhibition: A Comparative Review of PI3K/AKT/mTOR Pathway Inhibitors. Mol. Cancer Ther. 2014, 13, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Apsel, B.; Blair, J.A.; Gonzalez, B.; Nazif, T.M.; Feldman, M.E.; Aizenstein, B.; Hoffman, R.; Williams, R.L.; Shokat, K.M.; Knight, Z.A. Targeted polypharmacology: Discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat. Chem. Biol. 2008, 4, 691–699. [Google Scholar] [CrossRef]
- Powles, T.; Lackner, M.R.; Oudard, S.; Escudier, B.; Ralph, C.; Brown, J.E.; Hawkins, R.E.; Castellano, D.; Rini, B.I.; Staehler, M.D.; et al. Randomized Open-Label Phase II Trial of Apitolisib (GDC-0980), a Novel Inhibitor of the PI3K/Mammalian Target of Rapamycin Pathway, Versus Everolimus in Patients with Metastatic Renal Cell Carcinoma. J. Clin. Oncol. 2016, 34, 1660–1668. [Google Scholar] [CrossRef]
- Brown, J.R.; Hamadani, M.; Hayslip, J.; Janssens, A.; Wagner-Johnston, N.; Ottmann, O.; Arnason, J.; Tilly, H.; Millenson, M.; Offner, F.; et al. Voxtalisib (XL765) in patients with relapsed or refractory non-Hodgkin lymphoma or chronic lymphocytic leukaemia: An open-label, phase 2 trial. Lancet Haematol. 2018, 5, e170–e180. [Google Scholar] [CrossRef]
- Curigliano, G.; Shapiro, G.I.; Kristeleit, R.S.; Abdul Razak, A.R.; Leong, S.; Alsina, M.; Giordano, A.; Gelmon, K.A.; Stringer-Reasor, E.; Vaishampayan, U.N.; et al. A Phase 1B open-label study of gedatolisib (PF-05212384) in combination with other anti-tumour agents for patients with advanced solid tumours and triple-negative breast cancer. Br. J. Cancer 2023, 128, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Wise-Draper, T.M.; Moorthy, G.; Salkeni, M.A.; Karim, N.A.; Thomas, H.E.; Mercer, C.A.; Beg, M.S.; O’Gara, S.; Olowokure, O.; Fathallah, H.; et al. A Phase Ib Study of the Dual PI3K/mTOR Inhibitor Dactolisib (BEZ235) Combined with Everolimus in Patients with Advanced Solid Malignancies. Target. Oncol. 2017, 12, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Voss, M.H.; Gordon, M.S.; Mita, M.; Rini, B.; Makker, V.; Macarulla, T.; Smith, D.C.; Cervantes, A.; Puzanov, I.; Pili, R.; et al. Phase 1 study of mTORC1/2 inhibitor sapanisertib (TAK-228) in advanced solid tumours, with an expansion phase in renal, endometrial or bladder cancer. Br. J. Cancer 2020, 123, 1590–1598. [Google Scholar] [CrossRef]
- Wolin, E.; Mita, A.; Mahipal, A.; Meyer, T.; Bendell, J.; Nemunaitis, J.; Munster, P.N.; Paz-Ares, L.; Filvaroff, E.H.; Li, S.; et al. A phase 2 study of an oral mTORC1/mTORC2 kinase inhibitor (CC-223) for non-pancreatic neuroendocrine tumors with or without carcinoid symptoms. PLoS ONE 2019, 14, e0221994. [Google Scholar] [CrossRef]
- Chresta, C.M.; Davies, B.R.; Hickson, I.; Harding, T.; Cosulich, S.; Critchlow, S.E.; Vincent, J.P.; Ellston, R.; Jones, D.; Sini, P.; et al. AZD8055 Is a Potent, Selective, and Orally Bioavailable ATP-Competitive Mammalian Target of Rapamycin Kinase Inhibitor with In vitro and In vivo Antitumor Activity. Cancer Res. 2010, 70, 288–298. [Google Scholar] [CrossRef]
- Wen, P.Y.; Lee, E.Q.; Reardon, D.A.; Ligon, K.L.; Alfred Yung, W.K. Current clinical development of PI3K pathway inhibitors in glioblastoma. Neuro. Oncol. 2012, 14, 819–829. [Google Scholar] [CrossRef]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.; Li, T.; Ma, H.; Yang, Y.; Zhang, C.; Hai, L.; Liu, P.; Yuan, F.; Li, J.; Yi, L.; et al. Autophagy suppresses self-renewal ability and tumorigenicity of glioma-initiating cells and promotes Notch1 degradation. Cell Death Dis. 2018, 9, 1063. [Google Scholar] [CrossRef] [PubMed]
- Rodrik-Outmezguine, V.S.; Okaniwa, M.; Yao, Z.; Novotny, C.J.; McWhirter, C.; Banaji, A.; Won, H.; Wong, W.; Berger, M.; de Stanchina, E.; et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature 2016, 534, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Aksoy, O.; Wong, R.A.; Ilkhanizadeh, S.; Novotny, C.J.; Gustafson, W.C.; Truong, A.Y.-Q.; Cayanan, G.; Simonds, E.F.; Haas-Kogan, D.; et al. A Kinase Inhibitor Targeted to mTORC1 Drives Regression in Glioblastoma. Cancer Cell 2017, 31, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Kuroshima, K.; Yoshino, H.; Okamura, S.; Tsuruda, M.; Osako, Y.; Sakaguchi, T.; Sugita, S.; Tatarano, S.; Nakagawa, M.; Enokida, H. Potential new therapy of Rapalink-1, a new generation mammalian target of rapamycin inhibitor, against sunitinib-resistant renal cell carcinoma. Cancer Sci. 2020, 111, 1607–1618. [Google Scholar] [CrossRef]
- Wan, X.; Harkavy, B.; Shen, N.; Grohar, P.; Helman, L.J. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene 2007, 26, 1932–1940. [Google Scholar] [CrossRef]
- O’Shaughnessy, J.; Thaddeus Beck, J.; Royce, M. Everolimus-based combination therapies for HR+, HER2− metastatic breast cancer. Cancer Treat. Rev. 2018, 69, 204–214. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Glen, H.; Michaelson, M.D.; Molina, A.; Eisen, T.; Jassem, J.; Zolnierek, J.; Maroto, J.P.; Mellado, B.; et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: A randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 2015, 16, 1473–1482. [Google Scholar] [CrossRef]
- Colon-Otero, G.; Weroha, S.J.; Foster, N.R.; Haluska, P.; Hou, X.; Wahner-Hendrickson, A.E.; Jatoi, A.; Block, M.S.; Dinh, T.A.; Robertson, M.W.; et al. Phase 2 trial of everolimus and letrozole in relapsed estrogen receptor-positive high-grade ovarian cancers. Gynecol. Oncol. 2017, 146, 64–68. [Google Scholar] [CrossRef]
- Schwartz, G.K.; Tap, W.D.; Qin, L.-X.; Livingston, M.B.; Undevia, S.D.; Chmielowski, B.; Agulnik, M.; Schuetze, S.M.; Reed, D.R.; Okuno, S.H.; et al. Cixutumumab and temsirolimus for patients with bone and soft-tissue sarcoma: A multicentre, open-label, phase 2 trial. Lancet Oncol. 2013, 14, 371–382. [Google Scholar] [CrossRef]
- Crook, T.; Patil, D.; Gaya, A.; Plowman, N.; Limaye, S.; Ranade, A.; Bhatt, A.; Page, R.; Akolkar, D. Improved Treatment Outcomes by Using Patient Specific Drug Combinations in Mammalian Target of Rapamycin Activated Advanced Metastatic Cancers. Front. Pharmacol. 2021, 12, 631135. [Google Scholar] [CrossRef] [PubMed]
- Rathkopf, D.E.; Larson, S.M.; Anand, A.; Morris, M.J.; Slovin, S.F.; Shaffer, D.R.; Heller, G.; Carver, B.; Rosen, N.; Scher, H.I. Everolimus combined with gefitinib in patients with metastatic castration-resistant prostate cancer: Phase 1/2 results and signaling pathway implications. Cancer 2015, 121, 3853–3861. [Google Scholar] [CrossRef] [PubMed]
- Schram, A.M.; Gandhi, L.; Mita, M.M.; Damstrup, L.; Campana, F.; Hidalgo, M.; Grande, E.; Hyman, D.M.; Heist, R.S. A phase Ib dose-escalation and expansion study of the oral MEK inhibitor pimasertib and PI3K/MTOR inhibitor voxtalisib in patients with advanced solid tumours. Br. J. Cancer 2018, 119, 1471–1476. [Google Scholar] [CrossRef] [PubMed]
- Yardley, D.A. Combining mTOR Inhibitors with Chemotherapy and Other Targeted Therapies in Advanced Breast Cancer: Rationale, Clinical Experience, and Future Directions. Breast Cancer Basic Clin. Res. 2013, 7, BCBCR–S10071. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Bendell, J.C.; Papadopoulos, K.P.; Burris, H.A.; Patnaik, A.; Jones, S.F.; Rasco, D.; Cox, D.S.; Durante, M.; Bellew, K.M.; et al. A phase IB trial of the oral MEK inhibitor trametinib (GSK1120212) in combination with everolimus in patients with advanced solid tumors. Ann. Oncol. 2015, 26, 58–64. [Google Scholar] [CrossRef]
- Christopoulos, P.; Engel-Riedel, W.; Grohé, C.; Kropf-Sanchen, C.; von Pawel, J.; Gütz, S.; Kollmeier, J.; Eberhardt, W.; Ukena, D.; Baum, V.; et al. Everolimus with paclitaxel and carboplatin as first-line treatment for metastatic large-cell neuroendocrine lung carcinoma: A multicenter phase II trial. Ann. Oncol. 2017, 28, 1898–1902. [Google Scholar] [CrossRef] [PubMed]
- Simmons, J.K.; Michalowski, A.M.; Gamache, B.J.; DuBois, W.; Patel, J.; Zhang, K.; Gary, J.; Zhang, S.; Gaikwad, S.; Connors, D.; et al. Cooperative Targets of Combined mTOR/HDAC Inhibition Promote MYC Degradation. Mol. Cancer Ther. 2017, 16, 2008–2021. [Google Scholar] [CrossRef]
- Zhang, H.; Li, X.; Yang, Y.; Zhang, Y.; Wang, H.; Zheng, X.F.S. Significance and mechanism of androgen receptor overexpression and androgen receptor/mechanistic target of rapamycin cross-talk in hepatocellular carcinoma. Hepatology 2018, 67, 2271–2286. [Google Scholar] [CrossRef]
- Miyata, H.; Ashizawa, T.; Iizuka, A.; Kondou, R.; Nonomura, C.; Sugino, T.; Urakami, K.; Asai, A.; Hayashi, N.; Mitsuya, K.; et al. Combination of a STAT3 Inhibitor and an mTOR Inhibitor Against a Temozolomide-resistant Glioblastoma Cell Line. Cancer Genom. Proteom. 2017, 14, 83–92. [Google Scholar] [CrossRef]
- Soefje, S.A.; Karnad, A.; Brenner, A.J. Common toxicities of mammalian target of rapamycin inhibitors. Target. Oncol. 2011, 6, 125–129. [Google Scholar] [CrossRef]
- Hudes, G.; Carducci, M.; Tomczak, P.; Dutcher, J.; Figlin, R.; Kapoor, A.; Staroslawska, E.; Sosman, J.; McDermott, D.; Bodrogi, I.; et al. Temsirolimus, Interferon Alfa, or Both for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2007, 356, 2271–2281. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Escudier, B.; Oudard, S.; Hutson, T.E.; Porta, C.; Bracarda, S.; Grünwald, V.; Thompson, J.A.; Figlin, R.A.; Hollaender, N.; et al. Efficacy of everolimus in advanced renal cell carcinoma: A double-blind, randomised, placebo-controlled phase III trial. Lancet 2008, 372, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Hartford, C.M.; Desai, A.A.; Janisch, L.; Karrison, T.; Rivera, V.M.; Berk, L.; Loewy, J.W.; Kindler, H.; Stadler, W.M.; Knowles, H.L.; et al. A Phase I Trial to Determine the Safety, Tolerability, and Maximum Tolerated Dose of Deforolimus in Patients with Advanced Malignancies. Clin. Cancer Res. 2009, 15, 1428–1434. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, B.; Qazi, Y.; Wellen, J.R. Strategies for the management of adverse events associated with mTOR inhibitors. Transplant. Rev. 2014, 28, 126–133. [Google Scholar] [CrossRef]
- Willemsen, A.E.C.A.B.; Grutters, J.C.; Gerritsen, W.R.; van Erp, N.P.; van Herpen, C.M.L.; Tol, J. mTOR inhibitor-induced interstitial lung disease in cancer patients: Comprehensive review and a practical management algorithm. Int. J. Cancer 2016, 138, 2312–2321. [Google Scholar] [CrossRef]
- Busaidy, N.L.; Farooki, A.; Dowlati, A.; Perentesis, J.P.; Dancey, J.E.; Doyle, L.A.; Brell, J.M.; Siu, L.L. Management of Metabolic Effects Associated with Anticancer Agents Targeting the PI3K-Akt-mTOR Pathway. J. Clin. Oncol. 2012, 30, 2919–2928. [Google Scholar] [CrossRef]
- Peterson, D.E.; O’Shaughnessy, J.A.; Rugo, H.S.; Elad, S.; Schubert, M.M.; Viet, C.T.; Campbell-Baird, C.; Hronek, J.; Seery, V.; Divers, J.; et al. Oral mucosal injury caused by mammalian target of rapamycin inhibitors: Emerging perspectives on pathobiology and impact on clinical practice. Cancer Med. 2016, 5, 1897–1907. [Google Scholar] [CrossRef]
- Sofroniadou, S.; Kassimatis, T.; Goldsmith, D. Anaemia, microcytosis and sirolimus--is iron the missing link? Nephrol. Dial. Transplant. 2010, 25, 1667–1675. [Google Scholar] [CrossRef]
- Rizzieri, D.A.; Feldman, E.; DiPersio, J.F.; Gabrail, N.; Stock, W.; Strair, R.; Rivera, V.M.; Albitar, M.; Bedrosian, C.L.; Giles, F.J. A Phase 2 Clinical Trial of Deforolimus (AP23573, MK-8669), a Novel Mammalian Target of Rapamycin Inhibitor, in Patients with Relapsed or Refractory Hematologic Malignancies. Clin. Cancer Res. 2008, 14, 2756–2762. [Google Scholar] [CrossRef]
- Xu, J.; Tian, D. Hematologic toxicities associated with mTOR inhibitors temsirolimus and everolimus in cancer patients: A systematic review and meta-analysis. Curr. Med. Res. Opin. 2014, 30, 67–74. [Google Scholar] [CrossRef]
- Graham, J.; Wells, J.C.; Dudani, S.; Gan, C.L.; Donskov, F.; Lee, J.-L.; Kollmannsberger, C.K.; Meza, L.; Beuselinck, B.; Hansen, A.; et al. Outcomes of patients with advanced non-clear cell renal cell carcinoma treated with first-line immune checkpoint inhibitor therapy. Eur. J. Cancer 2022, 171, 124–132. [Google Scholar] [CrossRef]
- Moore, E.C.; Cash, H.A.; Caruso, A.M.; Uppaluri, R.; Hodge, J.W.; Van Waes, C.; Allen, C.T. Enhanced Tumor Control with Combination mTOR and PD-L1 Inhibition in Syngeneic Oral Cavity Cancers. Cancer Immunol. Res. 2016, 4, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Langdon, S.; Hughes, A.; Taylor, M.A.; Kuczynski, E.A.; Mele, D.A.; Delpuech, O.; Jarvis, L.; Staniszewska, A.; Cosulich, S.; Carnevalli, L.S.; et al. Combination of dual mTORC1/2 inhibition and immune-checkpoint blockade potentiates anti-tumour immunity. Oncoimmunology 2018, 7, e1458810. [Google Scholar] [CrossRef]
- Wainberg, Z.A.; Soares, H.P.; Patel, R.; DiCarlo, B.; Park, D.J.; Liem, A.; Wang, H.; Yonemoto, L.; Martinez, D.; Laux, I.; et al. Phase II trial of everolimus in patients with refractory metastatic adenocarcinoma of the esophagus, gastroesophageal junction and stomach: Possible role for predictive biomarkers. Cancer Chemother. Pharmacol. 2015, 76, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Doi, T.; Muro, K.; Boku, N.; Yamada, Y.; Nishina, T.; Takiuchi, H.; Komatsu, Y.; Hamamoto, Y.; Ohno, N.; Fujita, Y.; et al. Multicenter Phase II Study of Everolimus in Patients with Previously Treated Metastatic Gastric Cancer. J. Clin. Oncol. 2010, 28, 1904–1910. [Google Scholar] [CrossRef]
- Shen, Y.-C.; Li, C.-P.; Yen, C.-J.; Hsu, C.; Lin, Y.-L.; Lin, Z.-Z.; Chen, L.-T.; Su, W.-C.; Chao, Y.; Yeh, K.-H.; et al. Phase II Multicentered Study of Low-Dose Everolimus plus Cisplatin and Weekly 24-Hour Infusion of High-Dose 5-Fluorouracil and Leucovorin as First-Line Treatment for Patients with Advanced Gastric Cancer. Oncology 2014, 87, 104–113. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).