
Discovery of Novel Coumarin-Schiff Base Hybrids as Potential Acetylcholinesterase Inhibitors: Design, Synthesis, Enzyme Inhibition, and Computational Studies

 , , , , , , , and
, , , , , , , and
Abstract
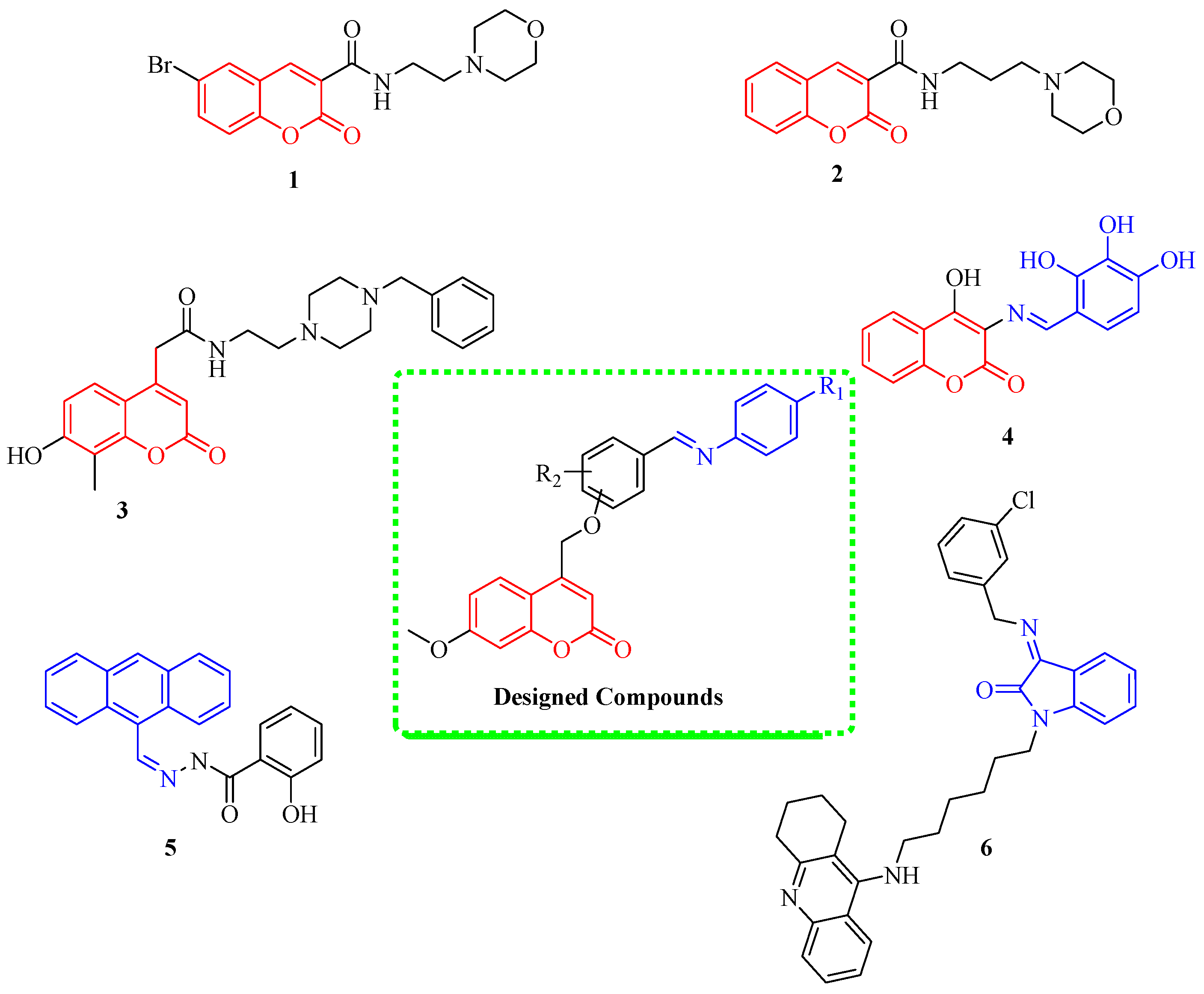
1. Introduction
2. Results and Discussion
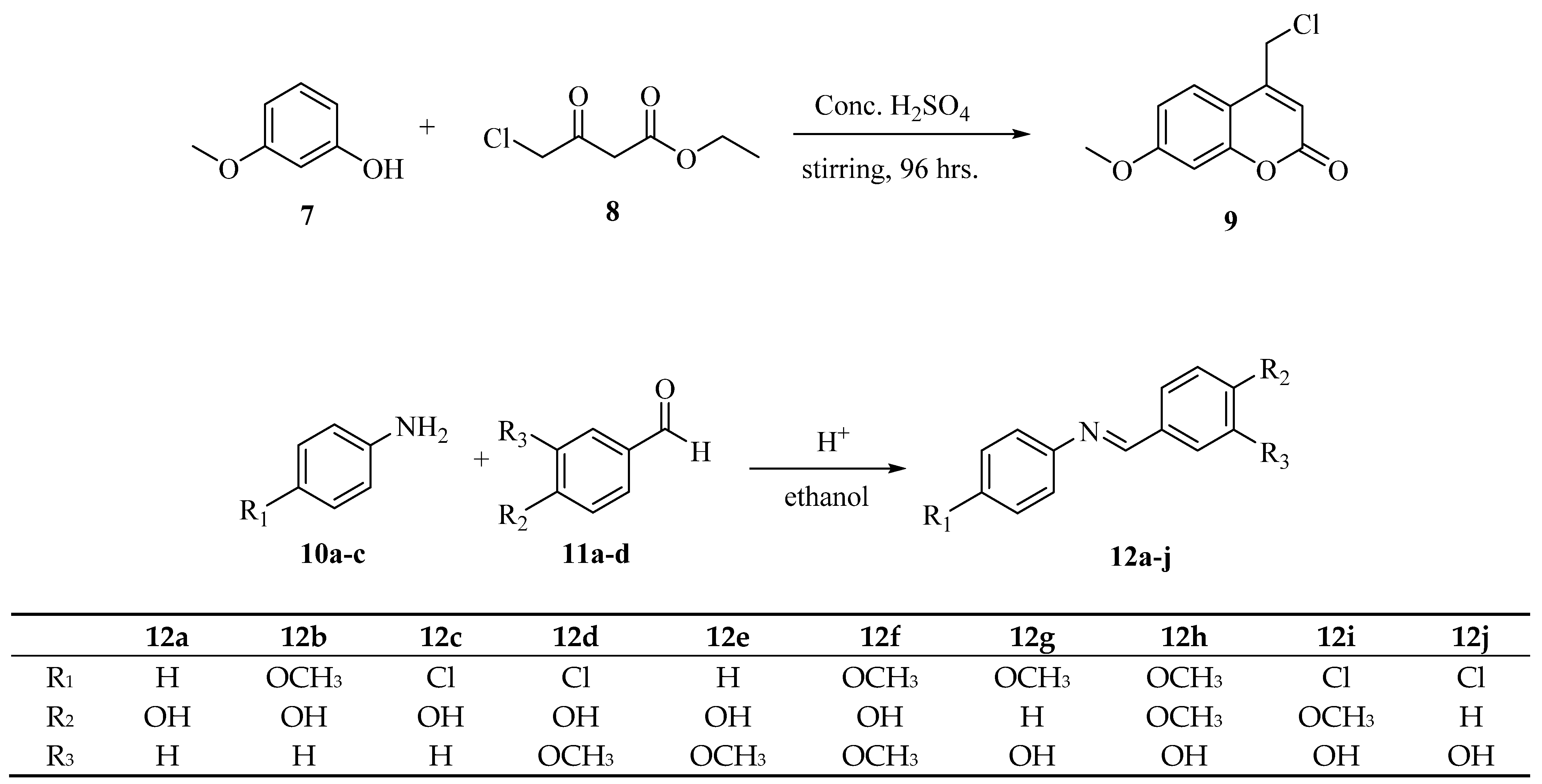
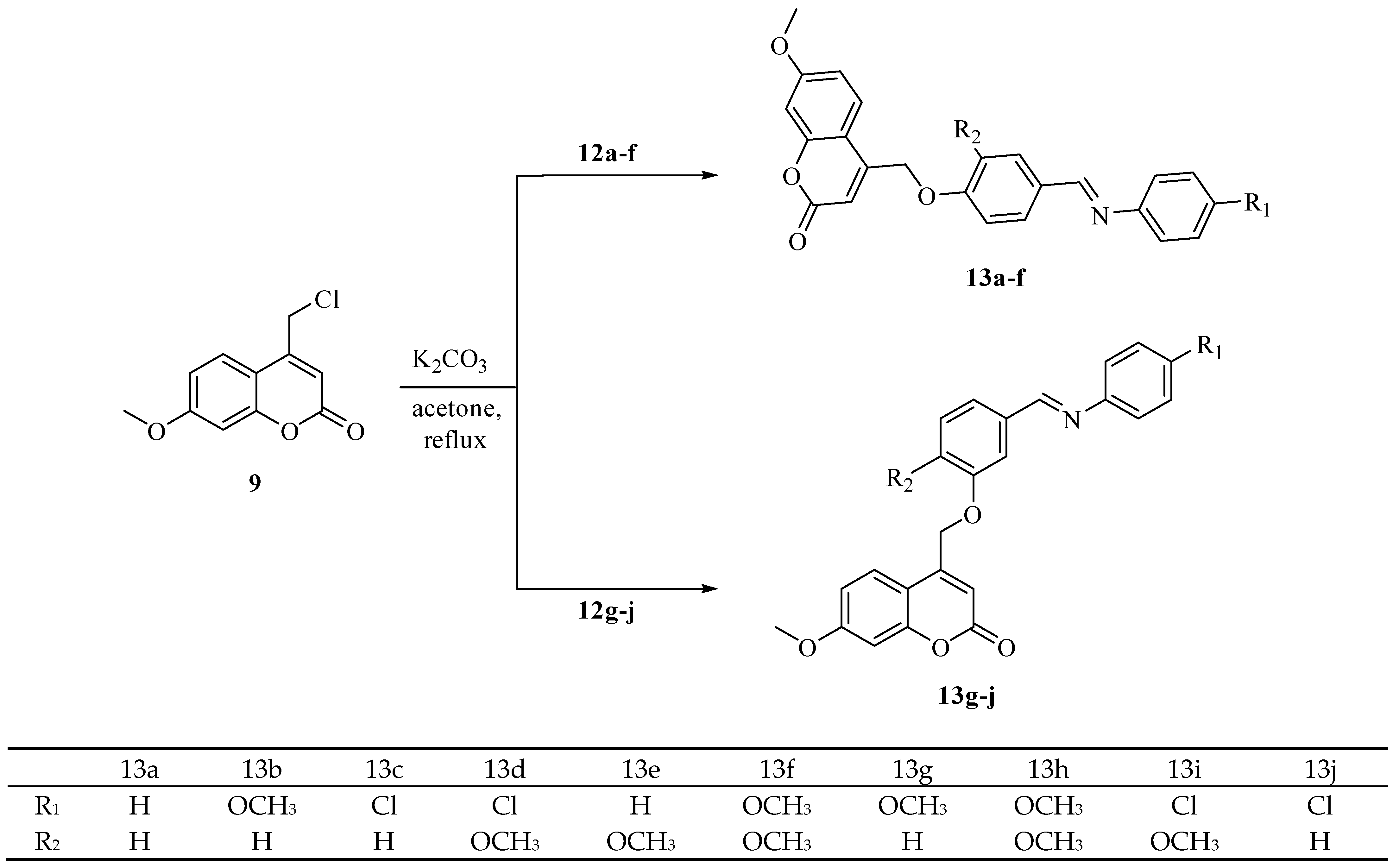
2.1. Chemistry
2.2. Anti-Acetylcholinesterase of Coumarin-Imine Hybrids (13a-j)
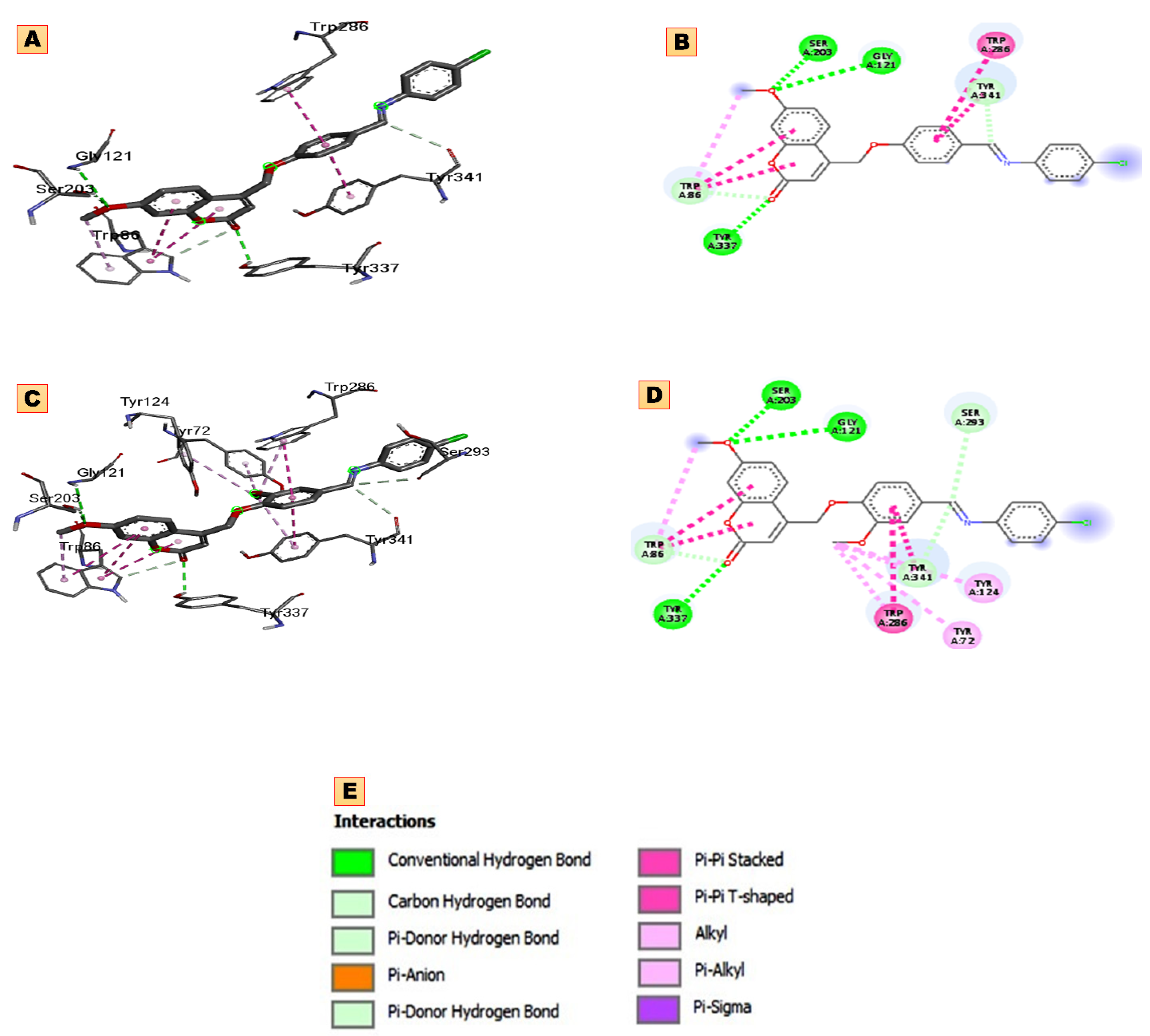
2.3. Molecular Docking
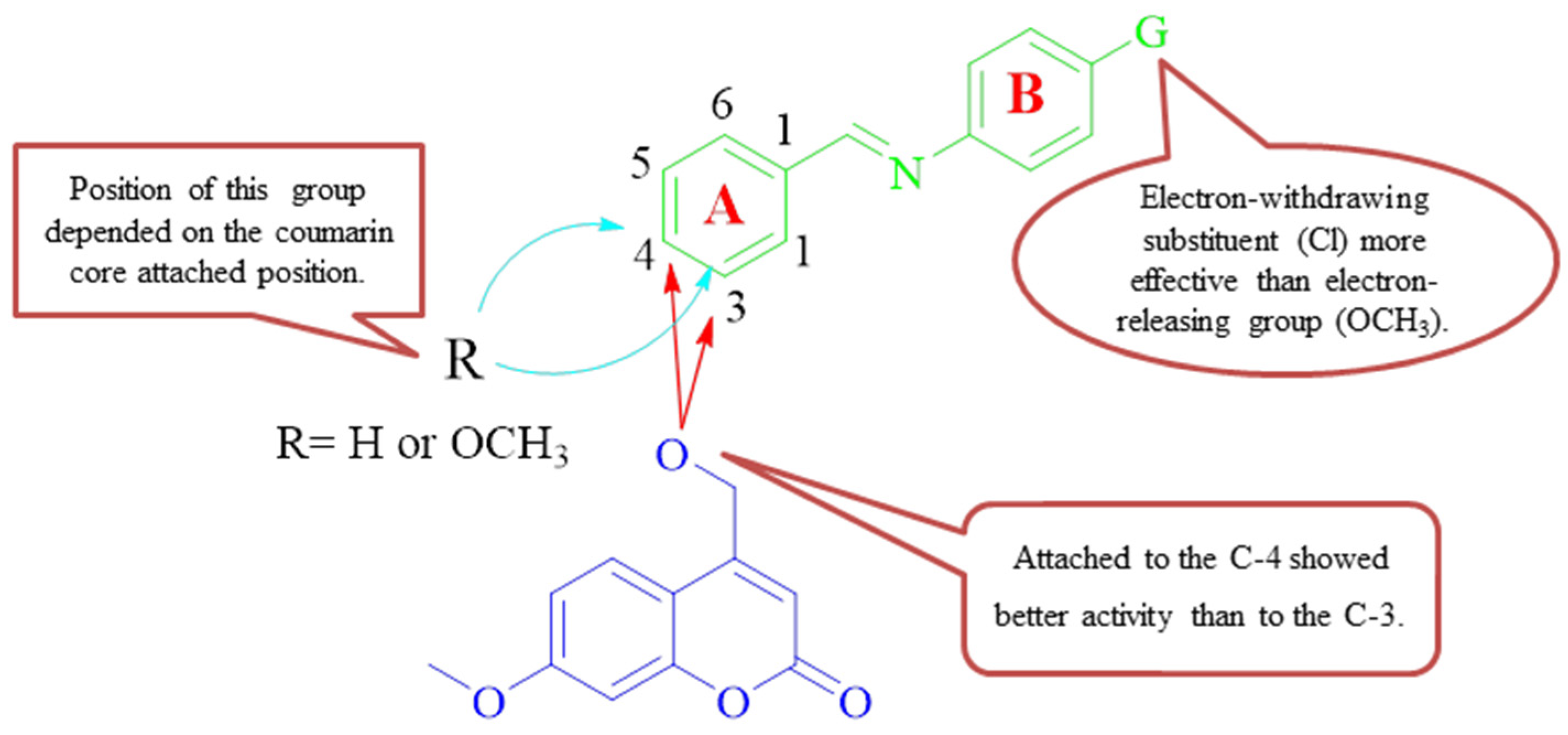
2.4. Structural Activity Relationships (SARs)
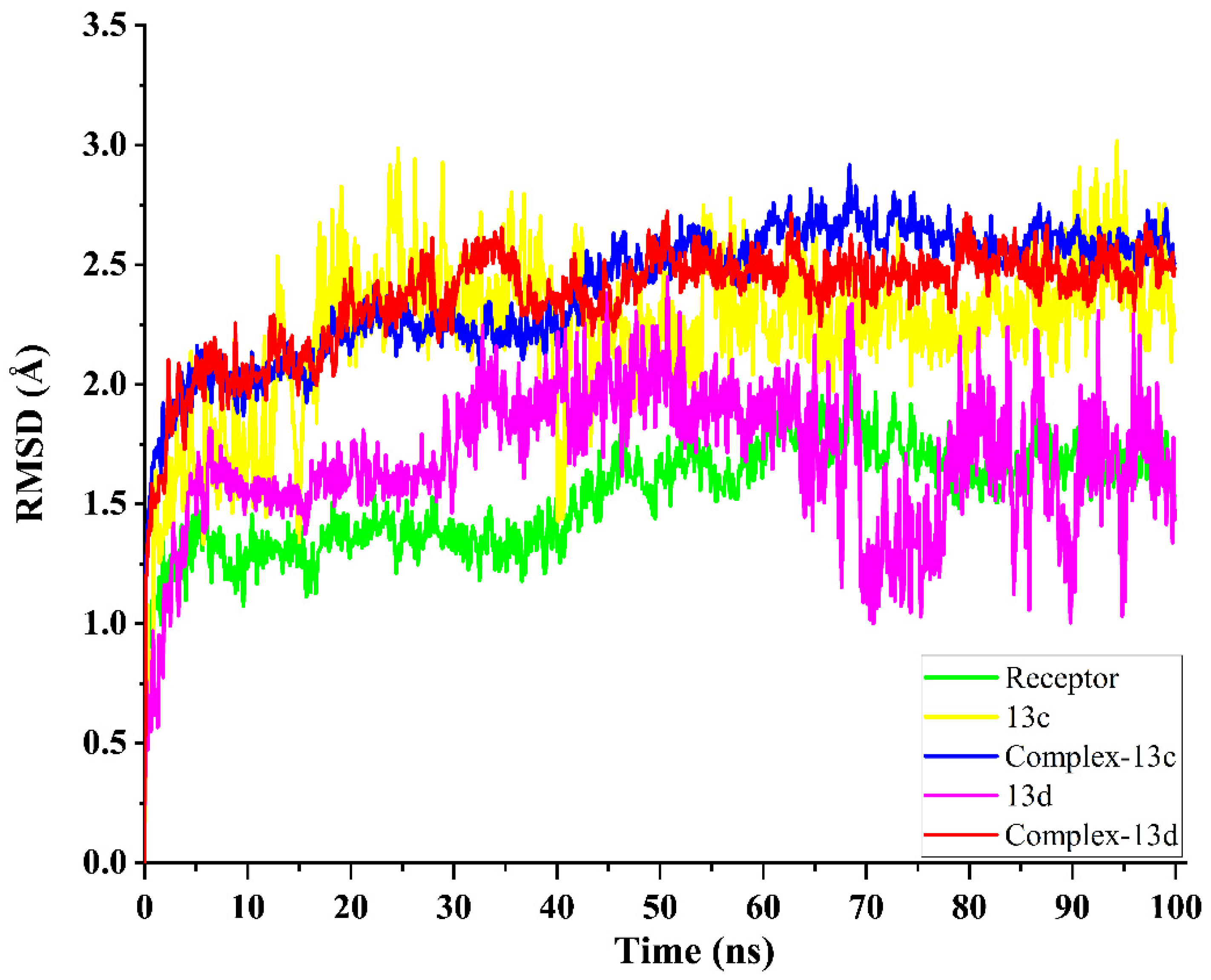
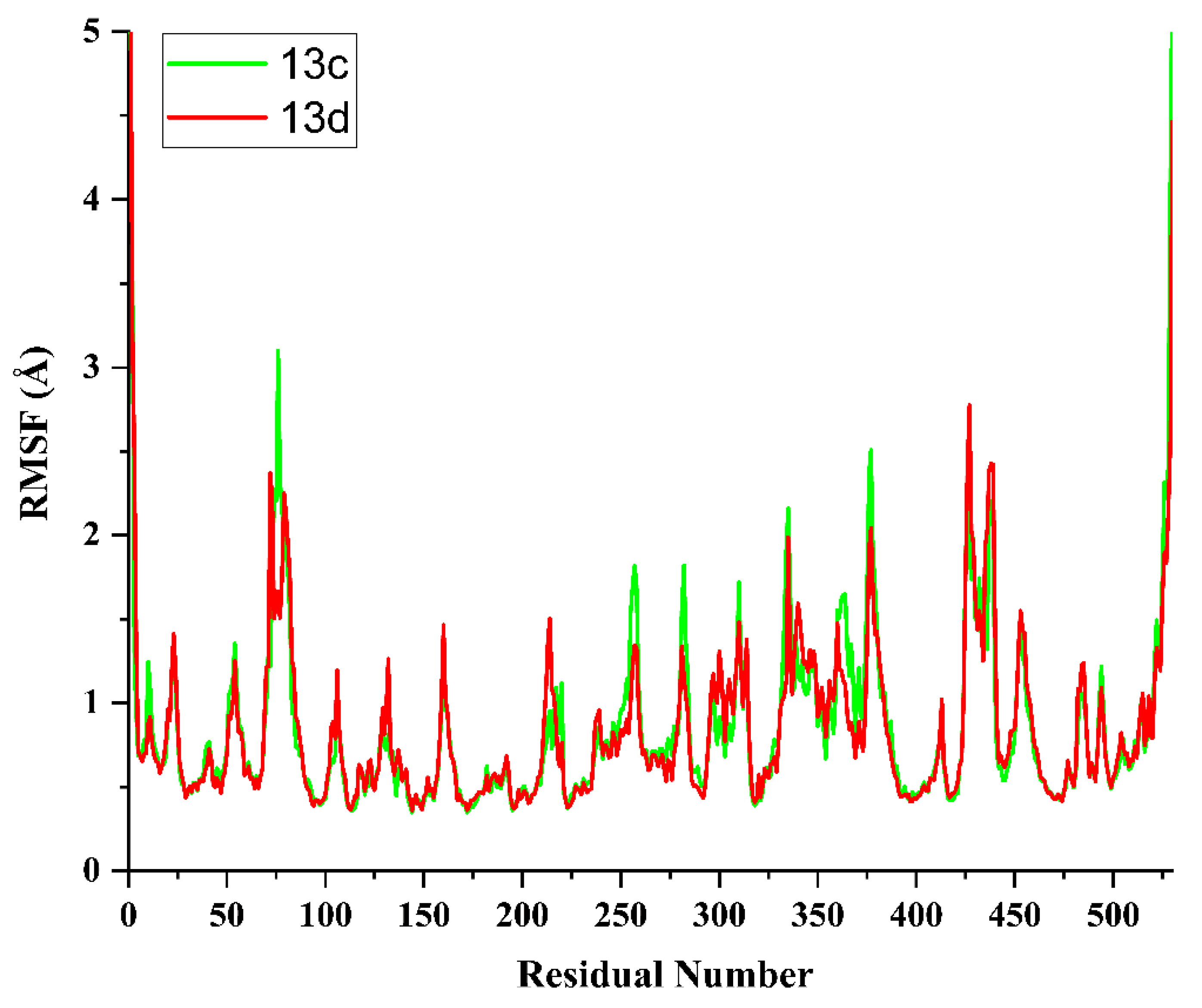
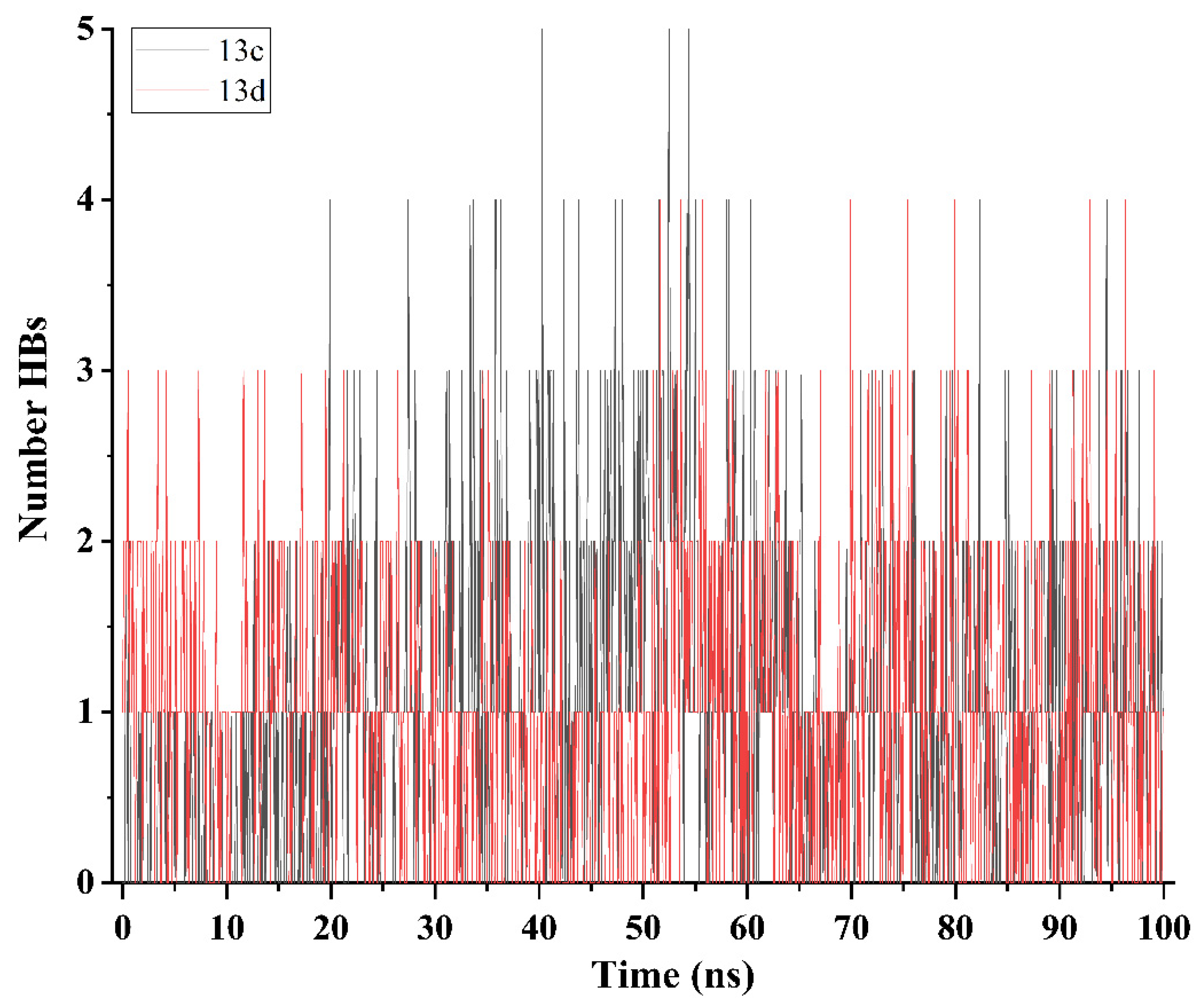
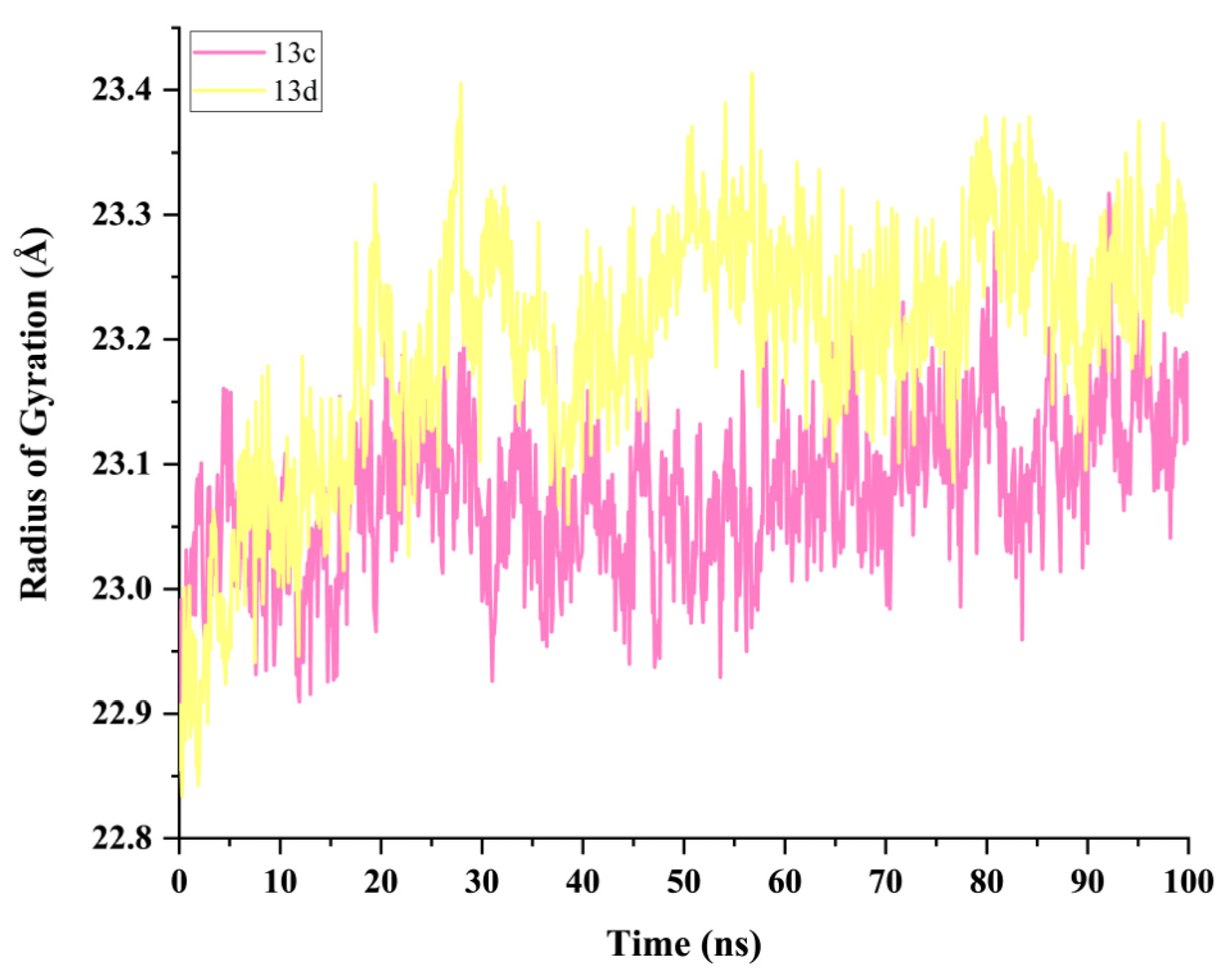
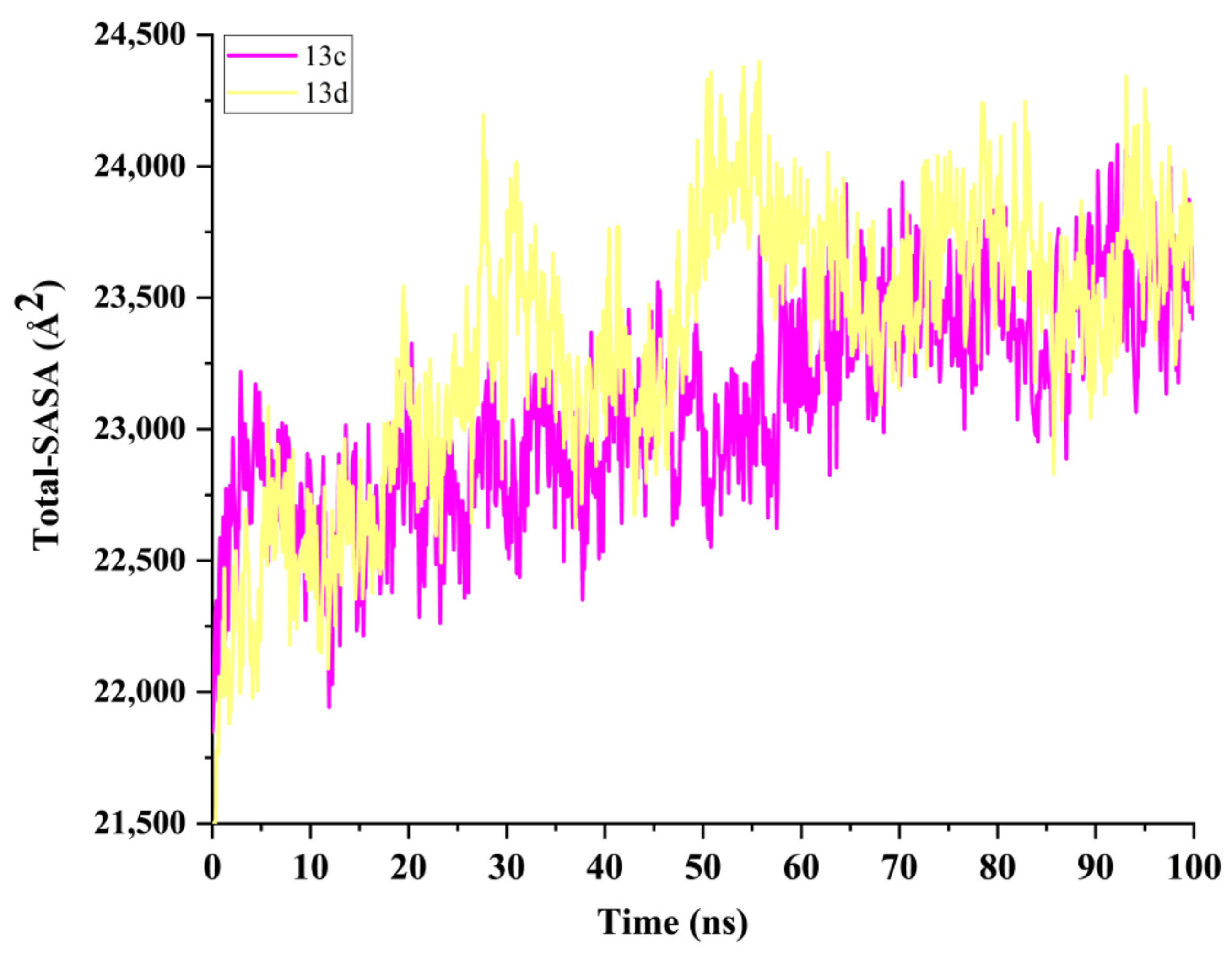
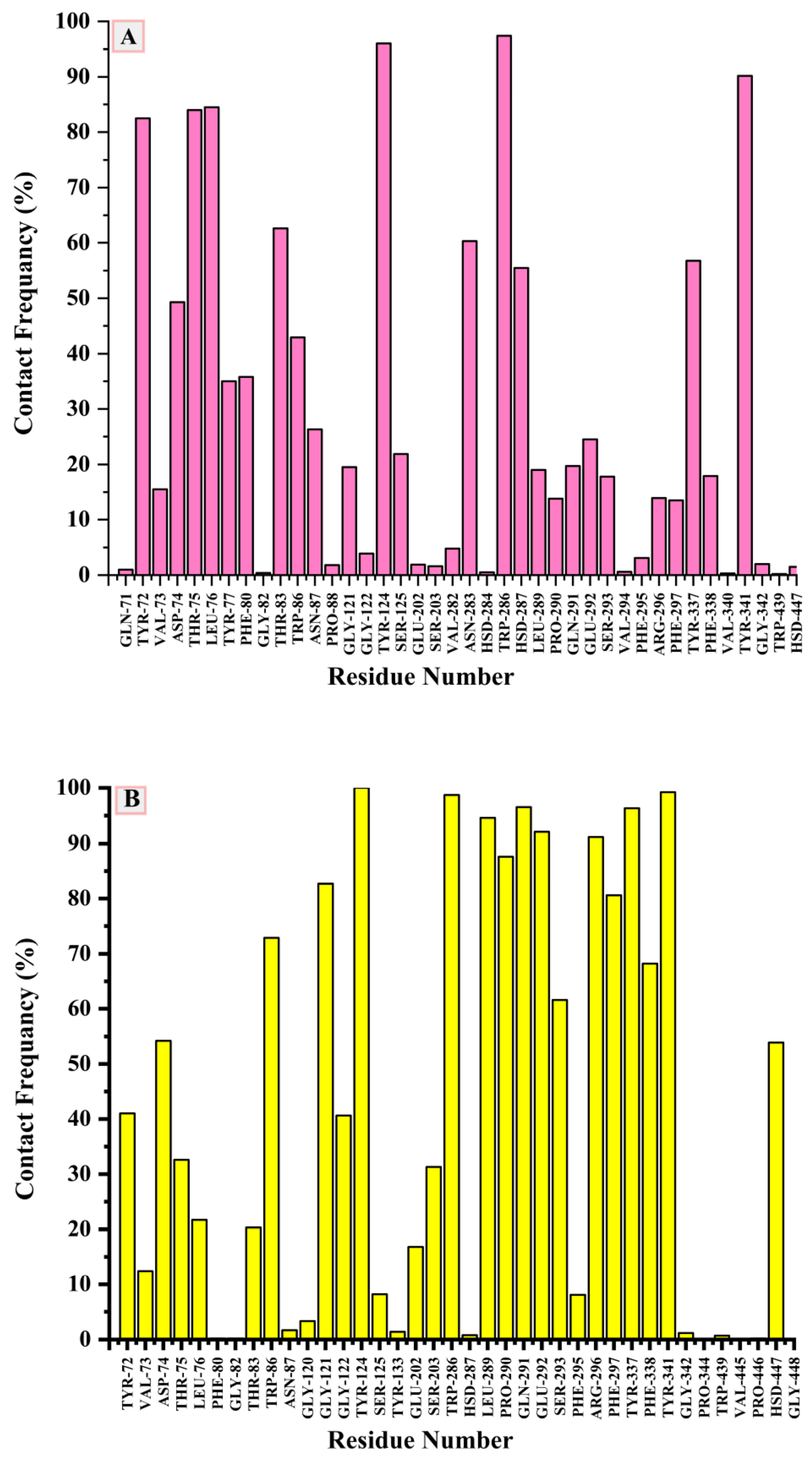
2.5. Molecular Dynamics Simulation and System Stability
2.6. Binding Free Energy by MM/GBSA Methods
2.7. Density Functional Theory (DFT)
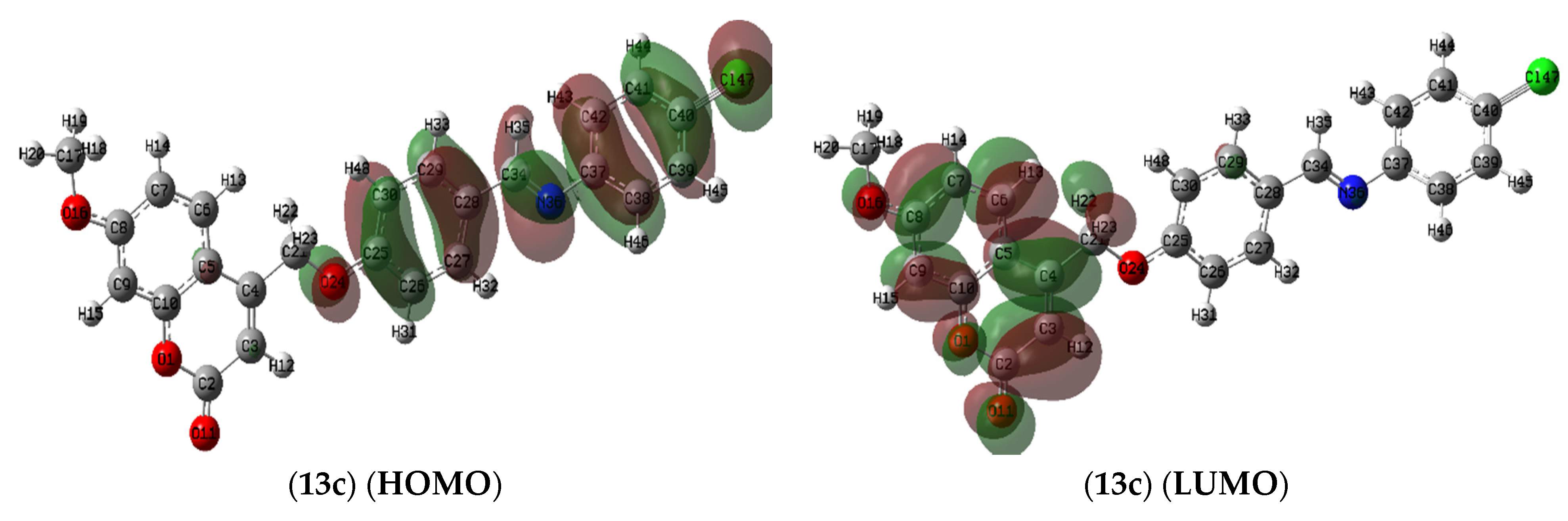
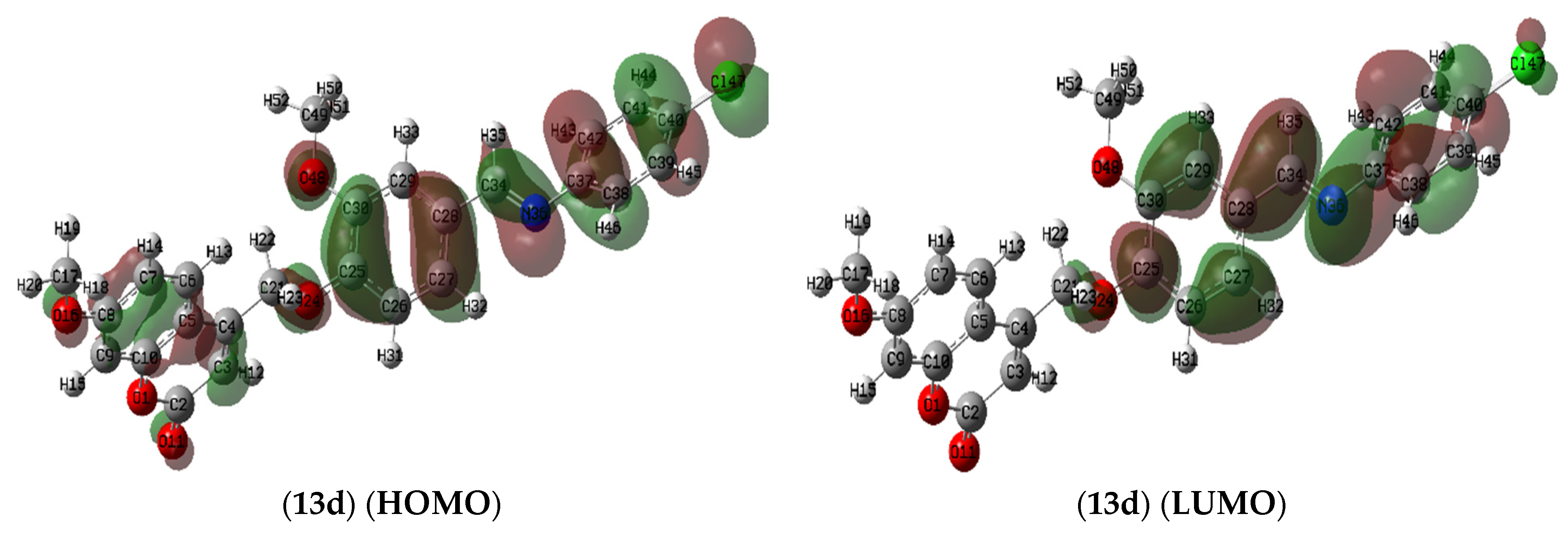
2.7.1. Molecule Orbital Calculations
Ground State Geometric Parameters (S281–S299)
Natural Charges and Natural Population
Frontier Molecular Orbitals (FMOs) Analysis
Global Reactivity Descriptors
Local Reactivity Descriptor
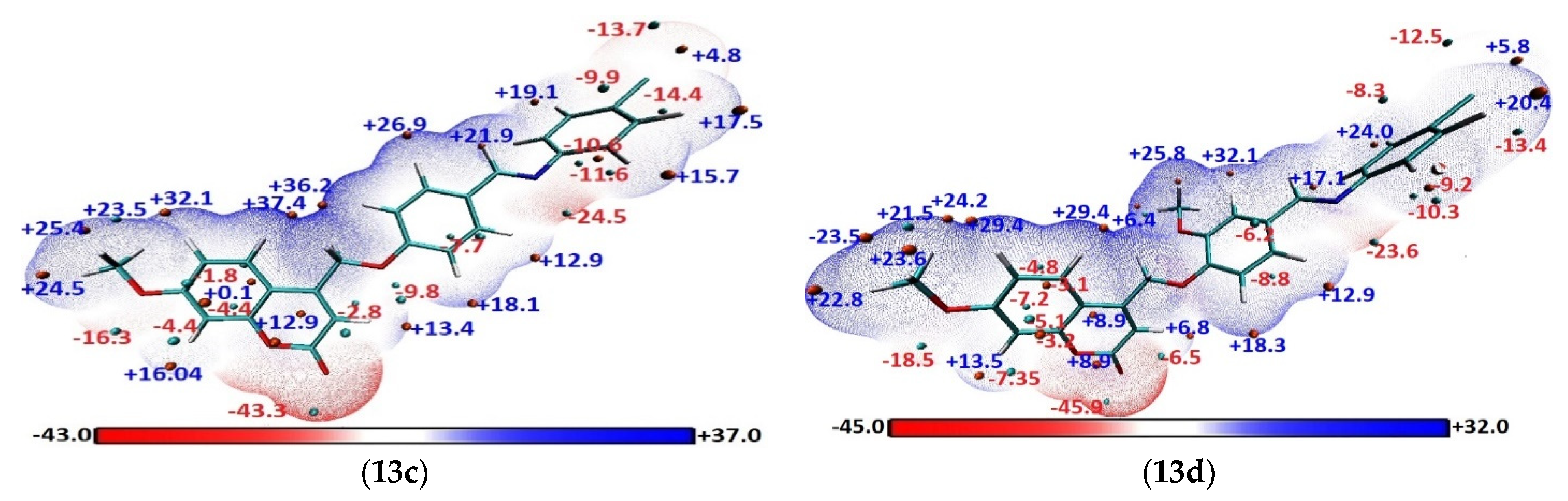
Molecular Electrostatic Potential (MEP)
3. Experimental
3.1. General Procedures
3.1.1. Synthesis of 4-(Chloromethyl)-7-methoxy-2H-chromen-2-One (9)
3.1.2. General Procedure of Synthesis of Schiff bases (12a-j)
(E)-4-((Phenylimino)methyl)phenol (12a)
(E)-4-(((4-Methoxyphenyl)imino)methyl)phenol (12b)
(E)-4-(((4-Chlorophenyl)imino)methyl)phenol (12c)
(E)-4-(((4-Chlorophenyl)imino)methyl)-2-methoxyphenol (12d) [58]
(E)-2-Methoxy-4-((phenylimino)methyl)phenol (12e)
(E)-2-Methoxy-4-(((4-methoxyphenyl)imino)methyl)phenol (12f)
(E)-3-(((4-Methoxyphenyl)imino)methyl)phenol (12g)
(E)-2-Methoxy-5-(((4-methoxyphenyl)imino)methyl)phenol (12h)
(E)-5-(((4-Chlorophenyl)imino)methyl)-2-methoxyphenol (12i) [63]
(E)-3-(((4-Chlorophenyl)imino)methyl)phenol (12j)
3.1.3. General Procedure of Synthesis of Coumarin-Schiff Base Hybrids (13a-j)
(E)-7-Methoxy-4-((4-((phenylimino)methyl)phenoxy)methyl)-2H-chromen-2-one (13a)
(E)-7-Methoxy-4-((4-(((4-methoxyphenyl)imino)methyl)phenoxy)methyl)-2H-chromen-2-one (13b)
(E)-4-((4-(((4-Chlorophenyl)imino)methyl)phenoxy)methyl)-7-methoxy-2H-chromen-2-one (13c)
(E)-4-((4-(((4-Chlorophenyl)imino)methyl)-2-methoxyphenoxy)methyl)-7-methoxy-2H-chromen-2-one (13d)
(E)-7-Methoxy-4-((2-methoxy-4-((phenylimino)methyl)phenoxy)methyl)-2H-chromen-2-one (13e)
(E)-7-Methoxy-4-((2-methoxy-4-(((4-methoxyphenyl)imino)methyl)phenoxy)methyl)-2H-chromen-2-one (13f)
(E)-7-Methoxy-4-((3-(((4-methoxyphenyl)imino)methyl)phenoxy)methyl)-2H-chromen-2-one (13g)
(E)-7-Methoxy-4-((2-methoxy-5-(((4-methoxyphenyl)imino)methyl)phenoxy)methyl)-2H-chromen-2-one (13h)
(E)-4-((5-(((4-Chlorophenyl)imino)methyl)-2-methoxyphenoxy)methyl)-7-methoxy-2H-chromen-2-one (13i)
(E)-4-((3-(((4-Chlorophenyl)imino)methyl)phenoxy)methyl)-7-methoxy-2H-chromen-2-one (13j)
3.2. Biological Evaluation of Hybrids against AChE
3.3. Molecular Docking Study
3.4. Molecular Dynamics Simulation (MDS)
3.5. Binding Energy Calculations
3.6. Geometry DFT Optimization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fancellu, G.; Chand, K.; Tomás, D.; Orlandini, E.; Piemontese, L.; Silva, D.F.; Cardoso, S.M.; Chaves, S.; Santos, M.A. Novel tacrine–benzofuran hybrids as potential multi-target drug candidates for the treatment of Alzheimer’s Disease. J. Enzym. Inhib. Med. Chem. 2020, 35, 211–226. [Google Scholar] [CrossRef]
- Australia, D.; Baker, S.; Banerjee, S. Alzheimer’s Disease International World Alzheimer Report 2019: Attitudes to Dementia; Alzheimer’s Disease International: London, UK, 2019. [Google Scholar]
- Marucci, G.; Buccioni, M.; Ben, D.D.; Lambertucci, C.; Volpini, R.; Amenta, F. Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology 2021, 190, 108352. [Google Scholar] [CrossRef]
- Hasan, A.H.; Amran, S.I.; Hussain, F.H.S.; Jaff, B.A.; Jamalis, J. Molecular Docking and Recent Advances in the Design and Development of Cholinesterase Inhibitor Scaffolds: Coumarin Hybrids. ChemistrySelect 2019, 4, 14140–14156. [Google Scholar] [CrossRef]
- Mohammadi-khanaposhtani, M.; Barazandeh Tehrani, M.; Rezaei, Z.; Asadi, M.A.; Behnammanesh, H.; Nadri, H.; Afsharirad, F.; Moradi, A.; Larijani, B.; Mahdavi, M. Design, synthesis, and cholinesterase inhibition assay of coumarin-3-carboxamide-N-morpholine hybrids as new anti-Alzheimer agents. Chem. Biodivers. 2019, 16, e1900144. [Google Scholar] [CrossRef]
- Kara, J.; Suwanhom, P.; Wattanapiromsakul, C.; Nualnoi, T.; Puripattanavong, J.; Khongkow, P.; Lee, V.S.; Gaurav, A.; Lomlim, L. Synthesis of 2-(2-oxo-2H-chromen-4-yl) acetamides as potent acetylcholinesterase inhibitors and molecular insights into binding interactions. Arch. Pharm. 2019, 352, e1800310. [Google Scholar] [CrossRef]
- Rahim, F.; Malik, F.; Ullah, H.; Wadood, A.; Khan, F.; Javid, M.T.; Taha, M.; Rehman, W.; Ur Rehman, A.; Khan, K.M. Isatin based Schiff bases as inhibitors of α-glucosidase: Synthesis, characterization, in vitro evaluation and molecular docking studies. Bioorg. Chem. 2015, 60, 42–48. [Google Scholar] [CrossRef]
- Zhai, L.; Jiang, Y.; Shi, Y.; Lv, M.; Pu, Y.-L.; Cheng, H.-L.; Zhu, J.-Y.; Yang, K.-W. Aromatic Schiff bases confer inhibitory efficacy against New Delhi metallo-β-lactamase-1 (NDM-1). Bioorg. Chem. 2022, 126, 105910. [Google Scholar] [CrossRef]
- Alam, A.; Ali, M.; Latif, A.; Rehman, N.U.; Saher, S.; Zainab; Faryal; Khan, A.; Ullah, S.; Ullah, O.; et al. Novel Bis-Schiff’s base derivatives of 4-nitroacetophenone as potent α-glucosidase agents: Design, synthesis and in silico approach. Bioorg. Chem. 2022, 128, 106058. [Google Scholar] [CrossRef]
- Rao, N.N.; Kishan, E.; Gopichand, K.; Nagaraju, R.; Ganai, A.M.; Rao, P.V. Design, synthesis, spectral characterization, DNA binding, photo cleavage and antibacterial studies of transition metal complexes of benzothiazole Schiff base. Chem. Data Collect. 2020, 27, 100368. [Google Scholar] [CrossRef]
- Hameed, A.; al-Rashida, M.; Uroos, M.; Abid Ali, S.; Khan, K.M. Schiff bases in medicinal chemistry: A patent review (2010-2015). Expert Opin. Ther. Pat. 2017, 27, 63–79. [Google Scholar] [CrossRef]
- Taha, M.; Ismail, N.H.; Imran, S.; Selvaraj, M.; Rahim, F. Synthesis of novel inhibitors of β-glucuronidase based on the benzothiazole skeleton and their molecular docking studies. RSC Adv. 2016, 6, 3003–3012. [Google Scholar] [CrossRef]
- Taha, M.; Ismail, N.H.; Jamil, W.; Rashwan, H.; Kashif, S.M.; Sain, A.A.; Adenan, M.I.; Anouar, E.H.; Ali, M.; Rahim, F.; et al. Synthesis of novel derivatives of 4-methylbenzimidazole and evaluation of their biological activities. Eur. J. Med. Chem. 2014, 84, 731–738. [Google Scholar] [CrossRef]
- Puchtler, H.; Meloan, S.N. On Schiff’s bases and aldehyde-fuchsin: A review from H. Schiff to R.D. Lillie. Histochemistry 1981, 72, 321–332. [Google Scholar] [CrossRef]
- Şahin, Ö.; Özmen Özdemir, Ü.; Seferoğlu, N.; Adem, Ş.; Seferoğlu, Z. Synthesis, characterization, molecular docking and in vitro screening of new metal complexes with coumarin Schiff base as anticholine esterase and antipancreatic cholesterol esterase agents. J. Biomol. Struct. Dyn. 2022, 40, 4460–4474. [Google Scholar] [CrossRef]
- Wang, Z.-M.; Xie, S.-S.; Li, X.-M.; Wu, J.-J.; Wang, X.-B.; Kong, L.-Y. Multifunctional 3-Schiff base-4-hydroxycoumarin derivatives with monoamine oxidase inhibition, anti-β-amyloid aggregation, metal chelation, antioxidant and neuroprotection properties against Alzheimer’s disease. RSC Adv. 2015, 5, 70395–70409. [Google Scholar] [CrossRef]
- Rahim, F.; Ullah, H.; Taha, M.; Wadood, A.; Javed, M.T.; Rehman, W.; Nawaz, M.; Ashraf, M.; Ali, M.; Sajid, M.; et al. Synthesis and in vitro acetylcholinesterase and butyrylcholinesterase inhibitory potential of hydrazide based Schiff bases. Bioorg. Chem. 2016, 68, 30–40. [Google Scholar] [CrossRef]
- Riazimontazer, E.; Sadeghpour, H.; Nadri, H.; Sakhteman, A.; Tüylü Küçükkılınç, T.; Miri, R.; Edraki, N. Design, synthesis and biological activity of novel tacrine-isatin Schiff base hybrid derivatives. Bioorg. Chem. 2019, 89, 103006. [Google Scholar] [CrossRef]
- Hasan, A.H.; Shakya, S.; Hussain, F.H.S.; Murugesan, S.; Chander, S.; Pratama, M.R.F.; Jamil, S.; Das, B.; Biswas, S.; Jamalis, J. Design, synthesis, anti-acetylcholinesterase evaluation and molecular modelling studies of novel coumarin-chalcone hybrids. J. Biomol. Struct. Dyn. 2023, 1–13. [Google Scholar] [CrossRef]
- Hasan, A.H.; Murugesan, S.; Amran, S.I.; Chander, S.; Alanazi, M.M.; Hadda, T.B.; Shakya, S.; Pratama, M.R.F.; Das, B.; Biswas, S.; et al. Novel thiophene Chalcones-Coumarin as acetylcholinesterase inhibitors: Design, synthesis, biological evaluation, molecular docking, ADMET prediction and molecular dynamics simulation. Bioorg. Chem. 2022, 119, 105572. [Google Scholar] [CrossRef]
- Hejchman, E.; Kruszewska, H.; Maciejewska, D.; Sowirka-Taciak, B.; Tomczyk, M.; Sztokfisz-Ignasiak, A.; Jankowski, J.; Młynarczuk-Biały, I. Design, synthesis, and biological activity of Schiff bases bearing salicyl and 7-hydroxycoumarinyl moieties. Mon. Für Chem. Chem. Mon. 2019, 150, 255–266. [Google Scholar] [CrossRef]
- Han, S.-H.; Yoshida, H.; Nobe, Y.; Fujiwara, M.; Kamizori, J.; Kikuchi, A.; Iwahori, F.; Abe, J. Molecular alignment and thermal stability of liquid-crystalline phases in binary mixtures of electron donor and acceptor. J. Mol. Struct. 2005, 735–736, 375–382. [Google Scholar] [CrossRef]
- Sılku, P.; Özkınalı, S.; Öztürk, Z.; Asan, A.; Köse, D.A. Synthesis of novel Schiff Bases containing acryloyl moiety and the investigation of spectroscopic and electrochemical properties. J. Mol. Struct. 2016, 1116, 72–83. [Google Scholar] [CrossRef]
- Nepali, K.; Lee, H.-Y.; Liou, J.-P. Nitro-Group-Containing Drugs. J. Med. Chem. 2019, 62, 2851–2893. [Google Scholar] [CrossRef]
- Wang, J.; Xie, X.-Q.; Hou, T.; Xu, X. Fast Approaches for Molecular Polarizability Calculations. J. Phys. Chem. A 2007, 111, 4443–4448. [Google Scholar] [CrossRef]
- Khaled, D.M.; Elshakre, M.E.; Noamaan, M.A.; Butt, H.; Abdel Fattah, M.M.; Gaber, D.A. A Computational QSAR, Molecular Docking and In Vitro Cytotoxicity Study of Novel Thiouracil-Based Drugs with Anticancer Activity against Human-DNA Topoisomerase II. Int. J. Mol. Sci. 2022, 23, 11799. [Google Scholar] [CrossRef] [PubMed]
- Andrasi, M.; Buglyo, P.; Zekany, L.; Gaspar, A. A comparative study of capillary zone electrophoresis and pH-potentiometry for determination of dissociation constants. J. Pharm. Biomed. Anal. 2007, 44, 1040–1047. [Google Scholar] [CrossRef]
- Elshakre, M.E.; Noamaan, M.A.; Moustafa, H.; Butt, H. Density Functional Theory, Chemical Reactivity, Pharmacological Potential and Molecular Docking of Dihydrothiouracil-Indenopyridopyrimidines with Human-DNA Topoisomerase II. Int. J. Mol. Sci. 2020, 21, 1253. [Google Scholar] [CrossRef] [PubMed]
- Belaidi, S.; Belaidi, H.; Bouzidi, D. Computational Methods Applied in Physical-Chemistry Property Relationships of Thiophene Derivatives. J. Comput. Theor. Nanosci. 2015, 12, 1737–1745. [Google Scholar] [CrossRef]
- Miller, J.N. Coumarin-6-sulphonyl chloride: A novel label in fluorimetry and phosphorimetry Part 1. Synthesis and Luminescence Properties. Anal. Chim. Acta 1989, 227, 145–153. [Google Scholar] [CrossRef]
- Mirzaei, S.; Eisvand, F.; Hadizadeh, F.; Mosaffa, F.; Ghasemi, A.; Ghodsi, R. Design, synthesis and biological evaluation of novel 5,6,7-trimethoxy-N-aryl-2-styrylquinolin-4-amines as potential anticancer agents and tubulin polymerization inhibitors. Bioorg. Chem. 2020, 98, 103711. [Google Scholar] [CrossRef]
- Machaba, K.E.; Mhlongo, N.N.; Soliman, M.E.S. Induced Mutation Proves a Potential Target for TB Therapy: A Molecular Dynamics Study on LprG. Cell Biochem. Biophys. 2018, 76, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Bai, Q.; Tan, S.; Xu, T.; Liu, H.; Huang, J.; Yao, X. MolAICal: A soft tool for 3D drug design of protein targets by artificial intelligence and classical algorithm. Brief. Bioinform. 2020, 22, 161. [Google Scholar] [CrossRef] [PubMed]
- Gnanaguru, K.; Ramasubbu, N.; Venkatesan, K.; Ramamurthy, V. A study on the photochemical dimerization of coumarins in the solid state. J. Org. Chem. 1985, 50, 2337–2346. [Google Scholar] [CrossRef]
- Anbukarasi, K.; Xavier, S.; Hasan, H.A.; Er, L.Y.; Jamalis, J.; Sebastian, S.; Periandy, S. DFT and Molecular Docking Analysis of Newly Synthesized Compound (2E)-3-[3-(Benzyloxy) Phenyl]-1-(4’-Chlorophe-Nyl)-2-Propen-1-One [Bpclpo]. Curr. Phys. Chem. 2023, 13, 1–38. [Google Scholar] [CrossRef]
- Günay, N.; Pir, H.; Avcı, D.; Atalay, Y. NLO and NBO Analysis of Sarcosine-Maleic Acid by Using HF and B3LYP Calculations. J. Chem. 2013, 2013, 712130. [Google Scholar] [CrossRef]
- Pearson, R.G. Absolute electronegativity and hardness correlated with molecular orbital theory. Proc. Natl. Acad. Sci. USA 1986, 83, 8440–8441. [Google Scholar] [CrossRef]
- Khan, S.A.; Rizwan, K.; Shahid, S.; Noamaan, M.A.; Rasheed, T.; Amjad, H. Synthesis, DFT, computational exploration of chemical reactivity, molecular docking studies of novel formazan metal complexes and their biological applications. Appl. Organomet. Chem. 2020, 34, e5444. [Google Scholar] [CrossRef]
- Parr, R.G. Density Functional Theory of Atoms and Molecules. Horizons of Quantum Chemistry; Fukui, K., Pullman, B., Eds.; Springer: Dordrecht, The Netherlands, 1980; pp. 5–15. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Roy, D.R. Update 1 of: Electrophilicity Index. Chem. Rev. 2007, 107, PR46–PR74. [Google Scholar] [CrossRef]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density functional approach to the frontier-electron theory of chemical reactivity. J. Am. Chem. Soc. 1984, 106, 4049–4050. [Google Scholar] [CrossRef]
- Liu, S.-B. Conceptual Density Functional Theory and Some Recent Developments. Acta Phys.-Chim. Sin. 2009, 25, 590–600. [Google Scholar]
- Contreras, R.R.; Fuentealba, P.; Galván, M.; Pérez, P. A direct evaluation of regional Fukui functions in molecules. Chem. Phys. Lett. 1999, 304, 405–413. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Giri, S. Stability, Reactivity, and Aromaticity of Compounds of a Multivalent Superatom. J. Phys. Chem. A 2007, 111, 11116–11121. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Morell, C.; Grand, A.; Toro-Labbé, A. New Dual Descriptor for Chemical Reactivity. J. Phys. Chem. A 2005, 109, 205–212. [Google Scholar] [CrossRef]
- Parthasarathi, R.; Padmanabhan, J.; Elango, M.; Subramanian, V.; Chattaraj, P.K. Intermolecular reactivity through the generalized philicity concept. Chem. Phys. Lett. 2004, 394, 225–230. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Local softness and chemical reactivity in the molecules CO, SCN− and H2CO. J. Mol. Struct. THEOCHEM 1988, 163, 305–313. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Naray-Szabo, G.; Ferenczy, G.G. Molecular Electrostatics. Chem. Rev. 1995, 95, 829–847. [Google Scholar] [CrossRef]
- Murray, J.S.; Politzer, P. The electrostatic potential: An overview. WIREs Comput. Mol. Sci. 2011, 1, 153–163. [Google Scholar] [CrossRef]
- Luque, F.J.; López, J.M.; Orozco, M. Perspective on “Electrostatic interactions of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects”. Theor. Chem. Acc. 2000, 103, 343–345. [Google Scholar] [CrossRef]
- Piloto, A.M.; Fonseca, A.S.C.; Costa, S.P.G.; Gonçalves, M.S.T. Carboxylic fused furans for amino acid fluorescent labelling. Tetrahedron 2006, 62, 9258–9267. [Google Scholar] [CrossRef]
- Huneck, S.; Schreiber, K.; Grimmecke, H.D. Schiffs bases and derived secondary amines as plant growth inhibitors. J. Plant Growth Regul. 1984, 3, 75–84. [Google Scholar] [CrossRef]
- Grammaticakis, P.; Texier, H. Contribution al’étude de l’absorption dans l’ultraviolet moyen et le visible de derivés fonctionnels azotés de quelques aldéhydes et cétones aromatiques. X.—Aniles (premier mémoire). Bull. Soc. Chim. Fr. 1971, 38, 1323–1330. [Google Scholar]
- Senier, A.; Forster, R.B. CCXXX.—Studies in phototropy and thermotropy. Part V. Polymorphic 4-hydroxybenzylideneamines produced by trituration and by the influence of sunlight. J. Chem. Soc. Trans. 1914, 105, 2462–2471. [Google Scholar] [CrossRef]
- Nagapandiselvi, P.; Baby, C.; Gopalakrishnan, R. A new Schiff base, (E)-4-((4-chlorophenylimino) methyl)-2-methoxyphenol: Crystal structure, thermal behavior, solid-state fluorescence, DFT calculations and FT NMR spectral analysis. J. Mol. Struct. 2014, 1056–1057, 110–120. [Google Scholar] [CrossRef]
- Chigurupati, S.; Muralidharan, S.; Cin, L.S.; Raser, W.Y.; Santhi, K.; Kesavanarayanan, K.S. Studying Newly Synthesized and Developed 4-Hydroxy-3-Methoxybenzaldehyde Schiff Bases by UV Spectrophotometry and High Performance Liquid Chromatography. Pharm. Chem. J. 2017, 50, 851–856. [Google Scholar] [CrossRef]
- Wheeler, A.S. The Condensation of Vanillin and Piperonal with Certain Aromatic Amines. J. Am. Chem. Soc. 1913, 35, 976–978. [Google Scholar] [CrossRef]
- Nakamura, M.; Komatsu, K.; Gondo, Y.; Ohta, K.; Ueda, Y. An Infrared Study of the C=N Stretching Frequency in N-Benzylideneaniline Derivatives. Chem. Pharm. Bull. 1967, 15, 585–592. [Google Scholar] [CrossRef]
- Al-Kahraman, Y.M.; Madkour, H.; Ali, D.; Yasinzai, M. Antileishmanial, antimicrobial and antifungal activities of some new aryl azomethines. Molecules 2010, 15, 660–671. [Google Scholar] [CrossRef]
- Sharma, K.P.; Reddi, R.S.B.; Bhattacharya, S.; Rai, R.N. Synthesis, crystal growth, structural and physicochemical studies of novel binary organic complex: 4-chloroaniline–3-hydroxy-4-methoxybenzaldehyde. J. Solid State Chem. 2012, 190, 226–232. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhang, D.; Ren, J.; Yang, M.; Li, S. Acetylcholinesterase inhibitory activity of the total alkaloid from traditional Chinese herbal medicine for treating Alzheimer’s disease. Med. Chem. Res. 2012, 21, 734–738. [Google Scholar] [CrossRef]
- Abdalla Ali, A.; Mhamad, S.A.; Hasan, A.H.; Ahmad, I.; Abdullah, S.A.; Jamil, S.; Patel, H.; Murugesan, S.; Jamalis, J. Synthesis, biological evaluation and molecular modeling studies of modulated benzyloxychalcones as potential acetylcholinesterase inhibitors. J. Biomol. Struct. Dyn. 2023, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hasan, H.A.; Yusof, S.M.F.; Kamarudin, A.N.; Murugesan, S.; Shakya, S.; Jamalis, J. Synthesis, Anti-acetylcholinesterase Evaluation, Molecular Docking and Molecular Dynamics Simulation of Novel Psoralen Derivatives. Curr. Org. Synth. 2023, 20, 1–17. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Salih, R.H.H.; Hasan, A.H.; Hussein, A.J.; Samad, M.K.; Shakya, S.; Jamalis, J.; Hawaiz, F.E.; Pratama, M.R.F. One-pot synthesis, molecular docking, ADMET, and DFT studies of novel pe evaluation and molecular modelling stuyrazolines as promising SARS-CoV-2 main protease inhibitors. Res. Chem. Intermed. 2022, 48, 4729–4751. [Google Scholar] [CrossRef]
- Hasan, A.H.; Hussen, N.H.; Shakya, S.; Jamalis, J.; Pratama, M.R.F.; Chander, S.; Kharkwal, H.; Murugesan, S. In silico discovery of multi-targeting inhibitors for the COVID-19 treatment by molecular docking, molecular dynamics simulation studies, and ADMET predictions. Struct. Chem. 2022, 33, 1645–1665. [Google Scholar] [CrossRef]
- Hussen, N.H.; Hasan, A.H.; Jamalis, J.; Shakya, S.; Chander, S.; Kharkwal, H.; Murugesan, S.; Ajit Bastikar, V.; Pyarelal Gupta, P. Potential inhibitory activity of phytoconstituents against black fungus: In silico ADMET, molecular docking and MD simulation studies. Comput. Toxicol. 2022, 24, 100247. [Google Scholar] [CrossRef]
- Hussen, H.N.; Hamid, J.S.; Sabir, N.M.; Hasan, H.A.; Mohammed, J.S.; Shali, A.K.A. Novel Penicillin Derivatives Against Selected Multiple-Drug Resistant Bacterial Strains: Design, Synthesis, Structural Analysis, in silico and in Vitro Studies. Curr. Org. Synth. 2023, 20, 1. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D., Jr. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Nosé, S.; Klein, M.L. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Grest, G.S.; Kremer, K. Molecular dynamics simulation for polymers in the presence of a heat bath. Phys. Rev. A 1986, 33, 3628–3631. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Frisch, M.J.; Frisch, H.B.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Revision E.01.; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Ulic, S.E.; Védova, C.O.D.; Hermann, A.; Mack, H.-G.; Oberhammer, H. Preparation and Properties of Trifluorothioacetic Acid- S-(trifluoromethyl)ester, CF 3C(O)SCF 3. J. Phys. Chem. A 2008, 112, 6211–6216. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.E.; Weinhold, F. Natural bond orbital analysis of near-Hartree–Fock water dimer. J. Chem. Phys. 1983, 78, 4066–4073. [Google Scholar] [CrossRef]
- Lin, Y.-S.; Li, G.-D.; Mao, S.-P.; Chai, J.-D. Long-Range Corrected Hybrid Density Functionals with Improved Dispersion Corrections. J. Chem. Theory Comput. 2013, 9, 263–272. [Google Scholar] [CrossRef]
- Salih, R.H.H.; Hasan, A.H.; Hussen, N.H.; Hawaiz, F.E.; Hadda, T.B.; Jamalis, J.; Almalki, F.A.; Adeyinka, A.S.; Coetzee, L.-C.C.; Oyebamiji, A.K. Thiazole-pyrazoline hybrids as potential antimicrobial agent: Synthesis, biological evaluation, molecular docking, DFT studies and POM analysis. J. Mol. Struct. 2023, 1282, 135191. [Google Scholar] [CrossRef]
- Keith, T.A.; Millam, J.M. GaussView, Version 6.1; Semichem, Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Chemcraft—Graphical software for visualization of quantum chemistry computations. Available online: https://www.chemcraftprog.com (accessed on 10 November 2022).
- Froimowitz, M. HyperChem: A software package for computational chemistry and molecular modeling. Biotechniques 1993, 14, 1010–1013. [Google Scholar]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Binding Energy (kcal/mol) | IC50 [µM] |
|---|---|---|
| 13a | −11.9 | 0.440 ± 0.016 |
| 13b | −11.7 | 0.466 ± 0.007 |
| 13c | −13.2 | 0.232 ± 0.011 |
| 13d | −13.2 | 0.190 ± 0.004 |
| 13e | −13.1 | 0.297 ± 0.006 |
| 13f | −12.7 | 0.365 ± 0.025 |
| 13g | −11.5 | 1.090 ± 0.058 |
| 13h | −10.6 | 1.175 ± 0.063 |
| 13i | −11.7 | 0.712 ± 0.044 |
| 13j | −12.3 | 0.651 ± 0.003 |
| GAL. | −9.6 | 1.142 ± 0.027 |
| Compound | Binding Energy (Kcal/mol) | Interactions | |||||
|---|---|---|---|---|---|---|---|
| H-Bond | Hydrophobic | Electrostatic or Other | |||||
| Alkyl | π-Alkyl | π-Sigma | π-π T-Shape | π-anion/π-Donor/Carbon H Bond | |||
| 13a | −11.9 | GLY121, GLY122, SER203, PHE295 | ALA204, TRP236, PHE297 | VAL294 | His447 | TYR337, TRP286, TYR341, PHE338 | |
| 13b | −11.7 | SER203, GLY122 | LEU289, ILE451, | VAL294, PHE338 | TRP286, TRP86, HIS447 | GLU202/TYP133 | |
| 13c | −13.2 | GLY121, SER203, TYR337 | TYR72, TYR124, TRP86, TRP286 | TRP286, TYR341, TRP86 | TRP86, TYR341 | ||
| 13d | −13.2 | GLY121, SER203, TYR337 | LEU289 | TRP286, TYR341, TRP86 | TRP86, TYR341, SER293 | ||
| 13e | −13.1 | GLY122, TYR133 | PRO88, PHE297, | VAL294 | VAL294 | PHE295, HIS447, GLY126, TYR124, SER125, GLN71, TYR72, TRP86 | |
| 13f | −12.7 | TYR133 | PHE297, ALA204, HIS447, PRO88, TRP286 | TRP86 | TRP86 | PHE295, GLY126, TYR124, SER125, GLN71, TYR72, ARG296, SER293, VAL294 | |
| 13g | −11.5 | TYR124 | VAL294 | TRP286, TYR337, PHE338 | TYR72, TYR124, TYR341 | ||
| 13h | −10.6 | SER203 | TRP286, TRP86, PHE297, LEU289, VAL294, TYR337 | VAL294 | TRP236 | TRP86, TYR341 | PHE295, TYR124, ARG296, SER293, HIS447 |
| 13i | −11.7 | TYR133 | PRO88, TYR387, TRP86 | TRP286, VAL294 | TRP86 | TYR124 | PHE295, GLY126, TYR124, SER125, GLN71, TYR72, HIS447 |
| 13j | −12.3 | TYR124, TYR337 | PRO88 | LEU289, TRP286 | VAL294 | PHE338, TRP286, TYR341 | ASP74/TRP86, GLN71, TYR341, |
| Compounds | Polarizability (A3) | Refractivity (A3) | Vol (A3) | Surface Area (Grid) A2 | HE (kcal/mol) | Log P | MW (DA) |
|---|---|---|---|---|---|---|---|
| 13a | 44.00 | 110.22 | 1111.03 | 668.68 | −11.09 | 4.53 | 385.42 |
| 13b | 46.47 | 116.68 | 1187.79 | 704.70 | −12.67 | 4.28 | 415.45 |
| 13c | 45.93 | 115.02 | 1156.57 | 692.72 | −10.72 | 5.05 | 419.86 |
| 13d | 48.40 | 121.48 | 1231.94 | 732.87 | −10.84 | 4.80 | 449.89 |
| 13e | 46.47 | 116.68 | 1188.57 | 708.71 | −11.21 | 4.28 | 415.45 |
| 13f | 48.94 | 123.14 | 1263.19 | 750.75 | −12.78 | 4.03 | 445.47 |
| 13g | 46.47 | 116.68 | 1187.79 | 704.73 | −12.52 | 4.28 | 415.45 |
| 13h | 48.94 | 123.14 | 1260.71 | 754.84 | −12.58 | 4.03 | 445.47 |
| 13i | 48.40 | 121.48 | 1230.12 | 732.60 | −10.62 | 4.80 | 449.89 |
| 13j | 45.93 | 115.02 | 1156.40 | 690.79 | −10.57 | 5.05 | 419.86 |
| Complexes | ΔΕVDW | ΔΕele + ΔGsol | ΔGbin |
|---|---|---|---|
| 13c-AChE | −44.913 | 12.267 | −32.645 ± 0.119 |
| 13d-AChE | −51.081 | 15.039 | −36.042 ± 0.121 |
| 13a | 13b | 13c | 13d | 13e | 13f | 13g | 13h | 13i | 13j | |
|---|---|---|---|---|---|---|---|---|---|---|
| O1 | −0.519 | −0.519 | −0.518 | −0.520 | −0.521 | −0.521 | −0.519 | −0.521 | −0.521 | −0.519 |
| C2 | 0.774 | 0.774 | 0.774 | 0.774 | 0.774 | 0.774 | 0.774 | 0.775 | 0.774 | 0.774 |
| C3 | −0.313 | −0.314 | −0.313 | −0.319 | −0.319 | −0.319 | −0.313 | −0.318 | −0.317 | −0.313 |
| C4 | 0.032 | 0.033 | 0.032 | 0.039 | 0.040 | 0.040 | 0.034 | 0.039 | 0.038 | 0.033 |
| C5 | −0.175 | −0.175 | −0.175 | −0.174 | −0.173 | −0.173 | −0.174 | −0.173 | −0.173 | −0.175 |
| C6 | −0.138 | −0.138 | −0.138 | −0.136 | −0.136 | −0.136 | −0.138 | −0.137 | −0.137 | −0.138 |
| C7 | −0.314 | −0.314 | −0.314 | −0.316 | −0.316 | −0.316 | −0.315 | −0.316 | −0.316 | −0.315 |
| C8 | 0.357 | 0.357 | 0.357 | 0.355 | 0.355 | 0.354 | 0.356 | 0.354 | 0.355 | 0.357 |
| C9 | −0.277 | −0.277 | −0.277 | −0.279 | −0.279 | −0.279 | −0.278 | −0.279 | −0.279 | −0.278 |
| C10 | 0.380 | 0.380 | 0.380 | 0.380 | 0.380 | 0.380 | 0.380 | 0.380 | 0.380 | 0.380 |
| O11 | −0.562 | −0.563 | −0.562 | −0.566 | −0.567 | −0.567 | −0.563 | −0.567 | −0.566 | −0.562 |
| O16 | −0.535 | −0.535 | −0.535 | −0.536 | −0.537 | −0.537 | −0.536 | −0.537 | −0.536 | −0.535 |
| C17 | −0.211 | −0.211 | −0.211 | −0.211 | −0.210 | −0.210 | −0.211 | −0.210 | −0.210 | −0.211 |
| C21 | −0.043 | −0.043 | −0.043 | −0.051 | −0.051 | −0.051 | −0.042 | −0.047 | −0.047 | −0.042 |
| O24 | −0.547 | −0.548 | −0.546 | −0.571 | −0.572 | −0.572 | −0.553 | −0.572 | −0.572 | −0.552 |
| C25 | 0.344 | 0.342 | 0.346 | 0.287 | 0.285 | 0.282 | 0.326 | 0.270 | 0.271 | 0.327 |
| C26 | −0.246 | −0.246 | −0.246 | −0.222 | −0.217 | −0.222 | −0.195 | −0.183 | −0.181 | −0.192 |
| C27 | −0.131 | −0.132 | −0.130 | −0.165 | −0.177 | −0.168 | −0.096 | −0.140 | −0.146 | −0.103 |
| C28 | −0.159 | −0.155 | −0.162 | −0.124 | −0.115 | −0.117 | −0.208 | −0.150 | −0.147 | −0.206 |
| C29 | −0.136 | −0.138 | −0.134 | −0.257 | −0.252 | −0.261 | −0.182 | −0.299 | −0.300 | −0.182 |
| C30 | −0.310 | −0.310 | −0.310 | 0.290 | 0.287 | 0.290 | −0.285 | 0.310 | 0.314 | −0.281 |
| C34 | 0.154 | 0.146 | 0.157 | 0.171 | 0.151 | 0.158 | 0.140 | 0.145 | 0.156 | 0.153 |
| N36 | −0.453 | −0.450 | −0.456 | −0.455 | −0.444 | −0.450 | −0.436 | −0.449 | −0.455 | −0.441 |
| C37 | 0.135 | 0.099 | 0.133 | 0.129 | 0.132 | 0.095 | 0.095 | 0.099 | 0.132 | 0.130 |
| C38 | −0.209 | −0.180 | −0.192 | −0.190 | −0.207 | −0.177 | −0.175 | −0.180 | −0.191 | −0.189 |
| C39 | −0.195 | −0.289 | −0.218 | −0.218 | −0.196 | −0.291 | −0.290 | −0.289 | −0.218 | −0.217 |
| C40 | −0.221 | 0.320 | −0.050 | −0.048 | −0.219 | 0.323 | 0.324 | 0.321 | −0.049 | −0.048 |
| C41 | −0.196 | −0.237 | −0.218 | −0.218 | −0.196 | −0.237 | −0.237 | −0.238 | −0.219 | −0.218 |
| C42 | −0.238 | −0.213 | −0.221 | −0.220 | −0.237 | −0.211 | −0.210 | −0.213 | −0.221 | −0.219 |
| A47 | 0.208 | −0.545 | −0.003 | −0.001 | 0.208 | −0.545 | −0.545 | −0.545 | −0.003 | −0.001 |
| A48 | 0.213 | 0.213 | 0.214 | −0.546 | −0.547 | −0.547 | 0.212 | −0.538 | −0.537 | 0.213 |
| Parameters | ET, au | EHOMO, au | ELUMO, au | Eg, eV | μ, D | I, eV | A, eV |
|---|---|---|---|---|---|---|---|
| 13a | −1281.51218 | −0.29732 | −0.00966 | 7.83 | 8.73 | 8.09 | 0.26 |
| 13b | −1396.03187 | −0.27977 | −0.00871 | 7.38 | 8.18 | 7.61 | 0.24 |
| 13c | −1741.11979 | −0.29937 | −0.01125 | 7.84 | 8.50 | 8.15 | 0.31 |
| 13d | −1855.63488 | −0.30068 | −0.01208 | 7.85 | 8.54 | 8.18 | 0.33 |
| 13e | −1396.02739 | −0.29860 | −0.00589 | 7.96 | 9.31 | 8.13 | 0.16 |
| 13f | −1510.54717 | −0.28213 | −0.00252 | 7.61 | 9.78 | 7.68 | 0.07 |
| 13g | −1396.03041 | −0.28300 | −0.00735 | 7.50 | 7.68 | 7.70 | 0.20 |
| 13h | −1510.54851 | −0.27929 | −0.00012 | 7.60 | 8.42 | 7.60 | 0.00 |
| 13i | −1855.63633 | −0.29807 | −0.00737 | 7.91 | 9.89 | 8.11 | 0.20 |
| 13j | −1741.11801 | −0.30477 | −0.01351 | 7.93 | 10.33 | 8.29 | 0.37 |
| Parameters | X, eV | η, eV | S, eV | V, eV | ω, eV | N, eV |
|---|---|---|---|---|---|---|
| 13a | 4.18 | 3.91 | 0.128 | −4.18 | 2.23 | −3.88 |
| 13b | 3.92 | 3.69 | 0.136 | −3.92 | 2.09 | −3.40 |
| 13c | 4.23 | 3.92 | 0.128 | −4.23 | 2.28 | −3.94 |
| 13d | 4.26 | 3.93 | 0.127 | −4.26 | 2.31 | −3.97 |
| 13e | 4.14 | 3.98 | 0.126 | −4.14 | 2.15 | −3.91 |
| 13f | 3.87 | 3.80 | 0.131 | −3.87 | 1.97 | −3.47 |
| 13g | 3.95 | 3.75 | 0.133 | −3.95 | 2.08 | −3.49 |
| 13h | 3.80 | 3.80 | 0.132 | −3.80 | 1.90 | −3.39 |
| 13i | 4.16 | 3.96 | 0.126 | −4.16 | 2.18 | −3.90 |
| 13j | 4.33 | 3.96 | 0.126 | −4.33 | 2.37 | −4.08 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasan, A.H.; Abdulrahman, F.A.; Obaidullah, A.J.; Alotaibi, H.F.; Alanazi, M.M.; Noamaan, M.A.; Murugesan, S.; Amran, S.I.; Bhat, A.R.; Jamalis, J. Discovery of Novel Coumarin-Schiff Base Hybrids as Potential Acetylcholinesterase Inhibitors: Design, Synthesis, Enzyme Inhibition, and Computational Studies. Pharmaceuticals 2023, 16, 971. https://doi.org/10.3390/ph16070971
Hasan AH, Abdulrahman FA, Obaidullah AJ, Alotaibi HF, Alanazi MM, Noamaan MA, Murugesan S, Amran SI, Bhat AR, Jamalis J. Discovery of Novel Coumarin-Schiff Base Hybrids as Potential Acetylcholinesterase Inhibitors: Design, Synthesis, Enzyme Inhibition, and Computational Studies. Pharmaceuticals. 2023; 16(7):971. https://doi.org/10.3390/ph16070971
Chicago/Turabian StyleHasan, Aso Hameed, Faruq Azeez Abdulrahman, Ahmad J. Obaidullah, Hadil Faris Alotaibi, Mohammed M. Alanazi, Mahmoud A. Noamaan, Sankaranarayanan Murugesan, Syazwani Itri Amran, Ajmal R. Bhat, and Joazaizulfazli Jamalis. 2023. "Discovery of Novel Coumarin-Schiff Base Hybrids as Potential Acetylcholinesterase Inhibitors: Design, Synthesis, Enzyme Inhibition, and Computational Studies" Pharmaceuticals 16, no. 7: 971. https://doi.org/10.3390/ph16070971
APA StyleHasan, A. H., Abdulrahman, F. A., Obaidullah, A. J., Alotaibi, H. F., Alanazi, M. M., Noamaan, M. A., Murugesan, S., Amran, S. I., Bhat, A. R., & Jamalis, J. (2023). Discovery of Novel Coumarin-Schiff Base Hybrids as Potential Acetylcholinesterase Inhibitors: Design, Synthesis, Enzyme Inhibition, and Computational Studies. Pharmaceuticals, 16(7), 971. https://doi.org/10.3390/ph16070971